

Abstract
We investigated the ways S100B, S100A1, S100A2, S100A4, and S100A6 bind to the different oligomeric forms of the tumor suppressor p53 in vitro, using analytical ultracentrifugation and multiangle light scattering. It is established that members of the S100 protein family bind to the tetramerization domain (residues 325-355) of p53 when it is uncovered in the monomer, and so binding can disrupt the tetramer. We found a stoichiometry of one dimer of S100 bound to a monomer of p53. We discovered that some S100 proteins could also bind to the tetramer. S100B bound the tetramer and also disrupted the dimer by binding monomeric p53. S100A2 bound monomeric p53 as well as tetrameric, whereas S100A1 only bound monomeric p53. S100A6 bound more tightly to tetrameric than to monomeric p53. We also identified an additional binding site for S100 proteins in the transactivation domain (1-57) of p53. Based on our results and published observations in vivo, we propose a model for the binding of S100 proteins to p53 that can explain both activation and inhibition of p53-mediated transcription. Depending on the concentration of p53 and the member of the S100 family, binding can alter the balance between monomer and tetramer in either direction.
The S100 family is a highly conserved group of more than 20 members of small, acidic calcium-binding proteins in vertebrates (1). They are called S100 because they remain soluble in 100% ammonium sulfate at neutral pH (2). S100 proteins are dimers or form higher oligomers (3, 4). They have intracellular functions such as the regulation of protein phosphorylation, the regulation of calcium homeostasis, cell survival, proliferation, and differentiation, as well as extracellular functions, for example, as attractors for leukocytes and macrophages, neurite outgrowth, or the induction of apoptosis (5-8). Further, the expression of several S100 proteins has been linked to metastasis (9) and different kinds of melanomas and carcinomas (8). Nevertheless, the molecular mechanism of action of the S100 proteins is not fully understood.
The tumor suppressor p53 is a crucial factor in the development of cancer. It acts as the central inducer of apoptosis and cell cycle arrest (10, 11). Posttranslational modifications and interaction with proteins regulate its activity (12-14). The interaction with the tumor suppressor protein p53 is a common feature of the S100 proteins (15-19). We previously demonstrated that S100 proteins generally bind to the tetramerization domain (residues 325-355) of p53, whereas only a subset can bind its negative regulatory domain (residues 367-393) (16, 20). S100B, S100A2, S100A4, and S100A6 have been reported to influence p53-mediated transcription, but the effect remains controversial because some studies show a stimulating effect, whereas others claim that S100 proteins inhibit the transcriptional activity of p53 (17-19, 21, 22). We previously showed that oligomerization of p53 weakens the binding to S100B and S100A4, and it was deduced that S100 proteins inhibit the oligomerization of p53, which causes the inhibitory effect on p53-mediated transcription (16, 20).
In this study, we analyzed the binding of five different S100 proteins to full-length p53 and also different oligomeric states of C-terminal fragments of p53 that consisted of the tetramerization (residues 325-355) and C-terminal domains (residues 360-393), some with mutations in the tetramerization domain that altered the oligomerization state. We used analytical size-exclusion chromatography (SEC),2 multiangle light scattering (MALS), and analytical ultracentrifugation (AUC) in vitro. We show that S100 family members differ in their ability to bind to the different oligomeric forms of p53. In addition, we found that some of the S100 proteins can bind p53 as a tetramer and identified the transactivation domain of p53 as another target site for S100 proteins. The in vitro data thus provide an explanation why S100 proteins have been found to activate as well as inhibit p53-mediated transcription.
MATERIALS AND METHODS
Plasmids, Protein Expression, and Purification—Plasmids used for the expression of S100A1, S100A2, S100A4, S100A6, and S100B and p53-(293-393) were as described (16, 20). pRSET plasmids of oligomerization-deficient mutants of p53-(293-393), respectively, p53-(293-393)-L344A and p53-(293-393)-L344P, were constructed by site-directed mutagenesis according to the QuikChange™ XL site-directed mutagenesis kit (Stratagene). The superstable full-length p53-variant (p53-QMFL) (23, 24) was expressed and purified as described previously (25, 26). The p53-QMFL with an additional tetracysteine motif Cys-Cys-Pro-Gly-Cys-Cys at the C terminus (p53-QMFL-FlAsH) was purified similarly and specifically labeled with FlAsH-EDT2 (Invitrogen), which becomes fluorescent upon binding to the tetracysteine motif.
