

Abstract
Linoleate (10R)-dioxygenase (10R-DOX) of Aspergillus fumigatus was cloned and expressed in insect cells. Recombinant 10R-DOX oxidized 18:2n-6 to (10R)-hydroperoxy-8(E),12(Z)-octadecadienoic acid (10R-HPODE; ∼90%), (8R)-hydroperoxylinoleic acid (8R-HPODE; ∼10%), and small amounts of 12S(13R)-epoxy-(10R)-hydroxy-(8E)-octadecenoic acid. We investigated the oxygenation of 18:2n-6 at C-10 and C-8 by site-directed mutagenesis of 10R-DOX and 7,8-linoleate diol synthase (7,8-LDS), which forms ∼98% 8R-HPODE and ∼2% 10R-HPODE. The 10R-DOX and 7,8-LDS sequences differ in homologous positions of the presumed dioxygenation sites (Leu-384/Val-330 and Val-388/Leu-334, respectively) and at the distal site of the heme (Leu-306/Val-256). Leu-384/Val-330 influenced oxygenation, as L384V and L384A of 10R-DOX elevated the biosynthesis of 8-HPODE to 22 and 54%, respectively, as measured by liquid chromatography-tandem mass spectrometry analysis. The stereospecificity was also decreased, as L384A formed the R and S isomers of 10-HPODE and 8-HPODE in a 3:2 ratio. Residues in this position also influenced oxygenation by 7,8-LDS, as its V330L mutant augmented the formation of 10R-HPODE 3-fold. Replacement of Val-388 in 10R-DOX with leucine and phenylalanine increased the formation of 8R-HPODE to 16 and 36%, respectively, whereas L334V of 7,8-LDS was inactive. Mutation of Leu-306 with valine or alanine had little influence on the epoxyalcohol synthase activity. Our results suggest that Leu-384 and Val-388 of 10R-DOX control oxygenation of 18:2n-6 at C-10 and C-8, respectively. The two homologous positions of prostaglandin H synthase-1, Val-349 and Ser-353, are also critical for the position and stereospecificity of the cyclooxygenase reaction.
Linoleate diol synthases (LDS)2 and linoleate 10R-DOX are fungal fatty acid dioxygenases of the myeloperoxidase gene family (1-3). LDS have dual enzyme activities and transform 18:2n-6 sequentially to 8R-HPODE in an 8R-dioxygenase reaction and to 5,8-, 7,8-, or 8,11-DiHODE in hydroperoxide isomerase reactions. These oxylipins affect sporulation, development, and pathogenicity of Aspergilli (4-6). Fatty acid dioxygenases of the myeloperoxidase gene family also occur in vertebrates, plants, and algae (7-9). The most thoroughly investigated vertebrate enzymes are ovine PGHS-1 and mouse PGHS-2 with known crystal structures (10-12). PGHS transforms 20:4n-6 to PGG2 in a cyclooxygenase and PGG2 to PGH2 in a peroxidase reaction. Aspirin and other nonsteroidal anti-inflammatory drugs inhibit the cyclooxygenase reaction. This is of paramount medical importance (13, 14), and PGHS-1 and -2 are commonly known as COX-1 and -2 (15). α-DOX occur in plants and algae, and biosynthesis of α-DOX in plants is elicited by pathogens (7). α-DOX oxidizes fatty acids to unstable (2R)-hydroperoxides, which readily break down nonenzymatically to fatty acid aldehydes and CO2 (7).
LDS, 10R-DOX, PGHS, and α-DOX oxygenate fatty acids to different products, but their oxygenation mechanisms have mechanistic similarities. Sequence alignment shows that many critical amino acid residues for the cyclooxygenase reaction are conserved in LDS, 10R-DOX, and α-DOX. These include the proximal histidine heme ligand, the distal histidine, and the catalytic important tyrosine (Tyr-385) of PGHS-1. The latter is oxidized to a tyrosyl radical, which initiates the cyclooxygenase reaction by abstraction of the pro-S hydrogen at C-13 of 20:4n-6 (16). In analogy, LDS and 10R-DOX catalyze stereospecific abstraction of the pro-S hydrogen at C-8 of 18:2n-6 (3), whereas α-DOX abstracts the pro-R hydrogen at C-2 of fatty acids (17). Site-directed mutagenesis of the conserved tyrosine homologues of Tyr-385 and proximal heme ligands abolishes the dioxygenase activities of 7,8-LDS and α-DOX (17, 18). The orientation of the substrate at the dioxygenation site differs. The carboxyl groups of fatty acids are positioned in a hydrophobic grove close to the tyrosine residue of α-DOX (19). In contrast, the ω ends of eicosanoic fatty acids are buried deep inside the cyclooxygenase channel so that C-13 lies in the vicinity of Tyr-385 (20). Several observations suggest that 18:2n-6 may also be positioned with its ω end embedded in the interior of 7,8-LDS of Gaeumannomyces graminis (18).
7,8-LDS of G. graminis and Magnaporthe grisea and 5,8-LDS of Aspergillus nidulans have been sequenced (5, 8, 21). Gene targeting revealed the catalytic properties of 5,8-LDS, 8,11-LDS, and 10R-DOX in Aspergillus fumigatus and A. nidulans (3). Homologous genes can be found in other Aspergilli spp. Alignment of the two 7,8-LDS amino acid sequences with 5,8-LDS, 8,11-LDS, and 10R-DOX sequences of five Aspergilli revealed several conserved regions with single amino acid differences between the enzymes with 8R-DOX and 10R-DOX activities, as illustrated by the selected sequences in Fig. 1. Leu-306, Leu-384, and Val-388 of 10R-DOX are replaced in 5,8- and 7,8-LDS by valine, valine, and leucine residues, respectively. Whether these amino acids are important for the oxygenation mechanism is unknown, and this is one topic of the present investigation. The predicted secondary structure of 10R-DOX suggests that Leu-384 of 10R-DOX can be present in an α-helix with Val-388 close to its border. This α-helix is homologous to helix 6 of PGHS-1, which contains Val-349 and Ser-353 at the homologous positions of Leu-384 and Val-388 (Fig. 1).
FIGURE 1.

Alignments of partial amino acid sequences of five heme containing fatty acid dioxgenases and a comparison of the predicted secondary structure of 10R-DOX with ovine PGHS-1. A, top, amino acids residues at the presumed peroxidase and hydroperoxide isomerase sites. The last two residues, His and Asn, are conserved in all myeloperoxidases (1). Middle and bottom, amino acid residues of the presumed dioxygenation sites are shown. Conserved residues in all sequences are in boldface, and mutated residues of 10R-DOX and/or 7,8-LDS are marked by an asterisk. B, alignment of partial amino acid sequences of 10R-DOX with ovine PGHS-1, and a secondary structure prediction of the 10R-DOX sequence. The secondary structure of 10R-DOX was predicted by PSIPRED (43) and the secondary structure of ovine PGHS-1 from its crystal structure (Protein Data Bank code 1diy; cf. Ref 19). In short, our first strategy for site-directed mutagenesis was to switch hydrophobic residues between the enzymes with 10R- and 8R-DOX activities and to assess the effects on the DOX and hydroperoxide isomerase activities (10R-DOX/7,8-LDS: Leu-306/Val-256, Leu-384/Val-330, Val-388/Leu-334, and Ala-426/Ile-375) and to switch one hydrophobic/charged residue (Ala-435/Glu-384). Only catalytically active pairs would provide clear information on their importance for the position of dioxygenation (e.g. L384V of 10R-DOX and V330L of 7,8-LDS, both of which were active). Unfortunately, replacements of 7,8-LDS often led to inactivation or very low activity (e.g. V330A, V330M, I375A, E384A). Our second strategy was to study replacements in two homologous positions of ovine PGHS-1 (Val-349 and Ser-353) with smaller and larger hydrophobic residues, i.e. at Leu-384 and Val-388 of 10R-DOX. Abbreviations used are as follows: oCOX-1, ovine cyclooxygenase-1; Af, A. fumigatus; Gg, G. graminis. The GenBank™ protein sequences were derived from P05979, EAL89712, AAD49559, EAL84400, and ACL14177. The amino acid sequences were aligned with the ClustalW algorithm (DNAStar).
