

Abstract
Mitochondrial dysregulation is strongly implicated in Parkinson disease. Mutations in PTEN-induced kinase 1 (PINK1) are associated with familial parkinsonism and neuropsychiatric disorders. Although overexpressed PINK1 is neuroprotective, less is known about neuronal responses to loss of PINK1 function. We found that stable knockdown of PINK1 induced mitochondrial fragmentation and autophagy in SH-SY5Y cells, which was reversed by the reintroduction of an RNA interference (RNAi)-resistant plasmid for PINK1. Moreover, stable or transient overexpression of wild-type PINK1 increased mitochondrial interconnectivity and suppressed toxin-induced autophagy/mitophagy. Mitochondrial oxidant production played an essential role in triggering mitochondrial fragmentation and autophagy in PINK1 shRNA lines. Autophagy/mitophagy served a protective role in limiting cell death, and overexpressing Parkin further enhanced this protective mitophagic response. The dominant negative Drp1 mutant inhibited both fission and mitophagy in PINK1-deficient cells. Interestingly, RNAi knockdown of autophagy proteins Atg7 and LC3/Atg8 also decreased mitochondrial fragmentation without affecting oxidative stress, suggesting active involvement of autophagy in morphologic remodeling of mitochondria for clearance. To summarize, loss of PINK1 function elicits oxidative stress and mitochondrial turnover coordinated by the autophagic and fission/fusion machineries. Furthermore, PINK1 and Parkin may cooperate through different mechanisms to maintain mitochondrial homeostasis.
Parkinson disease is an age-related neurodegenerative disease that affects ∼1% of the population worldwide. The causes of sporadic cases are unknown, although mitochondrial or oxidative toxins such as 1-methyl-4-phenylpyridinium, 6-hydroxydopamine (6-OHDA),3 and rotenone reproduce features of the disease in animal and cell culture models (1). Abnormalities in mitochondrial respiration and increased oxidative stress are observed in cells and tissues from parkinsonian patients (2, 3), which also exhibit increased mitochondrial autophagy (4). Furthermore, mutations in parkinsonian genes affect oxidative stress response pathways and mitochondrial homeostasis (5). Thus, disruption of mitochondrial homeostasis represents a major factor implicated in the pathogenesis of sporadic and inherited parkinsonian disorders (PD).
The PARK6 locus involved in autosomal recessive and early-onset PD encodes for PTEN-induced kinase 1 (PINK1) (6, 7). PINK1 is a cytosolic and mitochondrially localized 581-amino acid serine/threonine kinase that possesses an N-terminal mitochondrial targeting sequence (6, 8). The primary sequence also includes a putative transmembrane domain important for orientation of the PINK1 domain (8), a conserved kinase domain homologous to calcium calmodulin kinases, and a C-terminal domain that regulates autophosphorylation activity (9, 10). Overexpression of wild-type PINK1, but not its PD-associated mutants, protects against several toxic insults in neuronal cells (6, 11, 12). Mitochondrial targeting is necessary for some (13) but not all of the neuroprotective effects of PINK1 (14), implicating involvement of cytoplasmic targets that modulate mitochondrial pathobiology (8). PINK1 catalytic activity is necessary for its neuroprotective role, because a kinase-deficient K219M substitution in the ATP binding pocket of PINK1 abrogates its ability to protect neurons (14). Although PINK1 mutations do not seem to impair mitochondrial targeting, PD-associated mutations differentially destabilize the protein, resulting in loss of neuroprotective activities (13, 15).
Recent studies indicate that PINK1 and Parkin interact genetically (3, 16-18) to prevent oxidative stress (19, 20) and regulate mitochondrial morphology (21). Primary cells derived from PINK1 mutant patients exhibit mitochondrial fragmentation with disorganized cristae, recapitulated by RNA interference studies in HeLa cells (3).
Mitochondria are degraded by macroautophagy, a process involving sequestration of cytoplasmic cargo into membranous autophagic vacuoles (AVs) for delivery to lysosomes (22, 23). Interestingly, mitochondrial fission accompanies autophagic neurodegeneration elicited by the PD neurotoxin 6-OHDA (24, 25). Moreover, mitochondrial fragmentation and increased autophagy are observed in neurodegenerative diseases including Alzheimer and Parkinson diseases (4, 26-28). Although inclusion of mitochondria in autophagosomes was once believed to be a random process, as observed during starvation, studies involving hypoxia, mitochondrial damage, apoptotic stimuli, or limiting amounts of aerobic substrates in facultative anaerobes support the concept of selective mitochondrial autophagy (mitophagy) (29, 30). In particular, mitochondrially localized kinases may play an important role in models involving oxidative mitochondrial injury (25, 31, 32).
Autophagy is involved in the clearance of protein aggregates (33-35) and normal regulation of axonal-synaptic morphology (36). Chronic disruption of lysosomal function results in accumulation of subtly impaired mitochondria with decreased calcium buffering capacity (37), implicating an important role for autophagy in mitochondrial homeostasis (37, 38). Recently, Parkin, which complements the effects of PINK1 deficiency on mitochondrial morphology (3), was found to promote autophagy of depolarized mitochondria (39). Conversely, Beclin 1-independent autophagy/mitophagy contributes to cell death elicited by the PD toxins 1-methyl-4-phenylpyridinium and 6-OHDA (25, 28, 31, 32), causing neurite retraction in cells expressing a PD-linked mutation in leucine-rich repeat kinase 2 (40). Whereas properly regulated autophagy plays a homeostatic and neuroprotective role, excessive or incomplete autophagy creates a condition of “autophagic stress” that can contribute to neurodegeneration (28).
As mitochondrial fragmentation (3) and increased mitochondrial autophagy (4) have been described in human cells or tissues of PD patients, we investigated whether or not the engineered loss of PINK1 function could recapitulate these observations in human neuronal cells (SH-SY5Y). Stable knockdown of endogenous PINK1 gave rise to mitochondrial fragmentation and increased autophagy and mitophagy, whereas stable or transient overexpression of PINK1 had the opposite effect. Autophagy/mitophagy was dependent upon increased mitochondrial oxidant production and activation of fission. The data indicate that PINK1 is important for the maintenance of mitochondrial networks, suggesting that coordinated regulation of mitochondrial dynamics and autophagy limits cell death associated with loss of PINK1 function.
EXPERIMENTAL PROCEDURES
Plasmids—PINK1 pcDNA6.1 constructs C-terminally tagged with V5 (12) and amino-truncated ΔN-PINK1-3xFLAG in pAdTrack-CMV (14) were characterized previously. HA-tagged human cDNA for Parkin in the pReceiver vector was purchased from Genecopoeia (Germantown, MD). Mitochondrially targeted GFP (cytochrome oxidase VIII import sequence) and HA- or GFP-tagged wild-type and dominant negative K38A Drp1 in the EGFP-N1 backbone were provided by Dr. Stefan Strack (University of Iowa). The HA-Drp1 constructs simultaneously co-express shRNA specific for endogenous Drp1 (41). GFP-LC3 and RFP-LC3 were generously provided by Tamotsu Yoshimori (National Institute of Genetics, Japan) (42).
Generation of Custom PINK1 Polyclonal Antibody—A polyclonal antibody (C8830) recognizing a predicted exposed loop in kinase subdomain IX of human PINK1 was generated using the services of Invitrogen and characterized for specificity in immunoblot and fluorescent applications.
Generation of Stable PINK1-overexpressing and Knockdown Cell Lines—The SH-SY5Y cell line (American Type Culture Collection, Manassas, VA), a dopaminergic human neuroblastoma cell line, was maintained as described (31). For stable PINK1-overexpressing cell lines, SH-SY5Y cells were transfected with PINK1-3xFlag in pReceiver M14 (Genecopoeia) or empty pReceiver M14 (control) using Lipofectamine 2000 (0.10% final, Invitrogen) and selected with G418 (Invitrogen; 1 mg/ml in Dulbecco's modified Eagle's medium, 10% fetal bovine serum) for 24 days. Individual colonies were expanded in 800 μg/ml G418. Expression of PINK1-3xFlag was confirmed by immunoblotting with mouse anti-FLAG M2 antibody (1:5,000) (Sigma), and total PINK1 was detected using C8830 (1:2000).
