

Abstract
We report here a biochemical and structural characterization of domain 2 of the nonstructural 5A protein (NS5A) from the JFH1 Hepatitis C virus strain and its interactions with cyclophilins A and B (CypA and CypB). Gel filtration chromatography, circular dichroism spectroscopy, and finally NMR spectroscopy all indicate the natively unfolded nature of this NS5A-D2 domain. Because mutations in this domain have been linked to cyclosporin A resistance, we used NMR spectroscopy to investigate potential interactions between NS5A-D2 and cellular CypA and CypB. We observed a direct molecular interaction between NS5A-D2 and both cyclophilins. The interaction surface on the cyclophilins corresponds to their active site, whereas on NS5A-D2, it proved to be distributed over the many proline residues of the domain. NMR heteronuclear exchange spectroscopy yielded direct evidence that many proline residues in NS5A-D2 form a valid substrate for the enzymatic peptidyl-prolyl cis/trans isomerase (PPIase) activity of CypA and CypB.
Hepatitis C virus (HCV)4 is a small, positive strand, RNA-enveloped virus belonging to the Flaviviridae family and the genus Hepacivirus. With 120–180 million chronically infected individuals worldwide, hepatitis C virus infection represents a major cause of chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma (1). The HCV viral genome (∼9.6 kb) codes for a unique polyprotein of ∼3000 amino acids (recently reviewed in Refs. 2–4). Following processing via viral and cellular proteases, this polyprotein gives rise to at least 10 viral proteins, divided into structural (core, E1, and E2 envelope glycoproteins) and nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, NS5B). Nonstructural proteins are involved in polyprotein processing and viral replication. The set composed of NS3, NS4A, NS4B, NS5A, and NS5B constitutes the minimal protein component required for viral replication (5).
Cyclophilins are cellular proteins that have been identified first as CsA-binding proteins (6). As FK506-binding proteins (FKBP) and parvulins, cyclophilins are peptidyl-prolyl cis/trans isomerases (PPIase) that catalyze the cis/trans isomerization of the peptide linkage preceding a proline (6, 7). Several subtypes of cyclophilins are present in mammalian cells (8). They share a high sequence homology and a well conserved three-dimensional structure but display significant differences in their primary cellular localization and in abundance (9). CypA, the most abundant of the cyclophilins, is primarily cytoplasmic, whereas CypB is directed to the endoplasmic reticulum lumen or the secretory pathway. CypD, on the other hand, is the mitochondrial cyclophilin. Cyclophilins are involved in numerous physiological processes such as protein folding, immune response, and apoptosis and also in the replication cycle of viruses including vaccinia virus, vesicular stomatitis virus, severe acute respiratory syndrome (SARS)-coronavirus, and human immunodeficiency virus (HIV) (for review see Ref. 10). For HIV, CypA has been shown to interact with the capsid domain of the HIV Gag precursor polyprotein (11). CypA thereby competes with capsid domain/TRIM5 interaction, resulting in a loss of the antiviral protective effect of the cellular restriction factor TRIM5α (12, 13). Moreover, it has been shown that CypA catalyzes the cis/trans isomerization of Gly221-Pro222 in the capsid domain and that it has functional consequences for HIV replication efficiency (14–16). For HCV, Watashi et al. (17) have described a molecular and functional interaction between NS5B, the viral RNA-dependent RNA polymerase (RdRp), and cyclophilin B (CypB). CypB may be a key regulator in HCV replication by modulating the affinity of NS5B for RNA. This regulation is abolished in the presence of cyclosporin A (CsA), an inhibitor of cyclophilins (6). These results provided for the first time a molecular mechanism for the early-on observed anti-HCV activity of CsA (18–20). Although this initial report suggests that only CypB would be involved in the HCV replication process (17), a growing number of studies have recently pointed out a role for other cyclophilins (21–25).
In vitro selection of CsA-resistant HCV mutants indicated the importance of two HCV nonstructural proteins, NS5B and NS5A (26), with a preponderant effect for mutations in the C-terminal half of NS5A. NS5A is a large phosphoprotein (49 kDa), indispensable for HCV replication and particle assembly (27–29), but for which the exact function(s) in the HCV replication cycle remain to be elucidated. This nonstructural protein is anchored to the cytoplasmic leaflet of the endoplasmic reticulum membrane via an N-terminal amphipathic α-helix (residues 1–27) (30, 31). Its cytoplasmic sequence can be divided into three domains: D1 (residues 27–213), D2 (residues 250–342), and D3 (residues 356–447), all connected by low complexity sequences (32). D1, a zinc-binding domain, adopts a dimeric claw-shaped structure, which is proposed to interact with RNA (33, 34). NS5A-D2 is essential for HCV replication, whereas NS5A-D3 is a key determinant for virus infectious particle assembly (27, 35). NS5A-D2 and -D3, for which sequence conservation among HCV genotypes is significantly lower than for D1, have been proposed to be natively unfolded domains (28, 32). Molecular and structural characterization of NS5A-D2 from HCV genotype 1a has confirmed the disordered nature of this domain (36, 37).
As it is still not clear which cyclophilins are cofactors for HCV replication, and as mutations in HCV NS5A protein have been associated with CsA resistance, we decided to examine the interaction between both CypA and CypB and domain 2 of the HCV NS5A protein. We first characterized, at the molecular level, NS5A-D2 from the HCV JFH1 infectious strain (genotype 2a) and showed by NMR spectroscopy that this natively unfolded domain indeed interacts with both cyclophilin A and cyclophilin B. Our NMR chemical shift mapping experiments indicated that the interaction occurs at the level of the cyclophilin active site, whereas it lacks a precise localization on NS5A-D2. A peptide derived from the only well conserved amino acid motif in NS5A-D2 did interact with cyclophilin A but only with a 10-fold lower affinity than the full domain. We concluded from this that the many proline residues form multiple anchoring points, especially when they adopt the cis conformation. NMR exchange spectroscopy further demonstrated that NS5A-D2 is a substrate for the PPIase activities of both CypA and CypB. Both the NS5A/cyclophilin interaction and the PPIase activity of the cyclophilins on NS5A-D2 were abolished by CsA, underscoring the specificity of the interaction.
EXPERIMENTAL PROCEDURES
Sequence Analysis—Sequence analyses were performed using tools available at the Institut de Biologie et Chimie des Protéines (IBCP) network protein sequence analysis (NPSA) website (38). HCV NS5A sequences were retrieved from the European HCV Database (39). Multiple sequence alignments were performed with ClustalW (40) using default parameters. The repertoire of residues at each amino acid position and their frequencies observed in natural sequence variants were computed by the use of a program developed at the IBCP.5
Expression and Purification of Nonlabeled and 15N- and 15N,13C-Labeled NS5A-D2 (JFH1)—The synthetic sequence coding for domain 2 of the HCV NS5A protein from JFH1 strain (euHCVdb (39); GenBank™ accession number AB047639, genotype 2a) was introduced in the bacterial expression vector pT7.7 with a His6 tag (41). The resulting recombinant domain 2 of HCV NS5A (NS5A-D2; residues 248–341) has extra M- and -LQHHHHHH extensions at the N and C termini, respectively. The pT7–7-NS5A-D2 plasmid was introduced in Escherichia coli BL21(DE3) (Merck-Novagen, Darmstadt, Germany). Cells were grown at 37 °C in Luria-Bertani (LB) medium for nonlabeled protein or in a M9-based semi-rich medium (M9 medium supplemented with [15N]NH4Cl (1 g/liter), d-[13C6]glucose (2 g/liter) (for 13C labeling only), Isogro 13C,15N powder growth medium (1 g/liter, 10%; Sigma-Aldrich). At an A600 of ∼0.7, the protein production was induced with 0.4 mm isopropyl 1-thio-β-d-galactopyranoside (IPTG), and cells were harvested by centrifugation at 3.5 h post-induction. NS5A-D2 was first purified by Ni2+-affinity chromatography (HisTrap column, 1 ml, GE Healthcare Europe). Selected fractions were pooled, dialyzed against 20 mm Tris-Cl, pH 7.4, 2 mm EDTA, and then submitted to a second purification step by ion exchange chromatography (ResourceQ, 1 ml column, GE Healthcare Europe). Following SDS-PAGE analysis, NS5A-D2-containing fractions were selected and pooled. The protein was concentrated up to 340 μm with a Vivaspin 15 concentrator (cutoff, 5 kDa) (Satorius Stedim Biotech, Aubagne, France) while simultaneously exchanging the buffer against 20 mm NaH2PO4/Na2HPO4 pH 6.4, 30 mm NaCl, 1 mm DTT (or 1 mm Tris(hydroxypropyl)phosphine), 0.02% NaN3. After filtration (0.2 μm), NS5A-D2 aliquots were stored at -80 °C with a few Chelex 100 beads (Sigma-Aldrich).