The overall three-dimensional structures of myeloperoxidases are conserved. It is therefore conceivable that important residues for substrate binding in the cyclooxygenase channel of PGHS could be conserved in LDS and 10R-DOX. The three-dimensional structure of ovine PGHS-1 shows that Val-349 and Ser-353 are close to C-3 and C-4 of 20:4n-6, and residues in these positions can alter both position and stereospecificity of oxygenation (22-24). Replacement of Val-349 of PGHS-1 with alanine increased the biosynthesis of 11R-HETE, whereas V349L decreased the generation of 11R-H(P)ETE and increased formation of 15(R/S)-H(P)ETE (23, 25). V349I formed PGG2 with 15R configuration (22, 24). Replacement of Ser-353 with threonine reduced cyclooxygenase and peroxidase activities by over 50% and increased the biosynthesis of 11R-HPETE and 15S-HPETE 4-5 times (23).
There is little information on the hydroperoxide isomerase and peroxidase sites of LDS (18, 26), but the latter could be structurally related to the peroxidase site of PGHS. PGG2 and presumably 8R-HPODE bind to the distal side of the heme group, which can be delineated by hydrophobic amino acid residues (27). Val-291 is one of these residues, which form a dome over the distal heme side of COX-1. The V291A mutant retained cyclooxygenase and peroxidase activities (27). 5,8- and 7,8-LDS also have valine residues in the homologous position, whereas 8,11-LDS and 10R-DOX have leucine residues (Fig. 1). Whether these hydrophobic residues are important for the peroxidase activities is unknown.
In this study we decided to compare the two catalytic sites of 10R-DOX of A. fumigatus and 7,8-LDS (EC 1.13.11.44) of G. graminis (18). Our first aim was to find a robust expression system for 10R-DOX of A. fumigatus. The second objective was to determine whether C16 and C20 fatty acid substrates enter the oxygenation site of 10R-DOX “head” or “tail” first. Unexpectedly, we found that 10R-DOX oxygenated 20:4n-6 by hydrogen abstraction at both C-13 and C-10 with formation of two nonconjugated and four cis-trans-conjugated HPETEs. Our third objective was to investigate the structural differences between 10R-DOX and 7,8-LDS of G. graminis, which could explain that oxygenation of 18:2n-6 mainly occurred at C-10 and at C-8, respectively. The strategy for site-directed mutagenesis of 10R-DOX and 7,8-LDS is outlined in the legend to Fig. 1; an alignment of the amino acid sequences of 10R-DOX and 7,8-LDS is found in supplemental material.
EXPERIMENTAL PROCEDURES
Materials—Fatty acids (99%) were from Merck, Sigma, and Larodan and stored at 50-100 mm in ethanol (-20 °C). [2H8]20:4n-6 was a gift of the late Dr. van Dorp, and [2H4]18: 2n-6 (99%) was from Larodan. The RS and SR enantiomers of vernolic acid (99%) were from Larodan. Chemically competent Escherichia coli (Top10), mouse anti-His (C-terminal) antibody, InsectSelect System with pIZ/V5-His, Cellfectin, and phleomycin (Zeocin) were from Invitrogen. The enhanced avian RT-PCR kit, GSH, and mCPBA (85%) were from Sigma. Restriction enzymes were from New England Biolabs and Fermentas. Plasmid midi kit and QIAquick gel extraction kit were from Qiagen. Pre-stained protein ladder (Page-Ruler) for SDS-PAGE and CloneJet PCR cloning kit were from Fermentas. Phusion® DNA polymerase was from Finnzymes. ECL advanced Western blotting detection kit, dNTPs, and horseradish peroxidase-labeled anti-mouse IgG antibodies were from GE Healthcare. Taq and Pfu DNA polymerases were from Promega. Ex-Cell 420 insect serum-free medium was purchased from SAFC-Biosciences (Hampshire, UK). Gentamycin sulfate and Spodoptera frugiperda (Sf21) cells were obtained locally. Primers for site-directed mutagenesis were obtained from TIB Molbiol (Berlin, Germany), and other oligonucleotides were from CyberGene (Huddinge, Sweden). Sequencing was performed at Uppsala Genomic Center (Rudbeck Laboratories, Uppsala University). 10R-HODE was isolated from incubations of mycelia of Aspergilli (3) and 8R-HODE from incubations with M. grisea (21); the alcohols were purified by TLC as described (28). Racemic vernolic acid was obtained by epoxidation of 18:2n-6 with mCPBA (1.5 eq) in CH2Cl2 (29), purified by TLC (ethyl acetate/hexane, 1:1), and analyzed by LC-MS/MS. 10R-HODE and 8R-HODE were also oxidized with mCPBA, and the products were analyzed by LC-MS/MS. [8R-2H]18:2n-6 was synthesized as described (3). R and S stereoisomers of 12-, 11-, and 8-HETE were from Cayman. [2H8]15S-HETE and [2H4]13S-HPODE were prepared with soybean lipoxygenase (lipoxidase, type IV; Sigma), and 13R-HPODE, 13S-HPETE, and 15R-HPETE with manganese lipoxygenase (30) (supplemental material).
Cloning and Expression—Total RNA was isolated from nitrogen powder of A. fumigatus (Fres.), which was suspended in phenol (1 volume), and 0.1 m LiCl, 0.1 m Tris-HCl (pH 8.0), 10 mm EDTA, 1% SDS (1 volume), 4 m LiCl (1 volume) was added to aqueous phase, which was stored overnight at 4 °C. RNA was precipitated by centrifugation and purified by phenol/chloroform extraction. cDNA was generated by the enhanced avian RT-PCR kit (8). The cDNA of 10R-DOX was amplified by PCR in two partly overlapping fragments of 1447 bp (nucleotides 1-1443) and 2055 bp (nucleotides 1298-3366), respectively, covering the entire reading frame, using Phusion® DNA polymerase and two pairs of primers (forward 5′-cataatgttgcggaggttctc and reverse 5′-gaacgtgcgcttggatggatc, and forward 5′-ctagcgccaccgatgagaaatg and reverse 5′-gcttaggcatcgtactggacac). The fragments were ligated into pJET1/blunt (pJET1_10-DOX-(1-1443); pJET_10-DOX-(1298-3366)). A KpnI restriction site was introduced (ggtaccataATG) by PCR technology into pJET1_10-DOX-(1-1443). The two fragments were ligated sequentially (KpnI-BamHI and BamHI-XbaI) in pIZ/V5-His (generating pIZ_10-DOX). The stop codon was removed by site-directed mutagenesis so that the His tag of the expression plasmid came in-frame with expressed protein (yielding pIZ/V5-His_10-DOX); both plasmids were used for expression. The expression vector for 7,8-LDS, pIZ/V5-His_LDS, was described previously (26).
Site-directed Mutagenesis—Site-directed mutagenesis of pIZ_10-DOX and pIZ/V5-His_LDS was performed with Pfu DNA polymerase and oligonucleotides of 44-46 bases (supplemental Table 2S), as described by the QuikChange manual (Stratagene) with minor modifications (31). All constructs were confirmed by sequencing.
