

Abstract
We have determined the structure of the sarco(endo)plasmic reticulum
Ca2+-ATPase (SERCA) in an E2·Pi-like form
stabilized as a complex with  , an
ATP analog, adenosine 5′-(β,γ-methylene)triphosphate
(AMPPCP), and cyclopiazonic acid (CPA). The structure determined at 2.5Å
resolution leads to a significantly revised model of CPA binding when compared
with earlier reports. It shows that a divalent metal ion is required for CPA
binding through coordination of the tetramic acid moiety at a characteristic
kink of the M1 helix found in all P-type ATPase structures, which is expected
to be part of the cytoplasmic cation access pathway. Our model is consistent
with the biochemical data on CPA function and provides new measures in
structure-based drug design targeting Ca2+-ATPases, e.g.
from pathogens. We also present an extended structural basis of ATP modulation
pinpointing key residues at or near the ATP binding site. A structural
comparison to the Na+,K+-ATPase reveals that the
Phe93 side chain occupies the equivalent binding pocket of the CPA
site in SERCA, suggesting an important role of this residue in stabilization
of the potassium-occluded E2 state of Na+,K+-ATPase.
, an
ATP analog, adenosine 5′-(β,γ-methylene)triphosphate
(AMPPCP), and cyclopiazonic acid (CPA). The structure determined at 2.5Å
resolution leads to a significantly revised model of CPA binding when compared
with earlier reports. It shows that a divalent metal ion is required for CPA
binding through coordination of the tetramic acid moiety at a characteristic
kink of the M1 helix found in all P-type ATPase structures, which is expected
to be part of the cytoplasmic cation access pathway. Our model is consistent
with the biochemical data on CPA function and provides new measures in
structure-based drug design targeting Ca2+-ATPases, e.g.
from pathogens. We also present an extended structural basis of ATP modulation
pinpointing key residues at or near the ATP binding site. A structural
comparison to the Na+,K+-ATPase reveals that the
Phe93 side chain occupies the equivalent binding pocket of the CPA
site in SERCA, suggesting an important role of this residue in stabilization
of the potassium-occluded E2 state of Na+,K+-ATPase.
The Ca2+-ATPase from sarco(endo)plasmic reticulum of rabbit skeletal muscle (SERCA,5 isoform 1a) is a thoroughly studied member of the P-type ATPase family (1). SERCA possesses 10 transmembrane helices (M1 through M10) with both the N terminus and the C terminus facing the cytoplasmic side and three cytoplasmic domains, inserted in loops between M2 and M3 (A-domain) and between M4 and M5 (P- and N-domain) (2). The enzyme mediates the uptake of Ca2+ ions into the lumen of the sarcoplasmic reticulum (SR) after their release into the cytoplasm through calcium release channels during muscle contraction (3). SERCA, plasma membrane Ca2+-ATPase, and a third, Golgi-located secretory pathway Ca2+-ATPase are important factors in calcium and manganese homeostasis, transport, signaling, and regulation (4, 5).
Crystal structures of all major states in the reaction cycle of SERCA have
been determined. These include the Ca2E1·ATP
state (6,
7) with high affinity
Ca2+ binding sites accessible from the cytoplasmic side of the SR
membrane, the calcium-occluded
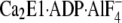 transition state (6), the open
E2P state with luminal facing ion binding sites that have low affinity for
Ca2+ and high affinity for protons
(8) and the proton-occluded
H2–3E2[ATP] state with a bound modulatory ATP
(9). This considerable amount
of structural information has turned the Ca2+-ATPase into a
valuable model system for studies on structural rearrangements that take place
during the catalytic cycle of P-type ATPases. SERCA is considered a promising
drug target in medical research, with a particular focus on prostate cancer
and infectious diseases. Several compounds have already been shown to bind and
inhibit SERCA by stabilizing the enzyme in a particular conformational state.
Thapsigargin (TG), cyclopiazonic acid (CPA), and 2,5-di-(tert-butyl)
hydroquinone (BHQ) stabilize an E2-like state, and 1,3-dibromo-2,4,6-tri
(methylisothiouronium)benzene stabilizes an E1-P-like conformation
(10–13).
CPA is a toxic indole tetramic acid first isolated from Penicillium
cyclopium (14) and later
found to be produced by Aspergillus versicolor and Aspergillus
flavus. Like TG, CPA specifically binds to and inhibits SERCA with
nanomolar affinity (15).
Indeed, CPA is widely used in biochemical and physiological studies on
Ca2+ signaling and muscle function, where it causes Ca2+
store depletion due to specific inhibition of Ca2+ reuptake by
SERCA. CPA and TG were originally proposed to bind to similar sites on SERCA
(16), but recent crystal
structures have shown a distinct site of interaction
(17,
18). Despite these structural
insights, a previously demonstrated magnesium dependence of CPA binding
(19) remained unexplained, and
opposing CPA binding modes were observed (see below).
transition state (6), the open
E2P state with luminal facing ion binding sites that have low affinity for
Ca2+ and high affinity for protons
(8) and the proton-occluded
H2–3E2[ATP] state with a bound modulatory ATP
(9). This considerable amount
of structural information has turned the Ca2+-ATPase into a
valuable model system for studies on structural rearrangements that take place
during the catalytic cycle of P-type ATPases. SERCA is considered a promising
drug target in medical research, with a particular focus on prostate cancer
and infectious diseases. Several compounds have already been shown to bind and
inhibit SERCA by stabilizing the enzyme in a particular conformational state.
