

Abstract
The small heat shock protein αB-crystallin interacts with N2B-Us, a large unique sequence found in the N2B element of cardiac titin. Using single molecule force spectroscopy, we studied the effect of αB-crystallin on the N2B-Us and its flanking Ig-like domains. Ig domains from the proximal tandem Ig segment of titin were also studied. The effect of wild type αB-crystallin on the single molecule force-extension curve was determined as well as that of mutant αB-crystallins harboring the dilated cardiomyopathy missense mutation, R157H, or the desmin-related myopathy mutation, R120G. Results revealed that wild type αB-crystallin decreased the persistence length of the N2B-Us (from ∼0.7 to ∼0.2 nm) but did not alter its contour length. αB-crystallin also increased the unfolding force of the Ig domains that flank the N2B-Us (by 51 ± 3 piconewtons); the rate constant of unfolding at zero force was estimated to be ∼17-fold lower in the presence of αB-crystallin (1.4 × 10-4 s-1 versus 2.4 × 10-3 s-1). We also found that αB-crystallin increased the unfolding force of Ig domains from the proximal tandem Ig segment by 28 ± 6 piconewtons. The effects of αB-crystallin were attenuated by the R157H mutation (but were still significant) and were absent when using the R120G mutant. We conclude that αB-crystallin protects titin from damage by lowering the persistence length of the N2B-Us and reducing the Ig domain unfolding probability. Our finding that this effect is either attenuated (R157H) or lost (R120G) in disease causing αB-crystallin mutations suggests that the interaction between αB-crystallin and titin is important for normal heart function.
αB-crystallin is a member of the small heat shock protein family that by inhibiting denaturation and aggregation of proteins functions as a molecular chaperone (1). Although αB-crystallin has been most intensively studied in the vertebrate eye lens, it is also found in many other tissues (2) with cardiac muscle expressing αB-crystallin at 3-5% of the total soluble protein (3). Up-regulation of αB-crystallin occurs in a number of cardiac disorders, including familial cardiac hypertrophy, and overexpression appears to protect the cardiac cell from ischemia reperfusion injury (for a review see Ref. 4). An important binding partner of αB-crystallin in cardiac muscle is titin (5, 6). Titin is a large filamentous protein that forms a continuous filament along the myofibril, with single titin molecules spanning from the edge to the middle of the sarcomere, a distance of ∼1 μm (7). The I-band region of titin is extensible and functions as a molecular spring that, when extended, develops force (8, 9). This force is an important determinant of the passive stiffness of the heart that determines the filling characteristics during the diastolic part of the heart cycle (10). The interaction between αB-crystallin and titin could be important for maintaining heart function, especially when stressed, such as during ischemia (5), warranting studies of the effect of αB-crystallin on the biomechanical properties of titin.
The molecular spring region of titin contains three distinct spring elements (7). The first element is the tandem Ig segment, consisting of serially linked Ig domains that form the so-called proximal tandem Ig segment (15 Ig domains) near the Z-disk of the sarcomere and a distal segment (22 Ig domains) near the A-band (11). The second spring element is the PEVK, a unique sequence that contains largely prolines, glutamates, valines, and lysines (11). The third element consists of a large unique sequence (in human 572 residues in size) named the N2B-Us; it is heart-specific and dominates the extension of titin near the upper limit of the physiological sarcomere length range (12). αB-crystallin appears to preferentially bind to the N2B-Us, although weak binding to Ig domains has also been detected (6). Previous studies have shown that αB-crystallin increases the unfolding force of Ig 91-98, a fragment that contains eight Ig domains from the distal tandem Ig segment of titin (6). However, the mechanical effect of αB-crystallin on the N2B-Us (its main binding partner in titin) has not been investigated.
The association between αB-crystallin and titin has prompted a search for disease causing mutations in αB-crystallin. This revealed in patients with dilated cardiomyopathy (DCM),2 a missense mutation, R157H, that affects an evolutionarily conserved amino acid residue; the mutation decreases the binding to the N2B domain without affecting distribution of the mutant crystallin protein in cardiomyocytes (13). In another disease, the desmin-related myopathy mutation R120G (14) decreases the binding of αB-crystallin to the N2B element and causes intracellular aggregates of the mutant protein (13).
