

Abstract
The RIG-I-like receptors (RLRs), RIG-I and MDA5, recognize single-stranded RNA with 5′ triphosphates and double-stranded RNA (dsRNA) to initiate innate antiviral immune responses. LGP2, a homolog of RIG-I and MDA5 that lacks signaling capability, regulates the signaling of the RLRs. To establish the structural basis of dsRNA recognition by the RLRs, we have determined the 2.0-Å resolution crystal structure of human LGP2 C-terminal domain bound to an 8-bp dsRNA. Two LGP2 C-terminal domain molecules bind to the termini of dsRNA with minimal contacts between the protein molecules. Gel filtration chromatography and analytical ultracentrifugation demonstrated that LGP2 binds blunt-ended dsRNA of different lengths, forming complexes with 2:1 stoichiometry. dsRNA with protruding termini bind LGP2 and RIG-I weakly and do not stimulate the activation of RIG-I efficiently in cells. Surprisingly, full-length LGP2 containing mutations that abolish dsRNA binding retained the ability to inhibit RIG-I signaling.
The innate immune response is the first line of defense against invading pathogens; it is the ubiquitous system of defense against microbial infections (1). Toll-like receptors (TLRs)3 and RIG-I (retinoic acid-inducible gene 1)-like receptors (RLRs) play key roles in innate immune response toward viral infection (2-5). Toll-like receptors TLR3, TLR7, and TLR8 sense viral RNA released in the endosome following phagocytosis of the pathogens (6). RIG-I-like receptors RIG-I and MDA5 detect viral RNA from replicating viruses in infected cells (3, 7, 8). Stimulation of these receptors leads to the induction of type I interferons (IFNs) and other proinflammatory cytokines, conferring antiviral activity to the host cells and activating the acquired immune responses (4, 9).
RIG-I discriminates between viral and host RNA through specific recognition of the uncapped 5′-triphosphate of single-stranded RNA (5′ ppp ssRNA) generated by viral RNA polymerases (10, 11). In addition, RIG-I also recognizes double-stranded RNA generated during RNA virus replication (7, 12). Transfection of cells with synthetic double-stranded RNA stimulates the activation of RIG-I (13, 14). Synthetic dsRNA mimics, such as polyinosinic-polycytidylic acid (poly(I·C)), can activate MDA5 when introduced into the cytoplasm of cells. Digestion of poly(I·C) with RNase III transforms poly(I·C) from a ligand for MDA5 into a ligand for RIG-I, suggesting that MDA5 recognizes long dsRNA, whereas RIG-I recognizes short dsRNA (15). Studies of RIG-I and MDA5 knock-out mice confirmed the essential roles of these receptors in antiviral immune responses and demonstrated that they sense different sets of RNA viruses (12, 16).
RIG-I and MDA5 contain two caspase recruiting domains (CARDs) at their N termini, a DEX(D/H) box RNA helicase domain, and a C-terminal regulatory or repressor domain (CTD). The helicase domain and the CTD are responsible for viral RNA binding, whereas the CARDs are required for signaling (3, 8). The current model of RIG-I activation suggests that under resting conditions RIG-I is in a suppressed conformation, and viral RNA binding triggers a conformation change that leads to the exposure of the CARDs for the recruitment of the downstream protein IPS-1 (also known as MAVS, Cardif, or VISA) (14, 17). Limited proteolysis of the RIG-I·dsRNA complex showed that RIG-I residues 792-925 of the CTD are involved in dsRNA and 5′ ppp ssRNA binding (14). The CTD of RIG-I overlaps with the C terminus of the previously identified repressor domain (18). The structures of RIG-I and LGP2 (laboratory of genetics and physiology 2) CTD in isolation have been determined by x-ray crystallography and NMR spectroscopy (14, 19, 20). A large, positively charged surface on RIG-I recognizes the 5′ triphosphate group of viral ssRNA (14, 19). RNA binding studies by titrating RIG-I CTD with dsRNA and 5′ ppp ssRNA suggested that overlapping sets of residues on this charged surface are involved in RNA binding (14). Mutagenesis of several positively charged residues on this surface either reduces or disrupts RNA binding by RIG-I, and these mutations also affect the induction of IFN-β in vivo (14, 19). However, the exact nature of how the RLRs recognize viral RNA and how RNA binding activates these receptors remains to be established.
LGP2 is a homolog of RIG-I and MDA5 that lacks the CARDs and thus has no signaling capability (21, 22). The expression of LGP2 is inducible by dsRNA or IFN treatment as well as virus infection (21). Overexpression of LGP2 inhibits Sendai virus and Newcastle disease virus signaling (21). When coexpressed with RIG-I, LGP2 can inhibit RIG-I signaling through the interaction of its CTD with the CARD and the helicase domain of RIG-I (18). LGP2 could suppress RIG-I signaling by three possible ways (23): 1) binding RNA with high affinity, thereby sequestering RNA ligands from RIG-I; 2) interacting directly with RIG-I to block the assembly of the signaling complex; and 3) competing with IKKi (IκB kinase ε) in the NF-κB signaling pathway for a common binding site on IPS-1. To elucidate the structural basis of dsRNA recognition by the RLRs, we have crystallized human LGP2 CTD (residues 541-678) bound to an 8-bp double-stranded RNA and determined the structure of the complex at 2.0 Å resolution. The structure revealed that LGP2 CTD binds to the termini of dsRNA. Mutagenesis and functional studies showed that dsRNA binding is likely not required for the inhibition of RIG-I signaling by LGP2.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification—DNAs encoding the C-terminal domains of human LGP2 (residues 541-678), RIG-I (residues 802-925), and MDA5 (residues 892-1017) were cloned into expression vector pET22b(+) (Novagen). All of the cloned DNA sequences were confirmed by plasmid DNA sequencing. The proteins were expressed in Escherichia coli strain BL21(DE3) by induction at A600 = 0.6-0.8 with 0.5 mm isopropyl-β-d-thiogalactoside overnight at 15 °C. The cells were lysed by sonication, and the proteins were purified by batch method using His-Select nickel affinity resin (Sigma-Aldrich) in a buffer containing 20 mm Tris, 150 mm NaCl, at pH 7.5 (buffer A). After incubation for 2 h, the nickel beads were collected and washed three times with 10 volumes of buffer A containing 10 mm imidazole, and the proteins were eluted with buffer A containing 250 mm imidazole. The proteins were further purified by gel filtration chromatography on a Superdex75 (1.6 × 60) column (GE Healthcare) eluted with buffer A. To form the complex with dsRNA, LGP2 CTD were mixed with an 8-bp dsRNA at a molar ratio of 1:1, and the 2:1 LGP2·dsRNA complex was purified by gel filtration chromatography on a Superdex75 (1.6 × 60) column. Mutants of full-length LGP2 and LGP2 CTD were generated using a QuikChange mutagenesis kit (Stratagene). The sequences of the mutants were confirmed by plasmid DNA sequencing. The mutant proteins were expressed and purified the same way as the native protein.