Thapsigargin (TG), cyclopiazonic acid (CPA), and 2,5-di-(tert-butyl)
hydroquinone (BHQ) stabilize an E2-like state, and 1,3-dibromo-2,4,6-tri
(methylisothiouronium)benzene stabilizes an E1-P-like conformation
(10–13).
CPA is a toxic indole tetramic acid first isolated from Penicillium
cyclopium (14) and later
found to be produced by Aspergillus versicolor and Aspergillus
flavus. Like TG, CPA specifically binds to and inhibits SERCA with
nanomolar affinity (15).
Indeed, CPA is widely used in biochemical and physiological studies on
Ca2+ signaling and muscle function, where it causes Ca2+
store depletion due to specific inhibition of Ca2+ reuptake by
SERCA. CPA and TG were originally proposed to bind to similar sites on SERCA
(16), but recent crystal
structures have shown a distinct site of interaction
(17,
18). Despite these structural
insights, a previously demonstrated magnesium dependence of CPA binding
(19) remained unexplained, and
opposing CPA binding modes were observed (see below).
Tetramic acids are synthesized naturally, and more than 150 natural derivatives have been isolated from bacterial and fungal species (reviewed in Ref. 20). Tetramic acids possessing a 3-acyl group have the ability to chelate divalent metal ions. For instance, tenuazonic acid from the fungus Phoma sorghina has been shown to form complexes with Ca2+ and Mg2+ (21), as well as heavier metals such as Cu(II), Ni(II), and Fe(III) (22).
Previously published crystallographic structures of the SERCA·CPA
complex (PDB ID 2O9J and 2EAS) demonstrated that CPA binds within the proposed
calcium access channel of SERCA. However, the structures did not reveal a role
for magnesium, and the orientation of CPA within this binding site differed in
the two studies (17,
18). To address these
ambiguities, we have determined the crystal structure of SERCA in complex with
 , AMPPCP (an ATP analog), and
Mn2+·CPA. The structure reveals novel insight into CPA
binding, which we find to be mediated by a divalent cation, as demonstrated by
means of the anomalous scattering properties of Mn2+. Further and
improved refinement using previously deposited data (PDB ID 2O9J and 2OA0), in
light of our new findings, also revealed a strong plausibility for a magnesium
ion bound at this site. Furthermore, we find a new configuration of the bound
AMPPCP nucleotide, addressing the modulatory role of ATP binding to the
E2·Pi occluded conformation of SERCA.
, AMPPCP (an ATP analog), and
Mn2+·CPA. The structure reveals novel insight into CPA
binding, which we find to be mediated by a divalent cation, as demonstrated by
means of the anomalous scattering properties of Mn2+. Further and
improved refinement using previously deposited data (PDB ID 2O9J and 2OA0), in
light of our new findings, also revealed a strong plausibility for a magnesium
ion bound at this site. Furthermore, we find a new configuration of the bound
AMPPCP nucleotide, addressing the modulatory role of ATP binding to the
E2·Pi occluded conformation of SERCA.
EXPERIMENTAL PROCEDURES
Protein Preparation and Crystallization—SERCA1a was prepared from SR vesicles isolated from rabbit fast twitch skeletal muscle (SERCA1a) and purified by extraction with deoxycholate, according to established protocols (23). To produce E2-stabilized protein, the purified membranes were solubilized in 20 mg/ml octaethyleneglycol mono-n-dodecyl ether (C12E8) in 85 mm MOPS-KOH (pH 6.8), 67 mm KCl, 17% glycerol (v/v), 1.5 mm EGTA, 2.8 mm MgCl2, 1.0 mm NaF, 1.0 mm AMPPCP, and 0.2 mm CPA. The solubilization was followed by ultracentrifugation, and the supernatant, with a protein concentration of ∼12 mg/ml, was used directly for crystallization experiments by the vapor diffusion method in hanging drops. Protein solution (80% supernatant, 20% 5 mm dithiothreitol) and crystallization buffer (14% polyethylene glycol 6000, 6% 2-methyl-2,4-pentanediol, 70 mm sodium acetate (pH 6.8), 10 mm MnCl2) were mixed in a ratio 1:1 and supplemented with 3 mm Zwittergent 3-12 as an additive. Large, single triangular-shaped crystals grew over 2 weeks at 19 °C. Crystals were cryoprotected by soaking in 20–30% ethylene glycol. The crystals were mounted in litholoops (molecular dimensions) and flash-cooled in liquid nitrogen.
Data Collection and Refinement—Diffraction data were
collected at 100 K on the end stations X06SA at the Swiss Light Source (SLS)
in Villigen, Switzerland and at I911-3 at MAX-lab in Lund, Sweden. The
diffraction data were processed and scaled with XDS
(24). Phases were obtained by
molecular replacement using the program PHASER
(25) and a search model of the
Ca2+-ATPase in the
 (CPA) form
(PDB ID 2O9J). Model building was performed using Coot
(26), and model refinement was
performed with phenix.refine
(27) for all models. For
anomalous map calculation and reflection file handling, programs from the CCP4
package were used (28). All
structural figures in this study were prepared with PyMOL (DeLano Scientific,
Palo Alto, CA).
(CPA) form
(PDB ID 2O9J). Model building was performed using Coot
(26), and model refinement was
performed with phenix.refine
(27) for all models. For
anomalous map calculation and reflection file handling, programs from the CCP4
package were used (28). All
structural figures in this study were prepared with PyMOL (DeLano Scientific,
Palo Alto, CA).