To generate stable PINK1-deficient lines, SH-SY5Y were transfected with PINK1 shRNA-encoding sequences (Origene, Rockville, MD) or control pRS shRNA vector and selected as above except that puromycin (0.6 μg/ml; Sigma) was used. To control for nonspecific or off-target effects, two series of PINK1 shRNA clones were generated, one targeting the N-terminal region (A series: CCAGGCTGGGCCGCAGGACCG) and the other a sequence in the kinase domain (D series: CCCCTCACCCCAACATCATCC). Individual colonies were screened by ΔΔCT analysis, using TaqMan quantitative reverse transcription-PCR probes for PINK1 and β-actin and the ABI 7500 real-time PCR system (Applied Biosystems, Foster City, CA). Decreased protein expression was confirmed by immunoblot analysis for PINK1 (C8830).
Immunofluorescence and Imaging—SH-SY5Y cells were formaldehyde-fixed and stained with C8830 (1:2000), mouse-anti-HA tag (1:1000; Covance) and mouse anti-mitochondrial antigen, 60 kDa (clone 113-1, 1:400; BioGenex, San Ramon, CA). Fixed cells were then incubated in the appropriate Alexa 488-donkey anti-rabbit (Molecular Probes, Eugene, CA) or Cy3-donkey anti-mouse secondary antibodies (1:400; Jackson ImmunoResearch Laboratories, West Grove, PA), and counter-stained with 1.25 μg/ml DAPI (Molecular Probes). Labeled cells were imaged at 25 °C using an IDX71 Olympus fluorescence microscope at a magnification of ×20 (0.45 numerical aperture) or ×40 (0.60 numerical aperture) and a DP70 microscope digital camera (Olympus America Inc., Melville, NY; excitation/emission filter, 490/520 nm; 541/572 nm).
Confocal Imaging—For mitophagy, GFP-LC3-transfected cells were stained with MitoTracker Red (100 nm, Molecular Probes) and imaged with an inverted FluoView 1000 laser-scanning confocal microscope at ×60 magnification (1.42 numerical aperture) using the linear sequential scan mode function (Olympus America; excitation/emission filter, 488/510 nm; 561/592 nm).
For three-dimensional reconstruction of mitochondria, 10-μm thick Z-stacks of 25-30 slices each were acquired using a FluoView 100 laser-scanning confocal microscope for at least 10-12 cells/condition. Three-dimensional projections were generated using Imaris 6.1 software (Bitplane Scientific Solutions, Zurich).
Cell Culture Treatments—Cells were treated with beef liver catalase (Roche Molecular Biochemicals; 260,000 units/ml) at 10 units/ml or manganese(III) tetrakis(4-benzoic acid)porphyrin (MnTBAP, Alexis Biochemicals, San Diego) at 100 μm. Bafilomycin (10 nm; Calbiochem) was used to inhibit autolysosomal degradation (43), and 0 .5 μm clasto-lactacystin β-lactone (Sigma) was used for 6 h to inhibit the proteasome.
Small interfering RNA (siRNA) for human Atg proteins were synthesized and characterized as described previously (31), and SH-SY5Y cells were transfected at 35% confluence with Atg8/LC3 siRNA, Atg7 siRNA, or the equivalent siControl nontargeting siRNA (Dharmacon, Lafayette, CO) 72 h prior to analysis (22). Mitochondria were isolated from SH-SY5Y cells as described previously (44).
Transmission Electron Microscopy—Cells were processed as described previously (31) and imaged on a JEOL JEM-1210 electron microscope.
Western Blot Analysis and Densitometry—Cell lysates (1.0% Triton X-100 with protease/phosphatase inhibitors) were resolved on 5-15% ammediol-buffered polyacrylamide gels and immunoblotted as described previously (31) using C8830 rabbit anti-PINK1 (1:2000), mouse anti-FLAG (1:2000; Sigma), rabbit anti-MAP-LC3 (5F10, 1:4000; Nanotools, San Diego), rabbit anti-GFP (1:1000; Invitrogen), mouse anti-mitochondrial antigen, 60 kDa (1:1000; Biogenex, San Ramon, CA), mouse anti-pyruvate dehydrogenase (1:1000 or 0.01 μg/ml; Molecular Probes), mouse anti-β-actin monoclonal antibody (1:5,000; Sigma), mouse anti-ATG7 (1:1000), anti-HA tag (1:1000; Covance, Princeton, NJ), rabbit anti-total ERK1/2 (1:30,000; Upstate Biotechnology, Lake Placid, NY), and anti-lactate dehydrogenase (1:1000; Chemicon/Millipore). Molecular weights were estimated by relative mobility plotted against the log molecular weight of kaleidoscope standards (Bio-Rad).
Cell Survival-Death Assay—Cell survival was measured using Alamar Blue (Trek Diagnostics, Cleveland, OH), at excitation 540 nm, emission 590 nm, in a Spectromax M2 microplate reader (Molecular Devices, Sunnyvale, CA). Cell death was quantified using propidium iodide (Molecular Probes; 1 μg/ml for 5-10 min at 37 °C) as described (25) and by counting the number of GFP-transfected cells containing fragmented, condensed, or pyknotic nuclei per epifluorescence micrograph.
Mitochondrial Superoxide Assays—Cells were stained with MitoSOX (Molecular Probes) as described previously (44). To quantify the average cellular integrated density of MitoSOX fluorescence, we used a Metamorph journal (Molecular Devices) designed by Simon Watkins of the Center for Biologic Imaging, which computes the integrated pixel intensity per cell. The average mean cellular integrated intensity of MitoSOX fluorescence per epifluorescence micrograph was normalized to cell number by counting the number of nuclei per field using the “count nuclei” function in the DRAQ5 (Biostatus Ltd., Leicestershire, UK) channel.
Quantification of GFP-LC3 Puncta, Mitophagy, and Mitochondrial Morphology—Mitophagy in GFP-LC3-transfected cells stained with MitoTracker Red was quantified as described previously (25). Automated quantification of the average cellular GFP-LC3 puncta as a measure of autophagy was performed employing a custom macro for NIH ImageJ software (version 1.39) as described previously (22, 25).
To quantify two parameters of mitochondrial morphology, another ImageJ macro was created for NIH Image (publically available for download from the ImageJ Wiki site; see supplemental Fig. S1 legend). The green channel of cells stained with MitoTracker Green FM (250 nm; Molecular Probes) or expressing mito-GFP was extracted to grayscale, inverted to show mitochondria-specific fluorescence as black pixels, and thresholded to optimally resolve individual mitochondria. The macro traces mitochondrial outlines using “analyze particles.” The mean area/perimeter ratio was employed as an index of mitochondrial interconnectivity, with inverse circularity used as a measure of mitochondrial elongation, as validated using well characterized mediators of mitochondrial fission and fusion (Fig. S1).
Statistics—Unless indicated otherwise, results are expressed as compiled means ± S.E. from at least three independent experiments. Multiple group comparisons were performed using one-way analysis of variance with Fisher's least significant difference. Values of p < 0.05 were considered significant.
RESULTS
Stable PINK1-overexpressing and -deficient Cell Lines—To investigate the biochemical mechanisms associated with perturbations in PINK1 function, we generated a panel of SH-SY5Y clonal cell lines that either stably overexpress or exhibit decreased expression of endogenous PINK1. On a 5-15% gradient gel, the rabbit C8830 antiserum labels a faint ∼66-kDa band and a strong, processed ∼50-kDa band of endogenous PINK1. In addition, the antibody recognizes a variably intense ∼35-kDa band and a ∼22-24-kDa band. Immunoreactive bands are effectively reduced by competition with the immunizing peptide (Fig. 1A, middle panel) or by shRNA to PINK1 (Fig. 1E). By immunofluorescence, C8830 yielded strong staining of endogenous PINK1 colocalizing with mitochondria (Fig. 1C, yellow) with light, diffuse staining in the cytoplasm (Fig. 1C, green).
FIGURE 1.