Circular Dichroism (CD)—CD spectra were recorded on a Chirascan dichrographe (Applied Photophysics, Surrey, UK) calibrated with (1S)-(+)-10-camphorsulfonic acid. Measurements were carried out at room temperature in a 0.1-cm path length quartz cuvette with protein concentrations ranging from 5 to 15 μm. Spectra were recorded in the 185–260 nm wavelength range at 0.5-nm increments and a 2-s integration time. Spectra were processed, base-line-corrected, and smoothed using Chirascan software. Spectral units were expressed as the molar ellipticity per residue using protein concentrations determined by measuring the UV light absorbance of tyrosine and tryptophan at 280 nm. The α-helix content was estimated using the method of Chen et al. (42).
Peptide Synthesis—A synthetic peptide (named PepD2) corresponding to residues 304–323 of NS5A (304GFPRALPAWARPDYNPPLVE323) was obtained from Neosystems (Strasbourg, France). The purity of the peptide was verified by high pressure liquid chromatography and mass spectrometry as greater than 95%.
Expression and Purification of Nonlabeled and 15N,13C-Labeled Cyclophilin B—Production and purification of recombinant human cyclophilin B in E. coli were done as described previously (43). Briefly, the pET15b-CypB plasmid was introduced into E. coli BL21(DE3) strain, recombinant bacteria were grown in LB medium (or in M9 medium supplemented with [15N]NH4Cl and [13C]glucose for labeled samples), and production was induced with 0.4 mm IPTG. Cyclophilin B was purified by ion exchange (SP Sepharose Fast Flow) and then by gel filtration (Superose 12 Prep Grade) chromatography. The purified and concentrated Cyclophilin B was stored at -80 °C.
Expression and Purification of Nonlabeled and 15N,13C-Labeled Cyclophilin A—Sequence coding for human CypA was amplified from the plasmid pKK233–2-CypA, kindly provided by Prof. Allain (UMR8576, CNRS-University of Sciences and Technologies of Lille, France), using the forward primer 3′-cttcatatggtcaaccccaccgtg-5′ and the reverse primer 5′-caaggatccttattcgagttgtcc-3′ and was then inserted in the pET15b plasmid (Merck-Novagen) between the NdeI and BamHI restriction sites. The pET15b-CypA plasmid, coding for a recombinant CypA with an N-terminal His tag, was introduced in E. coli BL21 (DE3). Cells were grown in M9 medium supplemented with [15N]NH4Cl or [15N]NH4Cl and [13C]glucose. When the culture reach an A600 =∼0.8, protein production was induced with 0.4 mm IPTG; cells were harvested 3 h after induction at 37 °C. Recombinant CypA was purified by Ni2+-affinity chromatography (HiTrap Chelating HP, GE Healthcare Europe). Finally, the protein was dialyzed against 50 mm NaH2PO4/Na2HPO4, pH 6.3, 20 mm NaCl, 2 mm EDTA, 1 mm DTT, concentrated, filtered (0.2 μm), and then stored at 4 °C.
NMR Data Collection and Assignments—Spectra were acquired on either a Bruker Avance 600 MHz equipped with a cryogenic triple resonance probe head or a Bruker Avance 800 MHz with a standard triple resonance probe (Bruker, Karlsruhe, Germany). The proton chemical shifts were referenced using the methyl signal of TMSP (sodium 3-trimethylsill-[2,2′,3,3′-d4]propionate) at 0 ppm. Spectra were processed with the Bruker TopSpin software package 1.3 and analyzed using the product plane approach developed in our laboratory (44).
Assignments of NS5A-D2 backbone resonances were achieved using two-dimensional 1H,15N HSQC and three-dimensional 1H,15N,13C HNCO, HNCACO, HNCACB, HNCOCACB, and HNCANNH spectra (45) acquired at 600 MHz on a 340 μm 15N, 13C-labeled NS5A-D2 sample at 298 K (Biological Magnetic Resonance Data Bank (BMRB) accession number 16165).
Assignments of the CypB spectrum were taken from our previous study (43). Assignments of CypA resonances were taken from the literature (46) and confirmed with a HNCACB spectrum acquired at 600 MHz on a 340 μm [15N,13C]CypA sample in 50 mm NaH2PO4/Na2PO4 pH 6.3, 40 mm NaCl, 2 mm EDTA, 1 mm DTT at 25 °C.
Interaction between NS5A-D2 and Cyclophilins—To study the interaction between NS5A-D2 and CypA or CypB, differentially labeled proteins (15N for NS5A-D2 and 15N,13C for CypA or CypB) were mixed at different molar ratios. The (1H,15N) plane of the HNCO spectrum thereby selects only for the 15N,13C-labeled protein component, whereas the HNCO spectrum with modified phases to select for the non-13C-labeled 15N nuclei (which we will further call the HN(noCO) spectrum (47)) was used for selection of the only-15N-labeled protein. The combined chemical shift perturbations following NS5A-D2 addition were calculated as shown in Equation 1, whereby δΔ(1HN) and δΔ(15N) are the chemical shift perturbations in the 1H and 15N dimensions, respectively.
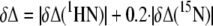 |
(Eq. 1) |
Cyclophilin PPIase Activity toward NS5A-D2—PPIase activity of CypA and CypB on NS5A-D2 were assessed using EXSY spectra, whereby the exchange was monitored on the proton resonance (in homonuclear 1H,1H spectra (48)) or on the 15N nucleus (in heteronuclear 1H,15N z-exchange spectra (49)). The ratio between the cis and trans populations for a given residue (pc and pt, respectively) was measured on the basis of a 1H,15N HSQC spectrum in the absence of any cyclophilin assuming that an exchange peak for this residue was observed.
1H, 1H EXSY spectra were acquired as 1H,1H planes from a three-dimensional 15N-edited NOESY-HSQC with different mixing times (50, 100, 200, and 400 ms) on a sample of 320 μm [15N,13C]NS5A-D2 and 40 μm [15N]CypB or -CypA in 20 mm NaH2PO4/Na2HPO4, pH 6.4, 30 mm NaCl, 0.02% NaN3, 1 mm DTT.
15N z-exchange spectra were recorded on an 800-MHz spectrometer with 0.88, 25, 50, 100, 200, 300, and 400-ms mixing times. PPIase activities were analyzed on a sample of 220 μm [15N]NS5A-D2 and 23 μm CypB or CypA in 20 mm NaH2PO4/Na2HPO4 pH 6.3, 30 mm NaCl, 0.02% NaN3, 1 mm DTT. Exchange rates were derived from a simplified version of the analytical form given in Ref. 38 by taking into account only the maximal intensity of the trans-cis exchange peak (Itc) and the trans diagonal peak (Itt). This procedure minimized any problems with exchange broadening of the cis diagonal peak due to the interaction with the Cyp and with significant proton overlap hindering the reliable integration of the weak off-diagonal peaks. The exchange rate (kexch), as a function of mixing time (MT), was determined by using a least-squares fitting procedure between the experimental data and the theoretical Equation 2 adapted from Ref. 15.
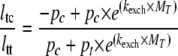 |
(Eq. 2) |
To confirm that the exchange peaks were due to the PPIase activity of cyclophilins, CsA was added into the sample, and a 1H, 15N z-exchange spectra was recorded with a 100-ms mixing time. PyMOL software was used for the molecular graphics (DeLano Scientific).