RESULTS
Indications for the Presence of a Divalent Cation—Early
studies on fungal toxins had demonstrated that the tetramic acids tend to
occur naturally as metal-chelate complexes
(29). This was confirmed in a
recent biochemical experiment demonstrating CPA binding to SERCA in an
Mg2+-dependent manner
(19). There is a striking
discrepancy from previous reports on how CPA is oriented in its binding pocket
within the putative calcium entry channel in SERCA. A structure by Toyoshima
and co-workers (18) shows the
tetramic acid moiety of CPA in a buried position, pointing toward
transmembrane segment M3 of SERCA (PDB entry 2EAS), whereas the Young group
(17) has presented a structure
with the tetramic acid group pointing outwards toward transmembrane segment M2
of SERCA (PDB entry 2O9J). The two structural models represent inhibitor
orientations deviating by roughly 180°. Being interested in identifying
the correct binding mode of CPA to SERCA and intrigued by the absence of a
divalent cation and proper planarity of the tetramic acid moiety in both
structures, we first resorted to further refinement using the structure factor
amplitudes deposited by the Young group
(17). Data from the Toyoshima
group (18) were not available.
We found the 2O9J structure to be substantiated by unbiased omit maps, but we
also noted a strong positive peak in the Fo -
Fc map located at the position of a water molecule
(numbered 2074 in PDB ID 2O9J) coordinated by O1 and O2 of the tetramic acid
moiety of CPA, and furthermore, noted that it displayed a substantially lower
B-factor when compared with the oxygens of the tetramic acid (∼55
versus ∼90 Å2). Given the recently demonstrated
magnesium dependence of CPA inhibition
(19) and the metal-chelating
properties of tetramic acids, we suspected that a divalent cation might occupy
this position. This assumption was supported by the fact that exchanging the
water molecule with Mg2+ and re-refining the model led to improved
B-factor consistency while also eliminating the positive peak of the
difference map. However, to resolve this issue with independent
crystallographic data, we identified crystallization conditions for the
 .
complex in the presence of manganese, with the aim of replacing the suspected
Mg2+ at the CPA site by Mn2+ displaying anomalous
scattering properties.
.
complex in the presence of manganese, with the aim of replacing the suspected
Mg2+ at the CPA site by Mn2+ displaying anomalous
scattering properties.
Crystallization of SERCA-CPA in the Presence of
Mn2+—The crystals were obtained by screening
against an in-house screen
(30) with sparse matrix
optimization in 24-well vapor diffusion hanging drop format followed by a
detergent optimization procedure developed for the
Na+,K+-ATPase crystals
(31). SERCA stabilized in the
 form crystallized in a new crystal form exhibiting
P212121 space group symmetry with two
molecules in the asymmetric unit. The crystals display favorable diffraction
properties with low mosaic spread (as low as 0.07°), and a 2.5 Å
resolution data set was collected (supplemental Table 1). The structure was
readily determined by molecular replacement using the structure of the Young
group (17) (PDB ID 2O9J, now
3FPB) as a search model with CPA omitted.
form crystallized in a new crystal form exhibiting
P212121 space group symmetry with two
molecules in the asymmetric unit. The crystals display favorable diffraction
properties with low mosaic spread (as low as 0.07°), and a 2.5 Å
resolution data set was collected (supplemental Table 1). The structure was
readily determined by molecular replacement using the structure of the Young
group (17) (PDB ID 2O9J, now
3FPB) as a search model with CPA omitted.
To investigate whether Mn2+ was bound, data were collected close to the manganese absorption edge at an energy of 7.74 keV (wavelength 1.6 Å). Several strong peaks were observed in the anomalous difference Fourier map at equivalent positions in both molecules of the asymmetric unit. A major peak at the CPA binding pocket corresponding to the suspected Mg2+ site at the tetramic acid, another peak between the α- and β-phosphates of the AMPPCP molecule in the nucleotide binding site between the N-, A-, and P domains (Fig. 1, A and B, and supplemental Fig. 1), and a peak in the P-domain K+ site indicating a bound K+ (supplemental Fig. 2) were found. Remaining peaks appeared at the positions of well ordered sulfur atoms.
FIGURE 1.

Interpretation of the SERCA·CPA complex. A, overall
structure of SERCA showing the N-domain (red), A-domain
(yellow), P-domain (purple), and transmembrane region
(light blue). A 2Fo - Fc
electron density map, contoured at 1σ, shows electron density both for
AMPPCP between the N- and A-domains and for the CPA binding site in the
Ca2+ entry channel. The Mn2+ ion is shown by a van der
Waals sphere representation (orange) both in the AMPPCP site and in
the CPA site. B, the AMPPCP binding site. The residues
Asn628, Arg678, and Lys205 are within
interacting distance of the γ-phosphate in AMPPCP, and Asp203
stabilizes Arg678. Hydrogen bonding to a water molecule
(W4) is colored blue and shown as dashed lines,
whereas other hydrogen-bonding networks are colored black.
Arg174 and Glu439 make a salt bridge linking the A- and
N-domains. The 2Fo - Fc map is
contoured at 1σ (blue mesh), covering the AMPPCPC molecule.