Analytical Size-exclusion Chromatography—Analytical gel
filtrations were performed using a GE Healthcare Superdex™ 75 analytical
gel filtration column with a flow rate of 0.7 ml/min or a Superose™ 6
10/300GL column with a flow of 0.4 ml/min. The proteins were buffer-exchanged
in physiological ionic strength buffer (25 mm Tris, pH 7.4, 10
mm CaCl2, 99.2 mm NaCl, and 1 mm
dithiothreitol), and 100 μl of protein sample at different concentrations
were injected. For experiments with full-length p53, the proteins were changed
into a buffer containing 20 mm Tris, pH 7.4, 150 mm
NaCl, 10% glycerol, 10 mm CaCl2, and 1 mm
dithiothreitol. To analyze the resulting peaks, we collected the eluted
protein with a fraction collector, and the fractions were concentrated with a
centrifugational filter and analyzed by SDS-PAGE. Alternatively, the SEC was
coupled to a DAWN HELEOS™ MALS instrument (Wyatt Technology) and an
Optilab™ rEX (Wyatt Technology). The on-line measurement of the
intensity of the Rayleigh scattering as a function of the angle as well as the
differential refractive index of the eluting peak in SEC can be used to
determine the weight average molar mass
(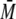 w) of eluted oligomers and
protein complexes (27), using
the ASTRA™ (Wyatt Technologies) software. The number average molar mass
(
w) of eluted oligomers and
protein complexes (27), using
the ASTRA™ (Wyatt Technologies) software. The number average molar mass
(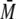 n) was also determined to
estimate the monodispersity of the peaks. All analyzed peak areas were
monodisperse
(
n) was also determined to
estimate the monodispersity of the peaks. All analyzed peak areas were
monodisperse
(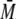 w/
w/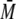 n
< 1.01).
n
< 1.01).
Fluorescence-monitored Analytical Ultracentrifugation—The AUC experiments were performed in a Beckman Coulter Optima XL-I ultracentrifuge with an Aviv fluorescence detection system (28) (FDS, Aviv Biomedical, Lakewood, NJ) in SedVel60K-FDS fluorescence velocity cells (Spin Analytical, Inc., Durham, NH). Proteins were buffer-exchanged in 20 mm Tris, pH 7.4, 150 mm NaCl, 10 mm CaCl2, 10% glycerol, 15 mm β-mercaptoethanol, 0.1% bovine serum albumin. The concentration of p53 was 250 nm with varying amounts of S100 protein. The samples were equilibrated in the cell for 3 h at 10 °C before centrifugation. The AUC runs were performed as described (26). Data were further analyzed using the SedFit software (29) to determine the sedimentation coefficient and to estimate the molecular mass of the protein complex.
Fluorescence Anisotropy—The peptide p53-(1-57) with a C-terminal Lys-methoxycoumarin as label was a gift from Dr. D. Teufel. Fluorescence anisotropy studies were performed at 20 °C with a Cary Eclipse Varian fluorescence spectrophotometer equipped with a Hamilton Microlab M dispenser. Reactions were carried out in physiological ionic strength buffer. Fluorescence anisotropy was measured with excitation at 328 nm and emission at 393 nm. 250 μl of S100 protein were titrated into 1 ml of 0.5 μm peptide. The titration data were corrected for the S100 protein dilution effect and analyzed with Kaleida-Graph (Synergy Software). The dissociation constant (Kd) was calculated with a quadratic fitting equation for a single site binding model, when necessary with the addition of a term accounting for linear drift (30). Each experiment was repeated three times.
RESULTS
Oligomerization of p53-(293-393) Variants—The state of
oligomerization of a protein will depend on the ratio of its concentration to
its dissociation constants. To study the binding to different oligomeric forms
of the p53 C terminus, we expressed and purified: p53-(293-393) recombinant
protein, which should be tetrameric under the experimental conditions; a
mutant of weakened interface that should tend to be dimeric,
p53-(293-393)-L344A (31); and
an even more weakened one that should tend to be monomeric,
p53-(293-393)-L344P (32). The
oligomerization state of proteins was monitored by analytical SEC combined
with MALS detection. The p53 variants have a large fraction of intrinsically
disordered structure (33) and
do not elute like globular proteins in a SEC; hence the relative molecular
weight and the oligomerization cannot be studied appropriately using globular
protein standards. MALS is a powerful tool to characterize the
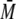 w of compounds showing
anomalous elution profiles in SEC
(34). The three individual
p53-(293-393) variants eluted with different retention times
(tr) in SEC in separate runs. The calculated
w of compounds showing
anomalous elution profiles in SEC
(34). The three individual
p53-(293-393) variants eluted with different retention times
(tr) in SEC in separate runs. The calculated
 w of ∼12, ∼24, and
∼46 kDa for the peaks in the elution profiles corresponded well to
p53-(293-393) in different oligomeric states
(Fig. 1 and
Table 1).
w of ∼12, ∼24, and
∼46 kDa for the peaks in the elution profiles corresponded well to
p53-(293-393) in different oligomeric states
(Fig. 1 and
Table 1).
FIGURE 1.

