

Abstract
We have previously shown that the binding of epidermal growth factor (EGF) to its receptor can best be described by a model that involves negative cooperativity in an aggregating system (Macdonald, J. L., and Pike, L. J. (2008) Proc. Natl. Acad. Sci. U. S. A. 105, 112–117). However, despite the fact that biochemical analyses indicate that EGF induces dimerization of its receptor, the binding data provided no evidence for positive linkage between EGF binding and dimer assembly. By analyzing the binding of EGF to a number of receptor mutants, we now report that in naive, unphosphorylated EGF receptors, ligand binding is positively linked to receptor dimerization but the linkage is abolished upon autophosphorylation of the receptor. Both phosphorylated and unphosphorylated EGF receptors exhibit negative cooperativity, indicating that mechanistically, cooperativity is distinct from the phenomenon of linkage. Nonetheless, both the positive linkage and the negative cooperativity observed in EGF binding require the presence of the intracellular juxtamembrane domain. This indicates the existence of inside-out signaling in the EGF receptor system. The intracellular juxtamembrane domain has previously been shown to be required for the activation of the EGF receptor tyrosine kinase (Thiel, K. W., and Carpenter, G. (2007) Proc. Natl. Acad. Sci. U. S. A. 104, 19238–19243). Our experiments expand the role of this domain to include the allosteric control of ligand binding by the extracellular domain.
The EGF2 receptor is a tyrosine kinase composed of an ∼620-amino-acid extracellular domain that recognizes and binds EGF, a single pass α-helical transmembrane domain, and an intracellular tyrosine kinase domain, encompassing roughly residues 685–950 (1). In addition, the receptor contains an ∼230-amino-acid-long C-terminal tail that contains the bulk of the sites of receptor autophosphorylation (2–4). An intracellular juxtamembrane domain of about 40 residues connects the transmembrane domain to the kinase domain and has been shown to be crucial in the allosteric activation of the EGF receptor kinase (5, 6).
In the membrane, the EGF receptor exists as a monomer, but a wealth of data indicate that the binding of EGF induces the formation of EGF receptor dimers (7–10). Dimerization appears to be mediated in large part by the extracellular domain of the receptor, which is comprised of four subdomains, designated I through IV. X-ray crystallography data suggest that in the absence of ligand, the extracellular domain is held in a closed configuration through the interaction of loops or arms that extend from the backs of subdomains II and IV (11). Upon binding of EGF, this intramolecular tether is released, allowing the receptor to adopt an open conformation in which EGF is tightly bound between subdomains I and III. In this configuration, the “dimerization arm” that was previously involved in tethering the receptor closed mediates the formation of a back-to-back EGF receptor dimer (12, 13).
Analyses of the binding of 125I-EGF to its receptor have invariably resulted in concave up Scatchard plots that have been interpreted as indicating the presence of two classes of EGF binding sites. However, we have recently used global analysis of the binding of 125I-EGF to cells expressing increasing levels of EGF receptors to show that EGF binding is best described by a model involving negative cooperativity in an aggregating system (14) (see Fig. 6). Ligand binding is negatively cooperative if the binding of ligand to the first site on a dimer reduces the affinity of the ligand for binding to the second site on the dimer.
FIGURE 6.

Model for the binding of EGF to its receptor. Circles represent receptor subunits. E represents a molecule of EGF. The equilibrium association constants are written above or beside the reaction to which they apply.
The concept of cooperativity only applies to existing dimers. It does not relate to the effect of ligand on the assembly or disassembly of those dimers. The effect of ligand on the formation of receptor dimers is captured in the concept of linkage (15, 16). If ligand binding is positively linked to dimer formation, then ligand promotes the assembly of receptor dimers. In a monomer-dimer equilibrium, positive linkage arises when a ligand binds with higher affinity to the first site on the dimer than to the monomer. Under these circumstances, the ligand will preferentially bind to the dimer, shifting the equilibrium in favor of the dimeric species. In the case of the EGF receptor, biochemical data suggest that EGF induces receptor dimerization; however, evidence for positive linkage in binding studies has been lacking.