C, transmembrane domain of SERCA with bound CPA. Three residues in
the transmembrane segment of SERCA are involved in polar interactions with
CPA: Gln56, Asp59, and Asn101. Both
Asn101 and Gln56 contribute with side chain and backbone
atoms to the coordination of the tetramic acid part of CPA. The side chain
oxygen of Gln56 participates in the coordination sphere of the
manganese atom. The anomalous difference Fourier map (orange mesh)
contoured at 10 σ identifies the Mn2+ ion coordinated at the
CPA-SERCA binding interface. D, SERCA structures with TG-Boc12-ADT
(green) (PDB ID 2BY4) and BHQ (blue) (PDB ID 2AGV) are
superimposed onto transmembrane helices 1–5 of our new structure (PDB ID
3FGO). TG-Boc12-ADT, BHQ, and CPA·Mn2+ (yellow) are
shown in stick representation. The drug pocket is viewed from the
cytoplasmic side of the SR membrane. E, a structural alignment
between the Na+, K+-ATPase (purple) and SERCA
(light blue), both stabilized with
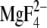 . The M1 kink region of the
Na+, K+-ATPase exhibits a significant structural
difference to SERCA. The intruding M1 loop is stabilized by Phe93
in the Na+,K+-ATPase, occupying the same binding pocket
as the indole moiety of CPA and the aromatic moiety of BHQ in SERCA.
. The M1 kink region of the
Na+, K+-ATPase exhibits a significant structural
difference to SERCA. The intruding M1 loop is stabilized by Phe93
in the Na+,K+-ATPase, occupying the same binding pocket
as the indole moiety of CPA and the aromatic moiety of BHQ in SERCA.
The Architecture of the CPA Binding Pocket—In agreement with the previously presented models, we find that the CPA binding pocket is located at a groove formed between transmembrane segments M1, M2, M3, and M4 of SERCA (Fig. 1, A and C). The quality of the electron density maps allowed us to unambiguously determine an orientation of the CPA molecule in accordance with the model of the Young group (17). The hydrophobic indole group of CPA thus sits in a wide hydrophobic groove between M3 and M4, whereas the tetramic acid moiety is involved in a network of polar interactions with residues on M1 and M2. The Mn2+ ion has replaced the probable Mg2+ located at this site in the previous structures (Figs. 1C and 2A). Gln56-Oε1 is the only side chain directly linked to the Mn2+ ion, whereas backbone carbonyls from Gln56, Asp59, and Asn101 coordinate two water molecules within the liganding sphere. A third water molecule is less apparent from the electron density maps and is probably interacting only with the Mn2+ ion (Figs. 1C and 2A). The octahedral coordination sphere of the Mn2+ ion is thus completed by the tetramic acid of CPA, the Gln56 side chain of SERCA, and three water molecules. The top water molecule (Fig. 1C, W1) is coordinated by the carbonyl oxygen from Asn101 and Gln56-Oε. The bottom water molecule (W2) is bifurcated between the carbonyl oxygen of Gln56 and Oδ2 from Asp59. The tetramic acid is likely to be in the non-protonated state (pKa ∼ 3) (32), with the negative charge distribution polarized toward the Mn2+ ion. The 9-carbonyl and the tertiary 8-amino group of CPA are both within weak hydrogen-bonding distance (3.6 Å) to Asn101-Nδ. The hydrogen on Asn101-NδH may act as hydrogen donor to the conjugated π-acceptor system above the tetramic ring. This type of π-HX interaction (Where HX is any hydrogen bond donor) is often observed in protein structures (33), with the indole ring of a tryptophan acting as the acceptor. It is unlikely that a hydrogen bond between Asp254-Oε and the indole nitrogen (position N-2) of CPA exists given their approximate distance of 5.4 Å (Fig. 2A). For a thorough description of the hydrophobic binding pocket for the indole group of CPA, please refer to Moncoq et al. (17).
FIGURE 2.

Binding pocket of Cyclopiazonic acid. A, topology of the CPA binding site. Conjugated doublebond systems of the CPA molecule are high lighted in green. Atom numbering in CPA is according to International Union of Pure and Applied Chemistry (IUPAC) nomenclature. B, fragment-based view of the CPA binding pocket. The drug binding pocket can be subdivided into three regions, each accommodating drug moieties of distinct chemical character. The polar region (designated A, highlighted in yellow) coordinates the acyl-tetramic acid moiety, with Gln56, Asp59, and Asn101 as main interacting residues. The coordinated M2+ occupies a central position in this interaction. The center of the pocket displays a preference for conjugated π-systems (B, highlighted in blue). BHQ binds to this part, superposing with CPA in this “B-pocket.” The relatively wide hydrophobic region of the pocket (C, highlighted in green) accommodates bulky, non-polar moieties. The decomposed analysis of this site proposes functional extensions or modifications to yield tailored high affinity drugs. For instance, an aliphatic chain at site D, bridging toward the TG binding site, and a suitable hydrogen-bonding partner to Asp254 (region E), may further improve drug affinity.
DISCUSSION
The Nucleotide Binding Site—In both structures (PDB ID 3FGO
and 3FPB), the nucleotide is not fully occupied, confirming an antagonistic
effect of CPA against ATP binding, which has already been observed in earlier
biochemical studies on CPA-SERCA interaction
(10,
34). These are the first
structures with AMPPCP present in the
H+2–3E2·Pi(CPA) state, with the
liberated phosphate group mimicked by
 . We find this configuration of
the bound AMPPCP to be different from the modulatory AMPPCP in the
H+2–3E2(TG) state, which is the dephosphorylated
state, where the α- and β-phosphates are bridged via a magnesium
ion to Glu439 (9).
The AMPPCP is, however, in an equivalent conformation as in
. We find this configuration of
the bound AMPPCP to be different from the modulatory AMPPCP in the
H+2–3E2(TG) state, which is the dephosphorylated
state, where the α- and β-phosphates are bridged via a magnesium
ion to Glu439 (9).
The AMPPCP is, however, in an equivalent conformation as in
 (8), but with our improved
resolution of 2.5 Å, we are able to make a more accurate description of
the nucleotide binding site. In our structure, the γ-phosphate of the
AMPPCP is in direct hydrogen-bonding distance to Lys205-NζH.