RESULTS
Sequence Analysis—We performed sequence analysis and structure predictions to assess the degree of conservation of the NS5A-D2 domains across the different strains and to identify potential essential amino acids (aa) and motifs. The aa repertoire deduced from the analysis of 21 HCV isolates of genotype 2a revealed that aa are strictly conserved in 70% of the sequence positions (denoted by asterisks in Fig. 1A). The apparent variability is limited at most positions because the observed residues exhibit similar physicochemical properties, as indicated both by the similarity pattern (Fig. 1A, colons and dots) as well as the hydropathic pattern, where the letters o, i, and n denote hydrophobic, hydrophilic, and neutral residues, respectively (see legend to Fig. 1A for details). The degree of conservation among different genotypes was investigated by ClustalW alignment of 27 reference sequences representative for the major HCV genotypes and subtypes (see legend to Fig. 1A). The aa repertoire derived from this alignment revealed an apparent high level of variability, except for some well conserved positions that are likely essential for the structure and/or function of NS5A-D2. However, despite this apparent variability, conservation of the hydropathic character at most positions indicates that the overall structure of NS5A-D2 is conserved among the different HCV genotypes. There are, however, some short variable stretches of sequences (underlined in Fig. 1A, bottom), which appear to be genotype-specific. Typically, a main sequence difference between genotypes is the four-aa deletion observed in genotype 2, including JFH1 (indicated by hyphens in Fig. 1A).
FIGURE 1.

Sequence analysis and biochemical characterization of NS5A-D2 domain 2 from HCV. A, amino acid repertoires. The NS5A-D2 aa 248–341 sequence from the HCV JFH1 strain of genotype 2a (GenBank™ accession number AB047639), which was used in this study, is indicated. Amino acids are numbered with respect to NS5A and the HCV JFH1 polyprotein (top row). The hyphens indicate the aa deletions compared with the sequence alignment of any genotypes (see below). The aa repertoire deduced from the ClustalW multiple alignments of 21 NS5A sequences of genotype 2a is shown at the top. Amino acids observed at a given position less than twice were not included. At the bottom is the aa repertoire of the 27 representative NS5A sequences from confirmed HCV genotypes and subtypes (listed with accession numbers in Table 1 in Ref. 71; see the European HCV Database for details). The degree of aa and physicochemical conservation at each position can be inferred from the extent of variability (with the observed aa listed in decreasing order of frequency from top to bottom) together with the similarity index according to ClustalW convention (asterisk, invariant; colon, highly similar; dot, similar (40)) and the consensus hydropathic pattern deduced from the consensus aa repertoire: o, hydrophobic position (Phe, Ile, Trp, Tyr, Leu, Val, Met, Pro, Cys); n, neutral position (Gly, Ala, Thr, Ser); i, hydrophilic position (Lys, Gln, Asn, His, Glu, Asp, Arg); v, variable position (i.e. when both hydrophobic and hydrophilic residues are observed at a given position). A position that is underlined in the hydropathic pattern for any reference genotypes (bottom) indicates the change of position status when compared with the hydropathic pattern for genotype 2a (middle). Secondary structure predictions of NS5A-D2 are indicated as helical (h), extended (e), undetermined (coil (c)), or ambiguous (?). Sec. Cons., consensus of protein secondary structures predictions for NS5A-D2 from JFH1 strain deduced from a large set of prediction methods available at the NPSA website, including DSC, HNNC, MLRC, PHD, Predator, SOPM, and SIMPA96 available at the NPSA website (see Ref. 38 and references therein). B, gel filtration analysis of NS5A-D2 was performed on a Superdex S200 column equilibrated in 50 mm sodium phosphate, pH 7.4, 1 mm Tris(2-carboxyethyl) phosphine hydrochloride with a flow rate of 0.5 ml/min. Elution volumes of globular protein standards are indicated by black arrows with the following corresponding molecular masses: 1, thyroglobulin (669,000 Da); 2, ferritin (44,0000 Da); 3, aldolase (158,000 Da); 4, conalbumin (75,000 Da); 5, ovalbumin (43,000 Da); 6, chymotrypsin (25,000 Da); 7, ribonuclease (13,700 Da); 8, vitamin B12 (1,355 Da). C, Far-UV circular dichroism analysis of 8 μm NS5A-D2 in 10 mm sodium phosphate, pH 7.4, 1 mm Tris(2-carboxyethyl) phosphine hydrochloride (solid line) complemented with 50% TFE (dotted line). The difference spectrum (alternating dashed line) was obtained by subtracting the latter spectrum from the former.
Molecular Characterization of HCV NS5A-D2 (JFH1)— NS5A-D2 is efficiently produced in a soluble form when recombinantly overexpressed in E. coli and could be purified to almost homogeneity (see supplemental Fig. 1). Despite an excellent agreement between expected (11,639 Da) and experimental mass as determined by mass spectroscopy, NS5A-D2 has an apparent molecular weight of ∼18 kDa by SDS-PAGE. This discrepancy is probably due to the primary aa sequence of NS5A-D2, which includes many acidic residues and prolines (50, 51). In gel filtration chromatography, the protein elutes at a volume corresponding to a ∼30-kDa globular protein, (Fig. 1B). Such a large apparent molecular weight in a gel filtration assay is commonly associated with natively unfolded proteins devoid of globular domain (52).
The structure of NS5A-D2 was further characterized by CD spectroscopy (Fig. 1C). In aqueous buffer, NS5A-D2 gave a complex spectrum with a large negative band around 198 nm and a shoulder in the 220–240 nm range, indicating a mixture of random coil structure with the presence of some poorly defined structures. To probe the potential conformational preference of NS5A-D2, we used TFE, which is known to stabilize the folding of peptidic sequences, especially those exhibiting an intrinsic propensity to adopt an α-helical structure (53). The addition of 50% TFE induced a limited structuration attributed to some α-helix formation. Indeed, the difference spectrum shown in Fig. 1C is consistent with a small amount of α-helical folding with a maximum at 192 nm and two minima at 208 and 222 nm. Assuming that the residue molar ellipticity at 222 nm is exclusively due to α-helix upon addition of TFE, a maximum of only about 6% α-helix content could be estimated, in agreement with the low level of α-helical structure predicted from aa sequence analysis (Fig. 1A).
The 1H,15N HSQC of NS5A-D2 (Fig. 2A) displays a narrow proton chemical shift range, limited to 1 ppm excluding three outlying peaks (Trp312, Ala313, and Arg326; see below). This low level of dispersion again points to the nonstructured nature of the polypeptide, at least when isolated in solution. Using triple resonance NMR spectroscopy on a doubly labeled NS5A-D2 sample and an in-house developed product plane-based assignment procedure (44), all backbone amide proton resonances could be assigned except for the 15 proline residues. The outlying peaks were assigned to Trp312, Ala313, and Arg326 (Fig. 2). 13CO, 13Cα, and 13Cβ resonances were assigned for 94 residues, and were used to probe the secondary structure content at a per-residue level in NS5A-D2. Carbon chemical shifts when compared with their values for the amino acid in a short unstructured peptide give a good indication of the secondary structure adopted by the amino acid in the full protein (54). Analysis of the chemical shift index (CSI) shows a majority of negative CSI values for 13Cα and 13CO, whereas the 13Cβ CSI values are generally positive (Fig. 2B) (54). Although this hints at an extended structure, the CSI consensus values are zero all along the NS5A-D2 sequence, confirming the absence of stable secondary structure elements even at the local level.
FIGURE 2.

NMR characterization of NS5A-D2 (JFH1). A, assigned 1H,15N HSQC spectrum of domain 2 of NS5A (JFH1). The smaller insert shows the spectrum region corresponding to the two tryptophan side chains. B, CSI analysis of NS5A-D2. NS5A-D2 13Cα, 13Cβ, and 13CO chemical shifts were analyzed using CSI software (54). The consensus CSI is zero along the complete sequence and therefore is not shown.
Next to the assigned peaks, and despite the high level of purity obtained by our two-step purification procedure (supplemental Fig. 1), numerous, less intense peaks could be observed in the 1H,15N HSQC spectrum (Fig. 2A). Corresponding to residues in the vicinity of a proline in the cis conformation, 32 of these minor peaks could be assigned in the same triple resonance spectra used for the initial assignment (minor forms will be named cis forms in the following). Although the high content of proline residues (15 prolines in the 94-aa fragment of NS5A-D2) sometimes led to ambiguity regarding the identity of the cis-Pro at the origin of the chemical shift difference, the presence of several minor peaks corresponding to various residues around a given proline allowed the assignment and quantification of the cis/trans ratio for a major fraction of the prolyl bonds (supplemental Table 1).