Characterization of PINK1-overexpressing and knockdown cell lines. A, cell lysates from stable pRec vector control SH-SY5Y cell line (C), a PINK1 knockdown stable cell line (A14), and a PINK1-overexpressing clonal cell line (#24) were resolved on a 5-15% ammediol-buffered gel and immunoblotted for PINK1 with C8830 antisera in the presence (middle panel) or absence (left panel) of blocking PINK1 peptide (100 μg/ml for 1 h). The blot was reprobed for FLAG tag (right panel) and for β-actin as loading control. Inset 1 shows a longer exposure of the film to better visualize full-length endogenous PINK1 relative to the overexpressed PINK1-3xFlag. Immunoreactive bands include full-length recombinant PINK1-3xFlag (band a′, ∼69 kDa), endogenous full-length (band a, ∼66 kDa) and a major processed band (band b, ∼50 kDa). One or two lower molecular mass processed forms (<39 kDa) were also observed. A processed form of PINK1-3xFlag was sometimes observed (∼60 kDa, supplemental Fig. S8); Inset 2 shows the accentuation of this processed band in cells treated with the proteasome inhibitor clasto-lactacystin β-lactone (band a″). Smaller processed PINK1 bands likely reflect further processing, potentially at the C-terminal end, as no smaller FLAG bands are observed. B, Western blots of mitochondrial (mito) and cytoplasmic (cyto) fractions derived from parental SH-SY5Y cells (W), a stable vector line (Ctrl), and PINK1-3xFlag clone #24 resolved on a 10% Tris-glycine gel and immunoblotted for FLAG, endogenous PINK1, mitochondrial P60, and lactate dehydrogenase (LDH). Similar results were obtained for clone 9 (data not shown). Asterisk, indicates a nonspecific band. Full-length recombinant PINK1-3xFlag (band a′), endogenous full-length (band a), and processed (band b) PINK1 are observed. C, epifluorescence image of SH-SY5Y cells double labeled for endogenous PINK1 (green) and 60-kDa human mitochondrial antigen (red) and counterstained for nuclei with DAPI (blue). D, PINK1 mRNA levels in clonal SH-SY5Y lines that express shRNA directed against the N-terminal (A series clones) or middle (D series clones) regions of PINK1. Data are normalized to β-actin mRNA as relative quantification values ± max and min. A control clone generated using pRS control vector (V series clones) served as the reference value. *, p < 0.001 versus V17. E, PINK1 immunoblot showing reduction of endogenous PINK1 in stable PINK1shRNA cell lines compared with a vector control cell lines, with β-actin serving as the loading control. The 50-kDa processed band (b) and two smaller processed bands (c and d) are observed in this blot (see supplemental Fig. S7 for uncropped version of the ECL film). Densitometry data (compiled from three independent experiments for each line) is shown below (*, p < 0.01 versus control cell line).
PINK1-overexpressing clone 24 (Fig. 1A) and clone 9 (Fig. S8) showed stable expression of PINK1 bearing a C-terminal 3xFlag tag. Both anti-FLAG monoclonal antibodies and C8830 antiserum recognize overexpressed full-length 3xFlag-tagged PINK1 (∼69 kDa) (Fig. 1A, left and right panels; Fig. S8). A fainter band representing full-length endogenous PINK1 (∼66 kDa) is also observed in both vector control and PINK1-3xFlag lines (Fig. 1A, and inset 1 showing longer exposure). PINK1-3xFlag is present in the mitochondrial and cytosolic fractions (Fig. 1B). A processed ∼60-kDa PINK1-3xFlag form (supplemental Fig. S8) was not consistently observed but increased in abundance upon inhibition of the proteasome with lactacystin (Fig. 1A, inset 2) or with MG132 (data not shown).
The efficiency of PINK1 knockdown in stable PINK1 shRNA clones was assessed at mRNA and protein levels by quantitative reverse transcription-PCR, Western blot analyses, and immunofluorescence (data not shown). PINK1 shRNA clones in both the A and D series efficiently reduced mRNA levels of PINK1 compared with vector control cell lines (Fig. 1D). All processed forms of PINK1 were reduced significantly in multiple PINK1 shRNA clones compared with vector control cell lines by Western blot (Fig. 1, A and E).
Loss of PINK1 Elicits Mitochondrial and Autophagolysosomal Alterations—Ultrastructural analysis revealed that stable vector control lines exhibited normal mitochondrial morphology and a near absence of AVs (Fig. 2A). Stable knockdown of PINK1 using independent short hairpin sequences (Fig. 2, B and C) elicited small, fragmented mitochondrial profiles with occasional enlarged, swollen appearing profiles. The number of cristae folds per μm of mitochondrial length was reduced in PINK1 knockdown cells (Fig. 2E), similar to ultrastructural observations in human PD/Lewy body disease substantia nigra neurons (4) and human PD cybrid cells (45). The average number of AVs and lysosomes per cell was markedly elevated compared with control vector cells (Fig. 2F), suggesting that loss of PINK1 promotes autophagy.
FIGURE 2.

Ultrastructural analysis of PINK1 knockdown and overexpressing cell
lines. A-D, electron micrographs of stable cell lines that
express empty shRNA vector control (A), A and D series PINK1 shRNAs
(B and C), or PINK1-3xFlag (D). m,
mitochondria; av, autophagic vacuoles; ly, lysosomes;
n, nuclei. Inset, enlarged mitochondria showing dense
cristae (scale bars:2 μm). E, the average number of
cristae folds/μm of mitochondria
(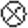 , p < 0.001
versus vector control; mean ± S.E., n = 80-150
mitochondria/condition). F, average number of AVs (autophagosomes and
autolysosomes) quantified per cell.
(, p < 0.05
versus vector control; mean ± S.E., n = 30
cells/condition).
, p < 0.001
versus vector control; mean ± S.E., n = 80-150
mitochondria/condition). F, average number of AVs (autophagosomes and
autolysosomes) quantified per cell.
(, p < 0.05
versus vector control; mean ± S.E., n = 30
cells/condition).
PINK1-overexpressing clones exhibited longer mitochondrial profiles with a higher density of cristae folds (Fig. 2, D and E), as well as some abnormally enlarged mitochondria with abundant cristae (Fig. 2D, inset). In contrast to the PINK1 knockdown cell lines, the presence of these abnormal mitochondria did not appear to elicit an autophagic response, as the average number of AVs per cell remained low in overexpressing cells (Fig. 2, D and F).
PINK1 Expression Modulates Mitochondrial Morphology—As mitochondrial networks are better analyzed by whole cell analysis than by electron microscopy, we utilized computer-automated image analysis to compare the morphologic effects of altered expression levels of PINK1 with those of known mediators of mitochondrial fission/fusion. The custom macro for NIH ImageJ traces individual mitochondria in an unbiased manner and computes indices of mitochondrial interconnectivity and mitochondrial elongation. It was validated by transfection with the fission protein dynamin-related protein (Drp1) and dominant negative Drp1 (Drp1-DN), well characterized molecular manipulations that result in increased mitochondrial fission and fusion, respectively (46, 47) (supplemental Fig. S1). Either transient or stable overexpression of wild-type PINK1 increased mitochondrial interconnectivity and elongation scores compared with vector control stable cells (Fig. 3, A, B, and D). Conversely, stable knockdown of PINK1 induced a fragmentation of the mitochondrial network compared with vector control cells (Fig. 3, A-C).
FIGURE 3.

Knockdown or overexpression of PINK1 alters mitochondrial
morphology. A, epifluorescence images of mitochondria labeled by
transient transfection of mito-GFP in vector control, stable PINK1 shRNA
lines, and a stable PINK1-overexpressing clone. Scale bar:20 μm.
B, three-dimensional reconstruction of Z-stacks of mitochondria
labeled with mito-GFP in vector control, PINK1 shRNA clones, and a
PINK1-overexpressing clone. C, quantitative image analysis of
mitochondrial interconnectivity and mitochondrial elongation applied to empty
vector control and PINK1shRNA clonal cells (representative of three
experiments; , p
< 0.02 versus control (Ctrl); mean ± S.E.,
n = 25-35 cells). D, quantification of mitochondrial
interconnectivity and mitochondrial elongation in a
PINK1-3xFlag-overexpressing cell line and in SH-SY5Y cells transiently
transfected with PINK1-WT-V5
(, p < 0.05
versus stable controls or empty vector; mean ± S.E.,
n = 25-35 cells/condition, three independent experiments).
As shorter, circular, mitochondrial profiles could reflect an altered spatial rearrangement of mitochondria, we performed three-dimensional reconstruction of Z-stacks of mito-GFP-expressing mitochondria (Fig. 3B). This confirmed that PINK1-deficient lines exhibit fragmented mitochondria compared with vector control cells, which showed a mixed profile of tubular and spherical mitochondria. Conversely, overexpression of PINK1 increased mitochondrial interconnectivity.