Mutational studies on Lys205 confirm a modulatory effect of ATP on
SERCA (Fig. 1B)
(35). Additional residues in
close contact to the γ-phosphate are Arg678 and
Asn628 in the P-domain. Arg678 is coordinating a water
molecule (Fig. 1B,
W4) that is bifurcated by the γ-phosphate, whereas
Asn628 and Asp203 make hydrogen bonds to
Arg678, presumably stabilizing its conformation Arg678
(Fig. 1B).
Arg560 is important for modulation of the ATP in the E2 to E1
transition (9). Mutation of
Arg560 has been shown not to affect the low affinity ATP
site in this particular functional transition state and is indeed found
not to interact with the AMPPCP
(36). Although
Arg678 has been proposed to be involved with the modulatory effect
of ATP in the E2·Pi occluded state and E2 state
(37), Asp203 has
been proposed to have influence on the transport rate and ATP hydrolysis
(23,
38). Thus, our structural data
confirm Asp203 as an important stabilizing residue of
Arg678. It is interesting that the stabilization of
Arg678 is also mediated by a direct hydrogen bond from
Asn628. This stabilizing triplet is also evident in the occluded
(8), but with our improved
resolution of 2.5 Å, we are able to make a more accurate description of
the nucleotide binding site. In our structure, the γ-phosphate of the
AMPPCP is in direct hydrogen-bonding distance to Lys205-NζH.
Mutational studies on Lys205 confirm a modulatory effect of ATP on
SERCA (Fig. 1B)
(35). Additional residues in
close contact to the γ-phosphate are Arg678 and
Asn628 in the P-domain. Arg678 is coordinating a water
molecule (Fig. 1B,
W4) that is bifurcated by the γ-phosphate, whereas
Asn628 and Asp203 make hydrogen bonds to
Arg678, presumably stabilizing its conformation Arg678
(Fig. 1B).
Arg560 is important for modulation of the ATP in the E2 to E1
transition (9). Mutation of
Arg560 has been shown not to affect the low affinity ATP
site in this particular functional transition state and is indeed found
not to interact with the AMPPCP
(36). Although
Arg678 has been proposed to be involved with the modulatory effect
of ATP in the E2·Pi occluded state and E2 state
(37), Asp203 has
been proposed to have influence on the transport rate and ATP hydrolysis
(23,
38). Thus, our structural data
confirm Asp203 as an important stabilizing residue of
Arg678. It is interesting that the stabilization of
Arg678 is also mediated by a direct hydrogen bond from
Asn628. This stabilizing triplet is also evident in the occluded
 stabilized structure (8), but
no water molecules were visible in the electron density. The interaction to
the γ-phosphate is mediated through a hydrogen-bonding network including
only water (Fig. 1B)
but no direct hydrogen bonds. A direct hydrogen bond is formed between
Glu439 and Arg174, linking the N- and A-domain, but
neither of these residues are involved in a direct interaction with the
modulatory ATP. The residues Lys492 and Phe478 have both
been shown to have a catalytic and a modulatory role (supplemental Fig. 1)
(39), and they are both within
van der Waals distance (<3.5 Å) of the ATP analog. The modulatory
effect observed on Ile188
(35) is not apparent. However,
Ile188 is within van der Waals distance of the AMPPCP. This
structure allows for the first accurate structural characterization of the
bound ATP and especially of the water-mediated hydrogen-bonding network around
the γ-phosphate in the modulatory state prior to dephosphorylation (E2P
→ E2).
stabilized structure (8), but
no water molecules were visible in the electron density. The interaction to
the γ-phosphate is mediated through a hydrogen-bonding network including
only water (Fig. 1B)
but no direct hydrogen bonds. A direct hydrogen bond is formed between
Glu439 and Arg174, linking the N- and A-domain, but
neither of these residues are involved in a direct interaction with the
modulatory ATP. The residues Lys492 and Phe478 have both
been shown to have a catalytic and a modulatory role (supplemental Fig. 1)
(39), and they are both within
van der Waals distance (<3.5 Å) of the ATP analog. The modulatory
effect observed on Ile188
(35) is not apparent. However,
Ile188 is within van der Waals distance of the AMPPCP. This
structure allows for the first accurate structural characterization of the
bound ATP and especially of the water-mediated hydrogen-bonding network around
the γ-phosphate in the modulatory state prior to dephosphorylation (E2P
→ E2).
The Role of the Divalent Cation—We find that a divalent metal (Mg2+ or Mn2+) is important for CPA interaction with SERCA, forming a SERCA·CPA·M2+ complex. The Mg2+/Mn2+ ion bound with CPA also interacts with residues of SERCA lining the putative ion access channel between M1 and M2 at the cytoplasmic membrane interface. This may hint at a transient Ca2+ site important for selectivity in the entry channel. An earlier finding that Ca2+ can compete with CPA and prevent inhibition, if added to SERCA before CPA (34), makes it tempting to speculate that the CPA·Mn2+ site indeed mimics a transient Ca2+ site along the entry pathway. It has been proposed based on transport experiments that CPA is able to uncouple ATP hydrolysis in SERCA in a Ca2+-dependent manner (40). These studies concluded that only one Ca2+ ion is bound in the presence of CPA. However, our present data suggest that under appropriate conditions, a Ca2+ ion could be stabilized in the entry channel with CPA, corresponding to the single Ca2+ ion detected (40). Metal chelation by the tetramic acid seems to be important for transport across membranes in biological tissues (20), and presumably, CPA may form a stable complex with a range of other divalent cations. The residues that are structurally important for CPA binding are conserved (supplemental Table 2). Homology models of two Ca2+-ATPases from Plasmodium falciparum (causative of malaria), pfATP4 and pfATP6, and of a putative Ca2+-ATPase from Mycobacterium tuberculosis (MTB) Locus tag: Rv1997 were generated with 3FGO as template, and all indicate that a similar mode of CPA binding is possible. In particular, the residues Gln56 and Asn101 that we find to be important for binding of the tetramic acid moiety are conserved. In support of such arguments, CPA inhibition has been observed for pfATP4 (41) and the Ca2+-ATPase LCA1 from tomato (Solanum lycopersicum) (42).