Crystallization, Data Collection, and Structure Determination—Purified LGP2·dsRNA complex was concentrated to ∼30 mg ml-1 in a buffer containing 20 mm Tris, 150 mm NaCl, and 4 mm dithiothreitol at pH 7.5. The complex was crystallized with 16-18% (w/v) PEG3350 in a buffer containing 0.2 m (NH4)2SO4, 0.1 m Tris-HCl at pH 8.5. The crystals were transferred stepwise from the mother liquor to a cryoprotectant containing 25% (v/v) glycerol and flash frozen in liquid nitrogen. The LGP2·dsRNA complex crystallized in space group C2, with cell dimensions: a = 116.46 Å, b = 54.19 Å, c = 67.20 Å, and β = 97.26°. The crystallographic asymmetric unit contains one 2:1 LGP2·dsRNA complex. Diffraction data were collected using a Rigaku RAXIS IV2+ image plate detector mounted on a Rigaku Micromax-007HF generator and processed with the HKL package (24). Statistics of data collection and refinement are shown in Table 1.
TABLE 1.
Data collection and refinement statistics
| LGP2/dsRNA | |
|---|---|
| Data collection | |
| Space group | C2 |
| Cell dimensions | |
| a, b, c (Å) | 116.46, 54.19, 67.20 |
| α, β, γ (°) | 90.00, 97.26, 90.00 |
| Resolution (Å) | 50-2.00 (2.07-2.00)a |
| Rmerge | 4.6 (33.6) |
| I/σI | 46.2 (5.1) |
| Completeness (%) | 99.6 (98.6) |
| Redundancy | 3.6 (3.5) |
| Refinement | |
| Resolution (Å) | 50-2.00 |
| No. reflections | 27234 |
| Rwork/Rfree | 21.3/25.4 |
| No. atoms | |
| Protein | 2167 |
| RNA | 340 |
| Zinc ion | 2 |
| Water | 215 |
| B factors | |
| Protein | 46.6 |
| RNA | 43.5 |
| Zinc ion | 51.5 |
| Water | 52.2 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.43 |
The values in parentheses are for highest resolution shell.
The crystal structure of the LGP2·dsRNA complex was determined by molecular replacement with MOLREP in the CCP4 suite (25) using the crystal structure of RIG-I CTD as search model (Protein Data Bank code 2QFB, chain A). The model was rebuilt using O (26). After several rounds of rebuilding and refinement with CNS (27), the electron density for the dsRNA became apparent. An 8-bp dsRNA from the TLR3·dsRNA complex structure (Protein Data Bank code 3CIY) was docked into the electron density map and rebuilt with O. The complex structure was refined by several rounds of positional, simulated annealing, and individual B-factor refinement using CNS followed by manual remodeling after each round of refinement.
RNA Binding Studies by Gel Filtration Chromatography—RNAs used in the binding studies were chemically synthesized by IDT (Coralville, IA) or by in vitro transcription using T7 RNA polymerase. The sequences of the RNAs are shown in supplemental Table S2. Double-stranded RNAs were generated by heating the ssRNA at 95 °C for 5 min and annealing at room temperature for 30 min. Each dsRNA was mixed with excess protein (RNA to protein molar ratio of 1:3), and 100 μl of samples were injected over a Superdex200 (10/300 GL) column (GE Healthcare) eluted with buffer A. The column was calibrated with a set of protein standards for gel filtration chromatography (Bio-Rad) to ensure accurate estimation of the molecular weight of the LGP2·dsRNA complexes.
RNA Binding Studies by Fluorescence Anisotropy—Cy3-labeled
RNA probes were heated to anneal with their complementary RNA to form dsRNA.
Fluorescence measurements were performed at room temperature using a
PerkinElmer Life Sciences luminescence spectrometer LS55. The excitation and
emission wavelengths are 540 and 565 nm, respectively, with an integration
time of 2 s and slit width of 5 nm. Purified LGP2, RIG-I, and MDA5 CTD were
titrated into the dsRNA in buffer A and mixed by magnetic stirring. Total
volume of protein added was less than 3% of the final sample volume. The
represented anisotropy values used to calculate the affinity are the average
of 10 measurements. The binding data were analyzed by nonlinear least square
fitting using KaleidaGraph software (Synergy Software, Reading, PA). The Hill
equation, ΔA = Bmax
Xn/(Xn +
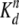 ) was used to determine the dissociation
constant (Kd). In this equation, ΔA is the
anisotropy change caused by the ligand binding, Bmax is
the maximum anisotropy change, X is the total concentration of the
input protein, and the exponential term n is the Hill coefficient.
Binding studies for the mutants of LGP2 CTD were carried out under similar
conditions.
) was used to determine the dissociation
constant (Kd). In this equation, ΔA is the
anisotropy change caused by the ligand binding, Bmax is
the maximum anisotropy change, X is the total concentration of the
input protein, and the exponential term n is the Hill coefficient.
Binding studies for the mutants of LGP2 CTD were carried out under similar
conditions.
Analytical Ultracentrifugation—To verify the stoichiometry of LGP2 binding to dsRNA, LGP2 CTD complexes with the 8-bp dsRNA, the 24-bp dsRNA, and the hairpin RNA were analyzed by sedimentation velocity. 400-μl samples in 20 mm Tris-HCl buffer at pH 7.5, 150 mm NaCl, 10 mm β-mercaptoethanol were spun overnight at 48,000 rpm at 20 °C in a Beckman XL-I using absorbance optics at 302 nm. The data were analyzed by the program Sedfit using the c(s) and c(M) models to determine differential sedimentation coefficient and apparent mass distributions, respectively. The 8-bp dsRNA·LGP2 complex gave an estimated molecular mass value similar to that of a 2:1 complex, whereas the hairpin RNA·LGP2 complex had an estimated molecular mass consistent with a 1:1 complex. The 24-bp dsRNA·LGP2 complex sedimented as a single peak, but the estimated mass could indicate either a 2:1 or a 2:2 complex. To resolve the stoichiometry, a sedimentation equilibrium experiment was performed on the 24-bp dsRNA·LGP2 complex. 100-μl samples of purified 24-bp dsRNA·LGP2 complex at concentrations of 10, 33.3, and 100 μm were spun at 17,000, 21,000, and 30,000 rpm at 20 °C until equilibrium was reached, and scanned at 297 and 305 nm. The data were trimmed using Win-Reedit and globally analyzed using WinNonlin. The global analysis of nine data sets revealed that the data could be described by a single species with a reduced buoyant molecular weight of 1.836 (at a speed of 17,000 rpm). To convert to experimental molecular weight, the partial specific volume must be known; for a protein·RNA complex this value corresponds to the weight average of each component in the sedimenting species. The partial specific volume for LGP2 CTD alone was calculated to be 0.7328 based on sequence using Sednterp, and that for the 24-bp dsRNA was calculated to be 0.5688 using the NucProt server. Both 2:1 and 2:2 complexes were considered, with weight average partial specific volumes of 0.6813 or 0.6566, respectively. The data were consistent with a 2:1 complex but were not consistent with either a 1:1 or a 2:2 complex.