Analytical SEC-MALS of p53-(293-393) variants. 100 μl of ∼250
μm wild-type (WT)(blue), L344A
(green) and L344P (red) variants eluted at different
tr from a SEC. The
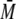 w determined by MALS
correspond to a tetramer, dimer, and monomer for the three individual
peaks.
w determined by MALS
correspond to a tetramer, dimer, and monomer for the three individual
peaks.
TABLE 1.
Size-exclusion chromatography of S100 proteins together with different p53(293-393) variants
The weight average molar mass (M̄w) of the dominant peak was determined by MALS.
|
S100
|
M̄wa
|
+p53
|
|||
|---|---|---|---|---|---|
| −p53 | WT | L344A | L344P | ||
| kDa | |||||
| −S100 | 45.8 ± 0.4 | 24.2 ± 0.1 | 11.8 ± 0.3 | ||
| S100A1 | 21.2 ± 1.0 | 49.3 ± 2.8 | 24.4 ± 1.8 | 28.7 ± 0.7 | |
| S100A2 | 22.8 ± 0.4 | 45.2 ± 2.2 | 23.0 ± 1.7 | 29.7 ± 0.6 | |
| S100A4 | 24.8 ± 0.1 | 45.7 ± 2.7 | 23.7 ± 1.1 | 24.5 ± 0.8b | |
| S100A6 | 20.5 ± 0.8 | 45.7 ± 0.9 | 24.1 ± 0.6 | 14.9 ± 0.7b | |
| S100B | 21.8 ± 0.2 | 78.7 ± 7.5 | 33.5 ± 3.1 | 32.1 ± 0.2 | |
S.E. of four measurements at different concentrations.
Small shift in retention time but no shift in M̄w.
S100 Proteins Form a Complex with the Monomeric Fragment of p53—S100A1, S100A2, S100A4, S100A6, and S100B were expressed and purified. All S100 proteins eluted as dimers (Table 1) under the experimental conditions at μm concentrations in SEC-MALS experiments and eluted with a tr between 17.5 and 18.5 min (data not shown).
S100 proteins were injected together with the p53-(293-393)-L344P variant
in SEC-MALS experiments. S100B, S100A2, and S100A1 (∼20 kDa) interacted
with the monomeric p53-(293-393)-L344P variant (∼12 kDa), resulting in a
peak (tr ∼16 min) with a
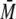 w of ∼30 kDa, which fits
to a complex of one dimer of S100 binding to one monomer of p53
(Table 1). No tight complex
formation was observed between the monomeric p53 variant and S100A4 and
S100A6. Despite a small shift in tr of the S100 peaks, the
w of ∼30 kDa, which fits
to a complex of one dimer of S100 binding to one monomer of p53
(Table 1). No tight complex
formation was observed between the monomeric p53 variant and S100A4 and
S100A6. Despite a small shift in tr of the S100 peaks, the
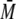 w of 24.5 and 14.9 kDa did
not correspond to a stable complex between S100A4 or S100A6 and the p53
monomer (Table 1) but instead
had only a weak interaction.
w of 24.5 and 14.9 kDa did
not correspond to a stable complex between S100A4 or S100A6 and the p53
monomer (Table 1) but instead
had only a weak interaction.
The stoichiometry of the complex of one dimer of S100 protein binding one
monomeric p53-(293-393)-L344P was also observed at different concentrations of
S100 (Fig. 2). Even with an
excess of S100 protein, the
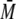 w of the resulting complex
was ∼30 kDa. No peak corresponding to a complex of a dimer of S100 binding
to two monomers with a
w of the resulting complex
was ∼30 kDa. No peak corresponding to a complex of a dimer of S100 binding
to two monomers with a  w of
∼44 kDa could be detected. Further, there was no complex present
corresponding to two dimers of S100 binding to a monomer of p53 (∼54
kDa).
w of
∼44 kDa could be detected. Further, there was no complex present
corresponding to two dimers of S100 binding to a monomer of p53 (∼54
kDa).
FIGURE 2.

SEC-MALS of S100B and p53-(293-393)-L344P at different concentrations. The samples contained 140 μm p53-(293-393)-L344P and 35 (blue), 70 (red), 280 (green), and 560 μm (purple) S100B. At all concentrations, one dimer of S100B (∼21 kDa) bound to one p53 monomer (∼12 kDa). Unbound S100B and p53-(293-393)-L344P eluted with a tr between 17 and 18 min.
S100B Disrupts the p53 Dimer to Form a Complex with the Monomeric
Fragment—S100B also bound to the tetramerization-deficient dimer
variant p53-(293-393)-L344A when injected together in SEC-MALS. The calculated
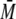 w of ∼33 kDa of the
eluted peak corresponded to a complex of one dimer of S100B (∼21 kDa)
binding a monomer of p53 (∼12 kDa)
(Fig. 3A).