Fragment-based Drug Design—CPA and BHQ bind to the same region in the entry channel. Despite the overall differences in the helical arrangement of M3 and M4, the cyclic ring systems of both BHQ and the isoindole moiety in CPA are accommodated at the same binding pocket (Figs. 1D and 2B). The binding pocket seems to accommodate three distinct interaction sites, with selectivity for specific chemical groups. First, the tetramic acid binds near the access channel entry site between M1 and M2, where Gln56 (M1) participates directly in metal coordination and Asp59 (M1) and Asn101 (M2) coordinate water molecules that interact with the metal (Fig. 2A). Second, a hydrophobic pocket provides selectivity for chemical groups with conjugated π-systems represented by the indole moiety of CPA and the hydroquinone of BHQ (Fig. 2B). Third, an extension of the same hydrophobic binding pocket between M3 and M5 can accommodate aliphatic moieties, as was demonstrated when the butanoyl group on O-8 of TG was replaced with an N-tert-butoxycarbonyl-12-aminododecanoyl (Boc12-ADT) group. The TG-Boc12-ADT derivative had similar inhibitory properties as TG itself, and the crystal structure of SERCA with TG-Boc12-ADT revealed the presence of the Boc12-ADT group extending from the TG binding side between helices M3, M5, and M7 into the binding pocket occupied by BHQ and CPA (43) (Fig. 1D). These observations invite new drug development strategies for derivatizing the indole group of CPA with the addition of aliphatic groups expanding toward the TG binding pocket (Fig. 2B) as one possible strategy to gain specificity for specific Ca2+-ATPase targets.
Comparison with Na+,K+-ATPase
Indicates a Structural Importance of Phe93—The recently
determined crystal structure of the potassium-bound
Na+,K+-ATPase was stabilized by
 , thus in an
E2·Pi like state presented here for SERCA. A structural
alignment of the Na+,K+-ATPase and SERCA was performed
on transmembrane helices 1, 2, and 4 that define the CPA binding site. The
structural alignment offers a clear explanation as to why CPA is unable to
inhibit Na+,K+-ATPase and the closely related
H+,K+-ATPase
(10). Residues in the kink
region of the M1 helix protrude into the equivalent site of the CPA binding
pocket, preventing CPA and the related BHQ from binding due to sterical
hindrance. The 90FGGF93 motif of the M1 is specific for
the Na+,K+-ATPase, and it was well defined from the
experimental electron density map of the Na+,K+-ATPase
structure (31). The presence
of the aromatic Phe93 side chain at the M1 kink binding pocket
replacing the hydroquinone of BHQ or the indole moiety of CPA indicates that
the pocket has a preference for compounds containing a conjugated π-system.
We speculate that this pocket is of functional importance in the
Na+,K+-ATPase, possibly in an auto-regulatory mechanism
where Phe93 prevents other ions and water from entering the buried
ion binding sites. The corresponding residue in rat has been investigated by
mutagenesis (Phe95) and was shown to be essential because COS cells
expressing the mutants F95A and F95R were not viable, whereas the F95Y and
F95L mutants showed decreased sodium affinity
(44). The role of the two
glycines leading up to Phe93 have also been investigated by
mutagenesis, indicating that both residues are important for Na+
and K+ binding, and thus, their mutation prevents proper reverse
transition into the E2P state
(45). We now offer a unifying
model for these mutagenesis data guided by the finding that a pocket with
similar chemical characteristics exists in both SERCA and
Na+,K+-ATPase. This observation points to
Phe93 of Na+,K+-ATPase as an intramolecular
determinant of E2 stabilization where the aromatic phenyl group blocks the
cytoplasmic access channel by a mechanism reminiscent of CPA or BHQ inhibition
of SERCA. This further suggests that the Mn2+ ion trapped in the
CPA complex with SERCA may have pinpointed a pre-entry site for
Ca2+ ions centered on Gln56 at the water-membrane
interface.
, thus in an
E2·Pi like state presented here for SERCA. A structural
alignment of the Na+,K+-ATPase and SERCA was performed
on transmembrane helices 1, 2, and 4 that define the CPA binding site. The
structural alignment offers a clear explanation as to why CPA is unable to
inhibit Na+,K+-ATPase and the closely related
H+,K+-ATPase
(10). Residues in the kink
region of the M1 helix protrude into the equivalent site of the CPA binding
pocket, preventing CPA and the related BHQ from binding due to sterical
hindrance. The 90FGGF93 motif of the M1 is specific for
the Na+,K+-ATPase, and it was well defined from the
experimental electron density map of the Na+,K+-ATPase
structure (31). The presence
of the aromatic Phe93 side chain at the M1 kink binding pocket
replacing the hydroquinone of BHQ or the indole moiety of CPA indicates that
the pocket has a preference for compounds containing a conjugated π-system.