Reporter Gene Assays—Actively growing HEK 293T cells were plated in CoStar White 96-well plates at 4.4 × 104 cells ml-1 for transfection. When the cells were ∼60-80% confluent, they were transfected with a mixture of Lipofectamine 2000 reagent (Invitrogen) and constant amounts of the reporter plasmids IFN-β Luc (30 ng) or pNiFty Luc (15 ng; Invivogen), which contain the firefly luciferase gene, phRL-TK (5 ng, Promega), and phRIG-I (0.5 ng; Invivogen). In the functional assays of LGP2, phLGP2 (1.0 ng; Invivogen) were cotransfected with the same amount of phRIG-I and the reporter plasmids as described above. The cells were incubated for 24 h to allow expression from the plasmids. The dsRNA ligands were then transfected into the cells at a final concentration of 0.5 or 0.2 μm as indicated in the figure legend. After 12 h of incubation, the cells were analyzed using the Dual Glo luciferase assay system reagents (Promega), quantifying luminescence with the FLU-Ostar OPTIMA Plate Reader (BMG Labtech.). The ratios of firefly luciferase over Renilla luciferase were plotted. The RNA ligands used were purified by preparative denaturing polyacrylamide gel electrophoresis or gel filtration chromatography.
Western Blot—293T cells transfected with wild type LGP2 or its mutants were lysed with the passive lysis buffer (Promega) and sonicated to degrade chromosomal DNA. Equal amounts of proteins from each sample, as determined by staining with Coomassie Blue, were separated on NuPAGE 4-12% (w/v) bistris gels (Invitrogen) and blotted onto polyvinylidene difluoride membrane. Affinity-purified rabbit antibody against human LGP2 (Proteintec) was used as primary antibody. The blots were developed with peroxidase-conjugated secondary antibodies and an ECL Plus Western blotting detection system (GE Healthcare).
RESULTS
Overall Structure of the LGP2·dsRNA Complex—The structure of LGP2 CTD in complex with dsRNA was determined by molecular replacement using the crystal structure of RIG-I CTD (19) as a search model. The crystallographic asymmetric unit contains a 2:1 complex between LGP2 CTD and the 8-bp dsRNA (Fig. 1A). The refined model includes residues 546-672 of LGP2 in molecule A, residues 544-678 of LGP2 plus a five-residue His tag in molecule B, and the 8-bp dsRNA. The complex exhibits pseudo 2-fold symmetry. The root mean square deviation between the two LGP2 CTD molecules is 0.45 Å. The following discussion is based on the structure of the complex between LGP2 (A) and the dsRNA caused by better defined electron density of the molecule. The structure of LGP2 CTD in the complex is shown in Fig. 1B.
FIGURE 1.

Structure of human LGP2 C-terminal domain in complex with an 8-bp dsRNA. A, two LGP2 CTD bound to the termini of an 8-bp blunt-ended dsRNA in the crystallographic asymmetric unit. The two protein molecules are shown as cyan (LGP2, A) and green (LGP2, B) ribbons. The dsRNA is shown as a stick model. The zinc ions in LGP2 CTD are shown as gray spheres. B, ribbon representation of the structure of LGP2 CTD. The dsRNA-binding surface of LGP2 is defined by the β-sheet containing β5 to β8, the loop connecting β5 to β6 (loop5-6), the hairpin containing β3 and β4, and the two loops connecting β5 to β6, and β9 to the C-terminal helix. The side chains of the four conserved cysteine residues coordinating with the zinc ion are shown as stick models. C, structure of LGP2 CTD bound to the 8-bp dsRNA. LGP2 CTD is colored as a rainbow from blue at the N terminus to red at the C terminus. The orientation of LGP2 is related to that of LGP2 in B by a rotation of ∼90° counter-clockwise along the z axis. D, electrostatic surface potential (ranging from blue = 10 kT/e to red = -10 kT/e) of LGP2 CTD showing the high degree of shape and charge complementarity between LGP2 CTD and the first turn of blunt-ended dsRNA. LGP2 CTD is in the same orientation as shown in C. E, stereo view of the superposition of the LGP2 CTD structure (blue) over the RIG-I CTD structure (green).
The 8-bp dsRNA binds to a deep groove between two LGP2 CTD molecules, making primary contacts with LGP2 through the two blunt ends (Fig. 1A). Apart from two hydrogen bonds that involve residue Lys650, the two protein molecules do not interact with each other, suggesting that formation of the protein dimer is not required for RNA binding. The dsRNA adopts a typical A-form structure with slight distortions at the two ends. The binding surfaces between LGP2 and dsRNA show a high degree of shape and charge complementarity (Fig. 1, C and D). LGP2 interacts with the phosphate backbone of the RNA through extensive electrostatic interactions and hydrogen bonding. The exposed terminal GC base pair interacts with LGP2 through extensive hydrophobic interactions. The total buried surface area at the LGP2 dsRNA interface is ∼1540 Å2, with major contributions from the first six nucleotides at the 5′ end of one RNA strand (buried surface area, ∼1020 Å2) and minor contributions from the two nucleotides from the 3′ end of the complementary strand (buried surface area, ∼520 Å2). The calculated shape correlation statistics (Sc, a measure of the degree that two contacting surfaces are geometrically matched) is 0.70, where an Sc value of 1.0 indicates a perfect fit (28). The buried surface area and shape complementarity between LGP2 and dsRNA are comparable with typical antibody·peptide antigen complexes, which have an average buried surface area of 1430 Å2 and an Sc of 0.75 (29).
Structure of LGP2 C-terminal Domain—Although the amino acid sequences of LGP2 and RIG-I CTD are only 25% identical (supplemental Fig. S1), the structures of LGP2 and RIG-I CTD are highly conserved (Fig. 1E). The root mean square deviation between the 102 conserved Cα atoms in the two proteins is only 0.90 Å. LGP2 CTD contains a three-stranded (β1, β2, and β9) antiparallel β-sheet near its N terminus and a four-stranded (β5, β6, β7, and β8) antiparallel β-sheet in the middle (Fig. 1B). The two β-sheets are connected by a β-hairpin formed by strands β3 and β4 and two short α-helices. Four conserved cysteine residues (Cys556, Cys559, Cys612, and Cys615) in the two loops connecting strands β1-β2 and β6-β7 make additional connections between the two β-sheets by coordinating a zinc ion (Fig. 1B). The eight residues near the C terminus of LGP2 form a well defined α-helix, whereas the corresponding helix in RIG-I is only four residues long. The RNA-binding site of LGP2 is located at the large concave surface defined by the β-sheet containing strands β5 to β8, the β-hairpin, and the three loops connecting β5 to β6, β8 to β9, and β9 to the C-terminal helix (Fig. 1, B and C).
The major difference between the crystal structure of LGP2 and RIG-I CTD occurs in the long loop (loop5-6) connecting strands β5 and β6 (Fig. 1E). NMR structure of RIG-I CTD showed that this loop is flexible (14). The structure of LGP2 CTD in isolation showed that this loop is not ordered (20). The ordered structure of this loop observed in the LGP2·dsRNA complex structure (Fig. 1E) is most likely due to the binding of dsRNA, especially the hydrophobic interactions with the two bases at the blunt end of the dsRNA.