In the present study, we used single molecule force spectroscopy and determined the contour length (CL; end-to-end length when stretched with infinite force) and persistence length (PL; a measure of the bending rigidity) of the N2B-Us. We also studied the unfolding force of Ig domains, those that flank the N2B-Us and those that make up the proximal tandem Ig segment. In addition, we investigated the effect of wild type and R157H and R120G αB-crystallin on the molecular mechanics of the N2B-Us, its flanking Ig domains, and the Ig domains in the proximal tandem Ig segment. Findings support that αB-crystallin functions as a chaperone that lowers the probability of Ig domain unfolding and the persistence length of the titin N2B-Us spring region. Importantly, this chaperone function is significantly reduced by the R157H mutation and abolished by the R120G mutation.
EXPERIMENTAL PROCEDURES
Construction of pET Plasmids—The N2B element corresponding to Ig24-Ig25-N2B-Us-Ig26 was amplified by PCR from total human cDNAs using Hot Taq polymerase (Bioron, Ludwigshafen) with primer pairs derived from the human cardiac titin N2B isoform (GenBank™ accession number X90568; residues 3410-4336). We similarly amplified segments coding for the proximal tandem Ig segment fragments Ig 1-8 and Ig 8-15 (accession number X90568; residues 2040-2749 and 2661-3389, respectively). Under “Results,” we also show previous findings on Ig 91-98 (accession number X90568; residues 5164-5875). The domain numbering system is based on that of Bang et al. (37); Ig 91-98 was previously known as Ig 27-I34. Wild type human αB-crystallin was expressed in Escherichia coli as previously described (6). Both disease-causing mutations (R120G and R157H, respectively) were introduced into the wild type αB-crystallin construct by PCR assembly mutagenesis. All PCR fragments were then cloned into pETM11; correct sequence was verified by sequence analysis using T7, T7 terminator, and internal primers.
Protein Expression and Purification—The pETM11 constructs were transformed into BL21(DE3) pLysS cells, and expressed proteins were purified from the soluble fraction on Ni2+-nitrilotriacetic acid columns with a His-Bind purification kit (Novagen, Madison, WI). Purified proteins were dialyzed into assay buffer (AB) (25 mm imidazole HCl (pH 7.4), 0.2 m KCl, 4 mm MgCl2, 1 mm EGTA, 0.01% NaN3, and 5 mm dithiothreitol), quick frozen, and stored at -80 °C until later use. For αB-crystallin experiments, we removed the His tag by treating proteins with tobacco etch virus protease and passing them again over Ni2+-nitrilotriacetic acid columns essentially as described (15).
Single Molecule Force Spectroscopy—Molecules were stretched using an atomic force microscope (MFP3D; Asylum Research, Santa Barbara, CA). Methods used followed those established in the field (6, 16-20). Briefly, 10 μl of protein in AB (concentrations as follows: N2B-Us, 250 μg/ml; Ig 1-8, 200 μg/ml; I8-15, 200 μg/ml) (for composition of AB, see above) was spotted on a cleaned glass microscope slide (experiments with mica- and gold-coated glass gave identical results). Unbound molecules were washed away with AB. Surface protein density was kept low to ensure a low probability for tethering to the atomic force microscope tip (∼1 in 100 attempts) to minimize the chance on stretching multiple molecules simultaneously. In the αB-crystallin experiments, we added 10 μl of αB-crystallin (2.5 mg/ml) to the absorbed N2B fragments on a glass microscope slide and incubated for 15 min. (In initial experiments, proteins were premixed before adding them to the glass slide; identical results were obtained.) Molecules were then stretched in AB by pressing the cantilever (MSCT-AUHW; ThermoMicroscopes) against the protein-coated surface and pulling the cantilever away at a constant pulling speed. Four speeds were used in most experiments: 500, 1000, 2000, and 4000 nm/s. Force versus displacement curves were collected in repeated stretch and release cycles. The displacement of the cantilever base was measured by using an integrated linear voltage differential transformer. Force (F) was obtained from cantilever bending (Δd) as follows,
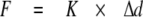 |
(Eq.1) |
where K represents cantilever stiffness. Stiffness was obtained for each unloaded cantilever by measuring its mean thermally driven vertical bending and applying the equipartition theorem as follows,
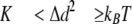 |
(Eq.2) |
where kB is Boltzmann's constant, and T is absolute temperature. Cantilever stiffness was ∼20 pN/nm, and root mean square force noise at the 0-20-kHz bandwidth used in this work was ∼15 pN. Force versus displacement curves were corrected for several factors to obtain force versus molecular end-to-end length. 1) The zero length, zero force data point was obtained from the force response that corresponded to the cantilever tip reaching (or departing from) the substrate surface. 2) Forces were corrected for base-line slope obtained from the force response of the displaced but unloaded cantilever. 3) The end-to-end length (z) of the tethered molecule was calculated by correcting the cantilever base displacement (s) with cantilever bending as follows,
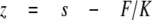 |
(Eq.3) |
As a result of cantilever bending during force development, imposing a constant velocity stretch on the cantilever base will result in a fluctuating velocity (due to sawtooth-like force trace) of the cantilever tip. When determining the velocity dependence of unfolding force (as in Fig. 4) the used velocities are the imposed velocities corrected for cantilever bending.