We speculate that this pocket is of functional importance in the
Na+,K+-ATPase, possibly in an auto-regulatory mechanism
where Phe93 prevents other ions and water from entering the buried
ion binding sites. The corresponding residue in rat has been investigated by
mutagenesis (Phe95) and was shown to be essential because COS cells
expressing the mutants F95A and F95R were not viable, whereas the F95Y and
F95L mutants showed decreased sodium affinity
(44). The role of the two
glycines leading up to Phe93 have also been investigated by
mutagenesis, indicating that both residues are important for Na+
and K+ binding, and thus, their mutation prevents proper reverse
transition into the E2P state
(45). We now offer a unifying
model for these mutagenesis data guided by the finding that a pocket with
similar chemical characteristics exists in both SERCA and
Na+,K+-ATPase. This observation points to
Phe93 of Na+,K+-ATPase as an intramolecular
determinant of E2 stabilization where the aromatic phenyl group blocks the
cytoplasmic access channel by a mechanism reminiscent of CPA or BHQ inhibition
of SERCA. This further suggests that the Mn2+ ion trapped in the
CPA complex with SERCA may have pinpointed a pre-entry site for
Ca2+ ions centered on Gln56 at the water-membrane
interface.
Supplementary Material
Acknowledgments
We express gratitude for beam line support by Thomas Ursby (MAX-lab) and Clemens Schulze-Briese, Anuschka Pauluhn, and Rouven Bingel-Erlenmeyer (Swiss Light Source) for making our experiments possible and to Anne-Marie Lund Winther and Anna Marie Nielsen for valuable discussions and technical support.
The atomic coordinates and structure factors (codes 3FGO, 3FPB and 3FPS) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported by a center of excellence grant from the Danish National Research Foundation and by a Hallas-Møller research stipend from the Novo Nordisk Foundation (to P. N.) and by grants from the Danish Medical Research Council, Aarhus University Research Foundation and Novo Nordisk Foundation and the “Fabrikant Vilhelm Pedersen og hustrus legat” (to J. V. M.).
The on-line version of this article (available at http://www.jbc.org) contains two supplemental tables and two supplemental figures.
Footnotes
The abbreviations used are: SERCA, sarco(endo)plasmic reticulum Ca2+-ATPase; SR, sarcoplasmic reticulum; TG, thapsigargin; CPA, cyclopiazonic acid; BHQ, 2,5-di-(tert-butyl) hydroquinone; AMPPCP, adenosine 5′-(β,γ-methylene)triphosphate; E1, ubiquitin-activating enzyme; E2, ubiquitin carrier protein; Boc12-ADT, N-tert-butoxycarbonyl-12-aminododecanoyl; MOPS, 4-morpholinepropanesulfonic acid; PDB, Protein Data Bank.
References
- 1.Moller, J. V., Juul, B., and le Maire, M. (1996) Biochim. Biophys. Acta 1286 1-51 [DOI] [PubMed] [Google Scholar]
- 2.Toyoshima, C., Nakasako, M., Nomura, H., and Ogawa, H. (2000) Nature 405 647-655 [DOI] [PubMed] [Google Scholar]
- 3.MacLennan, D. H. (1970) J. Biol. Chem. 245 4508-4518 [PubMed] [Google Scholar]
- 4.Van Baelen, K., Dode, L., Vanoevelen, J., Callewaert, G., De Smedt, H., Missiaen, L., Parys, J. B., Raeymaekers, L., and Wuytack, F. (2004) Biochim. Biophys. Acta 1742 103-112 [DOI] [PubMed] [Google Scholar]
- 5.Clapham, D. E. (2007) Cell 131 1047-1058 [DOI] [PubMed] [Google Scholar]
- 6.Sorensen, T. L., Moller, J. V., and Nissen, P. (2004) Science 304 1672-1675 [DOI] [PubMed] [Google Scholar]
- 7.Toyoshima, C., and Mizutani, T. (2004) Nature 430 529-535 [DOI] [PubMed] [Google Scholar]
- 8.Olesen, C., Picard, M., Winther, A. M., Gyrup, C., Morth, J. P., Oxvig, C., Moller, J. V., and Nissen, P. (2007) Nature 450 1036-1042 [DOI] [PubMed] [Google Scholar]
- 9.Jensen, A. M., Sorensen, T. L., Olesen, C., Moller, J. V., and Nissen, P. (2006) EMBO J. 25 2305-2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seidler, N. W., Jona, I., Vegh, M., and Martonosi, A. (1989) J. Biol. Chem. 264 17816-17823 [PubMed] [Google Scholar]
- 11.Moore, G. A., McConkey, D. J., Kass, G. E., O'Brien, P. J., and Orrenius, S. (1987) FEBS Lett. 224 331-336 [DOI] [PubMed] [Google Scholar]
- 12.Berman, M. C., and Karlish, S. J. (2003) Biochemistry 42 3556-3566 [DOI] [PubMed] [Google Scholar]
- 13.Hua, S., Xu, C., Ma, H., and Inesi, G. (2005) J. Biol. Chem. 280 17579-17583 [DOI] [PubMed] [Google Scholar]
- 14.Holzapfel, C. W. (1968) Tetrahedron 24 2101-2119 [DOI] [PubMed] [Google Scholar]