PINK1 Loss of Function Promotes Macroautophagy and Mitophagy—We utilized established fluorescent and biochemical measures of autophagy to study the effects of PINK1 knockdown on macroautophagy (22). Covalent lipidation of the ubiquitin-fold protein Atg8/microtubule-associated protein light chain 3 (LC3) is essential for macroautophagy induction (23). The unconjugated LC3-I form is diffuse in the cytosol, whereas phosphatidylethanolamine-conjugated LC3-II is localized to AVs and exhibits greater mobility by SDS-PAGE (48). Stable knockdown of PINK1 robustly increased the LC3-II to β-actin ratio in multiple PINK1 shRNA clones, indicating that loss of endogenous PINK1 promotes autophagy (Fig. 4A). The average number of AVs was determined in live cells transfected with GFP-LC3, a marker of earlier AVs, or RFP-LC3, which retains strong fluorescence in late AVs/autolysosomes (42). Knockdown of PINK1 increased the average cellular number of early AVs (Fig. 4B, left graph) and late AVs (Fig. 4B, right graph) and caused lysosomal expansion (supplemental Fig. S2), indicating that AV maturation and lysosomal fusion are intact in PINK1shRNA cell lines. Use of bafilomycin A, which inhibits autolysosomal degradation (43), confirmed intact autophagic flux in the PINK1-deficient lines (Fig. 4B).
FIGURE 4.

Knockdown of PINK1 increases macroautophagy and mitophagy.
A, LC3 Western blot demonstrating increase in LC3-II to β-actin
ratios indicative of elevated autophagy in PINK1shRNA clones compared with a
vector control clone or parental SH-SY5Y cells. B, the average number
of GFP-LC3 puncta quantified per cell in a vector control line and two PINK1
shRNA clonal cell lines transiently expressing GFP-LC3 (marker of early AVs
(left graph)) or RFP-LC3 (marker of later AVs (right graph))
in the presence (black bars) or absence (gray bars) of
bafilomycin to inhibit lysosomal degradation
(, p < 0.01
versus control cell line (Ctrl); n = 20-40 cells
quantified/condition; 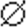 ,
p < 0.01 versus PINK1shRNA line in the absence of
bafilomycin; n = 30-40 cells analyzed/condition). C,
confocal microscopy of MitoTracker Red-labeled mitochondria in control and
three PINK1shRNA lines transiently expressing GFP-LC3 (green).
Scale bar:20 μm. D, quantitative analysis of
GFP-LC3/MitoTracker colocalization as index of mitophagy in vector control and
three PINK1 shRNA clonal lines. Bar graph shows means ± S.E.
from 45-90 cells/condition compiled from four experiments
(, p < 0.001
versus control cell line; n = 25-30 cells/condition).
E, computer-aided quantification of the percent of cellular area
occupied by mitochondria in control and PINK1 shRNA cells in the presence or
absence of bafilomycin A. Cells were transiently transfected with
mitochondrially targeted GFP and untargeted RFP to label the cellular
perimeters. (
,
p < 0.01 versus PINK1shRNA line in the absence of
bafilomycin; n = 30-40 cells analyzed/condition). C,
confocal microscopy of MitoTracker Red-labeled mitochondria in control and
three PINK1shRNA lines transiently expressing GFP-LC3 (green).
Scale bar:20 μm. D, quantitative analysis of
GFP-LC3/MitoTracker colocalization as index of mitophagy in vector control and
three PINK1 shRNA clonal lines. Bar graph shows means ± S.E.
from 45-90 cells/condition compiled from four experiments
(, p < 0.001
versus control cell line; n = 25-30 cells/condition).
E, computer-aided quantification of the percent of cellular area
occupied by mitochondria in control and PINK1 shRNA cells in the presence or
absence of bafilomycin A. Cells were transiently transfected with
mitochondrially targeted GFP and untargeted RFP to label the cellular
perimeters. ( , p
< 0.05 versus control cell line;
, p < 0.05
versus untreated PINK1shRNA cell lines; n = 25-35 cells
analyzed/condition) F, protein levels of mitochondrial pyruvate
dehydrogenase (PDH), reprobed for total ERK1/2 as a loading control.
G, computer-aided quantification of the integrated intensity of
immunoreactive bands specific for human mitochondrial antigen of 60 kDa
(mito-P60) relative to total ERK levels in a control cell line, three
PINK1shRNA cell lines, and a PINK1-3x-Flag-overexpressing line. The bar
graph shows means ± S.E. compiled from 3-4 Western blot
experiments for each cell line
(, p < 0.05
versus control cell line; n = 5-7 immunoreactive bands
analyzed/condition). Inset, representative Western blot showing
decreased level of mito-P60 in PINK1 shRNA cell compared with vector stable
control (Ctrl), reprobed for total ERK1/2 as a loading control.
, p
< 0.05 versus control cell line;
, p < 0.05
versus untreated PINK1shRNA cell lines; n = 25-35 cells
analyzed/condition) F, protein levels of mitochondrial pyruvate
dehydrogenase (PDH), reprobed for total ERK1/2 as a loading control.
G, computer-aided quantification of the integrated intensity of
immunoreactive bands specific for human mitochondrial antigen of 60 kDa
(mito-P60) relative to total ERK levels in a control cell line, three
PINK1shRNA cell lines, and a PINK1-3x-Flag-overexpressing line. The bar
graph shows means ± S.E. compiled from 3-4 Western blot
experiments for each cell line
(, p < 0.05
versus control cell line; n = 5-7 immunoreactive bands
analyzed/condition). Inset, representative Western blot showing
decreased level of mito-P60 in PINK1 shRNA cell compared with vector stable
control (Ctrl), reprobed for total ERK1/2 as a loading control.
PINK1 shRNA stable cell lines were transiently transfected with GFP-LC3 and analyzed for colocalization of GFP-LC3 with Mito-Tracker Red as an index of mitophagy (25, 49). We found that stable knockdown of PINK1 induced autophagic mitochondrial sequestration (Fig. 4, C and D) and delivery to lysosomes (supplemental Fig. S2) and decreased total cellular levels of mitochondria as assessed by immunoblotting for mitochondrial matrix (pyruvate dehydrogenase) and membrane (P60) proteins (Fig. 4, F and G). Quantitative analysis revealed about a 2-fold decrease in mitochondrial P60 levels in three shRNA clones, whereas overexpression of PINK1 showed an insignificant increase (Fig. 4G). Treating cells with bafilomycin A (Fig. 4E, supplemental Fig. S3B) or RNAi of Atg7 or Atg8/LC3B proteins (supplemental Fig. S3A) significantly reversed the loss of mitochondrial levels, indicating autophagic degradation of mitochondria in PINK1-deficient cells (Fig. 4E).
PINK1 Overexpression Reverses PINK1 Knockdown-induced Autophagy—6-OHDA induces autophagy/mitophagy (25). Whereas stable knockdown of PINK1 promoted autophagy, stable PINK1 overexpression suppressed 6-OHDA induced autophagy and mitophagy compared with stable vector control clones (Fig. 5, A and B).
FIGURE 5.

Increased expression of wild-type PINK1 inhibits autophagy elicited by
either 6-OHDA or PINK1 deficiency. A, macroautophagy in
PINK1-overexpressing cell lines analyzed by quantifying the average number of
GFP-LC3 puncta per cell in the presence of dH20 (vehicle) or 6-OHDA
(at 120 μm for 4 h; Sigma) as an inducer of autophagy
(, p < 0.05
versus untreated vector control cell line;
, p < 0.05
versus 6-OHDA-treated control cell line (Ctrl); means
± S.E., n = 20-35 cells quantified/condition). B,
mitophagy quantification in PINK1-overexpressing clones in the absence or
presence of 6-OHDA as an inducer of mitophagy (representative of three
experiments; , p
< 0.05 versus untreated control cell line;
, p < 0.05
versus 6-OHDA treated control cell line; 25-30 wells
quantified/condition). Restoration of PINK1 expression using the A series
RNAi-resistant ΔN-PINK1-3xFLAG plasmid suppresses autophagy in PINK1
shRNA line A14 as assessed by analysis of LC3 shift (C) and in line
A4 as assessed by GFP-LC3 puncta (D).
(, p < 0.0005
versus control cell line;
, p < 0.0005
versus empty vector transfected PINK1 shRNA; means ± S.E.,
n = 15-30 cells quantified/condition.)