FIGURE 4.

Unfolding force versus stretch speed in absence ofαB crystallin (control) and in the presence of WT, R120G, and R157H αB-crystallin. At each stretch speed, we determined the mean and S.E. of all force peaks. Stretch speed was corrected for cantilever bending (see “Experimental Procedures” for details). Results were fit with linear lines (control, y = -89 + 89x; WT αB-crystallin, y = -72 + 101x; R120G, y = -129 + 104x; R157H, y = -27 + 78x). See “Experimental Procedures” for additional details).
For analysis of force-extension curves, we fit data with the wormlike chain model (WLC) as follows (21),
 |
(Eq.4) |
where F represents force (pN), PL is the persistence length (nm), z is end-to-end extension (nm), CL is the contour length (nm), kB is Boltzmann's constant, and T is absolute temperature. The adjustable parameters are the PL and CL. We fit the initial trace of the force-extension curve of the N2B recombinant protein (A in Fig. 1A) with the WLC model and determined PL and CL of the N2B-Us. We also fit the force-extension curve between the first and second and between the second and third peaks, to determine the PL and CL of the N2B-Us, N2B-Us plus 1, and N2B-Us plus 2 unfolded Ig domains, respectively. The unfolding force of the three Ig domains was directly read from the force-extension traces. Ig 1-8 and Ig 8-15 were similarly studied, by fitting the force-extension traces leading up to force peaks with the WLC equation (providing PL and CL) and reading the unfolding forces directly from the force traces.
FIGURE 1.

Single molecule force spectroscopy of N2B-Us. A, example of force-extension curve of N2B molecule (stretch speed 1000 nm/s). Top, schematic of the N2B molecule. Blue, large unique sequence (N2B-Us); red, Ig domains. Bottom, as the molecule is stretched, force gradually increases at first and then more steeply (marked as phase A); when a force level of ∼150 pN is reached, force peaks appear. Broken lines, WLC fits. Right, schematic explaining main features of the force-extension curve(note that it is likely that any of the three Ig domains can unfold first). B, CL of the first three force peaks in the force-extension curves of molecules stretched at 500, 1000, 2000, and 4000 nm/s. CL of the first peak is assumed to be derived from the N2B-Us (∼195 nm), and the slopes of the lines are the average CL gain due to unfolding of single Ig domains (∼33 nm). Obtained values are independent of stretch speed. C, left, unfolding forcehistogram of the first unfolding peak (speed, 2000 nm/s). Right, average UF of peaks 1-3 at four stretch speeds. See “Experimental Procedures” for details.
To deduce the rate constant of unfolding at zero external force (Ku0) and the location of unfolding barrier, or the width of the unfolding potential along the unfolding reaction coordinate(ΔXu), we modeled the stretch rate dependence of unfolding force with a Monte Carlo simulation based on previously used simulation algorithms (22). Briefly, the mechanical behavior was simulated by superimposing domain unfolding/refolding kinetics on the WLC force-extension curve. To calculate WLC force, we considered the molecule as a serially linked chain containing segments with folded domains that each make a 5-nm contribution to the contour length and unfolded domains that add 29 nm to the contour length. The persistence length of unfolded domains was set at 0.2 nm, and that of the tandem Ig segment with folded domains was set at 12 nm. The force-dependent unfolding rate constant is given by the following,
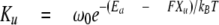 |
(Eq.5) |
where ω0 represents the attempt frequency set by Brownian dynamics of the chain (108 s-1), Ea is the unfolding activation energy. The width of the unfolding potential (ΔXu) and Ku at zero force (Ku0) were user-adjustable. From repeated Monte Carlo simulation trials, we computed the average unfolding force at different stretch rates. By fitting the calculated stretch rate dependence to that experimentally measured, we obtained estimates of Ku0 and ΔXu (for further details, see Ref. 23).