- 15.Goeger, D. E., Riley, R. T., Dorner, J. W., and Cole, R. J. (1988) Biochem. Pharmacol. 37 978-981 [DOI] [PubMed] [Google Scholar]
- 16.Ma, H., Zhong, L., Inesi, G., Fortea, I., Soler, F., and Fernandez-Belda, F. (1999) Biochemistry 38 15522-15527 [DOI] [PubMed] [Google Scholar]
- 17.Moncoq, K., Trieber, C. A., and Young, H. S. (2007) J. Biol. Chem. 282 9748-9757 [DOI] [PubMed] [Google Scholar]
- 18.Takahashi, M., Kondou, Y., and Toyoshima, C. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 5800-5805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montigny, C., Picard, M., Lenoir, G., Gauron, C., Toyoshima, C., and Champeil, P. (2007) Biochemistry 46 15162-15174 [DOI] [PubMed] [Google Scholar]
- 20.Schobert, R., and Schlenk, A. (2008) Bioorg. Med. Chem. 16 4203-4221 [DOI] [PubMed] [Google Scholar]
- 21.Steyn, P. S., and Rabie, C. J. (1976) Phytochemistry 15 1977-1979 [Google Scholar]
- 22.Lebrun, M. H., Duvert, P., Gaudemer, F., Gaudemer, A., Deballon, C., and Boucly, P. (1985) J. Inorg. Biochem. 24 167-181 [DOI] [PubMed] [Google Scholar]
- 23.Andersen, J. P., Lassen, K., and Moller, J. V. (1985) J. Biol. Chem. 260 371-380 [PubMed] [Google Scholar]
- 24.Kabsch, W. (1993) J. Appl. Crystallogr. 26 795-800 [Google Scholar]
- 25.McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C., and Read, R. J. (2005) Acta Crystallogr. Sect. D Biol. Crystallogr. 61 458-464 [DOI] [PubMed] [Google Scholar]
- 26.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 27.Adams, P. D., Grosse-Kunstleve, R. W., Hung, L. W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K., and Terwilliger, T. C. (2002) Acta Crystallogr. Sect. D Biol. Crystallogr. 58 1948-1954 [DOI] [PubMed] [Google Scholar]
- 28.Collaborative Computational Project, Number 4 (1994) Acta Crystallogr. Sect. D Biol. Crystallogr. 50 760-76315299374 [Google Scholar]
- 29.Gallagher, R. T., Richard, J. L., Stahr, H. M., and Cole, R. J. (1978) Mycopathologia 66 31-36 [DOI] [PubMed] [Google Scholar]
- 30.Sorensen, T. L., Olesen, C., Jensen, A. M., Moller, J. V., and Nissen, P. (2006) J. Biotechnol. 124 704-716 [DOI] [PubMed] [Google Scholar]
- 31.Morth, J. P., Pedersen, B. P., Toustrup-Jensen, M. S., Sorensen, T. L., Petersen, J., Andersen, J. P., Vilsen, B., and Nissen, P. (2007) Nature 450 1043-1049 [DOI] [PubMed] [Google Scholar]
- 32.Sobolev, V. S. (2005) J. AOAC Int. 88 1367-1370 [PubMed] [Google Scholar]
- 33.Steiner, T., and Koellner, G. (2001) J. Mol. Biol. 305 535-557 [DOI] [PubMed] [Google Scholar]
- 34.Soler, F., Plenge-Tellechea, F., Fortea, I., and Fernandez-Belda, F. (1998) Biochemistry 37 4266-4274 [DOI] [PubMed] [Google Scholar]
- 35.Clausen, J. D., McIntosh, D. B., Woolley, D. G., and Andersen, J. P. (2008) J. Biol. Chem. 283 35703-35714 [DOI] [PubMed] [Google Scholar]
- 36.Clausen, J. D., McIntosh, D. B., Vilsen, B., Woolley, D. G., and Andersen, J. P. (2003) J. Biol. Chem. 278 20245-20258 [DOI] [PubMed] [Google Scholar]
- 37.Clausen, J. D., McIntosh, D. B., Anthonisen, A. N., Woolley, D. G., Vilsen, B., and Andersen, J. P. (2007) J. Biol. Chem. 282 20686-20697 [DOI] [PubMed] [Google Scholar]
- 38.Kato, S., Kamidochi, M., Daiho, T., Yamasaki, K., Gouli, W., and Suzuki, H. (2003) J. Biol. Chem. 278 9624-9629 [DOI] [PubMed] [Google Scholar]
- 39.McIntosh, D. B., Woolley, D. G., Vilsen, B., and Andersen, J. P. (1996) J. Biol. Chem. 271 25778-25789 [DOI] [PubMed] [Google Scholar]
- 40.Martinez-Azorin, F. (2004) FEBS Lett. 576 73-76 [DOI] [PubMed] [Google Scholar]
- 41.Krishna, S., Woodrow, C., Webb, R., Penny, J., Takeyasu, K., Kimura, M., and East, J. M. (2001) J. Biol. Chem. 276 10782-10787 [DOI] [PubMed] [Google Scholar]
- 42.Johnson, N. A., Liu, F., Weeks, P. D., Hentzen, A. E., Kruse, H. P., Parker, J. J., Laursen, M., Nissen, P., Costa, C. J., and Gatto, C. (2009) Arch. Biochem. Biophys. 481 157-168 [DOI] [PubMed] [Google Scholar]
- 43.Sohoel, H., Jensen, A. M., Moller, J. V., Nissen, P., Denmeade, S. R., Isaacs, J. T., Olsen, C. E., and Christensen, S. B. (2006) Bioorg. Med. Chem. 14 2810-2815 [DOI] [PubMed] [Google Scholar]
- 44.Einholm, A. P., Andersen, J. P., and Vilsen, B. (2007) J. Biol. Chem. 282 23854-23866 [DOI] [PubMed] [Google Scholar]
- 45.Einholm, A. P., Toustrup-Jensen, M., Andersen, J. P., and Vilsen, B. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 11254-11259 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.