By analyzing the binding of 125I-EGF to cells expressing various EGF receptor mutants, we now report that in naive, unphosphorylated EGF receptors, ligand binding is, in fact, positively linked to receptor dimerization. Autophosphorylation of the EGF receptor abolishes the positive linkage that is present during the initial phase of the ligand binding reaction. Negative cooperativity is present in both the phosphorylated and the non-phosphorylated states of the receptor. Structure-function analyses demonstrate that both cooperativity and linkage are lost when the EGF receptor is truncated immediately after the transmembrane domain. However, both forms of regulation are restored in receptors that include the additional 40 amino acids that correspond to the intracellular juxtamembrane domain. These data expand the role of the intracellular juxtamembrane domain to include the allosteric regulation of EGF binding by the extracellular domain and demonstrate the presence of inside-out signaling in the EGF receptor system.
EXPERIMENTAL PROCEDURES
Construction of EGF Receptor Mutants and Plasmids—The V583D- and K721A-EGF receptor point mutants were constructed using the QuikChange mutagenesis kit (Stratagene). The c′698- and c′973-EGF receptors were generated using the QuikChange mutagenesis kit to introduce a stop codon immediately 3′ of the codon encoding Ala-698 or Phe-973 of the EGF receptor. The CC-EGF receptor (17) and the ΔC-EGF receptor (18) were generated as described previously. All mutants were completely sequenced and then cloned into the pBI Tet vector between the NheI and EcoRV sites as described (14). These mutants are shown schematically in supplemental Fig. 1.
Plasmids were co-transfected with pTK-Hyg into CHO-K1 Tet-on cells (Clontech) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Stable clones were isolated by selection in 400 μg/ml hygromycin. Stable cell lines were maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 100 μg/ml hygromycin, and 100 μg/ml G418. Cells were plated into 6-well dishes and treated with doses of doxycycline ranging from 20 to 1000 ng/ml 48 h prior to use.
125I-EGF Synthesis and Binding—EGF was purchased from Biomedical Technologies, Inc. (Stoughton, MA). 125I-EGF was synthesized using the oxidative ICl procedure of Doran and Spar (19).
Radioligand binding was performed in 6-well dishes using 3 ml/well Ham's F12 medium containing 3 mg/ml bovine serum albumin and 25 mm Hepes, pH 7.0, 20–40 pm 125I-EGF and increasing concentrations of unlabeled EGF. Cells were incubated overnight on ice. At the end of the incubation, cells were washed three times in Hanks' balanced salt solution, and the monolayers were dissolved in 1 n NaOH. The lysates were transferred to tubes and counted for 125I in a Beckman gamma counter. Nonspecific binding was determined by fitting the raw data to a competition binding model and using the fitted bottom value as nonspecific. All binding measurements were done in triplicate.
Fitting and analysis were done exactly as described previously (14). GraphPad Prism 4.0 was used to globally fit the data from multiple binding experiments to cells expressing increasing numbers of EGF receptors/cell to the equation (Equation 1)
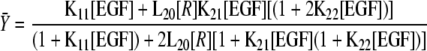 |
(Eq. 1) |
where [R] = concentration of unoccupied EGF receptor monomers (15). This can be calculated from the equation
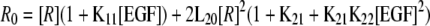 |
(Eq. 2) |
where R0 = total concentration of EGF receptors as derived by Wyman and Gill (16).
Receptor concentration is expressed as a density (mol of receptor/dm2) because the receptor is restricted to movement in the two dimensions of the cell membrane. Values for receptor density were derived from fluorescence correlation spectroscopy measurements of the number of green fluorescent protein-EGF receptors in a beam of known radius in CHO cells expressing a known level of green fluorescent protein-EGF receptors/cell (20). Therefore, the units for L20 are (mol/dm2)-1. As the units of L20 cancel out in all the mass action law equations, the units for the association constants for ligand binding events remain in m-1.
RESULTS
Effect of Tether Mutations on 125I-EGF Binding—For these experiments, all EGF receptor mutants were generated and inserted into a tet-inducible plasmid. CHO cells, which do not express endogenous EGF receptors or other ErbB family members, were stably transfected with these plasmids. This allowed us to control the level of EGF receptor expression by the addition of increasing concentrations of doxycycline.