Interaction between NS5A-D2 and Human Cyclophilins—As mutations in the C-terminal half of NS5A have been shown to confer CsA resistance for mutant HCV (26), we investigated the direct physical interaction between NS5A-D2 and cyclophilins. Although CypA is the prominent cytosolic isomerase (10, 25), the initial report of cyclophilins being involved in HCV replication suggests CypB as the corresponding partner (17). We therefore tested independently the interaction of NS5A-D2 with CypA and CypB. Finally, because we wanted to obtain, with a single sample, the chemical shift changes on both partners in order to map the mutual interaction surfaces, we mixed 15N-labeled NS5A-D2 and 15N,13C-labeled CypA or CypB and used the planes from the HN(CO) and HN(noCO) experiments to obtain subspectra displaying only the one or the other molecular entity (47).
Comparing the Cyp subspectra in the absence and presence of an equimolar quantity of NS5A-D2, we noted that only a limited number of CypA or CypB resonances was affected (supplemental Fig. 2). Beyond proving the existence of a direct physical interaction between both partners, mapping the chemical shifts on the Cyp primary sequences and then on their respective three-dimensional structures allowed us to define precisely the interaction sites (Fig. 3). For both CypA and CypB, the interaction site is centered on the active site for their isomerase activity, which coincides with the CsA binding surface and even extends somewhat beyond this direct CsA binding surface (Fig. 3). In agreement with this, the interaction was completely abolished in the presence of CsA, as the spectra of Cyp/CsA with or without NS5A-D2 were strictly identical (data not shown). To quantify the interaction strength between both partners, we titrated increasing amounts of unlabeled NS5A-D2 into samples of 15N-labeled CypA or CypB. Chemical shift changes of residues at the periphery of the binding site varied in a monotonous way from their free position toward the ligand saturated value, allowing the determination of KD values of 64 and 90 μm for CypA and CypB, respectively (Fig. 4, A–C). However, residues in the active site of both cyclophilins broadened with increasing NS5A-D2 concentrations, as if multiple interactions were present simultaneously. To confirm this unexpected observation, we repeated the titration experiment with a synthetic peptide (PepD2, residues 304–323 of NS5A) corresponding to the best conserved region of NS5A-D2 that simultaneously contains the motif 310PAWARP315 with the outlying 1H, 15N chemical shift values (see above). With this peptide, the titration behavior, when monitored on exactly the same residues of the active site of CypA, did not show the broadening observed with the full NS5A-D2 domain. On the other hand, saturation was much slower to reach, and we derived a 10-fold weaker binding with a KD value of 830 μm (Fig. 4, D–F). This all suggests that the D2 domain interacts in a distributed manner with the cyclophilin active site.
FIGURE 3.

Cyclophilin binding sites for NS5A-D2. The 1H and 15N combined chemical shift perturbations (δΔ) induced on the CypA (A) or CypB (B) spectra following NS5A-D2 addition in a 1:1 molar ratio were plotted along the cyclophilin primary sequences and on their respective three-dimensional molecular surfaces. Residues with combined chemical shift perturbations 0.02 ≤ δΔ ≤ 0.03 ppm are in yellow; 0.03 ≤ δΔ ≤ 0.05 ppm are in orange; and δΔ> 0.05 ppm are in red. For cyclophilin residues for which the proton amide resonances disappear due to important line broadening in the presence of NS5A-D2, a fixed δΔ value of 0.15 ppm was set. These residues are depicted by an open bar circled in red in the diagrams. The PPIase active site of cyclophilins is indicated by a dotted black arrow.
FIGURE 4.

Titration experiments between cyclophilins and NS5A-D2 or PepD2. Panels A, B, D, and E correspond to the superimposition of the 1H,15N HSQC spectra of CypA (or CypB, in B) acquired in the presence of increasing amounts of unlabeled NS5A-D2 (A, B, and D) or Pep-D2 (E) (PepD2 corresponds to residues 304–323 of NS5A-D2: 304GFPRALPAWARPDYNPPLVE323). Lys82 in CypA is equivalent to Arg90 in CypB and is at the periphery of the NS5A-D2 binding site. C, titration curves corresponding to experiments in A (black triangle) and B (gray diamonds). The 1H,15N combined chemical shift perturbations Δδ (in ppm) (Δδ = (δ(1H)2 + 0.2.δ(15N)2)½) were plotted as a function of the NS5A-D2:cyclophilin molar ratios. The dissociation constants (KD) were obtained by fitting the experimental data with the following equation: KD = [Cypfree]·[NS5A-D2free]/[Cyp:NS5A-D2]. D and E, Phe60 in CypA is directly in the binding site of NS5A-D2 and broadens when titrated with the D2 domain (D) but not with Pep-D2 (E). F, titration curve corresponding to experiments in E (black triangles).
To confirm this by direct observation on the NS5A-D2 spectrum, we compared the HN(noCO) subspectra of 15N-labeled NS5A-D2 alone with that of NS5A-D2 in the previous samples (supplemental Fig. 3). Concentrating first on the most intense peaks, which had all been mapped to their respective residue, next to a proline in the trans conformation, in the NS5A-D2 polypeptide we found a zone of significant spectral changes around the outlying peak of Trp312 (Fig. 5). Upon the addition of the cyclophilins, these peaks did not shift but rather broadened beyond detection. Line broadening occurs when the time scale of the exchange process is on the same order as that set by the frequency difference between the free and bound state. NMR line broadening of neighboring residues Arg302, Ser303, Ala311, Ala313, and Arg314 thereby was significantly more pronounced with CypA than with CypB (supplemental Figs. 3 and 5). Moreover, the amide proton resonances of residues Gly304, Ala308, Leu309 (see supplemental Fig. 3), Asp316, Tyr317, and Asn318 were unaffected in the presence of CypB, whereas they were no more detectable in the presence of CypA or broadened for Tyr317 (Figs. 5 and 6C). Among the numerous proline residues observed in NS5A-D2, only Pro310, Pro315, and Pro319, which are in the direct vicinity of this interaction region, are fully conserved in any genotypes (Fig. 1A).
FIGURE 5.

A major interaction site of NS5A-D2 with CypA and CypB. Each panel corresponds to the superposition of a 1H,15N HSQC spectrum acquired on NS5A-D2 alone (in blue) and of a 1H,15N plane obtained with the HNnoCO pulse sequence that specifically selects the NS5A-D2 subspectrum in a NS5A-D2/Cyp (1:1) sample (in red). The motif 310PAWARPDYNPP320 of NS5A-D2 interacts with CypA.
FIGURE 6.

Cis/trans isomerization of HCV NS5A-D2 X-Pro peptide bonds catalyzed by CypA and CypB. 1H,15N heteronuclear exchange spectra recorded at 800 MHz with mixing times of 0.88 ms (in blue) and 100 ms (in red) on [15N]NS5A-D2 samples (220 μm) with catalytic amounts of either CypA (A) or CypB (B) (23 μm). The NMR resonances (trans, cis, and the two exchange peaks) of NS5A-D2 residues for which the PPIase activity of a cyclophilin can be evidenced are connected by green dotted boxes. C, amino acid sequence of NS5A-D2 (JFH1) construction. Residues on which PPIase activities of CypA and CypB have been monitored are bold and shown in violet. The 15 proline residues of NS5A-D2 are indicated on a light gray background. The previously defined binding site of cyclophilins is boxed, and residues that are affected only by CypA binding (see Fig. 4 and under “Results”) are marked with asterisks. D, determination of the CypA (in black (Δ)) and CypB-catalyzed (in gray (×)) exchange rates toward the Val274-Pro275 NS5A-D2 peptide bond. Experimental data measured on Val274 for (Itc/Itt) (CypA (Δ); CypB (×)) were fitted to the theoretical Equation 2 (see “Experimental Procedures”) (CypA, black line; CypB, gray line). E, resulting exchange rates (kexch) of the cis/trans isomerization processes in NS5A-D2 catalyzed by the addition of catalytic amounts of either CypA or CypB.