Consequently, the dimer of p53-(293-393)-L344A was disrupted upon binding to
S100B. In contrast, S100A1, S100A2, S100A4, and S100A6 did not bind to the
dimer of p53 (Table 1). In
SEC-MALS elution profiles, the eluted peak with the highest
w of ∼33 kDa of the
eluted peak corresponded to a complex of one dimer of S100B (∼21 kDa)
binding a monomer of p53 (∼12 kDa)
(Fig. 3A).
Consequently, the dimer of p53-(293-393)-L344A was disrupted upon binding to
S100B. In contrast, S100A1, S100A2, S100A4, and S100A6 did not bind to the
dimer of p53 (Table 1). In
SEC-MALS elution profiles, the eluted peak with the highest
 w of ∼24 kDa was the
unbound p53 dimer.
w of ∼24 kDa was the
unbound p53 dimer.
FIGURE 3.

SEC-MALS of S100B and p53-(293-393) oligomers. A, 250
μm dimer p53-(293-393)-L344A (blue) and the complex of
200 μm p53 variant and 100 μm S100B (red)
differed in tr and
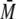 w determined by MALS.
B, the elution profile of 300 μm p53-(293-393) and the
complex of 300 μm p53 with 150 μm S100B differed
in tr and
w determined by MALS.
B, the elution profile of 300 μm p53-(293-393) and the
complex of 300 μm p53 with 150 μm S100B differed
in tr and
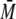 w. No additional peaks of
S100B in complex with lower oligomeric forms of p53 were detected.
WT, wild type.
w. No additional peaks of
S100B in complex with lower oligomeric forms of p53 were detected.
WT, wild type.
S100B Binds to the Tetramer of p53-(293-393)—S100B bound to
wild-type p53-(293-393) in SEC-MALS experiments
(Table 1 and
Fig. 3B). The
tr of the peak for p53-(293-393) shifted upon the addition
of S100B. The  w of ∼80
kDa of the elution peak would correspond best to two dimers of S100 (∼21
kDa) in a complex with a tetramer of p53 (∼46 kDa). Under the same
conditions, S100A1, S100A2, S100A4, and S100A6 did not form a complex with
tetrameric p53-(293-393). The
w of ∼80
kDa of the elution peak would correspond best to two dimers of S100 (∼21
kDa) in a complex with a tetramer of p53 (∼46 kDa). Under the same
conditions, S100A1, S100A2, S100A4, and S100A6 did not form a complex with
tetrameric p53-(293-393). The
 w of the dominating peak was
∼46 kDa, corresponding to unbound tetrameric p53-(293-393)
(Table 1). In addition, no
shift in tr for the eluted peaks was observed (data not
shown). For all S100 proteins, we did not detect additional peaks in the
elution profiles, indicating a disruption of the p53 tetramer upon binding,
not even after long incubation times of 48 h (data not shown).
w of the dominating peak was
∼46 kDa, corresponding to unbound tetrameric p53-(293-393)
(Table 1). In addition, no
shift in tr for the eluted peaks was observed (data not
shown). For all S100 proteins, we did not detect additional peaks in the
elution profiles, indicating a disruption of the p53 tetramer upon binding,
not even after long incubation times of 48 h (data not shown).
S100 Proteins Form a Complex with the Tetramer of Full-length
p53—The binding of S100 proteins to p53 was also studied by
fluorescence analytical ultracentrifugation with a thermodynamically
stabilized full-length p53-QMFL labeled with a FlAsH tag
(26). The concentration of p53
in the experiments was 250 nm. Further, an excess of S100 protein
was added to study the formation of complexes as well as a possible influence
on p53 tetramerization (Fig.