As we have reported previously (14) and as shown in Fig. 1, the binding of 125I-EGF to CHO cells expressing the wild type EGF receptor yields a family of saturation binding isotherms that shift from left to right as the number of EGF receptors/cell increases. Global fitting of the data from all five binding curves yielded the association constants given in the inset. The results are given as the association constants that were obtained from fitting the data. However, they are discussed throughout as dissociation constants (i.e. 1/KA) because this is the traditional measure used to describe hormone binding.
FIGURE 1.

Binding of 125I-EGF to cells expressing increasing levels of wild type EGF receptors. Cells were treated with increasing doses of doxycycline to induce EGF receptor expression. 125I-EGF binding isotherms were generated in cells expressing increasing levels of either receptor. All the binding isotherms from each cell line were globally fit to Equation 1 with only R0 varying among curves. Data points represent the mean ± S.D. of triplicate determinations. The solid lines represent the fitted curves through the data points of the same color. The r2 value of the global fit was 0.99. The numbers in the upper left indicate the number of receptors per cell in thousands.
The data are consistent with our previous findings (14) and indicate that the affinity of EGF for binding to the receptor monomer (K11) and the first site on the receptor dimer (K21) is similar and on the order of 300 pm. Thus, there is no preferential binding of ligand to the unoccupied dimer as compared with the monomer, i.e. no positive linkage.
The binding of EGF to the second site on the dimer (K22) is of significantly lower affinity (KD ∼5 nm), indicative of negative cooperativity. This is the reason the curves move from left to right with increasing receptor number. At higher receptor levels, more dimers are present, and so there are more of the low affinity sites present in the population.
X-ray crystallographic studies have shown that the unliganded EGF receptor exists in a closed, tethered configuration mediated by interactions between the dimerization arm in subdomain II and the tethering arm in subdomain IV (11). Mutation of Val-583 in subdomain IV to Asp was postulated to weaken the intramolecular tether and alter the binding of EGF (21). To determine whether this mutation affected any of the 125I-EGF binding parameters, we expressed this mutant in CHO cells. Fig. 2 shows the saturation binding isotherms for 125I-EGF binding to the V583D-EGF receptor in cells expressing increasing levels of the mutant receptor.
FIGURE 2.

Binding of 125I-EGF to cells expressing increasing levels of V583D-EGF receptors. Cells were treated with increasing doses of doxycycline to induce EGF receptor expression. 125I-EGF binding isotherms were generated in cells expressing increasing levels of either receptor. All the binding isotherms from each cell line were globally fit to Equation 1 with only R0 varying among curves. Data points represent the mean ± S.D. of triplicate determinations. The solid lines represent the fitted curves through the data points of the same color. The r2 value of the global fit was 0.98. The numbers in the upper left indicate the number of receptors per cell in thousands.
Like the wild type receptor, the binding isotherms for the V583D-EGF receptor shifted from left to right with increasing concentrations of receptors. As can be seen from the inset, the parameters obtained from the global fit of the data for the V583D-EGF receptor are very similar to those obtained for the wild type receptor. Thus, this mutation does not appear to significantly alter the ligand binding properties of the EGF receptor.
Effect of Receptor Phosphorylation on Ligand Binding—125I-EGF binding was assessed in CHO cells stably expressing the kinase-dead K721A-EGF receptor (Fig. 3A). In contrast to the situation observed in cells expressing wild type EGF receptor, the binding isotherms for the K721A-EGF receptor shifted from right to left as the receptor number increased. Global analysis of the data yielded fitted parameters (inset) that were significantly different from those for wild type EGF receptors.
FIGURE 3.

Binding of 125I-EGF to cells expressing increasing levels of A) K721A-EGF receptors or B) c′973-EGF receptors. Cells were treated with increasing doses of doxycycline to induce EGF receptor (EGFR) expression. 125I-EGF binding isotherms were generated in cells expressing increasing levels of either receptor. All the binding isotherms from each cell type were globally fit to Equation 1 with only R0 varying among curves. Data points represent the mean ± S.D. of triplicate determinations. The solid lines represent the fitted curves through the data points of the same color. The r2 value of the global fit was 0.98 for both the K721A-EGF receptor and the c′973-EGF receptor. The numbers in the upper left indicate the number of receptors per cell in thousands.