Other residues that had their peaks severely broadened upon the addition of the cyclophilins were Cys338 and Ala339, but these C-terminal residues are just upstream of the C-terminal His tag, making an interpretation of this interaction in the isolated D2 domain more difficult. Importantly, however, the signals assigned to the minor cis forms for all prolines almost completely disappeared in the spectra of the complexes. This indicates that individual peptides containing cis-prolyl bonds interact via these cis-prolines with the cyclophilins. We thus next investigated the peptidyl-prolyl cis/trans isomerase activity of CypA and CypB.
Enzymatic Activities of Cyclophilins on Domain 2 of HCV NS5A Protein—We first characterized the cyclophilin catalyzed peptidyl-prolyl cis/trans isomerization by homonuclear EXSY (48, 55). In this experiment, one visualizes as off-diagonal peaks those amide functions that have physically changed from the cis to trans (or vice versa) during the mixing delay (typically of the order of 100 ms). Without cyclophilins, the exchange rate of peptidyl-prolyl bonds, even in unstructured peptides, is too slow (kexch < 0.1 s-1) to lead to detectable exchange peaks in EXSY spectra. When adding the cyclophilin in catalytic amounts, we did detect several novel exchange peaks. However, the natively unfolded nature of NS5A-D2 and ensuing limited proton dispersion renders the assignment of these peaks extremely difficult on the sole basis of their proton chemical shift. Moreover, the proton chemical shift differences between trans and cis forms being often very limited, potential exchange peaks nearly coincide with the diagonal. The homonuclear EXSY experiments thus led us to conclude that both cyclophilins do isomerize distinct peptidyl-prolyl bonds within NS5A-D2 but without allowing the assignment of the peptidylprolyl bonds or evaluation of the catalytic efficacy.
To increase resolution and allow assignment of the individual processes, we performed a series of 15N z-exchange experiments (14, 15, 49, 56) on a 15N-labeled NS5A-D2 sample in the presence of catalytic amounts of CypA or CypB (1:10). The exchange between two conformations is now monitored at the level of the amide function (characterized by a 1H,15N correlation peak in the HSQC spectrum) rather than for the sole amide proton frequency as in the EXSY experiment. Heteronuclear exchange spectra were acquired at different mixing times: 0.88, 25, 50, 100, 200, 300 and 400 ms. At the shortest mixing time (0.88 ms), no exchange peaks connecting the major and minor peak of a given residue were visible, but the minor peaks did already broaden (Fig. 6 and supplemental Fig. 4), in agreement with our previous results on the 1:1 complexes. Broadening was more severe for the complex with CypA than for the one with CypB, pointing toward an equally stronger binding of CypA to those alternative anchoring points formed by the cis prolines in the NS5A-D2 sequence. Upon increasing the exchange interval (mixing time), additional connecting peaks could be observed and assigned for several residues (Fig. 6 and supplemental Fig. 4), thereby confirming their assignment and excluding the possibility that these minor peaks come from a degradation product or other molecular entity. Comparing the exchange spectra obtained with CypA and CypB, we found roughly the same set of additional peaks, suggesting that both Cyp have a similar activity toward the peptidyl-prolyl bonds in NS5A-D2. Both Cyp, moreover, lack a clear specificity, as PPIase-catalyzed exchange peaks could be assigned for residues in the vicinity of almost all of the 15 proline residues in the NS5A-D2 sequence. For Pro306, Pro319, and Pro320 only, we did not detect exchange peaks for residues in their direct neighborhood, but spectral overlap clearly limited our analysis of the process around these prolines. What does distinguish both cyclophilins, however, is the catalytic efficacy of the isomerization. Even with careful normalization of the enzyme content in NMR samples, the CypA-catalyzed exchange peaks were generally more intense than those obtained with CypB. Extracting a rate constant (kexch) from the buildup of the exchange peaks as a function of increasing exchange time (Fig. 6D), we found that the CypA-catalyzed exchange rates range from 14 s-1 for Gln331 to 61 s-1 for Met283, with a mean value of 29 s-1 as calculated over the 10 residues for which a reliable rate constant could be extracted (Fig. 6E). CypB as an enzyme is less effective, with exchange rates ranging from 3 s-1 for Leu277 to 31 s-1 for Gln331, and an average of 11 s-1 determined over the 14 NS5A-D2 residues for which the experimental data led to reliable curves. As was the case for the homonuclear EXSY spectra, the additional peaks connecting cis and trans conformers of a given residue disappeared upon the addition of CsA (supplemental Fig. 5).
DISCUSSION
NS5A is required in several steps of the HCV life cycle, including replication and infectious particle assembly (3, 27, 57), but its precise roles are still not known. Recent mutational analyses have shown that many residues of its D2 domain are essential for RNA replication (29), and several mutations in this domain were reported to confer resistance to CsA (26). However, the structural data required for further understanding of these observations are still limited.
We have chosen here to study the D2 domain in the context of the HCV genotype 2a (JFH1). This clone, isolated from a Japanese patient with a fulminant hepatitis, allows for infectious virus propagation in cell culture (58–60). All biochemical and biophysical characterization methods indicate that the isolated domain 2 of NS5A (JFH-1), when overproduced recombinantly and purified to homogeneity, is unstructured (Figs. 1 and 2). A similar increase in apparent molecular weight, random coil CD spectrum, and limited dispersion for the amide proton chemical shifts in the NMR spectrum were described previously for the genotype 1a NS5A-D2 domain, although the two domains share only 48% sequence identity (36, 37) (See supplemental Fig. 6). The NS5A-D2 domain thus belongs to the growing group of natively unstructured proteins that gain function upon interaction with their molecular partners (61, 62). Furthermore, when we used the carbon chemical shifts to detect potential structure at the local level (CSI strategy) (54), we could not detect even small stretches of stable secondary structure that might have gone undetected by the macroscopic approaches described above. However, the amide resonances of the Trp312 and Ala313 in the most conserved 310PAWARP315 motif resonate at an unusual proton and nitrogen frequency (Fig. 2A). As these anomalous chemical shifts were present equally in the genotype 1a NS5A-D2 domain (37), and as this Trp312-Ala313 segment, as well as Pro310 and Pro315, is fully conserved in all genotypes (Fig. 1A), we have synthesized a peptide centered on this motif and are currently pursuing a detailed NMR analysis to interpret the anomalous chemical shift values in structural terms.
Certain mutations in the D2 domain of NS5A confer resistance to CsA (26), a cyclic undecapeptide for which the primary target in the eukaryotic cell is members of the cyclophilin family (63). Cyclophilins are peptidy-prolyl cis/trans isomerases that are involved in the life cycle of several viruses. The best characterized example is CypA interacting with the capsid domain of the HIV Gag polyprotein precursor (12). For HCV, the implication of cyclophilins in the viral life cycle comes from the observation that the cyclophilin-specific inhibitor CsA has anti-HCV properties (18, 20, 23). In 2005, Watashi et al. (17) reported that CypB binds to the viral RdRp NS5B protein (genotype 1b) and regulates its RNA binding properties. However, Ishii et al. (22) reported that CypB does not regulate the RNA binding activity of NS5B in a JFH1 context (genotype 2a) and Robida et al. (24) have shown that there is no replication defect in a genotype 1b replicon system when CypB expression is abolished. Whereas these reports functionally link the cyclophilins to NS5B, a recent study indicates that the sensitivity of HCV for CsA depends not only on NS5B but equally (and even more) on NS5A (26). Finally, whereas the earlier reports mainly point to CypB as the modulator of the NS5A/B activity, recent results have questioned this, and the dependence of HCV replication on cyclophilin subtypes equally may vary with the genotype. Very recently, Yang et al. (25) have shown that CypA is an essential co-factor for numerous HCV genotypes, including genotypes 1a, 1b, and 2a (isolate JFH1).
In view of these conflicting reports, we used NMR spectroscopy to probe the interaction of NS5A-D2 (JFH1) with both CypB and CypA. Chemical shift perturbation experiments on both NS5A-D2·CypA and NS5A-D2·CypB complexes gave evidence for a direct physical interaction that is localized to the active site on the cyclophilins (Fig. 3). Important chemical shift perturbations have been measured for CypA Arg55, Phe60, Met61, Asn102, Phe113, and His126 residues and the equivalent residues on CypB, all previously shown to interact directly with a peptide substrate (64, 65). Titration experiments with the NS5A-D2 domain against both cyclophilins allowed us to quantify the interaction with KD values of 64 and 90 μm toward CypA and CypB, respectively. As the active site of the cyclophilins coincides with their CsA binding groove, we indeed found that CsA competes very efficiently with NS5A-D2 for binding to the cyclophilins. Its nanomolar affinity toward cyclophilins (66) causes a complete inhibition of the molecular interaction between NS5A-D2 and the cyclophilins.