Similarly, shRNA reversal studies showed that transient expression of ΔN-PINK1-3xFLAG, which lacks the sequence targeted by the A series PINK1-specific shRNA, completely reversed the increased autophagy observed in PINK1 knock-down lines (Fig. 5, C and D). ΔN-PINK1 is also fully active in protecting against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine/1-methyl-4-phenylpyridinium (MPP+) toxicity (14), which is associated with autophagic cell death (31).
Knockdown of PINK1 Induces Mitochondrial Superoxide, Which Regulates Fragmentation and Mitophagy—To study the mechanisms regulating the effects of PINK1 knockdown on mitochondrial function, we used a mitochondrially targeted fluorescent superoxide sensor. Stable knockdown of PINK1 significantly increased mitochondrial superoxide generation as measured by MitoSOX fluorescence, indicating that loss of PINK1 promotes mitochondrial ROS and dysfunction (Fig. 6, A and B). Moreover, catalase, which has been shown to affect both intracellular and extracellular hydrogen peroxide levels due to free diffusion of peroxide through the plasma membrane, and MnTBAP, a superoxide dismutase mimetic that distributes to mitochondria and cytosol (50), reversed mitochondrial morphology changes induced by loss of PINK1 to levels similar to those seen in vector control cells treated with antioxidants (Fig. 6, C and D). Transient expression of Drp1-DN, which reversed the effects of PINK1 knockdown on mitochondrial morphology, did not reduce MitoSOX fluorescence in PINK1shRNA cell lines (supplemental Fig. S4), suggesting that ROS lie upstream of mitochondrial fragmentation in PINK1-deficient cells.
FIGURE 6.

PINK1 knockdown elicits mitochondrial superoxide upstream of
mitochondrial fragmentation and autophagy in PINK1-deficient lines.
A, epifluorescence analysis of MitoSOX, a cell-permeable red
fluorescent mitochondrial superoxide indicator, in a control stable cell line
(Ctrl, left panel) and two PINK1 knockdown cell lines (right
panels). Nuclei were counterstained with DRAQ5 (blue). Scale
bar: 100 μm. B, bar graph shows compiled means ± S.E.
from three independent experiments with increases in the average MitoSOX
fluorescence per cell normalized to vector control cell line
(, p < 0.05
versus vector control cell line; n = 200-300 cells
analyzed/condition). C and D, quantification of
mitochondrial interconnectivity (C) and mitochondrial elongation
(D) in PINK1 knockdown clonal cell lines treated with catalase,
MnTBAP, or vehicle (untreated) for 24 h. (Representative of three independent
experiments; , p
< 0.05 versus control cell line; n = 25-35 cells analyzed
per condition.) E, representative epifluorescence micrograph of a
PINK1 knockdown cell line (A4) transiently expressing GFP-LC3 treated in the
presence or absence of antioxidant MnTBAP. Note that MnTBAP suppresses
autophagy in PINK1 knockdown cells. F, the average cellular number of
GFP-LC3 puncta in control or PINK1shRNA cells transiently expressing GFP-LC3
in the presence or absence of indicated antioxidants.
(, p < 0.05
versus untreated control cell line;
, p < 0.0001
versus respective untreated clones; representative bar graph
shows means ± S.E., n = 25-40 cells quantified/condition).
Stable shRNA PINK1 knockdown lines were transfected with GFP-LC3 and treated with catalase or MnTBAP to analyze the effects of antioxidants on autophagy induced by PINK1 loss. Only MnTBAP inhibited autophagy induced by PINK1 knockdown, showing also a significant effect on basal autophagy (Fig. 6, E and F). These results indicate that cellular ROS production is necessary for both mitochondrial fragmentation and autophagy but that superoxide (or its product, peroxynitrite) may be particularly important for autophagy induction.
Mitochondrial Fission Is Necessary for Mitochondrial Fragmentation and Mitophagy in PINK1-deficient Cells—We next assessed whether mitochondrial fragmentation observed in PINK1 shRNA cell lines was mediated by classic Drp1-dependent fission. We transiently transfected stable vector control and PINK1 shRNA lines with GFP-tagged Drp1-DN. This K38A substitution impaired the GTPase activity required for fission, resulting in increased mitochondrial interconnectivity in SH-SY5Y cells (supplemental Fig. S1). Transient expression of GFP-Drp1-DN significantly restored mitochondrial interconnectivity and length, indicating that classic Drp1-dependent fission is required for the mitochondrial remodeling observed in PINK1-deficient clones (Fig. 7, A and B). Interestingly, co-transfecting HA-tagged Drp1-DN with GFP-LC3 as a marker of autophagy resulted in decreased GFP-LC3 puncta and mitophagy in PINK-deficient clones to levels similar to those observed in wild-type control clones (Fig. 7, C and D). This indicates that the mitochondrial fission/fusion machinery regulates mitochondrial degradation induced by PINK1 deficiency.
FIGURE 7.

Mitochondrial changes in PINK1 knockdown cells require Drp1
activity. Control or PINK1shRNA stable clonal cell lines were
co-transfected with GFP-Drp1-DN and with a mitochondrially targeted RFP
(mito-RFP), and images were analyzed for mitochondrial interconnectivity
(A) and mitochondrial elongation (B) as described under
“Experimental Procedures.” (Representative of three independent
experiments; , p
< 0.005 versus control cell line;
, p < 0.05
versus respective empty vector-transfected PINK1shRNA cell line;
n = 25-35 cells quantified/condition.) Control or PINK1shRNA stable
lines were co-transfected with HA-tagged Drp1-DN and with GFP-LC3 to label
AVs. C and D, the average number of GFP-LC3 puncta
(C) or the percent colocalization of GFP-LC3 puncta with mitochondria
(D) by confocal microscopy and image analysis (representative of
three independent experiments;
, p < 0.01
versus control; ,
p < 0.05 versus empty vector-transfected A4 or D14;
n = 25-30 cells analyzed/condition).
Autophagic Machinery Is Necessary for Mitochondrial Fragmentation in PINK1 shRNA Cells but Not for Increased Mitochondrial ROS—We analyzed the effect of blocking autophagy on mitochondrial superoxide production and mitochondrial morphology using previously characterized siRNA targeting the essential autophagy proteins Atg7 and Atg8/LC3B (25, 31, 40). As expected, knockdown of these proteins inhibited autophagy in PINK1-deficient clones (Fig. 8A), but it had no effect on the increased mitochondrial superoxide, confirming that autophagy occurs downstream of mitochondrial oxidative stress (Fig. 8B). Interestingly, siRNA for either Atg8/LC3B or Atg7 also reversed the mitochondrial fragmentation observed in PINK1shRNA cell lines to levels similar to those found in cells with normal levels of endogenous PINK1 (Fig. 8, C and D). These data, combined with the differences in antioxidant sensitivity discussed above, suggest that autophagy is actively involved in remodeling mitochondria for clearance rather than serving a passive role downstream of fission. Thus, fission/fusion and autophagic machineries cooperate reciprocally in the regulation of pathologic mitochondrial remodeling and clearance induced by PINK1 deficiency.
FIGURE 8.

Autophagy is involved in mitochondrial morphology changes elicited by
PINK1 deficiency and plays a neuroprotective role. A, stable
vector control or PINK1shRNA cells co-transfected with the indicated siRNAs
and quantified for the number of GFP-LC3 puncta per cell as analyzed in a
representative experiment of two
(, p < 0.001
versus control cell line;
, p < 0.01
versus control siRNA; means ± S.E., n = 30-40 cells
quantified per condition). Inset shows a representative Western blot
showing effects of Atg7 or control siRNA. Note that knockdown of ATG7
decreases endogenous Atg7 levels and reduces the levels of activated LC3-II.