Expression of 10R-DOX, 7,8-LDS, and Their Mutants in Insect Cells—Plasmid-driven expression was performed by transfection of Sf21 cells with pIZ_10-DOX, pIZ/V5-His_10-DOX, and pIZ/V5-His_LDS, using Cellfectin as described (26). Cells were harvested after 48 h, suspended in lysis buffer (50 mm KHPO4 (pH 7.4), 1 mm EDTA, 1 mm GSH, 5% glycerol, 0.04% Tween 20), and sonicated (Branson Sonifier cell disrupter B15, maximum power 20%, three times with 5-s pulses at 4 °C). The cell debris was spun down (15,000 × g, 30 min; 4 °C), and the supernatant was used for enzyme assay. Expression of proteins with His tags was detected by Western blot analysis (26).
Enzyme Assay—Recombinant 10R-DOX, 7,8-LDS, and their mutants were incubated with 100 μm of unsaturated fatty acids for 30 min on ice. The reaction (0.2-0.5 ml) was terminated with ethanol (3-4 volumes), and an internal standard was added in some experiments, and proteins were precipitated by centrifugation. The metabolites were extracted on octadecyl silica (SepPak/C18), evaporated to dryness, and diluted in the mobile phase or methanol (20-40 μl), and 5-10 μl was subject to LC-MS/MS analysis. TPP was used to reduce hydroperoxides to alcohols. Oxygenation rates of 100 μm fatty acids by 10R-DOX and kinetic parameters of 18:2n-6 were determined in triplicate by LC-MS/MS (supplemental material).
HPLC-MS/MS Analysis—A multiple-stage linear ion trap mass spectrometer (LTQ, ThermoFisher) with the Surveyor MS pump and Surveyor autosampler was used with electrospray ionization and monitoring of negative ions. The electrospray needle was set at 4.5 kV; the temperature of the heated capillary was 315 °C, and PGF1α was used for tuning. Data were analyzed by the Xcalibur software. The RP-HPLC column contained octadecyl silica (5-μm, 150 × 2 mm) and was eluted with methanol/water/acetic acid, 80:20:0.01 or 75:25:0.01, at 0.3 ml/min (3, 28). For quantification we used the ratio of signal intensities of the carboxylate anions of the products and the internal standard ([2H4]13-HODE or [2H8]15-HETE) and measured the area under the peaks from experiments done in triplicate.
Epoxyalcohols and hydroxy fatty acids were separated on NP-HPLC (isopropyl alcohol/hexane/acetic acid, 3:97:0.02 and 2:98:0.02, respectively) at 0.5 ml/min, and the column effluent was mixed in a T junction with isopropyl alcohol/water, 3:2, at 0.3 ml/min, for MS/MS analysis (32). CP-HPLC-MS/MS analysis of 8-HODE and 8-, 11-, 12-, 13-, and 15-HETE was performed with Chiralcel® OB-H (250 × 4.6 mm; Daicel Chemical Industries, Ltd.; isopropyl alcohol/hexane/acetic acid, 5:95:0.01 (cf. Ref. 28). 10-HODE and 10-HETE were analyzed using Reprosil Chiral-NR (250 × 2 mm; 1.2-3% isopropyl alcohol in hexane with 0.01% acetic acid) (28). 10-HETE and 13-HETE were also analyzed on Chiralcel OD-H (250 × 4.6 mm; isopropyl alcohol, hexane, 0.01% acetic acid, 95:5:0.01), as the elution orders of the enantiomers are known on this column (33). Finally, stereoisomers of HODE and HETE were also resolved on amylose tris(3,5-dimethylphenylcarbonate) coated on silica (10 μm; 2 × 250 mm; Reprosil Chiral-AM) eluted at 0.15 ml/min with hexane/ethanol/acetic acid, 95:5:0.02.
RESULTS
Expression of 10R-DOX—The predicted sequence of 10R-DOX of A. fumigatus from genome analysis is available at GenBank™ (accession number XM_749316). We cloned 10R-DOX by RT-PCR and sequenced 3366 bp (GenBank™ accession number ACL14177). A comparison with the predicted gene sequence confirmed 13 exons and 12 short introns (supplemental material).
Recombinant 10R-DOX was successfully expressed in insect cells (Sf21) using the plasmid-driven expression method. Two different constructs were utilized, one with the native stop codon (pIZ_10-DOX) and one without but with 34 additional C-terminal amino acids (V5 epitope and His6 tag; pIZ/V5His_10-DOX; see supplemental material). Both recombinant enzymes appeared to oxygenate 18:2n-6 to the same products, as described below. Expression of 10R-DOX with the His tag was confirmed by Western blot analysis (Fig. 2). Low speed supernatant of untransformed Sf21 cells did not convert 18:2n-6 to any of these metabolites, and we could therefore use the 10R-DOX activity as evidence of enzyme induction.
FIGURE 2.
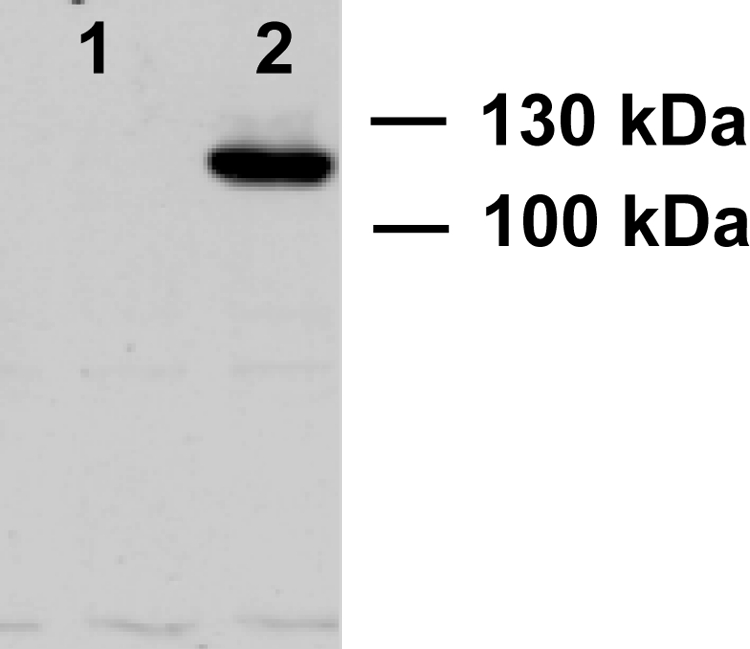
Western blot analysis of recombinant 10R-DOX with a C-terminal His tag. Lane 1, protein extract of untransformed Sf21 cells; lane 2, protein extract of Sf21 cells transformed with pIZ/V5His_10R-DOX for expression of 10R-DOX with a His tag. The Western blot signal was obtained at a slightly smaller apparent molecular weight than expected (∼130 kDa).
Substrate and Product Specificity of 10R-DOX—The natural substrates of 10R-DOX and LDS are likely C18 fatty acids (34). Recombinant 10R-DOX metabolized 18:2n-6 mainly to 10R-HODE (89.6%) and to 8R-HODE (10.4 ± 2.0% (S.D.), n = 11), as judged from LC-MS analysis. Incubation with [(8R)-2H]18:2-6 confirmed pro-S hydrogen abstraction at C-8, as described for native 10R-DOX (3) (data not shown). In addition, ODA (3) and small amounts of epoxyalcohols were formed, as described below. 18:3n-3 (n = 6) was oxidized to the same profile of metabolites as 18:2n-6 (91% 10-hydroxyoctadeca-8E,12Z,15Z-trienoic acid and 9% 8-hydroxyoctadeca-9Z,12Z,15Z-trienoic acid). The 12-13 double bonds of 18:2n-6 and 18:3n-3 appeared to be essential for the oxygenation at C-10, as 18:1n-9 (n = 4) was mainly oxidized at C-8 (96%) and only to small amounts of 10-hydroxyoctadecenoic acid.