4). The AUC of p53-QMFL-FlAsH gave two peaks, corresponding to a
tetramer of p53 and lower oligomers. The calculated
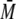 w of ∼178 kDa for the
p53-QMFL-FlAsH corresponded well with its theoretical
w of ∼178 kDa for the
p53-QMFL-FlAsH corresponded well with its theoretical
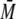 w of ∼176 kDa. Upon the
addition of S100 protein, an increase in the sedimentation coefficient
(measured at the top of the peak) could be observed for the tetramer as well
as for the lower oligomers of p53-QMFL-FlAsH. The increase in sedimentation
coefficient indicated a bigger size of the molecule, corresponding to a
complex formed between the labeled p53 and S100 protein. The addition of
excess S100B resulted in a calculated
w of ∼176 kDa. Upon the
addition of S100 protein, an increase in the sedimentation coefficient
(measured at the top of the peak) could be observed for the tetramer as well
as for the lower oligomers of p53-QMFL-FlAsH. The increase in sedimentation
coefficient indicated a bigger size of the molecule, corresponding to a
complex formed between the labeled p53 and S100 protein. The addition of
excess S100B resulted in a calculated
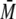 w of ∼268 kDa for the
complex using the same frictional coefficient as a fitting parameter as for
p53-QMFL-FlAsH alone. The estimated mass would correspond to four dimers of
S100B binding to a tetramer of p53. When the concentration of S100 was reduced
from 10 to 1 μm, a smaller shift in sedimentation coefficient
and increase in
w of ∼268 kDa for the
complex using the same frictional coefficient as a fitting parameter as for
p53-QMFL-FlAsH alone. The estimated mass would correspond to four dimers of
S100B binding to a tetramer of p53. When the concentration of S100 was reduced
from 10 to 1 μm, a smaller shift in sedimentation coefficient
and increase in 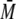 w to
∼199 kDa was detected, implying that the complex between S100B and the
tetramer of p53 was weak. Further, no relative increase in the fluorescence
signal for the lower oligomers when compared with the tetramer of QMFL-FlAsH
could be detected. A change in the signal distribution would indicate a
disruption of the p53-QMFL-FlAsH tetramer. Similar results were obtained for
S100A2 and S100A6. In contrast, only a minor increase in the sedimentation
coefficient and
w to
∼199 kDa was detected, implying that the complex between S100B and the
tetramer of p53 was weak. Further, no relative increase in the fluorescence
signal for the lower oligomers when compared with the tetramer of QMFL-FlAsH
could be detected. A change in the signal distribution would indicate a
disruption of the p53-QMFL-FlAsH tetramer. Similar results were obtained for
S100A2 and S100A6. In contrast, only a minor increase in the sedimentation
coefficient and  w was
detected for S100A1 and S100A4, implying a weaker interaction with
p53-QMFL-FlAsH (Fig. 4).
w was
detected for S100A1 and S100A4, implying a weaker interaction with
p53-QMFL-FlAsH (Fig. 4).
FIGURE 4.

Analytical AUC of p53 QMFL-FlAsH with S100 proteins. 250 nm of p53-QMFL-FlAsH and different amounts of S100 were run at 45,000 rpm. The sedimentation profile was fitted to a double Gaussian equation, and the Mr was calculated with SedFit.
To check the results, we performed analytical SEC with a Superose™ 6 column and injected p53-QMFL together with S100 proteins. The formation of a complex was monitored by SDS-PAGE analysis of the eluted peak. Upon the addition of S100B, S100A2, and S100A6, we saw a slight shift in the elution volume for the p53-QMFL peak (Fig. 5A). SDS-PAGE analysis of the eluted peaks revealed a co-elution of S100 proteins with p53-QMFL (Fig. 5B). The band corresponding to S100 proteins was faint when compared with the band for p53, which supports the idea that the complex between the p53 tetramer and S100 proteins was weak. No additional peaks corresponding to a complex of S100 proteins and lower oligomers of p53-QMFL could be observed. In contrast, no co-elution of S100A1 and S100A4 with p53-QMFL could be detected by SDS-PAGE (Fig. 5).
FIGURE 5.

SEC-PAGE of p53-QMFL and S100. A, the analytical gel filtrations of p53-QMFL in complex with different S100 proteins (in a ratio of 1 to 2) resulted in small tr shifts for S100B, S100A2, and S100A6. mAU, milliabsorbance units. B, the elution between 14 and 15 ml was collected, concentrated, and analyzed via SDS-PAGE. Lane 1, QMFL + S100B; lane 2, QMFL + S100A1; lane 3, QMFL + S100A2; lane 4, QMFL + S100A4; lane 5, QMFL + S100A6.
S100 Proteins Bind to the Transactivation Domain of p53—We noted that the binding properties for p53-(293-393) and full-length p53-QMFL were different for some of the S100 proteins (Table 2). In particular, for S100A6, we could not detect binding to the C terminus of p53-(293-393) by SEC-MALS but found a relatively tight interaction with full-length p53 by AUC and SEC-PAGE. We assumed, therefore, that there might be an additional binding site for S100 proteins within p53, and so we analyzed the binding to p53 core domain (residues 102-292) and to the transactivation domain p53-(1-57). We performed heteronuclear single quantum correlation experiments that could detect even weak binding events using 15N-labeled p53 core domain with S100B and S100A6, but no binding was observed (data not shown). In fluorescence anisotropy experiments with a labeled peptide of the transactivation domain p53-(1-57), we observed binding to S100 proteins with dissociation constants in the low μm to nm range (Table 3 and Fig. 6). S100A2 and S100A6 were the tightest binders with a Kd < 400 nm, whereas S100A1 and S100B bound ∼5-fold more weakly.
TABLE 2.
Summary of S100 proteins binding to p53
|
Protein
|
p53 full length
|
p53-(293-393)
|
||
|---|---|---|---|---|
| Tetramer | Dimer | Monomer | ||
| S100A1 | − | − | − | + |
| S100A2 | + | − | − | + |
| S100A4 | − | − | − | Weak |
| S100A6 | + | − | − | Weak |
| S100B | + | + | + | + |
TABLE 3.