Although the obtained KD values are comparable to the 15 μm dissociation constant that has been measured for CypA toward HIV Capsid (14, 67), one fundamental difference became clear from the observed broadening of the Cyp active site resonances upon increasing NS5A-D2 concentration. The CypA/HIV capsid interaction indeed has been localized to a single Gly221-Pro222 motif in the HIV capsid protein, whereas in the present case, it seems that many prolines can interact with the cyclophilins. When repeating the titration experiment with a peptide (PepD2, residues 304–323 of NS5A) containing only five out of the 15 proline residues in full-length NS5A-D2, the titration behavior proved more conventional but led to a 10-fold lower interaction strength. Other anchoring points thus contribute to the interaction with the intact D2 domain, and the line broadening observed for the cis proline-associated resonances, even upon addition of catalytic amounts of cyclophilins, suggests that the overall interaction strength comes from several anchorage points distributed over the NS5A-D2 sequence. The presence of multiple mutations in NS5A-D2 that confer CsA resistance to the HCV virus is in agreement with the absence of a single interaction hotspot on the D2 domain, but equally it suggests that a functional interaction requires a narrow window of Cyp concentration in the complex.
Both CypB and CypA bind a highly conserved motif in domain 2 of NS5A centered on the 310PAWARP315 sequence in the JFH1 HCV clone. Whereas CypB solely interacts with this hexapeptide, the motif recognized by CypA is, however, larger and corresponds to 304GFPRALPAWARPDYNPP320 (Figs. 5 and 6 and supplemental Fig. 3). Indeed NMR resonances of Gly304, Ala308, Leu309, Asp316, Tyr317, and Asn318 are affected only following CypA addition, whereas Ala311, Trp312, Ala313, and Arg314 resonances are perturbed in the presence of either cyclophilins. Although highly specific, this peptide does not contribute more than one-tenth of the interaction strength. The natural abundance 1H,15N HSQC spectrum acquired on this peptide mapped very well to the corresponding residues in the full-length NS5A-D2 sequence (data not shown), excluding a difference in structure and dynamics as the source of this discrepancy. Importantly, at least six residues in this NS5A-D2 motif recognized by CypA were shown previously to be essential for HCV replication in a subgenomic 1b replicon system (Con1 isolate) (29). In this genotype 1b isolate the motif is rather well conserved with only three amino acids substitution compared with genotype 2a (JFH1) (308KFPRAMPIWARPDYNPP324) (supplemental Fig. 6). The mutant M313A replicates with very low efficiency close to the detection limit, the mutant P314A was lethal, W316A was moderately impaired, mutant A317G yielded a small-colony phenotype, mutant Y321A was severely impaired in replication and also gave a small colony phenotype, and the P324A mutant was also lethal (residues highlighted in black in supplemental Fig. 6). These results from Tellinghuisen et al. (29), combined with ours showing a direct interaction of CypA with the corresponding region in NS5A-D2, and finally the finding that CypA is an essential co-factor for HCV replication (genotypes 1a, 1b, and 2a) (25) suggest that the replication defects of NS5A mutants might result from an altered interaction between the cyclophilin and NS5A-D2 in this zone.
Several groups have applied CsA treatment to virus-infected cell cultures to select for mutations that would confer resistance directly. A HCV replicon (genotype 1b) mutant bearing a mutation corresponding to Y317N in genotype 2a exhibited enhanced CsA resistance (26). This Tyr317 is just downstream of the identified motif and belongs to the binding site of CypA but not of CypB (Fig. 5). However, in the same study, six additional mutations were discovered in NS5A, of which four are located in domain 2, one in the low complexity sequence between D2 and D3, and another one in D3 (26). Mutations that have been identified in domain 2 (1b) (highlighted in gray in supplemental Fig. 6) correspond to the following residues in NS5A-D2 of genotype 2a (JFH1): Asp256, Val276, Leu280, and Met299. Therefore, they do not map directly to the above described conserved motif. These residues do however contribute to the interaction with the cyclophilins, as they are centered on the NS5A-D2 region for which highest efficiencies have been measured for the CypA-catalyzed cis/trans isomerization reactions (Fig. 6). Only Pro282 appears to be conserved in genotypes 1a and 1b (supplemental Fig. 6), arguing against the precise localization as the important factor for cyclophilin function in RNA replication. The presence of a well defined amount of cyclophilin at the NS5A-D2 surface seems to be required in order to confer functionality.
Our interaction experiments with a 1:1 molecular ratio between domain 2 of NS5A and cyclophilins showed a pronounced broadening of all resonances corresponding to residues in the vicinity of a cis-proline residue, leading us to investigate the peptidyl-prolyl cis/trans isomerase activity of the cyclophilins toward prolyl bonds in the NS5A-D2 domain. NMR exchange spectroscopy, previously used to characterize the CypA-catalyzed cis/trans isomerization of the Gly221-Pro222 peptide bond in the HIV capsid (14, 15), indeed provided direct evidence for the catalytic activity of the cyclophilins and allowed us to assign the effect to individual prolyl bonds. Because of the unstructured nature of NS5A-D2 and the resulting low proton amide dispersion, 1H,15N heteronuclear z-exchange spectroscopy (49) proved to be superior to 1H, 1H homonuclear EXSY spectroscopy and allowed us for the first time to prove in vitro that HCV NS5A-D2 is a substrate for the PPIase activity of at least two host cyclophilins (Fig. 6 and supplemental Fig. 4). Despite the fact that both CypA and CypB catalyze the cis/trans isomerization of the same NS5A-D2 X-Pro peptide bonds, they do not act with the same efficiency. Domain 2 of NS5A is a better substrate for CypA than for CypB, with a mean exchange rate (kexch) of 28.9 s-1 for CypA and only 11.1 s-1 for CypB. Every enzyme equally has its preferred sites, which do not necessarily coincide. The highest enzymatic efficiencies have been measured in the Glu271-Met283 region of NS5A-D2 for CypA, with maximal kexch values of 59 and 61 s-1 for Glu281 and Met283, respectively, which probably reflects the isomerization of the Glu281-Pro282 peptidyl-prolyl bond. The maximal activity of CypA in the N-terminal region of NS5A-D2 coincides with the localization of the majority of resistance conferring mutations. Together with the stronger affinity, this supports the dominant role for CypA in the infection process. This conclusion has been confirmed by HCV infection and replication assays using cell lines with stable knockdown of CypA and CypB.6 CypB displays more activity toward the C-terminal half of the NS5A-D2 domain, with an optimal activity toward the Gln331-Pro332 peptidyl-prolyl bond (kexch = 31 s-1) (Fig. 6). For comparison, Bosco et al. (14, 15) have found that with an enzyme:substrate ratio comparable to that used here, the CypA-catalyzed cis/trans isomerization of the Gly221-Pro222 HIV Capsid bond is characterized by a kexch value around 10 s-1. However, in their system, CypA specifically binds to and catalyzes cis/trans isomerization of Gly221-Pro222 over other Gly-Pro motifs in the HIV capsid. We show here that CypA is enzymatically active on almost all X-Pro NS5A-D2 sites, albeit with different efficiencies. The absence of specificity of CypA toward NS5A-D2 sites is possibly related to the unstructured character of the protein, as cyclophilins lack specificity toward peptide substrates (68).
The present structure-function study provides the first molecular basis for the further understanding of the resistance of HCV replication to CsA and analogues. As CsA abolishes the interaction between NS5A-D2 and CypA but also the PPIase activity of CypA toward this domain, we cannot conclude whether it is the binding, the catalytic activity, or even both of these that are involved in the HCV replication process (69). Indeed, cyclophilins may play biological roles either by catalyzing the cis/trans isomerization of a peptide bond, as for the tyrosine kinase Itk (70), or by interacting with an X-Pro motif that is no longer available for interaction with others partners, as is the case with HIV capsid with TRIM5α and CypA (12, 13). Further studies with NS5A-D2 and the cyclophilins in the presence of an interacting partner such as NS5B and/or RNA will be necessary in order to evaluate their precise role in the HCV life cycle.