Statistics—Data are presented as mean and S.E. Statistical comparisons used an unpaired two-tailed Student's t test (except for Table 4, where we used a paired t test) with p < 0.05 considered statistically significant.
TABLE 4.
Average unfolding force (pN) of Ig domains in Ig 1-8 and Ig 8-15 at pulling speed 1000 nm/s in the absence (control) or presence of αB-crystallin (wild type and R120G and R157H mutants)
The average unfolding force of each of the eight unfolding peaks was compared in a paired t test. The number of molecules per group was ∼25-30.
|
Control
|
αB-crystallin
|
|||
|---|---|---|---|---|
| Wild type | R120G | R157H | ||
| pN | pN | |||
| Ig1-8 | 221 ± 11 | 254 ± 13a | 224 ± 14 NSb | 248 ± 12a,c |
| Ig8-15 | 232 ± 12 | 263 ± 16a | 223 ± 10 NSb | 257 ± 13a,c |
p < 0.001 compared with control.
p < 0.001 compared with wild type αB-crystallin.
p < 0.001 compared with R120G.
RESULTS
The N2B-Us, a physiologically important spring element in cardiac titin (12), was recently found to be associated with αB-crystallin (6). To determine whether αB-crystallin affects the extensibility of the N2B-Us or the unfolding force of the Ig domains that flank the N2B-Us, we performed single molecule force spectroscopy. We used a recombinant protein in which the N2B-Us is flanked by the naturally occurring Ig domains, Ig 24/25 on one side and Ig26 at the other (see Fig. 1A, top). The force-extension curve of the construct was measured with an atomic force microscope specialized for pulling single molecules. A typical result is shown in Fig. 1A, bottom. As the molecule is stretched, force increases gradually at first and then more steeply until a force level of ∼150 pN is reached when force peaks start to appear. These force peaks are probably due to unfolding of Ig domains that lengthen the molecule (unfolding increases the domain length from ∼5 to ∼34 nm). Up to four peaks were seen, consistent with three Ig unfolding events and a fourth peak due to detachment of the molecule from its anchoring point(s). We analyzed those records in which at least three regularly spaced Ig-unfolding peaks appear in the force trace, ensuring that the whole molecule had been stretched, including the full N2B-Us. The initial trace leading up to the first unfolding peak (indicated by A in Fig. 1A) corresponds to the force-extension curve of the N2B-Us. We fit the initial trace with the WLC model (see “Experimental Procedures”) and determined the PL and the CL of the N2B-Us; we also fit the traces leading up to the second and third unfolding peaks and determined the PL and CL of those traces as well.
Fig. 1B shows the obtained CL at the four stretch speeds (500, 1000, 2000, and 4000 nm/s). Results show that the N2B-Us CL is independent of stretch speed and is ∼195 nm. This value is slightly less than expected (the CL of a 572-residue (N2B-Us) that is completely unfolded is ∼215 nm; also see “Discussion”). The slope of the linear fit to the CL versus peak number reflects the average CL gain that results from Ig domain unfolding. The slopes were independent of stretch velocity and, when ranked from low to high speed, are 34.5, 32.0, 33.0, and 32.0 nm (mean 32.8 ± 0.6 nm); these values are larger than expected (for details, see “Discussion”). Fig. 1C (left) shows an example of a UF histogram (first unfolding peak, stretch speed 2000 nm/s), and the data to the right show mean UF of all three peaks at four stretch speeds. At all speeds, UF progressively increases with peak number, reflecting different mechanical stabilities of the Ig domains. The slopes of the UF versus peak number were 19, 22, 24, and 25 pN/peak at the stretch speeds ranked from low to high. This UF increase can be compared with that previously measured for Ig 91-98 and Ig 65-70 fragments (18): ∼22 and ∼16 pN/peak, respectively. Thus, the N2B Ig domains have slopes that are similar to those of Ig 91-98. However, their UF is much lower than that of Ig 91-98. For example at a stretch speed of 1000 nm/s, UF of Ig 91-98 is 230-300 pN (18) (i.e. UF of Ig 24-26 domains is ∼100 pN less).