B, the average integrated intensity of MitoSOX fluorescence was
quantified in control or PINK1shRNA stable cell lines transiently transfected
with the indicated siRNAs (,
p < 0.01 versus control cell line (Ctrl), means
± S.E., n = 300-400 cells/condition). C and
D, quantification of mitochondrial interconnectivity (C) and
mitochondrial elongation (D) in PINK1 knockdown clonal lines
transfected with the indicated siRNAs (representative of three experiments;
, p < 0.05
versus control cell line transfected with control siRNA;
, p < 0.01
versus PINK1 A4 or D14 transfected with control siRNA; n =
25-30 cells/condition). E, basal cell survival (Alamar Blue assay) of
control and PINK1 shRNA clones (representative of four experiments;
, p < 0.005
versus control stable cell line; n = 8 wells
analyzed/condition). F, propidium iodide (PI) cell death
assay of control and PINK1 shRNA stable clonal cell lines
(, p < 0.05
versus control stable line; n = 800-1000 cells
analyzed/condition). G, cell survival of control or PINK1 shRNA
stable cell lines treated with bafilomycin or vehicle for 18 h (representative
of three experiments; ,
p < 0.05 versus control cell line/bafilomycin; n
= 4 wells analyzed/condition). H, cell survival of control or
PINK1shRNA cells transfected with Atg7 siRNA (representative of three
experiments; , p
< 0.05 versus control siRNA-treated clone A4;
, p < 0.005
versus control siRNA-treated clone D14; n = 6 wells
analyzed/condition).
Inhibition of Autophagy and Mitochondrial Fission Increases Cell Death in PINK1 Knockdown Cell Lines—Consistent with the view that loss of PINK1 is detrimental in neuronal cells (19), we found that knockdown of PINK1 induces a modest decrease in cell viability in SH-SY5Y cells (Fig. 8E), associated with an increase in cell death at 5 days in culture (Fig. 8F).
The potential role of autophagy in regulating cell viability in PINK1-deficient cells was investigated using siRNA to inhibit autophagy induction and a 24-h dose of bafilomycin to inhibit autophagic degradation. A further significant decrease in cell viability was observed in PINK1 shRNA cells treated with either bafilomycin (Fig. 8G) or Atg7 siRNA, the latter of which has no significant effect on the viability of control stable cell lines (Fig. 8H).
Likewise, mitochondrial fusion induced by GFP-Drp1-DN increased cell death in two PINK1shRNA cell lines, suggesting that mitochondrial fission is also a protective mechanism in the context of PINK1 deficiency, facilitating effective mitophagic responses (supplemental Fig. S5). These data implicate a beneficial role for autophagy in the context of PINK1 deficiency, presumably as a mechanism to remove damaged mitochondria.
Parkin Expression Further Potentiates Mitophagy and Cytoprotection in PINK1-deficient Cells—Given the recent observation that Parkin is recruited to damaged mitochondria and promotes autophagy (39), we studied the effects of Parkin in PINK1 knockdown cells. Immunofluorescence and live confocal microscopy showed that transient expression of HA-tagged Parkin, a 54-kDa fusion protein, in SH-SY5Y cells (Fig. 9B, inset) synergistically increased the number of GFP-LC3 puncta and the percent of GFP-LC3 puncta that colocalized with mitochondria per cell in PINK1 knockdown cell lines (Fig. 9A-C). On the other hand, transient expression of Parkin restored mitochondrial morphologic parameters to control levels (Fig. 9D). Cell death assays showed that transient expression of Parkin significantly reversed cell death induced by loss of PINK1 (Fig. 9E).
FIGURE 9.

Parkin promotes autophagy/mitophagy and restores mitochondria morphology
in PINK1 shRNA cell lines. A, representative epifluorescence
micrographs of control cells co-transfected with GFP-LC3 and vector
(left) and PINK1-deficient cell lines co-transfected with GFP-LC3 and
vector (middle) or HA tagged Parkin (right). Cells were fixed and
immunostained for HA-tag (red) and counterstained with DAPI to
visualize nuclei (blue). B, representative GFP-LC3 puncta
bar graph of control cell line and PINK1shRNA cell lines co-transfected with
GFP-LC3 and either empty vector or HA-Parkin
(, p < 0.0001
versus control cell line;
, p < 0.01
versus vector-transfected shRNA cell lines; n = 30-40 cells
analyzed per condition). Inset shows representative HA immunoblot for cells
transfected with HA tagged Parkin (black arrowhead). The predicted
molecular weight of HA-Parkin is 54kDa. C, mitophagy quantification
in control and PINK1 shRNA clones transfected with either empty vector or
HA-Parkin. (, p
< 0.02 versus control cell line;
, p < 0.05
versus PINK1shRNA cell lines; n = 30-40 cells analyzed per
condition). D, representative quantification of mitochondrial
interconnectivity in a control cell line and PINK1shRNA cell lines
co-transfected with mito-GFP and either empty vector or HA-Parkin
(, p < 0.002
versus control cell line;
, p < 0.05
versusPINK1shRNA cell lines; n = 25-30 cells analyzed per
condition). E, propidium iodide cell death assay of control and PINK1
shRNA stable clonal cell lines transfected with the indicated plasmids.
(, p < 0.001
versus control stable line;
, p < 0.05
versus PINK1shRNA cell lines; n = 800-1000 cells
analyzed/condition).
DISCUSSION
There are currently no therapies to slow the progression of PD and related diseases. Although the majority of cases are sporadic, the study of genes linked to rare parkinsonian families demonstrate convergent pathways involving oxidative stress, mitochondrial pathobiology, and protein aggregation (51, 52). In particular, studying the role of proteins implicated in autosomal recessive forms of PD (which generally show earlier ages of onset) holds promise for understanding the compensatory mechanisms that could serve to halt or delay disease progression. The present data indicate that PINK1 plays a physiological role in mitochondrial maintenance, suppressing mitochondrial oxidative stress, fission, and autophagy, and suggest that coordinated activation of fission, autophagy, and Parkin pathways are important protective cellular responses for the efficient removal of the damaged mitochondria.
Consistent with the concept that PINK1 is critical for mitochondrial integrity and homeostasis (2, 3, 6), we found that stable knockdown of PINK1 elevates mitochondrial superoxide and promotes mitochondrial fragmentation, macroautophagy and mitophagy, whereas overexpression of PINK1 stabilizes mitochondrial networks. These studies demonstrate a novel role of PINK1 in modulating autophagy. Superoxide appears to be a common mediator for PINK1 knockdown-induced mitochondrial fission, mitophagy, and macroautophagy. Reciprocal inhibition studies indicate that both the fission and autophagy machineries are essential for the mitochondrial remodeling that accompanies clearance of PINK1-deficient mitochondria and that this mitophagic response plays a protective role. In sum, our data implicate mitochondrial ROS as a key signal regulating coordinated activation of mitochondrial fission and autophagy to optimize clearance of abnormal mitochondria (supplemental Fig. S6).
Recently, Parkin has been shown to recognize and promote the autophagy of drug-depolarized mitochondria (39). Our data extend this observation to dysfunctional mitochondria caused by PINK1 deficiency. Specifically, Parkin amplifies the mitophagic response in PINK1 shRNA cell lines, restoring normal mitochondrial morphology and reducing cell death. As reversal of mitochondrial fission using Drp1-DN exacerbated cell death (supplemental Fig. S5), neuroprotection by Parkin is probably not simply due to the effect on mitochondrial morphology. Rather than a linear pathway of neuroprotection, these studies suggest additional mechanisms of complementation in which PINK1 stabilizes mitochondrial integrity and function, whereas Parkin senses damaged mitochondria for compensatory clearance.
Although our studies and other mammalian cell studies show mitochondrial fragmentation in PINK1-deficient cells (3), studies in Drosophila show the appearance of large diameter, perinuclear, fluorescence staining for mitochondria in mutant loss of function PINK1 flies (21, 53). A line of PINK1 knock-out mice also shows a subtle increase in larger mitochondrial profiles upon aging (54). Although most mitochondria are small and fragmented in PINK1-deficient human SH-SY5Y cells, we also saw occasional large, distended, mitochondrial profiles using electron microscopy (Fig. 2) and isolated larger spherical mitochondria by three-dimensional reconstruction (Fig. 3B); whether this reflects a direct or indirect effect remains unclear. Certainly the ability of Parkin to restore mitochondrial morphologic parameters in PINK1-deficient cells could be due directly to effects on fusion or indirectly through clearance of small, depolarized mitochondrial fragments isolated by fission. It has been suggested that there may be differences between vertebrate and invertebrate phyla (3, 19). It is also possible that fission observed in RNAi studies occurs in response to mitochondrial injury due to declining PINK1 function, which is not observed in neurons habituated to its complete absence. However, it is likely that both fission and fusion events are induced in response to stress and injury, but the predominant steady state result depends upon the balance of pathologic and compensatory mechanisms in a given cellular context.