Structural Basis of dsRNA Recognition by LGP2—Unlike TLR3 that binds primarily to the phosphate backbone of long dsRNA (30), LGP2 binds specifically to the ends of dsRNA (Fig. 2A and Table 2). The exposed terminal GC base pairs are recognized through hydrophobic interactions that involve residues Val632, Leu621, Val595, Ile597, Phe601, and Trp604 (Fig. 2, B and C, and Table 2). However, there are significant differences in the interactions between LGP2 CTD and the two RNA strands at each of the dsRNA terminus. The two hydroxyl groups of the ribose at the 3′ end of the two RNA strands interact with LGP2 through a network of five direct hydrogen bonds with the side chains of Glu573, His576, and Trp604 (Fig. 2B), as well as a solvent-mediated hydrogen bond with the backbone amine group of Lys602. The next nucleotide near the 3′ end (G7) interacts with LGP2 through electrostatic interactions between its phosphate group and the side chain of Lys599 (Fig. 2B and Table 2).
FIGURE 2.

Interactions between the LGP2 C-terminal domain and dsRNA. A, two views of the LGP2·dsRNA complex showing how LGP2 CTD interacts with the 3′ end (pink stick models, left panel) and 5′ end (yellow stick models, right panel) of the dsRNA. The protein is shown as a cyan ribbon, and the dsRNA is shown as a stick model. The zinc ion is shown as a gray sphere. Close-ups of regions in the gray boxes are shown in B and C. B, stereo close-up of the interactions between LGP2 CTD and the 3′ end of the 8-bp dsRNA. The complementary strand is shown in gray. Solvent molecules mediating hydrogen bonding between the protein and the RNA are shown as red spheres. C, stereo close-up of the interactions between the LGP2 CTD and the 5′ end of the 8-bp dsRNA.
TABLE 2.
Interactions between LGP2 CTD and the 8-bp dsRNA
HB, hydrogen bonds; VDW, van der Waals' contacts; HP, hydrophobic interactions; sHB, solvent mediated hydrogen bonds. The solvent distances to dsRNA atoms and protein atoms are shown. ES, electrostatic interactions.
| Nucleotide (ends) atoms | LGP2 CTD atoms | Distances | Type of interactions |
|---|---|---|---|
| Å | |||
| G1 (5′) | |||
| C-1′ | Val595 CG1 | 3.62 | HP |
| O-6 | Ile597 CG2 | 3.64 | HP |
| N-2 | Trp604 CH2 | 3.69 | VDW |
| O-2′ | Leu621 CB | 3.18 | VDW |
| N-7 | Val596 O | 2.99/3.30 | sHB |
| C2 (5′) | |||
| O-2′ | His576 ND1 | 2.83 | HB |
| O-2P | Gly620 CA | 3.40 | VDW |
| O-4′ | Leu621 CD2 | 3.51 | VDW |
| C-4′ | Val632 CG1 | 3.72 | VDW |
| O-1P | Lys634 NZ | 3.98 | ES |
| O-2′ | His577 N | 2.92/2.78 | sHB |
| O-3′ | Leu633 O | 3.25/2.73 | sHB |
| G3 (5′) | |||
| O-2′ | Thr575 CB | 3.35 | VDW |
| O-2P | Lys634 NZ | 3.53 | ES |
| O-1P | Val635 N | 3.05 | HB |
| O-1P | Trp652 N | 2.79/2.96 | sHB |
| O-3′ | Thr575 O | 2.96/2.89 | sHB |
| C4 (5) | |||
| O-2P | Lys651 CB | 3.64 | VDW |
| O-2P | Trp652 N | 3.58 | HB |
| O-2P | Ser653 OG | 2.54 | HB |
| O-2P | Ser653 N | 2.95 | HB |
| O-1P | Trp652 N | 2.74/2.96 | sHB |
| G5 (5′) | |||
| O-2P | Lys651 NZ | 2.96 | ES |
| O-1P | Arg654 NH2 | 3.24 | ES |
| C6 (5) | |||
| O-1P | Arg654 NH2 | 2.64/3.15 | sHB |
| G1 (3′) | |||
| C-8 | Arg636 NH1 | 3.39 | VDW |
| O-5′ | Arg636 NH1 | 3.51/2.94 | sHB |
| G7 (3′) | |||
| N-2 | His576 CE1 | 3.91 | VDW |
| O-2P | Lys599 NZ | 3.88 | ES |
| C8 (3′) | |||
| O-2′ | Glu573 OE2 | 2.50 | HB |
| O-3′ | Glu573 OE1 | 2.73 | HB |
| O-2′ | His576 NE2 | 2.87 | HB |
| N-4 | Lys599 CD | 3.73 | VDW |
| N-3 | Phe601 CZ | 3.46 | HP |
| O-2′ | Trp604 NE1 | 3.03 | HB |
| O-3′ | Lys602 N | 2.81/2.82 | sHB |
In contrast, the 5′ ends of the two RNA strands have more extensive interactions with LGP2. Six nucleotides at the 5′ end of the dsRNA interact with LGP2 CTD (Fig. 2C and Table 2). The 2′ hydroxyl group of C2 interacts with LGP2 through a direct hydrogen bond with the side chain of His576 and two solvent-mediated hydrogen bonds with the backbone amine group of His577 and the carbonyl group of Leu633. The side chain of Lys634 bridges the two phosphate groups of C2 and G3 through electrostatic interactions. The phosphate group of G3 also forms a direct hydrogen bond with the amine group of Val635 and a solvent-mediated hydrogen bond with the backbone amine group of Trp652. The nucleotide C4 binds LGP2 through a hydrogen bond with the side chain of Ser653 and a solvent-mediated hydrogen bond with the backbone amine group of Trp652. The phosphate group of G5 makes strong electrostatic interactions with the side chains of Lys651 and Arg654. Seven residues, Glu573, His576, Lys599, Gly620, Lys634, Lys651, and Trp652, involved in dsRNA binding by LGP2 are conserved in the sequences of human RIG-I and MDA5 (supplemental Fig. S1), suggesting that these residues should play similar roles in dsRNA recognition by RIG-I and MDA5.
The interaction between LGP2 CTD and dsRNA was further examined by molecular modeling based on the complex structure to elucidate how LGP2 CTD might interact with longer dsRNA. Superposition of 12- and 26-bp dsRNAs onto the complex structure (supplemental Fig. S2) showed that four positively charged residues, Arg636, Lys650, Lys651, and Arg654 of LGP2, could reach into the major grove of longer dsRNA and make electrostatic interactions with the phosphate backbone of dsRNA (supplemental Fig. S2). Consistent with this result, mutation of Lys651 in this cluster of positively charged residues to glutamic acid abolishes dsRNA binding by LGP2. The structure of B-form double-stranded DNA is unlikely to make similar electrostatic interactions because the overall shape of DNA does not match the shape of LGP2 dsRNA-binding surface, explaining why LGP2 does not bind DNA. The 2′ OH groups of dsRNA also make significant contributions to RNA binding by LGP2 CTD (Table 2). It is unlikely that LGP2 would bind to the middle of longer dsRNA because the loop between strands β5 and β6 would clash with the dsRNA (supplemental Fig. S2). The complex structure also suggests that dsRNA with 3′ overhangs may not bind LGP2 effectively because it will disrupt the hydrophobic interactions between the exposed base pair and the protein. However, dsRNA with 5′ overhangs would still bind LGP2 without clashing with the protein.