Fluorescence anisotropy of S100 proteins and p53(1-57)
| Protein | Kd |
|---|---|
| μm | |
| S100A1 | 1.91 ± 0.01 |
| S100A2 | 0.34 ± 0.05 |
| S100A4 | 0.76 ± 0.19 |
| S100A6 | 0.39 ± 0.10 |
| S100B | 1.99 ± 0.44 |
FIGURE 6.

Binding of S100 proteins to the transactivation domain of p53. S100 proteins were titrated to 0. 5 μm p53-(1-57)-Lys-methoxycoumarin in fluorescence anisotropy experiments.
DISCUSSION
We found that proteins of the S100 family bind tightly to the monomeric p53 fragment of residues 293-393 in a stoichiometry of one dimer of S100 binding per one p53 monomer. We discovered that a subset of S100 proteins can additionally bind tetrameric p53, and we noted that S100 proteins had different binding properties toward different oligomeric forms (Table 2). We also found a novel binding site common for all S100 proteins in the transactivation domain of p53-(1-57).
S100B and S100A2 bind tightly to the C-terminal domains of p53, residues 367-393, (16, 20). We found that these proteins were able to form a tight complex with a monomeric mutant of p53-(293-393), which was stable enough to be detected by SEC. No stable complex was observed with S100A4 and S100A6. This absence might have been a manifestation of their known weaker binding to p53 (16, 20) so that their complexes might not have been stable enough to remain intact during analytical gel filtration.
S100B was able to disrupt the dimeric mutant p53-(293-393)-L344A that has a weakened interface and bound p53 as a monomer. This confirmed that S100B is able to influence the oligomerization equilibrium of p53 as proposed previously (20). The equilibrium is shifted to the inactive monomeric side; thus the tight binding of the monomer of p53 and the disruption of the dimer in vitro explain the inhibitory effect of S100B on p53 activity as reported previously in transcriptional activation assays (21). It is possible on molecular mass data alone that the peak of ∼33 kDa is a monomer of S100B binding to a dimer of p53. However, the Kd for S100B dimerization is <500 pm (35), and our studies indicated that the dimer of S100 must be a dimer to bind p53 tightly.
S100A1, S100A2, S100A4, and S100A6 did not disrupt the oligomerization of p53. These proteins also had weaker affinities for p53-(293-393) than S100B in fluorescence anisotropy experiments (16, 20). S100B, S100A2, and S100A6 were also found to bind to p53 as a tetramer with a weak affinity. This binding mode is supported by fluorescence anisotropy studies that showed that the S100 proteins had an increasingly lower affinity the higher the concentration of p53 tetramerization domain rose (that is, the lower the concentration, the higher the fraction of lower oligomers). S100A2 and S100B also bind to the negative regulatory domain of p53-(367-393) (20). Consequently, the binding of S100B and S100A2 to the tetramer could result from the binding to the C terminus of p53.
On the other hand, we found that S100A6 bound to the full-length tetramer of p53, although it does not bind to the negative regulatory domain (16). We found that the S100 proteins bound to the N-terminal transactivation domain of p53. The tight binding of S100A6 to the N terminus explains why it was possible to detect binding to tetrameric full-length protein but not to p53-(293-393) constructs (Table 2). It would be interesting to study whether the binding to the N terminus, which has not been detected in previous pull-down studies (15, 17, 19), has a functional role and whether it has a synergistic effect on the binding to the C-terminal binding site of p53.
S100B, S100A2, and S100A6 activate p53-mediated transcription (17, 18, 22, 36). Activation of p53 cannot be explained by the previously reported influence of S100 on oligomerization of p53 in vitro but might be caused by binding of S100 proteins to the tetramer of p53. The binding to the tetramer could have a stabilizing effect or protect p53 from degradation.
Different S100 proteins bind p53 in different ways (Table 2). For example, only S100B is able to disrupt the dimer of p53. S100A1 is able to bind tightly to the monomer of p53 but not to the tetramer, whereas only binding to the tetramer but not to the monomer could be detected for S100A6.