The extensibility of the N2B construct was also studied in the presence of αB-crystallin, using wild type αB-crystallin and two mutated versions: R157H (found in familial DCM) and R120G (found in desmin-related myopathy). Examples of obtained force-extension curves are shown in Fig. 2. Fig. 2A shows a control force-extension curve and Fig. 2, B-D, shows force-extension curves in the presence of WT, R157H, and R120G αB-crystallin, respectively. No major differences in the force-extension curves are obvious from visual inspection. However, WLC analysis of the curves in the presence of αB-crystallin revealed significant changes in several of the parameters. The PL of the N2B-Us was significantly decreased by WT αB-crystallin, a finding that was consistent at the four stretch speeds (500, 1000, 2000, and 4000 nm/s) that were used in this work; see Fig. 3 and Table 1. The PL was ∼0.7 nm in the absence and ∼0.2 nm in the presence of WT αB-crystallin. The mutant αB-crystallins behaved differently from WT αB-crystallin. No effect was seen with the R120G mutant, whereas the R157H mutant showed a significant reduction in PL to ∼0.3 nm (Fig. 3). Comparing the PL in the presence of R157H with that of WT αB-crystallin showed that the R157H had an effect that was significantly less than obtained with WT αB-crystallin (Table 1, Footnote b).
FIGURE 2.

Examples of force-extension curves of N2B molecules in the absence of αB-crystallin (A) and in the presence of WT (B), R157H (C), and R120G (D) αB-crystallin.
FIGURE 3.

Effect of αB-crystallin on PL of N2B-Us. αB-crystallin reduces PL at all four pulling speeds. The effect is less with the R157H mutant and absent when using the R120G mutant.
TABLE 1.
Persistence length of N2B-Us (nm) as a function of pulling speed (nm/s) and the absence (control) or presence of αB-crystallin (wild type and R120G and R157H mutants)
Values in parenthesis were obtained at pH 6.4. All others were at pH 7.4. The number of molecules per group was ∼30-40.
|
Speed
|
Control
|
αB-crystallin
|
||
|---|---|---|---|---|
| Wild type | R120G | R157H | ||
| nm/s | nm | nm | ||
| 500 | 0.71 ± 0.19 | 0.21 ± 0.04a | 0.63 ± 0.05b | 0.31 ± 0.02c,d,e |
| 1000 | 0.70 ± 0.11 | 0.18 ± 0.02a | 0.69 ± 0.14b | 0.36 ± 0.02c,d,e |
| 2000 | 0.65 ± 0.05 | 0.20 ± 0.03a | 0.67 ± 0.16f | 0.29 ± 0.02a,f,g |
| (0.62 ± 0.12) | (0.17 ± 0.01)a | |||
| 4000 | 0.67 ± 0.13 | (0.18 ± 0.02)a | ||
p < 0.001 compared with control.
p < 0.001 compared with wild type αB-crystallin.
p < 0.01 compared with control.
p < 0.05 compared with wild type αB-crystallin.
p < 0.01 compared with R120G.
p < 0.01 compared with wild type αB-crystallin.
p < 0.001 compared with R120G.
The effect of αB-crystallin on the CL of the N2B-Us is shown in Table 2. Although the mean CL values were all lower in the presence of WT and R157H mutant αB-crystallins, these differences did not reach statistical significance. Thus, the main effect of αB-crystallin on the extensibility of the N2B-Us is on the PL, with a large reduction in the presence of WT αB-crystallin, a slight attenuation of the effect with the R157H mutant, and absence of the effect with the R120G mutant.
TABLE 2.