LGP2 Binds to the Blunt End of dsRNA—First, we examined the binding of blunt-ended dsRNA to LGP2, RIG-I, and MDA5 CTD by fluorescence anisotropy. The affinity of LGP2 CTD for a 10-bp and a 27-bp dsRNA are 105 nm and 1 μm, respectively (Fig. 3A). This result is comparable with the previously reported affinities of full-length LGP2 to these dsRNA (31). The difference in the affinities of the 10- and 27-bp dsRNA for LGP2 CTD is most likely due to the different kinetic properties of the association reaction. Short dsRNA has a better chance to be oriented properly for the binding reaction to occur. The affinities of RIG-I and MDA5 CTD for these two dsRNA are considerably lower than LGP2 CTD (Fig. 3A). To confirm that LGP2 binds to the ends of the dsRNA, we synthesized an 8-bp dsRNA, a short hairpin RNA, a 24-bp dsRNA, an siRNA-like dsRNA, and a dsRNA with 5′ overhangs of two adenylates (supplemental Table S1) and studied their binding properties with LGP2 CTD by gel filtration chromatography and analytical ultracentrifugation (Fig. 3 and Tables 3 and 4). We found that LGP2 CTD binds tightly to the 8- and 24-bp bluntended dsRNA and forms complexes of 2:1 stoichiometry (Fig. 3, B and D, and Table 3) with expected apparent molecular masses of 45.9 and 63.8 kDa, respectively. The small hairpin RNA, which has only one blunt end, binds to LGP2 at a stoichiometry of nearly 1:1 (Fig. 3C and Table 3). The expected apparent molecular mass of a 1:1 complex between the small hairpin RNA and LGP2 CTD is 35.4 kDa, very close to the observed molecular mass (32.0 kDa) of the complex (Table 3). Sedimentation velocity analysis confirmed that the 8-bp dsRNA forms a 2:1 complex and the hairpin RNA forms a 1:1 complex with LGP2 CTD (Fig. 3E and Table 4). Sedimentation equilibrium analysis of purified 24-bp dsRNA·LGP2 CTD complex demonstrated unambiguously that the stoichiometry between LGP2 and 24-bp dsRNA is 2:1 rather than 1:1 or 2:2 (Fig. 3F and Table 4).
FIGURE 3.

LGP2 C-terminal domain binds to the blunt ends of dsRNA. A, fluorescence anisotropy changes measured by titrating the CTD of LGP2, RIG-I, and MDA5 into Cy3-labeled 10- and 27-bp dsRNA. LGP2 CTD binding with the 10- and 27-bp dsRNA are represented by the black and red curves, respectively. RIG-I CTD binding to the 10- and 27-bp dsRNA is represented by the green and cyan curves, and MDA5 CTD by the blue and magenta curves, respectively. B, binding study of LGP2 CTD with the 8-bp dsRNA by gel filtration chromatography. Elution volumes of three protein standards of molecular masses 158, 44, and 17 kDa were shown above the chromatogram. The elution volume of a 670-kDa protein standard is 9.45 ml. C, binding study of LGP2 CTD with a 24-nucleotide hairpin RNA (shRNA) containing one blunt end. D, binding study of LGP2 CTD with a 24-bp dsRNA containing two blunt ends. E, sedimentation velocity of purified LGP2 CTD in complex with the 8-bp dsRNA, the small hairpin RNA, and the 24-bp dsRNA. F, sedimentation equilibrium of purified LGP2 CTD·24 bp dsRNA complex. The estimated molecular mass of a 2:1 complex between LGP2 CTD and the 24-bp dsRNA complex is ∼45 kDa, and the calculated molecular mass of the complex is 49.1 kDa. The upper panel shows the residuals of the fit of the sedimentation equilibrium data.
TABLE 3.
Apparent molecular masses of LGP2 CTD complexes with different forms of dsRNA determined by gel filtration chromatography
| Proteins, RNA, and protein/RNA complexes | Elution volume | Apparent molecular mass | Calculated molecular mass |
|---|---|---|---|
| ml | kDa | kDa | |
| LGP2 CTD | 17.36 | 16.73 | 16.830 |
| 8-bp dsRNA | 17.98 | 12.52 | 5.078 |
| Small hairpin RNA | 17.13 | 18.63 | 7.716 |
| 24-bp dsRNA | 16.08 | 30.43 | 15.426 |
| 24-bp 5′ ppp dsRNA | 15.70 | 36.31 | 15.666 |
| 5′ overhang 22-bp dsRNA | 16.18 | 29.04 | 14.171 |
| 3′ overhang 22-bp dsRNA | 15.86 | 33.73 | 14.171 |
| LGP2 CTD and 8-bp dsRNA | 15.74 | 35.67 | 21.908 (1:1), 38.738 (2:1)a |
| LGP2 CTD and small hairpin dsRNA | 15.97 | 32.03 | 24.546 (1:1), 41.376 (2:1) |
| LGP2 CTD and 24-bp dsRNA | 14.40 | 66.72 | 32.256 (1:1), 49.086 (2:1) |
| LGP2 CTD and 24-bp 5′ ppp dsRNA | 14.78 | 55.85 | 32.496 (1:1), 49.326 (2:1) |
| LGP2 CTD and 5′ overhang 22-bp dsRNA | 15.04 | 49.48 | 31.001 (1:1), 47.831 (2:1) |
| LGP2 CTD and 3′ overhang 22-bp dsRNA | 15.70 | 36.34 | 31.001 (1:1), 47.831 (2:1) |
Calculated molecular masses of the complexes with 1:1 and 2:1 stoichiometry.
TABLE 4.
Molecular masses of purified LGP2/dsRNA complexes determined by sedimentation velocity and sedimentation equilibrium
S* is the apparent sedimentation coefficient calculated
from the c(s) analysis by Sedfit ± standard deviation.
f/f0 is the frictional ratio, and Mest
is the estimated mass from the c(M) analysis in Sedfit. s is the
reduced buoyant molecular mass at the reference speed of 17,000 rpm.
Mcalc is the experimental molecular mass calculated from s
using a weight average 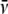 of
0.6813.
of
0.6813.