Based on the finding that proteins of the S100 family bind p53 as a monomer as well as a tetramer, we propose a model for the regulatory effect of S100 proteins on p53 where S100 can bind the monomer of p53 and inhibit its activity as well as to the tetramer with an activating effect (Fig. 7A). The model implies that the regulation of p53 activity by S100 proteins is complex and depends on: 1) the concentration of Ca2+ to induce binding to p53; 2) the particular S100 protein because all the S100 proteins seem to have different affinities for the monomer and the tetramer of p53; and 3) the concentration of p53 and the equilibrium and the exchange rates between its oligomers. When the concentration of p53 is lower than the Kd of ∼120 nm (26), as in for example unstressed cells (1-10 nm) (37), the monomer, dimer, and tetramer forms of p53 are present within the cell, and S100 is able to bind the monomer and significantly shift the oligomerization equilibrium toward the inactive form (Fig. 7C). In stressed cells, the concentration of p53 increases 5-1000-fold and consequently is within the range of the Kd for tetramerization or even higher. At concentrations much higher than the Kd, practically all of p53 is tetrameric, and S100 will mainly bind to the tetramer (Fig. 7E).
FIGURE 7.

Binding model of S100 and p53. A, proteins of the S100 family can bind p53 as a tetramer as well as a monomer. The different oligomeric forms of p53 are in equilibrium. S100 can have an activating function binding to the tetramer or an inhibiting effect binding to the monomer of p53. B, at low concentrations of p53 (below the Kd for tetramerization), there is a significant equilibrium between tetramer, dimer, and monomer of p53. C, the tight binding of S100 to the monomer will displace the oligomerization equilibrium (illustrated by the different lengths of the equilibrium arrows) in favor of the monomer and consequently inhibit p53 function. D, when the concentration of p53 is much higher than the Kd for tetramerization, almost all of p53 is present as a tetramer. E, under these circumstances, the tight binding of S100 to the p53 monomer will not significantly alter the tetramerization equilibrium, and the inhibiting effect is overwhelmed by the activating or stabilizing effect of S100 binding to the tetramer of p53.
The dual function of S100 could explain contradictory results about the influence of S100 proteins on p53. S100B was shown to have an inhibitory effect in one study (21), but in other studies, it was proposed to stimulate p53 activity (22, 36). S100A2 activates p53-mediated transcription, but the positive effect disappears when more DNA encoding S100A2 is transfected (17). According to our model, the two S100 proteins bind both the tetramer and the monomer of p53. Consequently, the results of in vivo studies might differ depending on the expression level of the proteins. S100A6 activates p53 (18), and according to our model, S100A6 binds to the tetramer of p53 but only weakly to the monomer of p53 when compared with the other S100 proteins.
Finally, the proposed model suggests that S100 proteins contribute to the fine regulation of p53 activity. In unstressed cells, the concentration of p53 is kept very low; thus a relatively high amount will be in the form of a monomer, which binds tightly to S100 proteins. Binding of S100 to p53 in low concentrations could therefore help to reduce any basal activity. On the other hand, at high concentrations of p53, S100 proteins can further stimulate its transcriptional activity by binding to the tetramer.
Supplementary Material
Acknowledgments
We thank Dr. C. M. Johnson for help with the experimental setup of SEC-MALS and critical reading of the manuscript and Caroline Blair for help with protein expression and purification.
This article was selected as a Paper of the Week.
Footnotes
The abbreviations used are: SEC, size-exclusion chromatography; MALS,
multiangle light scattering; AUC, analytical ultracentrifugation; QMFL,
superstable full-length p53 variant;
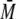 w, weight average molar
mass;
w, weight average molar
mass; 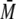 n, number average
molar mass.
n, number average
molar mass.
References
- 1.Donato, R. (2003) Microsc. Res. Tech. 60 540-551 [DOI] [PubMed] [Google Scholar]
- 2.Moore, B. W. (1965) Biochem. Biophys. Res. Commun. 19 739-744 [DOI] [PubMed] [Google Scholar]
- 3.Koch, M., Bhattacharya, S., Kehl, T., Gimona, M., Vasak, M., Chazin, W., Heizmann, C. W., Kroneck, P. M., and Fritz, G. (2007) Biochim. Biophys. Acta 1773 457-470 [DOI] [PubMed] [Google Scholar]