Autophagy plays a critical role in mitochondrial homeostasis in neurons (55). However, little is known about the signaling regulation of mitophagy. Mitochondrial fission induces mitophagy in some systems (56), and we found that loss of endogenous PINK1 promotes both mitochondrial fission and autophagy. Paradoxically, the autophagic machinery is not only necessary for delivery of mitochondria to lysosomes but also contributes to mitochondrial fragmentation (Fig. 8, C and D). Inhibition of autophagy would be expected to cause increased detection of fragmented, but uncleared, mitochondria if autophagy functioned solely as a passive clearance mechanism downstream of fission. In contrast, inhibition of the autophagy conjugation machinery reversed the fragmentation induced by PINK1 deficiency (Fig. 8, C and D). Interestingly, live cell imaging in hepatocytes demonstrates that autophagosomes can mediate partial sequestration of different portions (center and ends) of intact, elongated mitochondria (29). Thus, coordinated activation of fission and autophagic machineries contributes to mitochondrial remodeling for autophagic turnover in PINK1-deficient cells.
In PD/Lewy body disease post-mortem brain tissues there is mitochondrial autophagy associated with increased mitochondrial kinase phosphorylation (4). The intriguing observation that overexpression of PINK1 enhances interconnected mitochondrial networks supports the concept that mitochondrially localized kinases may play a key role in regulating mitochondrial dynamics. The recent observations that protein kinase A-dependent phosphorylation of Drp1 inhibits fission (41, 57) suggest that other Ser/Thr kinases may also regulate mitochondrial fission/fusion proteins, although neither of the identified PINK1 substrates is known to affect mitochondrial dynamics (58, 59).
As mitochondrial dysfunction is strongly implicated in the pathogenesis of PD, mitophagy may initially represent a compensatory mechanism to remove damaged mitochondria. But excessive mitophagy, unbalanced by appropriate regenerative biogenesis, may ultimately prove detrimental because neurons are dependent upon mitochondrial respiration (28). Autophagy up-regulation in this chronic model of PINK1 deficiency plays a cytoprotective role (Fig. 8, G and H), although the potential impact on neuritic/synaptic maintenance will require further study (40, 60).
In summary, RNAi and overexpression studies demonstrate an endogenous role for PINK1 in maintaining mitochondrial homeostasis by reducing mitochondrial oxidative stress, modulating mitochondrial dynamics, and suppressing autophagy. Autophagy elicited in PINK1-deficient cells is dependent upon increased superoxide and Drp1-dependent fission. Both fission and autophagy play an active role in mitochondrial remodeling elicited by PINK1 deficiency, and coordinated activation of fission and Parkin-enhanced mitophagy may serve to reduce toxicity associated with dysfunctional mitochondria in recessive PD families.
Supplementary Material
Acknowledgments
We thank Simon Watkins and the Center for Biological Imaging at the University of Pittsburgh for assistance with electron microscopy and MetaMorph. We thank Charlotte Diges, Amy Sartori, and Megan Derezinski for technical assistance and all of the investigators listed under “Experimental Procedures” for generously providing reagents.
This work was supported, in whole or in part, by National Institutes of Health Grants AG026389, DC009120, and NS053777 (to C. T. C.). This work was also supported by grants from the Pittsburgh Foundation, Emmerling Fund (to C. T. C.), the American Parkinson Disease Association (to S. M. K.), and a Veterans Administration Advanced Career Development Award (to S. M. K.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1-S8.
Footnotes
The abbreviations used are: 6-OHDA, 6-hydroxydopamine; AV, autophagic vacuole; Drp1, dynamin-related protein-1; Drp1-DN, dominant negative Drp1; LC3, microtubule-associated protein light chain 3; PD, Parkinson disease/parkinsonian disorder; PINK1, PTEN-induced kinase 1; ROS, reactive oxygen species; siRNA, small interfering RNA; RNAi, RNA interference; shRNA, short hairpin RNA; HA, hemagglutinin; GFP, green fluorescent protein; RFP, red fluorescent protein; DAPI, 4′,6-diamidino-2-phenylindole; ERK, extracellular signal-regulated kinase; MnTBAP, manganese(III) tetrakis(4-benzoic acid)porphyrin.
References
- 1.Bove, J., Prou, D., Perier, C., and Przedborski, S. (2005) NeuroRx 2 484-494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoepken, H. H., Gispert, S., Morales, B., Wingerter, O., Del Turco, D., Mulsch, A., Nussbaum, R. L., Muller, K., Drose, S., Brandt, U., Deller, T., Wirth, B., Kudin, A. P., Kunz, W. S., and Auburger, G. (2007) Neurobiol. Dis. 25 401-411 [DOI] [PubMed] [Google Scholar]
- 3.Exner, N., Treske, B., Paquet, D., Holmstrom, K., Schiesling, C., Gispert, S., Carballo-Carbajal, I., Berg, D., Hoepken, H. H., Gasser, T., Kruger, R., Winklhofer, K. F., Vogel, F., Reichert, A. S., Auburger, G., Kahle, P. J., Schmid, B., and Haass, C. (2007) J. Neurosci. 27 12413-12418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu, J.-H., Guo, F., Shelburne, J., Watkins, S., and Chu, C. T. (2003) Brain Pathol. 13 473-481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abeliovich, A., and Flint Beal, M. (2006) J. Neurochem. 99 1062-1072 [DOI] [PubMed] [Google Scholar]
- 6.Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., Ali, Z., Del Turco, D., Bentivoglio, A. R., Healy, D. G., Albanese, A., Nussbaum, R., Gonzalez-Maldonado, R., Deller, T., Salvi, S., Cortelli, P., Gilks, W. P., Latchman, D. S., Harvey, R. J., Dallapiccola, B., Auburger, G., and Wood, N. W. (2004) Science 304 1158-1160 [DOI] [PubMed] [Google Scholar]
- 7.Valente, E. M., Salvi, S., Ialongo, T., Marongiu, R., Elia, A. E., Caputo, V., Romito, L., Albanese, A., Dallapiccola, B., and Bentivoglio, A. R. (2004) Ann. Neurol. 56 336-341 [DOI] [PubMed] [Google Scholar]
- 8.Zhou, C., Huang, Y., Shao, Y., May, J., Prou, D., Perier, C., Dauer, W., Schon, E. A., and Przedborski, S. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 12022-12027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silvestri, L., Caputo, V., Bellacchio, E., Atorino, L., Dallapiccola, B., Valente, E. M., and Casari, G. (2005) Hum. Mol. Genet. 14 3477-3492 [DOI] [PubMed] [Google Scholar]
- 10.Sim, C. H., Lio, D. S., Mok, S. S., Masters, C. L., Hill, A. F., Culvenor, J. G., and Cheng, H. C. (2006) Hum. Mol. Genet. 15 3251-3262 [DOI] [PubMed] [Google Scholar]
- 11.Deng, H., Jankovic, J., Guo, Y., Xie, W., and Le, W. (2005) Biochem. Biophys. Res. Commun. 337 1133-1138 [DOI] [PubMed] [Google Scholar]
- 12.Petit, A., Kawarai, T., Paitel, E., Sanjo, N., Maj, M., Scheid, M., Chen, F., Gu, Y., Hasegawa, H., Salehi-Rad, S., Wang, L., Rogaeva, E., Fraser, P., Robinson, B., St George-Hyslop, P., and Tandon, A. (2005) J. Biol. Chem. 280 34025-34032 [DOI] [PubMed] [Google Scholar]
- 13.Wang, H. L., Chou, A. H., Yeh, T. H., Li, A. H., Chen, Y. L., Kuo, Y. L., Tsai, S. R., and Yu, S. T. (2007) Neurobiol. Dis. 28 216-226 [DOI] [PubMed] [Google Scholar]
- 14.Haque, M. E., Thomas, K. J., D'Souza, C., Callaghan, S., Kitada, T., Slack, R. S., Fraser, P., Cookson, M. R., Tandon, A., and Park, D. S. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 1716-1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beilina, A., Van Der Brug, M., Ahmad, R., Kesavapany, S., Miller, D. W., Petsko, G. A., and Cookson, M. R. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 5703-5708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark, I. E., Dodson, M. W., Jiang, C., Cao, J. H., Huh, J. R., Seol, J. H., Yoo, S. J., Hay, B. A., and Guo, M. (2006) Nature 441 1162-1166 [DOI] [PubMed] [Google Scholar]