|
Sample
|
Sedimentation velocity
|
Sedimentation equilibrium
|
|||
|---|---|---|---|---|---|
| S* | f/f0 | Mest | s | Mcalc | |
| g/mol | cm−2 | g/mol | |||
| 2:1 LGP2:dsRNA (8 bp) | 2.98 ± 0.29 | 1.35 | 38,100 | ||
| 2:1 LGP2:dsRNA (24 bp) | 3.98 ± 0.56 | 1.39 | 58,300 | 1.8355 (1.7098-1.9612) | 44,850 (41,780-47,920) |
| 1:1 LGP2:RNA (hairpin) | 2.75 ± 0.43 | 1.18 | 27,800 | ||
A prediction of the structure and the asymmetric recognition of the two strands of the dsRNA is that dsRNA with 3′ overhangs will be more affected for LGP2 binding because hydrophobic interactions between the exposed base pair and the protein will be disrupted (Fig. 2A). However, dsRNA with 5′ overhangs would still bind LGP2 without clashing with the protein. To test this idea directly, we generated a siRNA-like dsRNA that contained two 3′ overhang nucleotides and a 22-bp dsRNA with 5′ overhangs and studied their binding with LGP2 CTD by gel filtration chromatography. We found that these siRNA-like dsRNAs did not bind the CTD of LGP2, whereas a 22-bp dsRNA with 5′ overhangs retained binding to LGP2 (Table 3). However, the stoichiometry between LGP2 and the 22-bp dsRNA with 5′ overhang is close to 1:1 (expected apparent molecular mass, 45.8 kDa) instead of 2:1 (expected apparent molecular mass, 62.5). This is most likely because of the lower affinity of dsRNA with 5′ overhangs for LGP2 compared with blunt-ended dsRNA. To confirm that LGP2 also binds naturally synthesized RNA, we generated a 24-bp dsRNA with 5′ triphosphates using ssRNA synthesized by in vitro transcription. Gel filtration chromatography showed LGP2 CTD binds this 24-bp dsRNA at a stoichiometry of 1:1 instead of 2:1 (Table 3). The observed molecular mass of the complex is 55.9 kDa, very close to the expected apparent molecular mass for a 1:1 complex (53.0 kDa). The only structural difference between the chemically synthesized RNA and the in vitro transcribed RNA is that the latter contains a 5′ triphosphate group, which reduced its affinity for LGP2 CTD in a similar way as dsRNA with 5′ overhangs.
Double-stranded RNA with Blunt Termini Binds and Activates RIG-I—Our structural and binding studies showed that LGP2 binds dsRNA with blunt ends or 5′ overhangs but does not bind dsRNA with 3′ overhangs. To test whether the blunt ends of dsRNA are required for RNA recognition by RIG-I, we expressed and purified the RIG-I CTD and conducted binding studies with different forms of dsRNA. In these studies, RIG-I CTD preferentially binds dsRNA with blunt ends but not dsRNA with 3′ or 5′ overhangs (supplemental Fig. S3). Similarly, the CTD of MDA5 only binds dsRNA with blunt ends (data not shown). To test whether the blunt ends of dsRNA are required for the activation of RIG-I in vivo, we conducted NF-κB and IFN-β reporter assays using different forms of dsRNA (supplemental Fig. S4). Consistent with results from the in vitro binding studies, these assays showed that RIG-I was activated by short 24- and 27-bp dsRNA containing blunt ends but was not activated by dsRNA of similar length with 5′ or 3′ overhangs (dsR22 5′ over, dsR22 3′ over, and 27UU2 in supplemental Fig. S4). Interestingly, dsRNA with only one blunt end (27UU) could induce RIG-I signaling similar to RNA with two blunt ends (supplemental Fig. S4). These results indicate that the blunt terminus of dsRNA is most likely a common structural feature recognized by the RLRs.
Effects of Mutations in LGP2 CTD on dsRNA Binding—The contributions of key residues in LGP2 CTD to dsRNA binding were examined by mutagenesis and binding studies using gel filtration chromatography and fluorescence anisotropy assays. Mutation of Glu573 to alanine reduced the affinity of LGP2 for a 10-bp dsRNA from 105 nm to 2.13 μm (Fig. 4, A and B, and supplemental Fig. S5), demonstrating that the hydrogen bonding network between Glu573 and the 3′ end of the dsRNA plays a key role in dsRNA binding. Mutation of Ile597 to serine reduced the affinity of LGP2 for the 10-bp dsRNA by almost 6-fold (Fig. 4, A and B, and supplemental Fig. S5), suggesting that hydrophobic interactions between the exposed bases and LGP2 CTD are important for dsRNA binding. The E573A and I597S mutants of LGP2 CTD bind the 8-bp dsRNA at a stoichiometry of 1:1 instead of 2:1 (Fig. 4B and supplemental Fig. S5), most likely because of their lower affinities to the dsRNA compared with the wild type protein. Mutation of Lys634, Lys651, or both Lys634 and Arg636 to negatively charged glutamate residues abolished dsRNA binding by LGP2 (Fig. 4A and supplemental Fig. S5), indicating that electrostatic interactions play primary roles in dsRNA recognition by LGP2.
FIGURE 4.

Double-stranded RNA binding is not required for the regulation of RIG-I signaling by LGP2. A, binding studies of LGP2 CTD mutants with a 10-bp dsRNA. Fluorescence anisotropy changes measured by titrating wild type and mutants of LGP2 CTD into Cy3-labeled 10-bp dsRNA. B, summary of dsRNA binding studies of LGP2 CTD mutants by fluorescence anisotropy and gel filtration chromatography. Stoichiometries of the complexes formed between LGP2 CTD and the 8-bp dsRNA are shown in parentheses. C, Western blot analysis of LGP2 and its mutants expressed in the 293T cells. Wt corresponds to wild type LGP2; L-Zn is the double mutant C556A/C559A. D, RNA binding is not required for the inhibition of RIG-I signaling by LGP2. Luciferase assay was performed using HEK 293T cells expressing RIG-I and LGP2 or its mutants at a ratio of 1:2. The data shown are the ratios of the firefly luciferase under the NF-κB promoter versus Renilla luciferase driven from a constitutive herpesvirus thymidine kinase promoter. The white bars correspond to uninduced cells, and the gray bars correspond to cells induced with dsR27 (0.2 μm). LGP2 containing mutations K634E or K651E that abolish dsRNA binding still suppressed RIG-I signaling at levels comparable with that of the wild type protein.
The dsRNA- and ssRNA-binding surface of RIG-I CTD overlaps with the dsRNA-binding surface of LGP2 CTD (Fig. 1E). Mutations K858A/K861A and K888A/K907A in full-length RIG-I caused impaired dsRNA and ssRNA binding and abolished RIG-I signaling in response to dsRNA and 5′ ppp ssRNA (14). Lys888 and Lys907 are conserved in the RLRs and correspond to Lys634 and Lys651 in human LGP2 (supplemental Fig. S1), indicating that these residues play crucial roles in dsRNA binding by the RLRs. Residues Lys858 and Lys861 of RIG-I are not conserved in LGP2 and may play a role in recognizing the 5′ triphosphates of dsRNA or ssRNA.
RNA Binding Is Not Required for the Suppression of RIG-I Signaling by LGP2—Mutations K634E and K651E that disrupted LGP2 CTD binding to dsRNA offer the possibility to test whether dsRNA binding by LGP2 is required to regulate RIG-I signaling. These mutations were constructed in full-length LGP2. Another mutant with substitutions of two of the cysteine residues (Cys556 and Cys559) that coordinate the zinc ion in the LGP2 CTD was also tested. All of these mutant proteins were expressed at levels comparable with WT LGP2 in transiently transfected 293T cells (Fig. 4C). The cells were transfected to express RIG-I together with LGP2 or its mutants and induced with a 27-bp dsRNA. An 8.7-fold induction of RIG-I signaling above the background was observed with luciferase reporter driven from the NF-κB promoter element (Fig. 4D). In the presence of WT LGP2, RIG-I signaling was reduced to 3.3-fold, whereas the C556A/C559A mutant lost the ability to suppress RIG-I signaling (Fig. 4D). Surprisingly, LGP2 mutants K634E and K651E coexpressed with RIG-I inhibited RIG-I signaling at a level comparable with that of WT LGP2 (Fig. 4D). Similar results were obtained with the luciferase reporter driven from the IFN-β promoter (data now shown). These results demonstrated that dsRNA binding is most likely not required for the suppression of RIG-I signaling by LGP2.