Contour length of N2B-Us (nm) as a function of pulling speed (nm/s) and the absence (control) or presence of αB-crystallin (wild type and R120G and R157H mutants)
Values in parenthesis were obtained at pH 6.4. All others were at pH 7.4. The number of molecules per group was ∼30-40.
|
Speed
|
Control
|
αB-crystallin
|
||
|---|---|---|---|---|
| Wild type | R120G | R157H | ||
| nm/s | nm | nm | ||
| 500 | 189 ± 4 | 184 ± 3a | 195 ± 14b | 182 ± 8c |
| 1000 | 191 ± 3 | 189 ± 3a | 192 ± 12b | 185 ± 5c |
| 2000 | 190 ± 3 (193 ± 12) | 184 ± 3a (188 ± 6)a | 194 ± 8b | 184 ± 7c |
| 4000 | 193 ± 5 | 179 ± 5a | ||
Comparison versus control. Not significant.
Comparison versus wild type αB-crystallin. Not significant.
Comparison versus R120G. Not significant.
We also studied the effect of αB-crystallin on Ig domain UF. WT αB-crystallin significantly increased the forces of all three unfolding peaks and at all stretch speeds that were used (Table 3). The R157H mutant also significantly increased the UF of all three peaks, although the effect was less than for the WT protein (under several stretch conditions, the difference between R157H and WT αB-crystallin was significant (see Table 3, Footnote b)). No effect was seen in the presence of R120G (Table 3). We also combined all three force peaks and determined their mean UF and plotted results against stretch speed. Fig. 4 shows that UF is greatly increased by WT αB-crystallin; the increase is attenuated when using the R157H mutant and is absent with the R120G mutant. We modeled the stretch rate dependence of UF with a Monte Carlo simulation based on previously used simulation algorithms (see “Experimental Procedures” for details) and estimated the rate constant of unfolding at zero external force (Ku0) and width of the unfolding potential along the unfolding reaction coordinate (ΔXu). The measured UFs were well simulated (error = 3.8 ± 1.3%) with ΔXu = 0.2 nm and Ku0 = 1.4 × 10-4 and 2.4 × 10-3 s-1 in the presence and absence of WT αB-crystallin, respectively.
TABLE 3.
Unfolding force (pN) of lg domains as a function of pulling speed (nm/s) in the absence (control) or presence of αB-crystallin (wild type and R120G and R157H mutants)
Values in parenthesis were obtained at pH 6.4. All others were at pH 7.4. The number of molecules per group was ∼30-40.
|
Speed
|
Peak
|
Control
|
αB-crystallin
|
||
|---|---|---|---|---|---|
| Wild type | R120G | R157H | |||
| nm/s | pN | pN | pN | ||
| 500 | 1 | 127 ± 8 | 168 ± 10a | 124 ± 9b | 158 ± 9a,c |
| 2 | 154 ± 8 | 184 ± 10a | 144 ± 10b | 180 ± 9c,d | |
| 3 | 165 ± 13 | 216 ± 21d | 163 ± 22e | 203 ± 24f | |
| 1000 | 1 | 147 ± 7 | 203 ± 8g | 159 ± 9b | 177 ± 5a,b,f |
| 2 | 177 ± 8 | 220 ± 9g | 180 ± 9b | 201 ± 6d,e,f | |
| 3 | 191 ± 16 | 258 ± 18a | 212 ± 16e | 232 ± 9d | |
| 2000 | 1 | 175 ± 5 (156 ± 9) | 236 ± 10g (215 ± 7)g | 170 ± 8b | 199 ± 6a,b,f |
| 2 | 201 ± 5 (192 ± 10) | 252 ± 10g (245 ± 6)g | 201 ± 8h | 245 ± 7 NSa,c | |
| 3 | 223 ± 6 (210 ± 20) | 276 ± 17a (264 ± 12d) | 231 ± 17 | 263 ± 14d | |
| 4000 | 1 | 202 ± 8 | 246 ± 12a | ||
| 2 | 225 ± 9 | 273 ± 14a | |||
| 3 | 252 ± 15 | 321 ± 25d | |||
p < 0.01 compared with control.
p < 0.01 compared with wild type αB-crystalline.
p < 0.01 compared with R120G.
p < 0.05 compared with control.
p < 0.05 compared with wild type.
p < 0.05 compared with R120G.
p < 0.001 compared with control.
p < 0.001 compared with wild type.
The CL gain that results from Ig domain unfolding was determined from the slope of the CL versus peaks number (as in Fig. 1B). In the presence of WT αB-crystallin the CL gain was 35, 35, 34, and 32 nm at 500, 1000, 2000, and 4000 nm/s stretch velocities, respectively. These values are higher than measured without αB-crystallin (mean values 34.0 ± 0.7 nm versus 32.8 ± 0.6 nm), but the difference did not reach significance.