The oxidation of 100 μm 18:2n-6 was linear for at least 30 min on ice. The apparent Km was 0.05 mm, as judged from triplicate LC-MS analysis with [2H8]15S-HETE as an internal standard with monitoring of the area under the carboxylate anions of the reconstructed ion chromatograms (supplemental material). By this method, 100 μm 18:3n-3 (n = 3) was found to be oxidized as rapidly as 100 μm 18:2n-6 (n = 3). 18:1n-9 (n = 2) was oxidized at a rate of ∼40% of 18:2n-6. Metabolites of 18:3n-6 could not be detected.
We next examined whether fatty acids are bound tail or head first to the active site, as this is important information for comparison with 7,8-LDS and PGHS. The results are summarized in Table 1. Both 16:1n-7 and C20 fatty acids were oxygenated by 10R-DOX in agreement with a tail first model. The double bonds of C18 and C20 fatty acids influenced the relative oxygenation at C-8/C-10 and C-10/C-12, respectively (Table 1). 20:3n-3 (n = 2) was mainly oxidized at C-10 (n-11) and 20:4n-6 (n = 6) and at C-12 (n-9), whereas 20:2n-6 (n = 2) was oxidized equally at C-10 (n-11) and C-12 (n-9).
TABLE 1.
Oxidation of unsaturated fatty acids and vernolic acids at the n-9 and n-11 positions by recombinant 10R-DOX
The following abbreviations are used: HEDE, hydroxyeicosadienoic acid; HETrE, hydroxyeicosatrienoic acid; HHME, hydroxyhexadecenoic acid; HOME, hydroxyoctadecenoic acid; 10-HOTrE, 10-hydroxyoctadeca-8E,12Z,15Z-trienoic acid; 8-HOTrE, 8-hydroxyoctadeca-9Z,12Z,15Z-trienoic acid.
| Substrates | Hydroxy metabolitesa | |||
|---|---|---|---|---|
| % | % | |||
| 16:1n-7 | 8-HHME | 81 | 10-HHME | 19 |
| 18:1n-9Z | 8-HOME | 96 | 10-HOME | 4% |
| 18:2n-6 | 8-HODE | 10 | 10-HODE | 90 |
| 18:3n-3 | 8-HOTrE | 9 | 10-HOTrE | 91 |
| 20:2n-6 | 10-HEDE | 53 | 12-HEDE | 47 |
| 20:3n-3 | 10-HETrE | 73 | 12-HETrE | 27 |
| 20:4n-6b | 10-HETE | 29 | 12-HETE | 71 |
| 12S(13R)-epoxy-9Z-18:1 | 12S(13R)-Epoxy-8R-hydroxy-9Z-18:1 | 80% | ||
| 12R(13S)-epoxy-9Z-18:1 | 12R(13S)-Epoxy-10R-hydroxy-8E-18:1 | >95 | ||
Biosynthesis of the metabolites was estimated from the ion intensities of characteristic ions during HPLC-MS/MS analysis.
20:4n-6 was also transformed to 8-, 11-, 13-, and 15-HETE (Fig. 3B).
Hydrogen abstraction also occurred at C-13 of 20:2n-6 and 20:4n-6, as judged from formation of cis-trans-conjugated metabolites (supplemental material). Untransformed insect cells did not oxidize 20:4n-6. The profile of HETEs first suggested nonenzymatic formation, but MS/MS analysis revealed that 10-HETE and 13-HETE were formed and in the same order of magnitude as cis-trans-conjugated HETEs, e.g. 15-HETE (Fig. 3A). We confirmed biosynthesis from exogenous 20:4n-6 by experiments with [2H8]20:4n-6.
FIGURE 3.

HPLC-MS/MS analysis of HETEs formed by recombinant 10R-DOX and 20:4n-6. A, RP-HPLC-MS/MS analysis. Top trace, total iron current of m/z 319 → full scan; 13-, 10-, and 15-HETEs were detected by MS/MS analysis with monitoring characteristic ions (13-HETE, m/z 193; 10-HETE, m/z 181; 15-HETE, m/z 175) (44, 45), as shown, and identified by their MS/MS spectra (data not shown). B, NP-HPLC separation and MS/MS analysis (m/z 319 → full scan) of HETEs formed by 10R-DOX. The HETEs eluted as marked.
The products were separated by NP-HPLC into six HETEs (Fig. 3B). Steric analysis showed that the S stereoisomers of 8-, 10-, 11-, and 13-HETE were formed in access over the R stereoisomers (Fig. 4). This was also confirmed by CP-HPLC analysis using Chiralcel® OD-H (with known elution order of the enantiomers of 10- and 13-HETE (33)), Reprosil Chiral-AM, and by preparation of 13S-HPETE (supplemental material). The corresponding HPETEs were also detected in some experiments, but significant amounts 5-H(P)ETE and 9-H(P)ETE could not be detected. The rate of total oxidation of 100 μm 20:4n-6 was estimated after reduction of hydroperoxides to alcohols to be ∼50% of the rate of oxidation of 100 μm 18:2n-6. We conclude that hydrogen abstraction occurred at C-10 and C-13 of 20:4n-6, and this could account for formation of all six metabolites (cf. Ref. 33).
FIGURE 4.

CP-HPLC-MS/MS analysis of metabolites formed by 10R-DOX from 20:4n-6. A, analysis of 10-, 8-, and 12-HETE. Top trace, analysis of 10-HETE (Reprosil Chiral-NR column); middle and bottom traces, analysis of 8- and 12-HETE (Chiralcel® OB-H column), respectively. The enantiomers of 10-HETE were resolved in the reverse order on Chiralcel® OD-H (cf. Ref. 33). B, analysis of 13-HETE (top), 11-HETE (middle), and 15-HETE (bottom) on Chiralcel® OB-H. MS/MS analysis (m/z 311 → characteristic ions of each HETE as follows: 13-, 10-, and 15-HETE, as in Fig. 3; 8-HETE, m/z 155; 11-HETE; m/z 167; 12-HETE, m/z 179) (44, 45). The enantiomers of the HETEs in A and B (except 10-HETE) were also separated on Reprosil Chiral-AM with the same results.
Site-directed Mutagenesis of the Dioxygenation Site of 10R-DOX—We first examined the importance of Leu-384 for the 10R-DOX activity, as 7,8-LDS with 8R-DOX activity has a valine residue in this position (Fig. 1). Replacement of leucine with two smaller hydrophobic residues, valine or alanine, resulted in an augmented formation of 8-HPODE in comparison with 10-HPODE, as judged from analysis of the corresponding alcohols. The relative biosynthesis of 8-HODE (8-HODE/(10-HODE + 8-HODE)) was 10.4 ± 2.0% (S.D.) for the recombinant enzyme, 22.1 ± 2.2% for the L384V mutant (n = 6; p < 0.05, sign test), and 53.7 ± 5.0% for the L384A mutant (n = 6; p < 0.05), as judged from LC-MS/MS analysis (Fig. 5A).
FIGURE 5.

Relative biosynthesis of 8R-HODE by 10R-DOX and 7,8-LDS. A, biosynthesis by recombinant 10R-DOX, L384M, L384V, and L384A. B, biosynthesis by recombinant 10R-DOX and by V388L and V388F. C, biosynthesis by recombinant 7,8-LDS and by V330L. The amino acids at studied positions are inserted over the bars. Untransfected insect cells transformed 18:2n-6 to negligible amounts of HODE (data not shown).
The L384A mutant changed the stereochemistry at C-8 and C-10. CP-HPLC-MS/MS analysis showed that L384A formed the R and S enantiomers of 8-HODE and 10-HODE in a ratio of ∼3:2 (Fig. 6), whereas native and recombinant 10R-DOX form both products with >95% R configuration (28). Replacement of Leu-384 with methionine increased the relative biosynthesis of 8-HODE significantly (n = 6, p < 0.05; sign test), although only with 3-4% units. Finally, we prepared the L384F mutant. The relative biosynthesis of 8-HODE was 48 ± 0.06% (n = 3). This mutant did apparently not shield C-8 for oxygenation. NP- and CP-HPLC separation and MS/MS showed that with this bulky replacement significant amounts of racemic 9- and 13-HODE also appeared to be formed (about 20% of 10-HODE); 10- and 8-HODE retained their R chirality (>90% R).