DISCUSSION
The structure of LGP2 CTD in complex with the 8-bp dsRNA showed that LGP2 binds to the blunt ends of dsRNA. Binding studies showed that LGP2 CTD also binds dsRNA with 5′ overhangs, but not dsRNA with 3′ overhangs. It has been demonstrated that RIG-I recognizes both 5′ ppp ssRNA and dsRNA (10, 11, 15), signatures of viral RNA. We speculate that RIG-I binds to the end of 5′ ppp ssRNA and dsRNA in a manner similar to how LGP2 associates with dsRNA. Consistent with this speculation, two overlapping sets of residues involved in RNA binding by RIG-I CTD are mapped onto the RNA-binding surface of LGP2 when the two structures are superimposed. If RIG-I also binds to the blunt ends of dsRNA, a long dsRNA would be less efficient in stimulating RIG-I activation, unless it is cleaved to generate a larger number of termini. Consistent with this hypothesis, it has been shown that RIG-I senses short dsRNA, and RNase III digestion can transform poly(I·C) from a MDA5 ligand into a RIG-I ligand (15, 32).
The cellular ligands for RIG-I might include products of ribonucleases specific for dsRNA in addition to 5′ ppp ssRNA. It has been shown that the activation of antiviral ribonuclease, RNase L, by 2′,5′-linked oligoadenylate produces small RNA cleavage products that initiate IFN production (32). Our binding studies showed that siRNA-like dsRNA with 3′ overhangs did not bind the CTD of LGP2 or RIG-I and are poor ligands for the induction of RIG-I signaling (Table 3 and supplemental Figs. S3 and S4). Our results are consistent with a report showing that synthetic dsRNA of 21-27 nucleotides in length activates interferon secretion when they did not contain the two 3′ overhanging nucleotides (13). In addition, dsRNA with 3′ overhangs, but not dsRNA with blunt ends, were unwound by RIG-I and did not stimulate the induction of IFNs (14).
Although our binding studies demonstrated that MDA5 CTD exhibits lower affinities for a 24-bp dsRNA than an 10-bp dsRNA (Fig. 3A), it was shown MDA5 recognizes long dsRNA of several kb length or poly(I·C) without RNase III treatment (15). The nature of MDA5 binding to dsRNA remains unclear. If MDA5 binds to the end of dsRNA in a manner similar to that of LGP2, short dsRNA should be good ligand for MDA5 as well. Our preliminary structural studies of MDA5 showed that the structure of MDA5 CTD is very similar to LGP2 and RIG-I, and similar binding surfaces are involved in dsRNA recognition by all the three proteins.4 The maximum length of dsRNA that can be covered by full-length RIG-I, MDA5, or LGP2 is likely 25-30 bp (31, 33), so it is not clear why MDA5 is specifically activated by long dsRNA of several kilobases in length. Although MDA5 shares higher sequence identity with LGP2 than RIG-I, MDA5 activation was not suppressed by its CTD or LGP2, suggesting a different mechanism of activation compared with RIG-I (18).
The structure of LGP2 CTD·dsRNA complex and binding studies with dsRNA of different lengths showed that LGP2 CTD binds to the two termini of dsRNA, forming complexes of 2:1 stoichiometry. Purified full-length LGP2 only forms dimers with dsRNA longer than 21 bp, and no higher order complex has been observed (31). Binding studies of RIG-I and MDA5 CTD with different forms of dsRNA also showed that only dsRNA with blunt ends bind to these two proteins efficiently (supplemental Fig. S3). However, RIG-I and MDA5 CTD only form 1:1 complexes with the 8-bp dsRNA at low concentrations, and a mixture of 1:1 and 2:1 complexes at higher concentrations (data not shown). The different stoichiometry of the RIG-I and MDA5 complexes is most likely due to the lower affinity of these two proteins to the dsRNA compared with LGP2. Consistent with this, mutants of LGP2 CTD with reduced affinities to the 8-bp dsRNA also forms 1:1 rather than 2:1 complexes at low concentrations (supplemental Fig. S5). It has been suggested that ligand induced dimerization and conformation change induces RIG-I activation (19). However, we did not observe extensive interactions between the two protein molecules in the LGP2 CTD·dsRNA complex, and when LGP2 CTD binds longer dsRNA, such as the 24-bp dsRNA, the two protein molecules are unlikely to interact with each other. Molecular modeling of the LGP2·dsRNA complex according to the structure of the complex derived by electron microscopy also shows the CTD likely binds to the termini of the dsRNA with the helicase domain makes additional contributions to binding (31). Although similar blunt end binding is likely involved in dsRNA recognition by RIG-I, we speculate that ligand-induced dimerization is not absolutely needed for the activation of RIG-I. The conformation of the dimers that would form between RIG-I and dsRNA could also be affected by the properties of the ligands (33).
An intriguing possibility presented by the recognition of LGP2 to the two ends of blunt-ended dsRNA is that this recognition could allow for a response to the concentration of viral dsRNAs independent of the lengths of the RNAs. Viral genomes and replication intermediates can range in length from less than 1 kb to up to ∼30 kb. LGP2, and likely RIG-I, may thus respond in the same way to these ligands until they are processed by cellular ribonucleases. Because LGP2 exhibits higher affinity for dsRNA than RIG-I, it has been suggested that LGP2 might compete with RIG-I for ligand binding and thus suppress the activation of RIG-I (22, 31). However, substitutions in the LGP2 CTD that abolished dsRNA binding in vitro did not affect the ability of full-length LGP2 to suppress RIG-I activation by dsRNA (Fig. 4D). These results suggest that direct interaction between LGP2 and RIG-I might play a dominant role for the suppression of RIG-I activation by LGP2. It is also possible that LGP2 regulates the activation of RIG-I through direct interaction with IPS-1 to block the assembly of the signaling complex (23, 34). Future structural studies of full-length RIG-I or LGP2 in isolation and in complex with longer RNA molecules should reveal additional mechanisms of RLR activation and regulation.
Supplementary Material
Acknowledgments
We thank Lance Ferguson, Allison Cockrell for critical reading of the manuscript, and Kanchan Bhardwaj for help with the fluorescence anisotropy experiments.
This work is dedicated to the memory of professor Yongxing Wang, a great teacher, a good friend, and a renowned historian of medieval Chinese history at Peking University, China.
This work was supported, in whole or in part, by National Institutes of Health Grant 1RO1AI073335 (to C. K.). This work was also supported by Robert Welch Foundation Grant A-1687 (to P. L.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and supplemental Figs. S1-S5.
Footnotes
The abbreviations used are: TLR, Toll-like receptor; CTD, C-terminal domain; 5′ ppp ssRNA, 5′-triphosphorylated single-stranded RNA; dsRNA, double-stranded RNA; RLR, RIG-I-like receptor; IFN, interferon; CARD, caspase recruiting domain; bis-tris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol; siRNA, small interfering RNA.