- 4.Ostendorp, T., Leclerc, E., Galichet, A., Koch, M., Demling, N., Weigle, B., Heizmann, C. W., Kroneck, P. M., and Fritz, G. (2007) EMBO J. 26 3868-3878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donato, R. (2001) Int. J. Biochem. Cell Biol. 33 637-668 [DOI] [PubMed] [Google Scholar]
- 6.Hiratsuka, S., Watanabe, A., Aburatani, H., and Maru, Y. (2006) Nat. Cell Biol. 8 1369-1375 [DOI] [PubMed] [Google Scholar]
- 7.Kriajevska, M., Bronstein, I. B., Scott, D. J., Tarabykina, S., Fischer-Larsen, M., Issinger, O., and Lukanidin, E. (2000) Biochim. Biophys. Acta 1498 252-263 [DOI] [PubMed] [Google Scholar]
- 8.Salama, I., Malone, P. S., Mihaimeed, F., and Jones, J. L. (2008) Eur. J. Surg. Oncol. 34 357-364 [DOI] [PubMed] [Google Scholar]
- 9.Davies, M. P., Rudland, P. S., Robertson, L., Parry, E. W., Jolicoeur, P., and Barraclough, R. (1996) Oncogene 13 1631-1637 [PubMed] [Google Scholar]
- 10.Levine, A. J. (1997) Cell 88 323-331 [DOI] [PubMed] [Google Scholar]
- 11.Vousden, K. H. (2000) Cell 103 691-694 [DOI] [PubMed] [Google Scholar]
- 12.Appella, E., and Anderson, C. W. (2000) Pathol. Biol. (Paris) 48 227-245 [PubMed] [Google Scholar]
- 13.Braithwaite, A. W., Del Sal, W., and Lu, X. (2006) Cell Death Differ. 13 984-993 [DOI] [PubMed] [Google Scholar]
- 14.Lavin, M. F., and Gueven, N. (2006) Cell Death Differ. 13 941-950 [DOI] [PubMed] [Google Scholar]
- 15.Baudier, J., Delphin, C., Grunwald, D., Khochbin, S., and Lawrence, J. J. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 11627-11631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernandez-Fernandez, M. R., Rutherford, T. J., and Fersht, A. R. (2008) Protein Sci. 17 1663-1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mueller, A., Schafer, B. W., Ferrari, S., Weibel, M., Makek, M., Hochli, M., and Heizmann, C. W. (2005) J. Biol. Chem. 280 29186-29193 [DOI] [PubMed] [Google Scholar]
- 18.Slomnicki, L. P., Nawrot, B., and Lésniak, W. (2008) Int. J. Biochem. Cell Biol. 41 784-790 [DOI] [PubMed] [Google Scholar]
- 19.Grigorian, M., Andresen, S., Tulchinsky, E., Kriajevska, M., Carlberg, C., Kruse, C., Cohn, M., Ambartsumian, N., Christensen, A., Selivanova, G., and Lukanidin, E. (2001) J. Biol. Chem. 276 22699-22708 [DOI] [PubMed] [Google Scholar]
- 20.Fernandez-Fernandez, M. R., Veprintsev, D. B., and Fersht, A. R. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 4735-4740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin, J., Blake, M., Tang, C., Zimmer, D., Rustandi, R. R., Weber, D. J., and Carrier, F. (2001) J. Biol. Chem. 276 35037-35041 [DOI] [PubMed] [Google Scholar]
- 22.Scotto, C., Delphin, C., Deloulme, J. C., and Baudier, J. (1999) Mol. Cell. Biol. 19 7168-7180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joerger, A. C., Allen, M. D., and Fersht, A. R. (2004) J. Biol. Chem. 279 1291-1296 [DOI] [PubMed] [Google Scholar]
- 24.Nikolova, P. V., Henckel, J., Lane, D. P., and Fersht, A. R. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 14675-14680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ang, H. C., Joerger, A. C., Mayer, S., and Fersht, A. R. (2006) J. Biol. Chem. 281 21934-21941 [DOI] [PubMed] [Google Scholar]
- 26.Rajagopalan, S., Jaulent, A. M., Wells, M., Veprintsev, D. B., and Fersht, A. R. (2008) Nucleic Acids Res. 36 5983-5991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trathnigg, B. (1995) Prog. Polym. Sci. 20 615-650 [Google Scholar]
- 28.MacGregor, I. K., Anderson, A. L., and Laue, T. M. (2004) Biophys. Chem. 108 165-185 [DOI] [PubMed] [Google Scholar]
- 29.Schuck, P. (2000) Biophys. J. 78 1606-1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teufel, D. P., Freund, S. M., Bycroft, M., and Fersht, A. R. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 7009-7014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lomax, M. E., Barnes, D. M., Hupp, T. R., Picksley, S. M., and Camplejohn, R. S. (1998) Oncogene 17 643-649 [DOI] [PubMed] [Google Scholar]
- 32.Mateu, M. G., and Fersht, A. R. (1998) EMBO J. 17 2748-2758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bell, S., Klein, C., Muller, L., Hansen, S., and Buchner, J. (2002) J. Mol. Biol. 322 917-927 [DOI] [PubMed] [Google Scholar]
- 34.Kim, C., Morel, M. H., Beuve, J. S., Guilbert, S., Collet, A., and Bonfils, F. (2008) J. Chromatogr. A 1213 181-188 [DOI] [PubMed] [Google Scholar]
- 35.Drohat, A. C., Nenortas, E., Beckett, D., and Weber, D. J. (1997) Protein Sci. 6 1577-1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scotto, C., Deloulme, J. C., Rousseau, D., Chambaz, E., and Baudier, J. (1998) Mol. Cell. Biol. 18 4272-4281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chene, P. (2001) Curr. Med. Chem. Anti-cancer Agents 1 151-161 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.