Supplementary Material
Acknowledgments
We gratefully acknowledge RD-Biotech (Besançon, France) for the cloning and initial expression and purification tests for NS5A-D2, Guillaume Blanc and Jennifer Molle for technical assistance, Michel Becchi for the mass spectroscopy measurements, Christophe Combet for bioinformatics support. CD experiments were performed on the platform “Production et Analyse de Protéines” of the IFR 128 BioSciences Gerland-Lyon Sud. The NMR facility used in this study was funded by the Région Nord-Pas de Calais (France), the CNRS, the Universities of Lille 1 and Lille 2, and the Institut Pasteur de Lille.
The 1H,15N, and 13C backbone resonances for this protein are available in the Biological Magnetic Resonance Data Bank under BMRB accession number 16165.
This work was supported by the French Centre National de la Recherche Scientifique (CNRS) and the Universities of Lille and Lyon and by grants from the French National Agency for Research on AIDS and Viral Hepatitis and the European Commission (VIRGIL Network of Excellence on Antiviral Drug Resistance).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–6 and Table 1.
Footnotes
The abbreviations used are: HCV, hepatitis C virus; aa, amino acid; CsA, cyclosporin A; Cyp, cyclophilin; EXSY, exchange spectroscopy; HIV, human immunodeficiency virus; HSQC, heteronuclear single quantum correlation; NMR, nuclear magnetic resonance; NOESY, nuclear Overhauser enhancement spectroscopy; NS5A, nonstructural protein 5A; PPIase, peptidylprolyl cis/trans isomerase; TFE, 2,2,2-trifluoroethanol; IPTG, isopropyl 1-thio-β-d-galactopyranoside; DTT, dithiothreitol; NPSA, network protein sequence analysis; CSI, chemical shift index.
F. Dorkeld, C. Combet, F. Penin, and G. Deléage, unpublished data.
A. Kaul and R. Bartenschlager, unpublished results.
References
- 1.National Institutes of Health (2002) Hepatology 36 Suppl. 1, S2-S20 [Google Scholar]
- 2.Appel, N., Schaller, T., Penin, F., and Bartenschlager, R. (2006) J. Biol. Chem. 281 9833-9836 [DOI] [PubMed] [Google Scholar]
- 3.Moradpour, D., Penin, F., and Rice, C. M. (2007) Nat. Rev. 5 453-463 [DOI] [PubMed] [Google Scholar]
- 4.Tellinghuisen, T. L., Evans, M. J., von Hahn, T., You, S., and Rice, C. M. (2007) J. Virol. 81 8853-8867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lohmann, V., Korner, F., Koch, J., Herian, U., Theilmann, L., and Bartenschlager, R. (1999) Science 285 110-113 [DOI] [PubMed] [Google Scholar]
- 6.Handschumacher, R. E., Harding, M. W., Rice, J., Drugge, R. J., and Speicher, D. W. (1984) Science 226 544-547 [DOI] [PubMed] [Google Scholar]
- 7.Schreiber, S. L. (1991) Science 251 283-287 [DOI] [PubMed] [Google Scholar]
- 8.Barik, S. (2006) Cell. Mol. Life Sci. 63 2889-2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergsma, D. J., Eder, C., Gross, M., Kersten, H., Sylvester, D., Appelbaum, E., Cusimano, D., Livi, G. P., McLaughlin, M. M., and Kasyan, K. (1991) J. Biol. Chem. 266 23204-23214 [PubMed] [Google Scholar]
- 10.Watashi, K., and Shimotohno, K. (2007) Drug Target Insights 1 9-18 [PMC free article] [PubMed] [Google Scholar]
- 11.Luban, J., Bossolt, K. L., Franke, E. K., Kalpana, G. V., and Goff, S. P. (1993) Cell 73 1067-1078 [DOI] [PubMed] [Google Scholar]
- 12.Luban, J. (2007) J. Virol. 81 1054-1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sokolskaja, E., Berthoux, L., and Luban, J. (2006) J. Virol. 80 2855-2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bosco, D. A., Eisenmesser, E. Z., Pochapsky, S., Sundquist, W. I., and Kern, D. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 5247-5252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bosco, D. A., and Kern, D. (2004) Biochemistry 43 6110-6119 [DOI] [PubMed] [Google Scholar]
- 16.Bukovsky, A. A., Weimann, A., Accola, M. A., and Gottlinger, H. G. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 10943-10948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watashi, K., Ishii, N., Hijikata, M., Inoue, D., Murata, T., Miyanari, Y., and Shimotohno, K. (2005) Mol. Cell 19 111-122 [DOI] [PubMed] [Google Scholar]
- 18.Nakagawa, M., Sakamoto, N., Enomoto, N., Tanabe, Y., Kanazawa, N., Koyama, T., Kurosaki, M., Maekawa, S., Yamashiro, T., Chen, C. H., Itsui, Y., Kakinuma, S., and Watanabe, M. (2004) Biochem. Biophys. Res. Commun. 313 42-47 [DOI] [PubMed] [Google Scholar]
- 19.Tanabe, Y., Sakamoto, N., Enomoto, N., Kurosaki, M., Ueda, E., Maekawa, S., Yamashiro, T., Nakagawa, M., Chen, C. H., Kanazawa, N., Kakinuma, S., and Watanabe, M. (2004) J. Infect. Dis. 189 1129-1139 [DOI] [PubMed] [Google Scholar]
- 20.Watashi, K., Hijikata, M., Hosaka, M., Yamaji, M., and Shimotohno, K. (2003) Hepatology 38 1282-1288 [DOI] [PubMed] [Google Scholar]
- 21.Flisiak, R., Horban, A., Gallay, P., Bobardt, M., Selvarajah, S., Wiercinska-Drapalo, A., Siwak, E., Cielniak, I., Higersberger, J., Kierkus, J., Aeschlimann, C., Grosgurin, P., Nicolas-Metral, V., Dumont, J. M., Porchet, H., Crabbe, R., and Scalfaro, P. (2008) Hepatology 47 817-826 [DOI] [PubMed] [Google Scholar]
- 22.Ishii, N., Watashi, K., Hishiki, T., Goto, K., Inoue, D., Hijikata, M., Wakita, T., Kato, N., and Shimotohno, K. (2006) J. Virol. 80 4510-4520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakagawa, M., Sakamoto, N., Tanabe, Y., Koyama, T., Itsui, Y., Takeda, Y., Chen, C. H., Kakinuma, S., Oooka, S., Maekawa, S., Enomoto, N., and Watanabe, M. (2005) Gastroenterology 129 1031-1041 [DOI] [PubMed] [Google Scholar]
- 24.Robida, J. M., Nelson, H. B., Liu, Z., and Tang, H. (2007) J. Virol. 81 5829-5840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang, F., Robotham, J. M., Nelson, H. B., Irsigler, A., Kenworthy, R., and Tang, H. (2008) J. Virol. 82 5269-5278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernandes, F., Poole, D. S., Hoover, S., Middleton, R., Andrei, A. C., Gerstner, J., and Striker, R. (2007) Hepatology 46 1026-1033 [DOI] [PubMed] [Google Scholar]
- 27.Appel, N., Zayas, M., Miller, S., Krijnse-Locker, J., Schaller, T., Friebe, P., Kallis, S., Engel, U., and Bartenschlager, R. (2008) PLoS Pathog. 4 e1000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Penin, F., Dubuisson, J., Rey, F. A., Moradpour, D., and Pawlotsky, J. M. (2004) Hepatology 39 5-19 [DOI] [PubMed] [Google Scholar]
- 29.Tellinghuisen, T. L., Foss, K. L., Treadaway, J. C., and Rice, C. M. (2008) J. Virol. 82 1073-1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brass, V., Bieck, E., Montserret, R., Wolk, B., Hellings, J. A., Blum, H. E., Penin, F., and Moradpour, D. (2002) J. Biol. Chem. 277 8130-8139 [DOI] [PubMed] [Google Scholar]