P. Li, unpublished results.
References
- 1.Janeway, C. A., Jr., and Medzhitov, R. (2002) Annu. Rev. Immunol. 20 197-216 [DOI] [PubMed] [Google Scholar]
- 2.Akira, S., Uematsu, S., and Takeuchi, O. (2006) Cell 124 783-801 [DOI] [PubMed] [Google Scholar]
- 3.Yoneyama, M., and Fujita, T. (2007) J. Biol. Chem. 282 15315-15318 [DOI] [PubMed] [Google Scholar]
- 4.Pichlmair, A., and Reis e Sousa, C. (2007) Immunity 27 370-383 [DOI] [PubMed] [Google Scholar]
- 5.Thompson, A. J., and Locarnini, S. A. (2007) Immunol. Cell Biol. 85 435-445 [DOI] [PubMed] [Google Scholar]
- 6.Akira, S., and Takeda, K. (2004) Nat. Rev. Immunol. 4 499-511 [DOI] [PubMed] [Google Scholar]
- 7.Yoneyama, M., Kikuchi, M., Natsukawa, T., Shinobu, N., Imaizumi, T., Miyagishi, M., Taira, K., Akira, S., and Fujita, T. (2004) Nat. Immunol. 5 730-737 [DOI] [PubMed] [Google Scholar]
- 8.Takeuchi, O., and Akira, S. (2008) Curr. Opin. Immunol. 20 17-22 [DOI] [PubMed] [Google Scholar]
- 9.Akira, S., Takeda, K., and Kaisho, T. (2001) Nat. Immunol. 2 675-680 [DOI] [PubMed] [Google Scholar]
- 10.Hornung, V., Ellegast, J., Kim, S., Brzozka, K., Jung, A., Kato, H., Poeck, H., Akira, S., Conzelmann, K. K., Schlee, M., Endres, S., and Hartmann, G. (2006) Science 314 994-997 [DOI] [PubMed] [Google Scholar]
- 11.Pichlmair, A., Schulz, O., Tan, C. P., Naslund, T. I., Liljestrom, P., Weber, F., and Reis e Sousa, C. (2006) Science 314 997-1001 [DOI] [PubMed] [Google Scholar]
- 12.Kato, H., Takeuchi, O., Sato, S., Yoneyama, M., Yamamoto, M., Matsui, K., Uematsu, S., Jung, A., Kawai, T., Ishii, K. J., Yamaguchi, O., Otsu, K., Tsujimura, T., Koh, C. S., Reis e Sousa, C., Matsuura, Y., Fujita, T., and Akira, S. (2006) Nature 441 101-105 [DOI] [PubMed] [Google Scholar]
- 13.Marques, J. T., Devosse, T., Wang, D., Zamanian-Daryoush, M., Serbinowski, P., Hartmann, R., Fujita, T., Behlke, M. A., and Williams, B. R. (2006) Nat. Biotechnol. 24 559-565 [DOI] [PubMed] [Google Scholar]
- 14.Takahasi, K., Yoneyama, M., Nishihori, T., Hirai, R., Kumeta, H., Narita, R., Gale, M., Jr., Inagaki, F., and Fujita, T. (2008) Mol. Cell 29 428-440 [DOI] [PubMed] [Google Scholar]
- 15.Kato, H., Takeuchi, O., Mikamo-Satoh, E., Hirai, R., Kawai, T., Matsushita, K., Hiiragi, A., Dermody, T. S., Fujita, T., and Akira, S. (2008) J. Exp. Med. 205 1601-1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gitlin, L., Barchet, W., Gilfillan, S., Cella, M., Beutler, B., Flavell, R. A., Diamond, M. S., and Colonna, M. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 8459-8464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoneyama, M., and Fujita, T. (2008) Immunity 29 178-181 [DOI] [PubMed] [Google Scholar]
- 18.Saito, T., Hirai, R., Loo, Y. M., Owen, D., Johnson, C. L., Sinha, S. C., Akira, S., Fujita, T., and Gale, M., Jr. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 582-587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui, S., Eisenacher, K., Kirchhofer, A., Brzozka, K., Lammens, A., Lammens, K., Fujita, T., Conzelmann, K. K., Krug, A., and Hopfner, K. P. (2008) Mol. Cell 29 169-179 [DOI] [PubMed] [Google Scholar]
- 20.Pippig, D. A., Hellmuth, J. C., Cui, S., Kirchhofer, A., Lammens, K., Lammens, A., Schmidt, A., Rothenfusser, S., and Hopfner, K. P. (2009) Nucleic Acids Res. 10.1093/nar/gkp059 [DOI] [PMC free article] [PubMed]
- 21.Rothenfusser, S., Goutagny, N., DiPerna, G., Gong, M., Monks, B. G., Schoenemeyer, A., Yamamoto, M., Akira, S., and Fitzgerald, K. A. (2005) J. Immunol. 175 5260-5268 [DOI] [PubMed] [Google Scholar]
- 22.Yoneyama, M., Kikuchi, M., Matsumoto, K., Imaizumi, T., Miyagishi, M., Taira, K., Foy, E., Loo, Y. M., Gale, M., Jr., Akira, S., Yonehara, S., Kato, A., and Fujita, T. (2005) J. Immunol. 175 2851-2858 [DOI] [PubMed] [Google Scholar]
- 23.Komuro, A., Bamming, D., and Horvath, C. M. (2008) Cytokine 43 350-358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 25.Collaborative Computational Project, number 4 (1994) Acta Crystallogr. D Biol. Crystallogr. 50 760-76315299374 [Google Scholar]
- 26.Jones, T. A., and Kjeldgaard, M. (1997) Methods Enzymol. 277 173-208 [DOI] [PubMed] [Google Scholar]
- 27.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. D Biol. Crystallogr 54 905-921 [DOI] [PubMed] [Google Scholar]
- 28.Lawrence, M. C., and Colman, P. M. (1993) J. Mol. Biol. 234 946-950 [DOI] [PubMed] [Google Scholar]
- 29.Li, P., Huey-Tubman, K. E., Gao, T., Li, X., West, A. P., Jr., Bennett, M. J., and Bjorkman, P. J. (2007) Nat. Struct. Mol. Biol. 14 381-387 [DOI] [PubMed] [Google Scholar]
- 30.Liu, L., Botos, I., Wang, Y., Leonard, J. N., Shiloach, J., Segal, D. M., and Davies, D. R. (2008) Science 320 379-381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murali, A., Li, X., Ranjith-Kumar, C. T., Bhardwaj, K., Holzenburg, A., Li, P., and Kao, C. C. (2008) J. Biol. Chem. 283 15825-15833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malathi, K., Dong, B., Gale, M., Jr., and Silverman, R. H. (2007) Nature 448 816-819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ranjith-Kumar, C. T., Murali, A., Dong, W., Srisathiyanarayanan, D., Vaughan, R., Ortiz-Alacantara, J., Bhardwaj, K., Li, X., Li, P., and Kao, C. C. (2009) J. Biol. Chem. 284 1155-1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Komuro, A., and Horvath, C. M. (2006) J. Virol. 80 12332-12342 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.