In the wild type EGF receptor, the affinity of EGF for binding to the monomer was approximately the same as its affinity for binding to the first site on the dimer. By contrast, in the K721A-EGF receptor, EGF bound to the monomer with an affinity of ∼6 nm but bound to the first site on the receptor dimer with an affinity of ∼20 pm. Thus, in the kinase-dead EGF receptor mutant, EGF binds with 300-fold higher affinity to the first site on the dimer as compared with the monomer. Thus, there is positive linkage between ligand binding and receptor dimerization. This is why the curves shift from right to left with increasing receptor number. At higher concentrations of receptor, more dimers are present, and hence there are more of the high affinity sites (the unoccupied dimer) to which EGF can bind. Negative cooperativity is still present in the K721A-EGF receptor as the affinity of EGF for binding to the second site on the dimer was ∼500 pm, about 25-fold lower than that for binding to the first site on the dimer.
Positive linkage between EGF binding and receptor dimerization was also observed in an EGF receptor truncated at residue 973, just beyond the kinase domain. This receptor is catalytically active, but because it lacks the C-terminal tail, cannot undergo autophosphorylation in this region (22, 23). As shown in Fig. 3B, the 125I-EGF binding isotherms of the c′973-EGF receptor mutant were very similar to those obtained for the K721A-EGF receptor; they shifted from right to left with increasing numbers of receptors/cell. The fitted parameters were also very similar to those observed for the kinase-dead K721A-EGF receptor and indicate the presence of strong positive linkage along with negative cooperativity in the C-terminally truncated EGF receptor.
Role of the Intracellular Juxtamembrane Domain in Linkage and Cooperativity—The observation that both positive linkage and negative cooperativity were present in the kinase-dead K721A-EGF receptor indicates that kinase activity is not required for these allosteric effects on EGF binding. To determine whether the cytoplasmic domain was required for this regulation, an EGF receptor mutant was generated in which the entire intracellular domain of the receptor had been deleted (ΔC-EGF receptor). The 125I-EGF saturation binding isotherms for this mutant are shown in Fig. 4. Although there was a small shift from right to left in the position of the binding curves with increasing levels of ΔC-EGF receptor expression, it was markedly reduced as compared with what was seen in the kinase-dead or c′973-EGF receptor mutants. Global analysis of the binding data yielded parameters that indicate a significant loss in both cooperativity and linkage in this mutant. EGF bound with only a 4-fold higher affinity to the first site on the dimer as compared with the monomer. This difference is statistically significant but substantially less than the ∼300-fold difference seen in the K721A- and c′973-EGF receptors that contained the cytoplasmic domain. In addition, the affinity of EGF for binding to the second site on the dimer is only ∼2-fold lower than its affinity for binding to the first site on the dimer, a difference that is not statistically significant. These data indicate that both linkage and cooperativity are largely eliminated by removal of the intracellular domain.
FIGURE 4.

Binding of 125I-EGF to cells expressing increasing levels of the ΔC-EGF receptor. Cells were treated with increasing doses of doxycycline to induce EGF receptor (EGFR) expression. 125I-EGF binding isotherms were generated in cells expressing increasing levels of either receptor. All the binding isotherms from each cell line were globally fit to Equation 1 with only R0 varying among curves. Data points represent the mean ± S.D. of triplicate determinations. The solid lines represent the fitted curves through the data points of the same color. The r2 value of the global fit was 0.99 and was significantly better (p < 0.05) than the fit to a single KA model. The numbers in the upper left indicate the number of receptors per cell in thousands.
Recent studies have implicated the intracellular juxtamembrane domain of the EGF receptor in the allosteric activation of the kinase (5, 6). To determine the role of this domain in the ligand binding properties of the receptor, a mutant was generated in which the EGF receptor was truncated at residue 698. This produces a receptor that contains the extracellular and transmembrane domains plus the intracellular juxtamembrane domain but lacks the catalytic core of the kinase domain (24) and the entire C-terminal tail.
As shown in Fig. 5A, the binding of 125I-EGF to the c′698-EGF receptor generated a family of saturation binding isotherms that shifted from right to left as receptor levels increased. Consistent with this pattern, the fitted parameters indicate that this mutant exhibits significant positive linkage; the affinity of EGF for binding to the first site on the receptor dimer is ∼6000-fold higher than the affinity of EGF for binding to the receptor monomer. In addition, the mutant shows significant negative cooperativity with EGF exhibiting a 30-fold lower affinity for binding to the second site on the receptor dimer (1.5 nm) as compared with the first site on the dimer (50 pm). These data suggest that the juxtamembrane domain is necessary and sufficient to confer allosteric regulation of ligand binding on the EGF receptor.