FIGURE 6.

Chiral-HPLC-MS/MS analysis of metabolites formed by the L384A mutant of 10R-DOX. A, analysis of 8-HODE by separation of the 8S and 8R enantiomers on Chiralcel® OB-H (28). B, analysis of 10-HODE by separation of the 10S and 10R enantiomers on Reprosil Chiral-NR (28).
The position of Val-388 was also of catalytic importance. V388L increased the relative biosynthesis of 8-HODE from 10.4 ± 2 to 16.2 ± 1.1% (n = 5; Fig. 5B and supplemental material) and V388F increased it to 34 ± 1.5% (n = 3). NP-HPLC-MS/MS analysis showed that the latter also increased the relative formation of 9- and 13-HODE (about 80% of 10-HODE), suggesting that this bulky residue may have profoundly changed the oxygenation site. CP-HPLC showed that 9- and 13-HODE were racemic; 8- and 10-HODE retained R chirality (>90% R).
We also prepared the 10R-DOX double mutant, L384A/V388L. This mutant formed almost the same relative amounts of 8R-HODE (49 ± 7%; n = 4) as the L384A mutant. Steric analysis showed that 8- and 10-HODE were both formed with reduced stereospecificity (supplemental material). We conclude that amino acids in the positions 384 and 388 can be crucial for the position of dioxygenation and can affect the absolute configuration of 8- and 10-HPODE.
Comparison with Site-directed Mutagenesis of 7,8-LDS—Val-330 is homologue to Leu-384 of 10R-DOX (Fig. 1). V330L oxidized 18:2n-6 at C-10 5.5% and at C-8 94.5 ± 0.3% (S.D.; n = 6; p < 0.05), whereas recombinant 7,8-LDS consistently formed 98.5 ± 0.3% 8-HODE (Fig. 5C). Replacement of Val-330 with a bigger hydrophobic residue, methionine, reduced the dioxygenase activity so that only traces of 8R-HODE could be detected (data not shown; n = 7). Replacement with a smaller hydrophobic residue, alanine, resulted in an inactive enzyme (n = 6; supplemental material).
Leu-334 is homologue to Val-388 of 10R-DOX (Fig. 1). Mutation of Leu-334 to valine in 7,8-LDS reduced the dioxygenase activity below the detection limit (n = 6).
Alignment of 7,8-LDS and 10R-DOX sequences around the proximal heme ligand showed several conserved differences. Ile-375 and Glu-384 were conserved in the 7,8-LDS sequences but replaced with Ala in 10R-DOX sequences (Fig. 1). These residues could conceivably be important for the position of oxygenation. I375A of 7,8-LDS (n = 3) had virtually no dioxygenase activity, whereas E384A (n = 5) formed only traces of 8-HODE and 10-HODE and in the same ratio as the native enzyme (LC-MS/MS analysis). These positions were therefore not further investigated. Satisfactory expression of recombinant 7,8-LDS was confirmed by Western blot analysis of all mutants described above (supplemental material).
Detection of EAS Activity of 10R-DOX—Recombinant 10R-DOX metabolized 18:2n-6 to ∼90% 10R-HPODE and ∼10% of 8R-HPODE, as estimated by LC-MS/MS analysis (Fig. 7A). In addition, more polar products were also formed, albeit in small amounts (∼1%). This may nevertheless be of interest. Recombinant 7,8-LDS has weak hydroperoxide isomerase activity, and the hydroperoxide isomerase activity of purified native 7,8-LDS varies considerably (2). This could also apply to 10R-DOX. The minor metabolites were therefore identified. The reconstructed ion chromatogram (Fig. 7B; MS/MS analysis with monitoring of m/z 311 → full scan) revealed elution of metabolites after 4 min (peak I) and a smaller amounts of metabolites on its right shoulder (peak II). They were further separated by NP-HPLC into one major and several minor metabolites (Fig. 8).
FIGURE 7.

MS/MS analysis of the oxidation of 18:2n-6 by recombinant 10R-DOX. A, reconstructed ion chromatogram from RP-HPLC-MS/MS analysis of hydroxy fatty acids (m/z 295 → full scan) with monitoring m/z 157 and m/z 183 for 8R-HODE and 10R-HODE, respectively. NL 1E6, normalized ion intensity to 1 × 106. B, reconstructed ion chromatogram from RP-HPLC-MS/MS analysis (m/z 311 → full scan) of dioxygenated metabolites. Peak I, 12(13)-epoxy-10R-hydroxy-8E-18:1, peak II, 8(9)-epoxy-10R-hydroxy-12Z-18:1 and 9(10)-epoxy-8R-hydroxy-12Z-18:1; and peak III, HPODE. C, MS/MS spectrum of carboxylate anion of 12(13)-epoxy-10R-hydroxy-8E-18:1. The inset shows formation of characteristic fragments.
FIGURE 8.

LC-MS/MS analysis of metabolites formed from 18:2n-6 and vernolic acids by 10R-DOX. A, NP-HPLC-MS/MS analysis (m/z 311→ full scan) of metabolites of 18:2n-6; peaks I and II contained 8(9)-epoxy-10-hydroxy-12Z-18:1; peak III contained 9(10)-epoxy-8-hydroxy-8E-18:1 (the second isomer of this compound eluted between peaks IV and V); and peaks IV and V contained stereoisomers of 12(13)-epoxy-10R-hydroxy-8E-18:1. B, RP-HPLC-MS/MS analysis (m/z 311→ full scan) of metabolites formed from vernolic acids by 10R-DOX. Top trace shows that (-)-vernolic acid was oxidized to a major metabolite, 12R(13S)-epoxy-10R-hydroxy-8E-18:1. The inset shows that small amounts of a stereoisomer (peak I) was also formed, but 12R(13S)-epoxy-10R-hydroxy-8E-18:1 eluted after this isomer in peak II (cf. peaks IV and V in A). The bottom trace shows RP-HPLC analysis of metabolites formed (+)-vernolic acid, and the main product was 12S(13R)-epoxy-8R-hydroxy-9Z-18:1 (supplemental material). These results suggested that the material in peaks IV and V of the chromatogram in A consisted of 12S(13R)-epoxy-10R-hydroxy-8E-18:1 and 12R(13S)-epoxy-10R-hydroxy-8E-18:1, respectively.
The MS/MS spectrum (m/z 311 → full scan) of the major metabolite showed signals, for example at m/z 293 (A- -18), m/z 275 (A- -2 × 18), m/z 267(A- -44), m/z 211 (possibly -OOC-(CH2)6-CH=CH-CH(OH)-CH=CH2), m/z 183 (possibly -OOC-(CH2)6-CH=CH-COH), m/z 181 (A- -130, possibly C=CH-CH(O-)-CH2-CH-(-O-)-CH-C5H11), m/z 155 (183-28, possibly loss of CO) and in the lower mass range at m/z 139 (possibly 183-44), and m/z 137 (Fig. 7C). The spectrum of the U-13C-labeled metabolite supported the fragmentation mechanism with informative signals at m/z 192 (likely 181 + 11), m/z 193 (183 + 10), m/z 164 (155 + 9), and m/z 148 and m/z 146. The fragmentation suggested that 12(13)-epoxy-10R-hydroxy-8E-18:1 was formed; based on the stereospecificity of 10R-DOX, we can assume that the hydroxyl group has 10R configuration.