- 17.Park, J., Lee, S. B., Lee, S., Kim, Y., Song, S., Kim, S., Bae, E., Kim, J., Shong, M., Kim, J. M., and Chung, J. (2006) Nature 441 1157-1161 [DOI] [PubMed] [Google Scholar]
- 18.Yang, Y., Gehrke, S., Imai, Y., Huang, Z., Ouyang, Y., Wang, J. W., Yang, L., Beal, M. F., Vogel, H., and Lu, B. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 10793-10798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wood-Kaczmar, A., Gandhi, S., Yao, Z., Abramov, A. S., Miljan, E. A., Keen, G., Stanyer, L., Hargreaves, I., Klupsch, K., Deas, E., Downward, J., Mansfield, L., Jat, P., Taylor, J., Heales, S., Duchen, M. R., Latchman, D., Tabrizi, S. J., and Wood, N. W. (2008) PLoS ONE 3 e2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang, D., Qian, L., Xiong, H., Liu, J., Neckameyer, W. S., Oldham, S., Xia, K., Wang, J., Bodmer, R., and Zhang, Z. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 13520-13525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poole, A. C., Thomas, R. E., Andrews, L. A., McBride, H. M., Whitworth, A. J., and Pallanck, L. J. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 1638-1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chu, C. T., Plowey, E. D., Dagda, R. K., Hickey, R. W., Cherra, S. J., III, Clark, R. S. B. (2009) Methods Enzymol. 453 217-249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mizushima, N., Ohsumi, Y., and Yoshimori, T. (2002) Cell Struct. Funct. 27 421-429 [DOI] [PubMed] [Google Scholar]
- 24.Gomez-Lazaro, M., Bonekamp, N. A., Galindo, M. F., Jordan, J., and Schrader, M. (2008) Free Radic. Biol. Med. 44 1960-1969 [DOI] [PubMed] [Google Scholar]
- 25.Dagda, R. K., Zhu, J., Kulich, S. M., and Chu, C. T. (2008) Autophagy 4 770-782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirai, K., Aliev, G., Nunomura, A., Fujioka, H., Russell, R. L., Atwood, C. S., Johnson, A. B., Kress, Y., Vinters, H. V., Tabaton, M., Shimohama, S., Cash, A. D., Siedlak, S. L., Harris, P. L., Jones, P. K., Petersen, R. B., Perry, G., and Smith, M. A. (2001) J. Neurosci. 21 3017-3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nixon, R. A., Wegiel, J., Kumar, A., Yu, W. H., Peterhoff, C., Cataldo, A., and Cuervo, A. M. (2005) J. Neuropathol. Exp. Neurol. 64 113-122 [DOI] [PubMed] [Google Scholar]
- 28.Cherra, S. J., and Chu, C. T. (2008) Future Neurol. 3 309-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim, I., Rodriguez-Enriquez, S., and Lemasters, J. J. (2007) Arch. Biochem. Biophys. 462 245-253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tolkovsky, A. M., Xue, L., Fletcher, G. C., and Borutaite, V. (2002) Biochimie (Paris) 84 233-240 [DOI] [PubMed] [Google Scholar]
- 31.Zhu, J. H., Horbinski, C., Guo, F., Watkins, S., Uchiyama, Y., and Chu, C. T. (2007) Am. J. Pathol. 170 75-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu, C. T., Zhu, J., and Dagda, R. (2007) Autophagy 3 663-666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hara, T., Nakamura, K., Matsui, M., Yamamoto, A., Nakahara, Y., Suzuki-Migishima, R., Yokoyama, M., Mishima, K., Saito, I., Okano, H., and Mizushima, N. (2006) Nature 441 885-889 [DOI] [PubMed] [Google Scholar]
- 34.Rideout, H. J., Lang-Rollin, I., and Stefanis, L. (2004) Int. J. Biochem. Cell Biol. 36 2551-2562 [DOI] [PubMed] [Google Scholar]
- 35.Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G., Scaravilli, F., Easton, D. F., Duden, R., O'Kane, C. J., and Rubinsztein, D. C. (2004) Nat. Genet. 36 585-595 [DOI] [PubMed] [Google Scholar]
- 36.Komatsu, M., Wang, Q. J., Holstein, G. R., Friedrich, V. L., Jr., Iwata, J., Kominami, E., Chait, B. T., Tanaka, K., and Yue, Z. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 14489-14494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jennings, J. J., Jr., Zhu, J. H., Rbaibi, Y., Luo, X., Chu, C. T., and Kiselyov, K. (2006) J. Biol. Chem. 281 39041-39050 [DOI] [PubMed] [Google Scholar]
- 38.Zhang, Y., Qi, H., Taylor, R., Xu, W., Liu, L. F., and Jin, S. (2007) Autophagy 3 337-346 [DOI] [PubMed] [Google Scholar]
- 39.Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008) J. Cell Biol. 183 795-803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plowey, E. D., Cherra, S. J., III, Liu, Y. J., and Chu, C. T. (2008) J. Neurochem. 105 1048-1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cribbs, J. T., and Strack, S. (2007) EMBO Rep. 8 939-944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kimura, S., Noda, T., and Yoshimori, T. (2007) Autophagy 3 452-460 [DOI] [PubMed] [Google Scholar]
- 43.Yamamoto, A., Tagawa, Y., Yoshimori, T., Moriyama, Y., Masaki, R., and Tashiro, Y. (1998) Cell Struct. Funct. 23 33-42 [DOI] [PubMed] [Google Scholar]
- 44.Kulich, S. M., Horbinski, C., Patel, M., and Chu, C. T. (2007) Free Radic. Biol. Med. 43 372-383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trimmer, P. A., Swerdlow, R. H., Parks, J. K., Keeney, P., Bennett, J. P., Jr., Miller, S. W., Davis, R. E., and Parker, W. D., Jr. (2000) Exp. Neurol. 162 37-50 [DOI] [PubMed] [Google Scholar]
- 46.Smirnova, E., Griparic, L., Shurland, D. L., and van der Bliek, A. M. (2001) Mol. Biol. Cell 12 2245-2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karbowski, M., Lee, Y. J., Gaume, B., Jeong, S. Y., Frank, S., Nechushtan, A., Santel, A., Fuller, M., Smith, C. L., and Youle, R. J. (2002) J. Cell Biol. 159 931-938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., Kominami, E., Ohsumi, Y., and Yoshimori, T. (2000) EMBO J. 19 5720-5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rodriguez-Enriquez, S., Kim, I., Currin, R. T., and Lemasters, J. J. (2006) Autophagy 2 39-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patel, M. N. (2003) Aging Cell 2 219-222 [DOI] [PubMed] [Google Scholar]
- 51.Betarbet, R., Sherer, T. B., Di Monte, D. A., and Greenamyre, J. T. (2002) Brain Pathol. 12 499-510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dawson, T. M., and Dawson, V. L. (2003) Science 302 819-822 [DOI] [PubMed] [Google Scholar]
- 53.Yang, Y., Ouyang, Y., Yang, L., Beal, M. F., McQuibban, A., Vogel, H., and Lu, B. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 7070-7075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gautier, C. A., Kitada, T., and Shen, J. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 11364-11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Larsen, K. E., and Sulzer, D. (2002) Histol. Histopathol. 17 897-908 [DOI] [PubMed] [Google Scholar]
- 56.Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., Stiles, L., Haigh, S. E., Katz, S., Las, G., Alroy, J., Wu, M., Py, B. F., Yuan, J., Deeney, J. T., Corkey, B. E., and Shirihai, O. S. (2008) EMBO J. 27 433-446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang, C. R., and Blackstone, C. (2007) J. Biol. Chem. 282 21583-21587 [DOI] [PubMed] [Google Scholar]
- 58.Plun-Favreau, H., Klupsch, K., Moisoi, N., Gandhi, S., Kjaer, S., Frith, D., Harvey, K., Deas, E., Harvey, R. J., McDonald, N., Wood, N. W., Martins, L. M., and Downward, J. (2007) Nat. Cell Biol. 9 1243-1252 [DOI] [PubMed] [Google Scholar]
- 59.Pridgeon, J. W., Olzmann, J. A., Chin, L. S., and Li, L. (2007) PLoS Biol. 5 e172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang, Y., Fukui, K., Koike, T., and Zheng, X. (2007) Eur. J. Neurosci. 26 2979-2988 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.