FIGURE 5.

Binding of 125I-EGF to cells expressing increasing levels of c′698-EGF receptors (A) or CC-EGF receptors (B). Cells were treated with increasing doses of doxycycline to induce EGF receptor (EGFR) expression. 125I-EGF binding isotherms were generated in cells expressing increasing levels of either receptor. All the binding isotherms from each cell line were globally fit to Equation 1 with only R0 varying among curves. Data points represent the mean ± S.D. of triplicate determinations. The solid lines represent the fitted curves through the data points of the same color. The r2 value of the global fit for the c′698-EGF receptor was 0.98. The data for the CC-EGF receptor were significantly better fit (p < 0.05) by a single KA model with an r2 value of the global fit of 0.98. The numbers in the upper left indicate the number of receptors per cell in thousands.
The importance of the juxtamembrane domain in cooperativity and linkage was confirmed by results obtained with an additional EGF receptor mutated in the juxtamembrane domain. We have recently reported the signaling properties of an EGF receptor (the CC-EGF receptor) in which residues 647 and 650 at the extreme N terminus of the intracellular juxtamembrane domain were converted to cysteines, allowing palmitoylation of the receptor at these sites (17). This mutant was significantly impaired in terms of signaling, exhibiting only ∼20% as much receptor autophosphorylation as wild type EGF receptor. To further analyze this mutant, we generated 125I-EGF binding isotherms in cells expressing levels of CC-EGF receptors ranging from 12,000 to 950,000 receptors/cell. As can be seen in Fig. 5B, all binding isotherms were well fit by a single curve corresponding to the binding of ligand to a single class of sites with an affinity of ∼600 pm. Thus, palmitoylation within the juxtamembrane domain completely abrogated both cooperativity and linkage in EGF binding.
DISCUSSION
It has been recognized for many years that Scatchard analyses of the binding of 125I-EGF to its receptor yield concave up plots (21, 25–29). Such plots were interpreted as indicating the existence of two classes of EGF binding sites. However, using global analysis of 125I-EGF binding data from cells expressing varying levels of EGF receptors, we have recently demonstrated that the heterogeneity in EGF binding affinities is the result of negative cooperativity in an aggregating system (14). The experiments reported here apply this method to provide further information on the allosteric regulation of EGF binding and on the contribution of the intracellular portion of the EGF receptor to the ligand binding properties of the extracellular domain.
Our findings distinguish between the related phenomena of linkage and cooperativity. Linkage occurs in an oligomerizing system, such as the EGF receptor, in which the binding of the ligand can affect the position of a pre-existing monomer-dimer equilibrium (Fig. 6). If the affinity of a ligand for the dimeric form of the receptor (K21) is higher than for the monomeric form of the receptor (K11), ligand will preferentially bind to the former species and promote dimerization of the receptor. In this situation, the system is said to exhibit positive linkage between binding and dimerization. Cooperativity refers to a system in which the subunits are already dimerized. A system is negatively cooperative if binding of ligand to the first subunit of the dimer (K21) decreases the affinity with which ligand binds to the second subunit in the dimer (K22).
Both our previous (14) and our current analyses of the wild type EGF receptor indicate the presence of negative cooperativity in EGF binding but show no linkage between ligand binding and receptor dimerization. This is paradoxical as it has been well documented that binding of EGF leads to dimerization of its receptor (7–10). The present findings provide an explanation for the apparent lack of linkage between EGF binding and receptor dimer formation.
Both the kinase-dead K721A-EGF receptor and the c′973 EGF receptor exhibited positive linkage, i.e. the binding of EGF to the unoccupied receptor dimer was of higher affinity than the binding of EGF to the receptor monomer. We have previously reported a similar phenotype for the L680N-EGF receptor (14) that contains a mutation in the asymmetric dimer interface that prevents activation of the EGF receptor kinase (6). The K721A-EGF receptor is kinase-dead. The L680N-EGF receptor is catalytically competent, but the kinase domain cannot be activated by ligand. The c′973-EGF receptor is kinase-active but lacks the C-terminal tail that contains the bulk of the sites for EGF receptor autophosphorylation. The common feature of all these receptors is that they are not autophosphorylated in response to ligand binding. This suggests that in the unphosphorylated receptor, the binding of EGF is positively linked to receptor dimerization.