12(13)-Epoxy-10R-hydroxy-8E-18:1 was separated by NP-HPLC into a major (peak IV, 80-90%) and minor stereoisomer (Fig. 8A, peak V). To investigate the absolute configuration of these isomers, we used vernolic acids as substrates of 10R-DOX (Fig. 8B; Table 1). 12R(13S)-Epoxy-9Z-18:1 was oxidized at C-10 to 12R(13S)-epoxy-10R-hydroxy-8E-18:1 (>95%), whereas 12S(13R)-epoxy-9Z-18:1 acid was mainly transformed to ∼80% 12S(13R)-epoxy-8-hydroxy-9Z-18:1 and ∼20% 12S(13R)-epoxy-10R-hydroxy-8E-18:1 (Fig. 8B). The former eluted as the second isomer during NP-HPLC (inset chromatogram in Fig. 8B). Because the stereoisomer formed by recombinant 10R-DOX likely has R configuration at C-10, we conclude that the major isomer consisted of 12S(13R)-epoxy-10R-hydroxy-8E-18:1. Recombinant 10R-DOX thus possessed EAS activity, but hydroperoxide isomerase activity was not detected.
The minor products (Fig. 8, peaks I-III) were epoxyalcohols derived from 8R-HPODE and 10R-HPODE. Because of Payne rearrangement in the mass spectrometer, 9(10)-epoxy-8-hydroxy-12Z-18:1 and 8(9)-epoxy-10-hydroxy-12Z-18:1 compounds will give identical mass spectra (32). 12(13)-Epoxy-10R-hydroxy-8E-18:1 and 8(9)-epoxy-10R-hydroxy-12Z-18:1 were obtained by epoxidation of 10R-HODE and 12(13)-epoxy-8R-hydroxy-9Z-18:1 and 9(10)-epoxy-8R-hydroxy-12Z-18:1 by epoxidation of 8R-HODE. The two isomers of 8(9)-epoxy-10-hydroxy-18:1 eluted before the isomers of 9(10)-epoxy-8-hydroxy-18:1 on NP-HPLC (supplemental material). The material of peak II (cf. Fig. 7B) was resolved into these four compounds by NP-HPLC, and the pairs of stereo isomers were formed in equal amounts and thus appeared to be racemic. The MS/MS spectra (m/z 311 → full scan) of the 8(9)-epoxy-10R-hydroxy-12Z-18:1 compounds were as reported previously (32).
12S(13R)-epoxy-10R-hydroxy-8E-18:1 co-elutes with 5,8-DiHODE on RP-HPLC, and many ions are identical in their MS/MS spectra. Re-examination of products formed from 18:2n-6 by mycelia of A. fumigatus and A. nidulans confirmed that 12(13)-epoxy-10-hydroxy-18:1 was formed.3
Site-directed Mutagenesis of the Distal Heme Side of 10R-DOX—Leu-306 of 10R-DOX is homologous to Val-291 of PGHS, which is one of the hydrophobic residues forming the dome over the distal heme region of PGHS (27). Valine residues of 7,8-LDS and 5,8-LDS are found in the homologous position of Leu-306 (Fig. 1). We hypothesized that mutation of Leu-306 to smaller hydrophobic residues might influence the EAS activity or lead to biosynthesis of DiHODE, but neither 5,8-DiHODE nor 7,8-DiHODE could be detected, and the EAS activity appeared to be unchanged. The L306A and L306V mutants may nevertheless affect the oxygenation, as the relative biosynthesis of 8R-HODE appeared to be slightly lowered (5.2 ± 1.2% (n = 5) and 6.7 ± 1.4% (n = 5), respectively; supplemental material), but this was not further investigated.
DISCUSSION
We have expressed 10R-DOX and studied its 10R-DOX and 8R-DOX activities with different fatty acid substrates and by site-directed mutagenesis. Our study revealed two important amino acids for the 10R-DOX and 8R-DOX activities, Leu-384 and Val-388. Our results showed similarities with replacement of the two homologous amino acids of active site of PGHS-1 (Val-349 and Ser-353). Recombinant 10R-DOX lacked hydroperoxide isomerase activity but possessed low EAS activity with biosynthesis of a novel oxylipin, 12S(13R)-epoxy-10R-hydroxy-18:1. In analogy with PGHS and LDS, 10R-DOX thus possesses dual enzyme activities.
We first examined 10R-DOX with different substrates to determine the orientation of the substrates at the active site. Oxygenation of 18:2n-6 and 18:3n-3 mainly occurred at C-10 (position n-9; ∼90%) but also at C-8 (position n-11; ∼10%). The oxygenation of C16-C20 fatty acids suggests that they enter the site of oxygenation tail first, as hydrogen abstraction apparently occurred at the n-11 position of C18 fatty acids and the n-9 position of C16:1n-7 (Table 1). Insertion of molecular oxygen then occurred at positions n-11 and n-9.
The C20 fatty acids were subject to hydrogen abstraction both at C-10 and C-13, as judged from the metabolites reduced to alcohols. 20:4n-6 was transformed by 10R-DOX to four cistrans-conjugated HETEs (8S-, 11S-, 12-, and 15-HETE) and to two bis-allylic metabolites, 13-HETE (∼65% S) and 10-HETE (∼65% S; the stereo configuration in analogy with 10R-DOX products of 18:2n-6; the designation S is because of the nomenclature rules). 10- and 13-HETE are acid-labile and can be transformed to 8-, 11-, 12- and 15-HETE nonenzymatically (33). Prostaglandin and leukotriene biosynthesis has been described by A. fumigatus (9, 35), but the mechanism of biosynthesis has not been identified. 10R-DOX may contribute to prostaglandin biosynthesis, but oxygenation of 20:4n-6 generated the 11S-peroxyl radical, which can be isomerized to isoprostanes but not to the classical prostaglandins (36).
Why does 10R-DOX favor C-10 of 18:2n-6 for oxygenation after hydrogen abstraction at C-8? It seems reasonable that C-8 is in proximity of Tyr-427 (cf. Fig. 1 and Fig. 9), which abstracts the pro-S hydrogen and forms the carbon-centered radical with electron density over C-8 to C-10. Dioxygen apparently then has only limited access to C-8 and will react with C-10. Conversely, the oxygen channel of LDS may lead to C-8 with little access to C-10. Can this difference be explained by substitution of single amino acids during evolution?
FIGURE 9.

An oxygenation model of 10R-DOX based on the conserved motif Tyr-Leu-(Xaa)3-Val, the presumed catalytically important Tyr residue for hydrogen abstraction, and a tentative oxygenation channel. Hydrogen is likely abstracted by Tyr-427, and O2 reacts with the carbon-centered radical at C-10 (∼90%) and at C-8 (∼10%). The radical at C-8 is partly shielded from O2 by Leu-384, but smaller hydrophobic residues (valine and alanine) in this position increase the oxygenation at C-8. Conversely, replacement of Val-388 with larger hydrophobic residues (leucine and phenylalanine) increase oxygenation at C-8, possibly by shielding C-10 from O2. The bulky phenylalanine residue may also induce other steric effects, as both L384F and V388F increased the relative biosynthesis of 8-, 9-, and 13-HODE to 10-HODE. The corresponding conserved motif of PGHS is Tyr-Val-(Xaa)3-Ser, and the three amino acids are important for the cyclooxygenase reaction. The LDS motif is Tyr-Val-(Xaa)3-Leu. Replacement of Tyr-329 with phenylalanine reduces the catalytic activity of 7,8-LDS (18), whereas V330L increases the relative 10R-DOX activity, albeit modestly. Unfortunately, V330M, V330A, and L334V of 7,8-LDS were inactive. These mutants emphasize the steric importance of this motif for catalytic activity.