- 31.Penin, F., Brass, V., Appel, N., Ramboarina, S., Montserret, R., Ficheux, D., Blum, H. E., Bartenschlager, R., and Moradpour, D. (2004) J. Biol. Chem. 279 40835-40843 [DOI] [PubMed] [Google Scholar]
- 32.Tellinghuisen, T. L., Marcotrigiano, J., Gorbalenya, A. E., and Rice, C. M. (2004) J. Biol. Chem. 279 48576-48587 [DOI] [PubMed] [Google Scholar]
- 33.Huang, L., Hwang, J., Sharma, S. D., Hargittai, M. R., Chen, Y., Arnold, J. J., Raney, K. D., and Cameron, C. E. (2005) J. Biol. Chem. 280 36417-36428 [DOI] [PubMed] [Google Scholar]
- 34.Tellinghuisen, T. L., Marcotrigiano, J., and Rice, C. M. (2005) Nature 435 374-379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tellinghuisen, T. L., Foss, K. L., and Treadaway, J. (2008) PLoS Pathog. 4 e1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang, Y., Kang, C. B., and Yoon, H. S. (2006) Mol. Cells 22 13-20 [PubMed] [Google Scholar]
- 37.Liang, Y., Ye, H., Kang, C. B., and Yoon, H. S. (2007) Biochemistry 46 11550-11558 [DOI] [PubMed] [Google Scholar]
- 38.Combet, C., Blanchet, C., Geourjon, C., and Deleage, G. (2000) Trends Biochem. Sci. 25 147-150 [DOI] [PubMed] [Google Scholar]
- 39.Combet, C., Garnier, N., Charavay, C., Grando, D., Crisan, D., Lopez, J., Dehne-Garcia, A., Geourjon, C., Bettler, E., Hulo, C., Le Mercier, P., Bartenschlager, R., Diepolder, H., Moradpour, D., Pawlotsky, J. M., Rice, C. M., Trepo, C., Penin, F., and Deleage, G. (2007) Nucleic Acids Res. 35 Database Suppl., D363-D366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994) Nucleic Acids Res. 22 4673-4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cortay, J. C., Negre, D., Scarabel, M., Ramseier, T. M., Vartak, N. B., Reizer, J., Saier, M. H., Jr., and Cozzone, A. J. (1994) J. Biol. Chem. 269 14885-14891 [PubMed] [Google Scholar]
- 42.Chen, Y. H., Yang, J. T., and Chau, K. H. (1974) Biochemistry 13 3350-3359 [DOI] [PubMed] [Google Scholar]
- 43.Hanoulle, X., Melchior, A., Sibille, N., Parent, B., Denys, A., Wieruszeski, J. M., Horvath, D., Allain, F., Lippens, G., and Landrieu, I. (2007) J. Biol. Chem. 282 34148-34158 [DOI] [PubMed] [Google Scholar]
- 44.Verdegem, D., Dijkstra, K., Hanoulle, X., and Lippens, G. (2008) J. Biomol. NMR 42 11-21 [DOI] [PubMed] [Google Scholar]
- 45.Grzesiek, S., Bax, A., Hu, J. S., Kaufman, J., Palmer, I., Stahl, S. J., Tjandra, N., and Wingfield, P. T. (1997) Protein Sci. 6 1248-1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ottiger, M., Zerbe, O., Guntert, P., and Wuthrich, K. (1997) J. Mol. Biol. 272 64-81 [DOI] [PubMed] [Google Scholar]
- 47.Golovanov, A. P., Blankley, R. T., Avis, J. M., and Bermel, W. (2007) J. Am. Chem. Soc. 129 6528-6535 [DOI] [PubMed] [Google Scholar]
- 48.Kaplan, J. L., and Fraenkel, G. (1980) NMR of Chemically Exchanging Systems, Academic Press, New York
- 49.Farrow, N. A., Zhang, O., Forman-Kay, J. D., and Kay, L. E. (1994) J. Biomol. NMR 4 727-734 [DOI] [PubMed] [Google Scholar]
- 50.Huang, L., Sineva, E. V., Hargittai, M. R., Sharma, S. D., Suthar, M., Raney, K. D., and Cameron, C. E. (2004) Protein Expression Purif. 37 144-153 [DOI] [PubMed] [Google Scholar]
- 51.Kieliszewski, M. J., Leykam, J. F., and Lamport, D. T. (1990) Plant Physiol. 92 316-326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tompa, P. (2002) Trends Biochem. Sci. 27 527-533 [DOI] [PubMed] [Google Scholar]
- 53.Buck, M. (1998) Q. Rev. Biophys. 31 297-355 [DOI] [PubMed] [Google Scholar]
- 54.Wishart, D. S., and Sykes, B. D. (1994) J. Biomol. NMR 4 171-180 [DOI] [PubMed] [Google Scholar]
- 55.Kern, D., Drakenberg, T., Wikstrom, M., Forsen, S., Bang, H., and Fischer, G. (1993) FEBS Lett. 323 198-202 [DOI] [PubMed] [Google Scholar]
- 56.Kern, D., Eisenmesser, E. Z., and Wolf-Watz, M. (2005) Methods Enzymol. 394 507-524 [DOI] [PubMed] [Google Scholar]
- 57.Macdonald, A., and Harris, M. (2004) J. Gen. Virol. 85 2485-2502 [DOI] [PubMed] [Google Scholar]
- 58.Pietschmann, T., Kaul, A., Koutsoudakis, G., Shavinskaya, A., Kallis, S., Steinmann, E., Abid, K., Negro, F., Dreux, M., Cosset, F. L., and Bartenschlager, R. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 7408-7413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wakita, T., Pietschmann, T., Kato, T., Date, T., Miyamoto, M., Zhao, Z., Murthy, K., Habermann, A., Krausslich, H. G., Mizokami, M., Bartenschlager, R., and Liang, T. J. (2005) Nat. Med. 11 791-796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhong, J., Gastaminza, P., Cheng, G., Kapadia, S., Kato, T., Burton, D. R., Wieland, S. F., Uprichard, S. L., Wakita, T., and Chisari, F. V. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 9294-9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dyson, H. J., and Wright, P. E. (2005) Nat. Rev. Mol. Cell Biol. 6 197-208 [DOI] [PubMed] [Google Scholar]
- 62.Gunasekaran, K., Tsai, C. J., Kumar, S., Zanuy, D., and Nussinov, R. (2003) Trends Biochem. Sci. 28 81-85 [DOI] [PubMed] [Google Scholar]
- 63.Liu, J., Farmer, J. D., Jr., Lane, W. S., Friedman, J., Weissman, I., and Schreiber, S. L. (1991) Cell 66 807-815 [DOI] [PubMed] [Google Scholar]
- 64.Zhao, Y., and Ke, H. (1996) Biochemistry 35 7362-7368 [DOI] [PubMed] [Google Scholar]
- 65.Zhao, Y., and Ke, H. (1996) Biochemistry 35 7356-7361 [DOI] [PubMed] [Google Scholar]
- 66.Mikol, V., Kallen, J., and Walkinshaw, M. D. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 5183-5186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yoo, S., Myszka, D. G., Yeh, C., McMurray, M., Hill, C. P., and Sundquist, W. I. (1997) J. Mol. Biol. 269 780-795 [DOI] [PubMed] [Google Scholar]
- 68.Harrison, R. K., and Stein, R. L. (1990) Biochemistry 29 3813-3816 [DOI] [PubMed] [Google Scholar]
- 69.Fischer, G., Tradler, T., and Zarnt, T. (1998) FEBS Lett. 426 17-20 [DOI] [PubMed] [Google Scholar]
- 70.Brazin, K. N., Mallis, R. J., Fulton, D. B., and Andreotti, A. H. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 1899-1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Simmonds, P., Bukh, J., Combet, C., Deleage, G., Enomoto, N., Feinstone, S., Halfon, P., Inchauspe, G., Kuiken, C., Maertens, G., Mizokami, M., Murphy, D. G., Okamoto, H., Pawlotsky, J. M., Penin, F., Sablon, E., Shin, I. T., Stuyver, L. J., Thiel, H. J., Viazov, S., Weiner, A. J., and Widell, A. (2005) Hepatology 42 962-973 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.