In the wild type receptor, ligand-induced dimerization quickly leads to receptor activation and autophosphorylation. It is this state that is captured in the binding assays as they are long term assays designed to report on equilibrium binding constants. Phosphorylation of the receptor dimer apparently leads to a decrease in its affinity for EGF. By contrast, the phosphorylation reaction appears to generate a forward “pull” on the binding of EGF to the monomer as the affinity of EGF for wild type receptor monomers is higher than that for monomers that cannot be phosphorylated. Together, these effects serve to equalize the apparent affinity of EGF for unoccupied receptor monomers and dimers in the wild type receptor. This masks the positive linkage that is present in the kinetically early phases of ligand binding. Because the three kinase-compromised mutants (K721A-, L680N-, and c′973-EGF receptors) cannot undergo autophosphorylation, they continue to exhibit positive linkage in the long term equilibrium binding assays. Thus, the answer to the conundrum regarding the apparent lack of positive linkage between ligand binding and EGF receptor dimerization is phosphorylation. Prior to phosphorylation, the EGF receptor shows positive linkage. However, once the receptor undergoes autophosphorylation, the positive linkage is lost (Fig. 6).
How autophosphorylation of the receptor leads to a change in receptor affinity is not known. Studies with the purified cytoplasmic domain of the EGF receptor have indicated that the C-terminal tail adopts a more extended conformation following phosphorylation (30–32). In addition, we have recently demonstrated a phosphorylation-dependent conformational change in the full-length EGF receptor in live cells using luciferase fragment complementation imaging (33). Thus, the change in ligand binding affinity may occur in response to an alteration in the conformation of the cytoplasmic domain of the receptor. This could be due to the phosphorylation event itself, the binding of SH2- and PTB domain-containing proteins to the receptor, or the binding of other proteins that become activated as a result of receptor stimulation. Regardless of the molecular trigger, it appears that information regarding alterations within the intracellular domain is transmitted to the extracellular domain to effect a change in ligand binding affinity. This implies that there is inside-out signaling in the EGF receptor system.
Unlike linkage, the negative cooperativity observed within the EGF receptor dimer is not affected by phosphorylation of the receptor. Phosphorylation reduces the affinity of EGF for binding to both the first site and the second site on the dimer by about an order of magnitude. As a result, the wild type and kinase-compromised receptors exhibit a similar degree of negative cooperativity. As cooperativity appears to be independent of phosphorylation status, it is likely that it occurs via a mechanism that is distinct from that involved in generating positive linkage.
Structurally, both negative cooperativity and positive linkage appear to be dependent on the presence of the juxtamembrane domain. In the absence of the entire cytoplasmic domain of the receptor (the ΔC-EGF receptor), both positive linkage and negative cooperativity were essentially eliminated. Simply adding back the intracellular juxtamembrane domain (the c′698-EGF receptor) restored both positive linkage and negative cooperativity to near normal levels. Because the presence of the juxtamembrane domain of the receptor modulates the affinity of the receptor for ligand, it is clear that information relating to the presence and/or position of this intracellular domain is transmitted to the extracellular domain. These data provide further evidence of inside-out signaling in the EGF receptor.
It is possible that the position of the juxtamembrane domain controls the rotation or tilt of the transmembrane helix, that in turn, changes the conformations accessible to the extracellular domain. The observation that tying the juxtamembrane domain to the membrane via palmitoylation (the CC-EGF receptor) ablates both cooperativity and linkage is consistent with the hypothesis that the relative orientation of the transmembrane and intracellular juxtamembrane domains is important for the allosteric regulation of ligand binding to the EGF receptor.