LDS and 10R-DOX can be aligned with 42-48% amino acid identity. A conserved motif, Tyr-Val-(Xaa)3-Leu, which occurs in two 7,8-LDS and five 5,8-LDS sequences, corresponds to the conserved motif Tyr-Leu-(Xaa)3-Val in 10R-DOX sequences of five Aspergilli (and to Tyr-Val-(Xaa)3-Ser in PGHS). We found that these leucine and valine residues were critical for the oxygenation at C-10 and C-8 of 18:2n-6. The L384V and L384A mutants of 10R-DOX increased the relative oxygenation at C-8 from 10% (native recombinant 10R-DOX) to 22 and 53%, respectively, and led to biosynthesis of 8- and 10-HODE with less R stereoselectivity. Conversely, replacement of the homologue Val-330 of 7,8-LDS by a leucine residue increased the oxygenation at C-10 by 7,8-LDS 3-fold, from 2 to 6%.
Val-388 of 10R-DOX also affected oxygenation. Replacement of Val-388 with leucine (the homologue in 7,8-LDS) and with phenylalanine increased the relative oxygenation at C-8 from 10-16 and 36%, respectively. The bulky phenylalanine residue as position 388 may have additional steric effects as judged from prominent formation of other products (9- and 13-HODE), and this was also the case with L384F.
The results of our replacement experiments with 10R-DOX can be visualized in a model of the conserved motif Tyr-Leu-(Xaa)3-Val and a tentative oxygen channel with access to C-10 (90%) and partially to C-8 (10%) of 18:2n-6 (Fig. 9). Leu-384 could be partly shielding C-8 for oxygen, and Val-388 could delineate the channel near C-10. Replacement of Leu-384 with smaller hydrophobic residues (valine, alanine) increased oxygen access to C-8, whereas replacement of Val-388 with larger hydrophobic residues (leucine and phenylalanine) shielded C-10 and thus augmented the relative oxygenation at C-8. L384F was expected to yield mainly 10R-DOX activity, but this bulky residue may have induced other structural changes. Three-dimensional structural analysis will be needed to accommodate all information into a comprehensive model. It is therefore interesting to compare these results with replacements of the corresponding residues of PGHS, valine and serine, where three-dimensional information is available.
Replacement of Val-349 of PGHS-1 affects the stereospecific oxygenation of C-11 and C-15 of 20:4n-6, and replacement of Ser-353 influences both the cyclooxygenase and peroxidase activities (22, 23, 37, 38). Previous work has also demonstrated that the tyrosine residue in this motif, Tyr-(Leu/Val)-(Xaa)3-(Val/Leu/Ser), is important for oxygenation by 7,8-LDS and by PGHS (18, 38). This tyrosine residue is in close contact with Tyr-385 of PGHS-1. The corresponding residues of 10R-DOX are Tyr-383 and Tyr-427 (Fig. 9). We conclude that residues of the conserved motif Tyr-(Leu/Val)-(Xaa)3-(Val/Leu/Ser) can be important for correct positioning of 18:2n-6 and 20:4n-6 for oxygenation by 10R-DOX and PGHS.
10R-DOX also possessed weak EAS activity and oxidized 18:2n-6 to 12S(13R)-epoxy-10R-hydroxy-8Z-18:1 with 80-90% stereospecificity. Biosynthesis of 12(13)-epoxy-10-hydroxy-8Z-18:1 was also detected in mycelia of A. fumigatus and may therefore be an inherent property of the enzyme. An intriguing phenomenon is the variable hydroperoxide isomerase activities of native and recombinant 7,8-LDS (2, 18, 26). The EAS activity of native 10R-DOX is therefore worthy of additional studies.
The peroxidase site of PGHS forms a hydrophobic dome over the distal site of the heme with the sequence Val-Phe-Gly-Leu-(Leu/Val) (27). Replacement of Val-291 with alanine had only little effect on PGHS, as judged from its cyclooxygenase and peroxides activity (27). The Val-291 homologue of 10R-DOX is Leu-306, as judged from the alignment illustrated in Fig. 1. We found that L306A and L306V retained the EAS activity of recombinant 10R-DOX. It seems possible that some of the other hydrophobic residues could be important for the peroxidase and EAS activities.
What are the biological functions of 10R-HPODE and related compounds? Recent gene deletion studies suggest that 10R-DOX of A. fumigatus can affect germination, the size of conidia, and influence the pathogenicity in cellular models of aspergillosis (4, 6, 39). Oxygenation of 20:4n-6 by 10R-DOX is of interest in this context. We could detect ODA from incubations of 18:2n-6 with recombinant 10R-DOX and mycelia of A. fumigatus (3). ODA, and the volatile 1-octen-3-ol, can likely be formed nonenzymatically from 10R-HPODE. Mushrooms can form 10S-HPODE (3, 40), and 10S-HPODE can be transformed to ODA and to 1-octen-3-ol enzymatically (40, 41). The latter gives a characteristic mushroom flavor and elicited conidiation in Trichoderma spp. (42).
In summary, we have expressed 10R-DOX with EAS activity and found that its 10R- and 8R-dioxygenase activities can be modified in a logical way by replacements of two hydrophobic residues in a common conserved motif of 10R-DOX, 7,8-LDS, and PGHS, viz. Tyr-(Leu/Val)-(Xaa)3-(Val/Leu/Ser). Replacement of hydrophobic resides in this motif also changed or abolished the 8R-DOX activity of 7,8-LDS.
Supplementary Material
This work was supported by VR Medicine Grant 03X-06523, FORMAS Grant 222-2005-1733, and by Knut and Alice Wallenberg Foundation Grant 2004.0123.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables 1S and 2S and Figs. 1S-11S.
Footnotes
The abbreviations used are: LDS, linoleate diol synthase; 18:1, octadecenoic acid; COX, cyclooxygenase; CP, chiral phase; DiHODE, dihydroxyoctadecadienoic acid; DOX, dioxygenase; EAS, epoxyalcohol synthase; HETE, hydroxyeicosatetraenoic acid; HODE, hydroxyoctadecadienoic acid; HPETE, hydroperoxyeicosatetraenoic acid; HPODE, hydroperoxyoctadecadienoic acid; LC-MS, liquid chromatography-mass spectrometry; mCPBA, m-chloroperoxybenzoic acid; ODA, 10-oxo-8E-decenoic acid; PG, prostaglandin; PGHS, prostaglandin H synthase; NP, normal phase; RP, reversed phase; HPLC, high pressure liquid chromatography; RT, reverse transcription.
U. Garscha and E. H. Oliw, unpublished observations.
References
- 1.Daiyasu, H., and Toh, H. (2000) J. Mol. Evol. 51 433-445 [DOI] [PubMed] [Google Scholar]
- 2.Su, C., and Oliw, E. H. (1996) J. Biol. Chem. 271 14112-14118 [DOI] [PubMed] [Google Scholar]
- 3.Garscha, U., Jerneren, F., Chung, D., Keller, N. P., Hamberg, M., and Oliw, E. H. (2007) J. Biol. Chem. 282 34707-34718 [DOI] [PubMed] [Google Scholar]
- 4.Tsitsigiannis, D. I., and Keller, N. P. (2006) Mol. Microbiol. 59 882-892 [DOI] [PubMed] [Google Scholar]
- 5.Tsitsigiannis, D. I., Zarnowski, R., and Keller, N. P. (2004) J. Biol. Chem. 279 11344-11353 [DOI] [PubMed] [Google Scholar]
- 6.Dagenais, T. R., Chung, D., Giles, S. S., Hull, C. M., Andes, D., and Keller, N. P. (2008) Infect. Immun. 76 3214-3220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamberg, M., Ponce de Leon, I., Rodriguez, M. J., and Castresana, C. (2005) Biochem. Biophys. Res. Commun. 338 169-174 [DOI] [PubMed] [Google Scholar]
- 8.Hörnsten, L., Su, C., Osbourn, A. E., Garosi, P., Hellman, U., Wernstedt, C., and Oliw, E. H. (1999) J. Biol. Chem. 274 28219-28224 [DOI] [PubMed] [Google Scholar]
- 9.Tsitsigiannis, D. I., Bok, J. W., Andes, D., Nielsen, K. F., Frisvad, J. C., and Keller, N. P. (2005) Infect. Immun. 73 4548-4559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malkowski, M. G., Ginell, S. L., Smith, W. L., and Garavito, R. M. (2000) Science 289 1933-1937 [DOI] [PubMed] [Google Scholar]
- 11.Malkowski, M. G., Thuresson, E. D., Lakkides, K. M., Rieke, C. J., Micielli, R., Smith, W. L., and Garavito, R. M. (2001) J. Biol. Chem. 276 37547-37555 [DOI] [PubMed] [Google Scholar]
- 12.Kiefer, J. R., Pawlitz, J. L., Moreland, K. T., Stegeman, R. A., Hood, W. F., Gierse, J. K., Stevens, A. M., Goodwin, D. C., Rowlinson, S. W., Marnett, L. J., Stallings, W. C., and Kurumbail, R. G. (2000) Nature 405 97-101 [DOI] [PubMed] [Google Scholar]
- 13.Marnett, L. J., Rowlinson, S. W., Goodwin, D. C., Kalgutkar, A. S., and Lanzo, C. A. (1999) J. Biol. Chem. 274 22903-22906 [DOI] [PubMed] [Google Scholar]
- 14.Kurumbail, R. G., Stevens, A. M., Gierse, J. K., McDonald, J. J., Stegeman, R. A., Pak, J. Y., Gildehaus, D., Miyashiro, J. M., Penning, T. D., Seibert, K., Isakson, P. C., and Stallings, W. C. (1996) Nature 384 644-648 [DOI] [PubMed] [Google Scholar]
- 15.Smith, W. L., DeWitt, D. L., and Garavito, R. M. (2000) Annu. Rev. Biochem. 69 145-182 [DOI] [PubMed] [Google Scholar]
- 16.Tsai, A., Hsi, L. C., Kulmacz, R. J., Palmer, G., and Smith, W. L. (1994) J. Biol. Chem. 269 5085-5091 [PubMed] [Google Scholar]
- 17.Koeduka, T., Matsui, K., Akakabe, Y., and Kajiwara, T. (2002) J. Biol. Chem. 277 22648-22655 [DOI] [PubMed] [Google Scholar]
- 18.Garscha, U., and Oliw, E. H. (2008) FEBS Lett. 582 3547-3551 [DOI] [PubMed] [Google Scholar]
- 19.Koszelak-Rosenblum, M., Krol, A. C., Simmons, D. M., Goulah, C. C., Wroblewski, L., and Malkowski, M. G. (2008) J. Biol. Chem. 283 24962-24971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garavito, R. M., Malkowski, M. G., and DeWitt, D. L. (2002) Prostaglandins Other Lipid Mediat. 68-69 129-152 [DOI] [PubMed] [Google Scholar]
- 21.Cristea, M., Osbourn, A. E., and Oliw, E. H. (2003) Lipids 38 1275-1280 [DOI] [PubMed] [Google Scholar]
- 22.Schneider, C., Boeglin, W. E., Prusakiewicz, J. J., Rowlinson, S. W., Marnett, L. J., Samel, N., and Brash, A. R. (2002) J. Biol. Chem. 277 478-485 [DOI] [PubMed] [Google Scholar]
- 23.Thuresson, E. D., Lakkides, K. M., and Smith, W. L. (2000) J. Biol. Chem. 275 8501-8507 [DOI] [PubMed] [Google Scholar]
- 24.Valmsen, K., Boeglin, W. E., Jarving, I., Schneider, C., Varvas, K., Brash, A. R., and Samel, N. (2004) Eur. J. Biochem. 271 3533-3538 [DOI] [PubMed] [Google Scholar]
- 25.Harman, C. A., Rieke, C. J., Garavito, R. M., and Smith, W. L. (2004) J. Biol. Chem. 279 42929-42935 [DOI] [PubMed] [Google Scholar]
- 26.Garscha, U., and Oliw, E. (2008) Biochem. Biophys. Res. Commun. 373 579-583 [DOI] [PubMed] [Google Scholar]
- 27.Liu, J., Seibold, S. A., Rieke, C. J., Song, I., Cukier, R. I., and Smith, W. L. (2007) J. Biol. Chem. 282 18233-18244 [DOI] [PubMed] [Google Scholar]
- 28.Garscha, U., and Oliw, E. H. (2007) Anal. Biochem. 367 238-246 [DOI] [PubMed] [Google Scholar]
- 29.Oliw, E. H. (1983) J. Chromatogr. 275 245-259 [DOI] [PubMed] [Google Scholar]
- 30.Cristea, M., and Oliw, E. H. (2007) J. Lipid Res. 48 890-903 [DOI] [PubMed] [Google Scholar]
- 31.Cristea, M., Engström, Å., Su, C., Hörnsten, L., and Oliw, E. H. (2005) Arch. Biochem. Biophys. 434 201-211 [DOI] [PubMed] [Google Scholar]
- 32.Oliw, E. H., Garscha, U., Nilsson, T., and Cristea, M. (2006) Anal. Biochem. 354 111-126 [DOI] [PubMed] [Google Scholar]
- 33.Brash, A. R., Boeglin, W. E., Capdevila, J. H., Yeola, S., and Blair, I. A. (1995) Arch. Biochem. Biophys. 321 485-492 [DOI] [PubMed] [Google Scholar]
- 34.Tsitsigiannis, D. I., and Keller, N. P. (2007) Trends Microbiol. 15 109-118 [DOI] [PubMed] [Google Scholar]
- 35.Noverr, M. C., Toews, G. B., and Huffnagle, G. B. (2002) Infect. Immun. 70 400-402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milne, G. L., Yin, H., and Morrow, J. D. (2008) J. Biol. Chem. 283 15533-15537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider, C., and Brash, A. R. (2000) J. Biol. Chem. 275 4743-4746 [DOI] [PubMed] [Google Scholar]
- 38.Thuresson, E. D., Lakkides, K. M., Rieke, C. J., Sun, Y., Wingerd, B. A., Micielli, R., Mulichak, A. M., Malkowski, M. G., Garavito, R. M., and Smith, W. L. (2001) J. Biol. Chem. 276 10347-10357 [DOI] [PubMed] [Google Scholar]
- 39.Tsitsigiannis, D. I., Kowieski, T. M., Zarnowski, R., and Keller, N. P. (2004) Eukaryot. Cell 3 1398-1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akakabe, Y., Matsui, K., and Kajiwara, T. (2005) Biosci. Biotechnol. Biochem. 69 1539-1544 [DOI] [PubMed] [Google Scholar]
- 41.Wurzenberger, M., and Grosch, W. (1984) Biochim. Biophys. Res. Commun. 794 18-24 [Google Scholar]
- 42.Nemcovic, M., Jakubikova, L., Viden, I., and Farkas, V. (2008) FEMS Microbiol. Lett. 284 231-236 [DOI] [PubMed] [Google Scholar]
- 43.Jones, D. T. (1999) J. Mol. Biol. 292 195-202 [DOI] [PubMed] [Google Scholar]
- 44.Bylund, J., Ericsson, J., and Oliw, E. H. (1998) Anal. Biochem. 265 55-68 [DOI] [PubMed] [Google Scholar]
- 45.Murphy, R. C., Barkley, R. M., Zemski Berry, K., Hankin, J., Harrison, K., Johnson, C., Krank, J., McAnoy, A., Uhlson, C., and Zarini, S. (2005) Anal. Biochem. 346 1-42 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.