Eight of the first 13 residues (645–657) of the intracellular juxtamembrane domain are either arginines or lysines. Hence, this segment is highly positively charged. It has been hypothesized that this sequence may interact with the negatively charged head groups of the lipids in the membrane bilayer, leading to the association of this sequence with the membrane (1, 34). By contrast, Aifa et al. (35, 36) have proposed that a negatively charged stretch of amino acids from residues 979–991 in the C-terminal tail interacts with the basic residues in the juxtamembrane domain of the EGF receptor. As residues 979–991 are absent in the c′698- and c′973-EGF receptors, both of which exhibit negative cooperativity and positive linkage, these negatively charged residues cannot be involved in the allosteric regulation of EGF binding affinity. We therefore favor the interpretation that the juxtamembrane domain supports negative cooperativity and positive linkage by interacting directly with the membrane or possibly by interacting in trans with the juxtamembrane domain of another EGF receptor. Additional experimental work will be necessary to test this hypothesis and determine how such interactions could modulate the conformation of the extracellular domain.
In summary, our data demonstrate the existence of positive linkage between ligand binding and receptor dimerization only in the unphosphorylated EGF receptor. This finding provides an explanation for the failure of equilibrium binding experiments on wild type receptors to uncover evidence for EGF-induced receptor dimer formation. Negative cooperativity is retained in the receptor regardless of its phosphorylation status, demonstrating that it is mechanistically distinct from the phenomenon of linkage. Nonetheless, both linkage and cooperativity require the presence of the intracellular juxtamembrane domain. The fact that amino acid sequences and conformational changes within the intracellular domain modulate receptor binding affinity indicates that there is inside-out signaling in the EGF receptor system. The intracellular juxtamembrane domain has previously been shown to be required for the activation of the EGF receptor tyrosine kinase (5, 6). Our experiments expand the role of this domain to include the allosteric control of ligand binding, further underscoring the importance of the juxtamembrane domain in the regulation of EGF receptor function.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM064491 and R01GM082824 (to L. J. P.).
The on-line version of this article (available at http://www.jbc.org) contains one supplemental figure.
This article was selected as a Paper of the Week.
Footnotes
The abbreviations used are: EGF, epidermal growth factor; CHO, Chinese hamster ovary.
References
- 1.Ullrich, A., Coussens, L., Hayflick, J. S., Dull, T. J., Gray, A., Tam, A. W., Lee, J., Yarden, Y., Libermann, T. A., Schlessinger, J., Downward, J., Mayes, E. L. V., Whittle, N., Waterfield, M. D., and Seeburg, P. H. (1984) Nature 309 418-425 [DOI] [PubMed] [Google Scholar]
- 2.Downward, J., Parker, P., and Waterfield, M. D. (1984) Nature 311 483-485 [DOI] [PubMed] [Google Scholar]
- 3.Hsuan, J. J., Totty, N., and Waterfield, M. D. (1989) Biochem. J. 262 659-663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Margolis, G., Li, N., Koch, A., Mohammadi, M., Hurwitz, D., Zilberstein, A., Ullrich, A., Pawson, T., and Schlessinger, J. (1990) EMBO J. 9 4375-4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thiel, K. W., and Carpenter, G. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 19238-19243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang, X., Gureasko, J., Shen, K., Cole, P. A., and Kuriyan, J. (2006) Cell 125 1137-1149 [DOI] [PubMed] [Google Scholar]
- 7.Clayton, A. H. A., Walker, F., Orchard, S. G., Henderson, C., Ruchs, D., Rothacker, J., Nice, E. C., and Burgess, A. W. (2005) J. Biol. Chem. 280 30392-30399 [DOI] [PubMed] [Google Scholar]
- 8.Cochet, C., Kashles, O., Chambaz, E. M., Borrello, I., King, C. R., and Schlessinger, J. (1988) J. Biol. Chem. 263 3290-3295 [PubMed] [Google Scholar]
- 9.Liu, P., Sudhaharan, T., Koh, R. M. L., Hwang, L. C., Ahmed, S., Maruyama, I. N., and Wohland, T. (2007) Biophys. J. 93 684-698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yarden, Y., and Schlessinger, J. (1987) Biochemistry 26 1443-1451 [DOI] [PubMed] [Google Scholar]
- 11.Ferguson, K. M., Berger, M. B., Mendrola, J. M., Cho, H.-S., Leahy, D. J., and Lemmon, M. A. (2003) Mol. Cell 11 507-517 [DOI] [PubMed] [Google Scholar]
- 12.Garrett, T. P. J., McKern, N. M., Lou, M., Elleman, T. C., Adams, T. E., Lovrecz, G. O., Zhu, H.-J., Walker, F., Frenkel, M. J., Hoyne, P. A., Jorissen, R. N., Nice, E. C., Burgess, A. W., and Ward, C. W. (2002) Cell 110 763-773 [DOI] [PubMed] [Google Scholar]
- 13.Ogiso, H., Ishitani, R., Nureki, O., Fukai, S., Yamanaka, M., Kim, J.-H., Saito, K., Sakamoto, A., Inoue, M., Shirouzu, M., and Yokoyama, S. (2002) Cell 110 775-787 [DOI] [PubMed] [Google Scholar]
- 14.Macdonald, J. L., and Pike, L. J. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 112-117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong, I., and Lohman, T. M. (1995) Methods Enzymol. 259 95-127 [DOI] [PubMed] [Google Scholar]
- 16.Wyman, J., and Gill, S. J. (1990) Binding and Linkage: Functional Chemistry of Biological Macromolecules, pp. 203-236, University Science Books, Mill Valley, CA
- 17.Macdonald-Obermann, J. L., and Pike, L. J. (2009) Biochemistry 48 2505-2513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macdonald, J. L., Li, Z., Wu, W., and Pike, L. J. (2006) Biochim. Biophys. Acta 1763 870-878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doran, D. M., and Spar, I. L. (1980) J. Immunol. Methods 39 155-163 [DOI] [PubMed] [Google Scholar]
- 20.Saffarian, S., Li, Y., Elson, E. L., and Pike, L. J. (2007) Biophys. J. 93 1021-1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walker, F., Orchard, S. G., Jorissen, R. N., Hall, N. E., Zhang, H.-H., Hoyne, P. A., Adams, T. E., Johns, T. G., Ward, C. W., Nice, E. C., and Burgess, A. W. (2004) J. Biol. Chem. 279 22387-22398 [DOI] [PubMed] [Google Scholar]
- 22.Chen, W. S., Lazar, C. S., Lund, K. A., Welsh, J. B., Chang, C.-P., Walton, G. M., Der, C. J., Wiley, H. S., Gill, G. N., and Rosenfeld, M. G. (1989) Cell 59 33-43 [DOI] [PubMed] [Google Scholar]
- 23.Wells, A., Welsh, J. B., Lazar, C. S., Wiley, H. S., Gill, G. N., and Rosenfeld, M. G. (1990) Science 247 962-964 [DOI] [PubMed] [Google Scholar]
- 24.Hanks, S. K., and Quinn, A. M. (1991) Methods Enzymol. 200 38-62 [DOI] [PubMed] [Google Scholar]
- 25.King, A. C., and Cuatrecasas, P. (1982) J. Biol. Chem. 257 3053-3060 [PubMed] [Google Scholar]
- 26.Magun, B. E., Matrisian, L. M., and Bowden, G. T. (1980) J. Biol. Chem. 255 6373-6381 [PubMed] [Google Scholar]
- 27.Mattoon, D., Klein, P., Lemmon, M. A., Lax, I., and Schlessinger, J. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 923-928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rees, A. R., Gregoriou, M., Johnson, P., and Garland, P. B. (1984) EMBO J. 3 1843-1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shoyab, M., DeLarco, J. E., and Todaro, G. J. (1979) Nature 279 387-391 [DOI] [PubMed] [Google Scholar]
- 30.Cadena, D. L., Chan, C.-L., and Gill, G. N. (1994) J. Biol. Chem. 269 260-265 [PubMed] [Google Scholar]
- 31.Lee, N. Y., Hazlett, T. L., and Koland, J. H. (2006) Protein Sci. 15 1142-1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee, N. Y., and Koland, J. H. (2005) Protein Sci. 14 2793-2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang, K. S., Ilagan, M. X. G., Piwnica-Worms, D., and Pike, L. J. (2008) J. Biol. Chem. 284 7474-7482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McLaughlin, S., Smith, S. O., Hayman, M. J., and Murray, D. (2005) J. Gen. Physiol. 126 41-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aifa, S., Aydin, J., Nordvall, G., Lundstrom, I., Svensson, S. P. S., and Hermanson, O. (2005) Exp. Cell Res. 302 108-114 [DOI] [PubMed] [Google Scholar]
- 36.Aifa, S., Miled, N., Frikha, F., Aniba, M. R., Svensson, S. P. S., and Rebai, A. (2006) Proteins 62 1036-1043 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.