Considering that the effect of binding of αB-crystallin to titin might be most pronounced under conditions of ischemia (4), we also performed experiments at pH 6.4, which is known to exist during ischemia/reperfusion injury of the heart (for a recent review, see Ref. 24). Results are shown in Tables 1, 2, 3 (values in parenthesis). No pH dependence of the effect of αB-crystallin on the N2B-Us and its flanking Ig domains was seen.
The effect of αB-crystallin on Ig domains from the proximal tandem Ig segment was also studied. Two fragments were used: one consisting of Ig 1-8 and a second of Ig 8-15 (see Fig. 5, top). When these proteins were stretched, a total of up to nine force peaks were seen (Fig. 5, A and B), probably reflecting unfolding of eight Ig domains plus a final detachment event. Plotting the UF versus peak number showed a progressively increasing UF (Fig. 6A), indicating that the domains have different mechanical stabilities. The slopes of UF versus peak number were (ranked from low to high velocity) 11.6, 12.8, and 12.4 pN/peak for Ig 1-8 and 12.3, 13.1, and 12.8 pN/peak for Ig 8-15. The slightly higher UF values of Ig 8-15 (shift between solid and broken lines in Fig. 6A is 8, 10, and 5 pN at 500, 1000, and 2000 nm/s, respectively) indicate a minor gradient in mechanical stability of Ig domains in the direction of Ig1 to Ig15. However, the differences in force levels are small (a continuous stability gradient from one end of the proximal tandem Ig to the other would result in a nonoverlapping UF range (i.e. the broken and solid lines would then be shifted by at least ∼100 pN). Thus results indicate that domains with different stabilities are nearly randomly distributed within the proximal tandem Ig segment (Ig 1-15). The UF of all peaks in Ig 1-8 and Ig 8-15 were averaged and plotted versus stretch velocity in Fig. 7A. The UF has a velocity dependence (slope of lines) that is similar to that of Ig 24-26, but UF values of Ig 1-15 are nearly 50 pN higher than for Ig 24-26. Interestingly, in the presence of αB-crystallin, UF of Ig 24-26 closely matches that of Ig 1-15 (Fig. 7A). Finally, a comparison with results from Ig 91-98, a fragment from the distal tandem Ig (for its location, see schematic at top of Fig. 5A) shows that Ig 1-15 has significantly lower UFs and a different stretch velocity dependence.
FIGURE 5.

Force-extension curve of Ig 1-8 and Ig 8-15 fragments. Top, location of fragments in spring region of cardiac titin (N2B isoform). An electron micrograph is also shown of a cardiac sarcomere (36) labeled with antibodies that demarcate the N2B-Us, PEVK, and tandem Ig segments; superimposed is a schematic of two titin molecules. A-D, force-extension curves show regularly separated force peaks that were fit with the WLC equation (broken lines). See “Experimental Procedures” for additional details.
FIGURE 6.

A, UF of Ig 1-8 and Ig 8-15 at three stretch speeds. (Mean force levels of typically 30 molecules are shown.) Linear fits show that UF is ∼5-10 pN higher in Ig 8-15. B, CL of each of the eight unfolding peaks in Ig 1-8 and Ig 8-15. Results largely overlap. Results were fit with linear lines (r2 of each line was >0.99), shown as solid lines for Ig 1-8 and broken lines for Ig 8-15. The inset shows the mean and S.E. of all slopes of Ig 1-8 and Ig 8-15 (shown as Ig 1-15) and of Ig 24-26 (from Fig. 1B). C, effect of αB-crystallin on UF of Ig 1-8 (stretch speed, 1000 nm/s). UF is significantly increased by αB-crystallin. Lines, control, y = 173 + 12.9x; αB-crystallin, y = 188 + 14.7x. control, 31 molecules; αB-crystallin, 34 molecules. D, effect of αB-crystallin on UF of Ig 8-15 (stretch speed, 1000 nm/s). UF is significantly increased by αB-crystallin. Lines, control, y = 173 + 13.1x; αB-crystallin, y = 195 + 13.1x. control, 22 molecules; αB-crystallin, 25 molecules.
FIGURE 7.

A, UF versus log stretch speed of all domains in Ig 1-15, Ig 24-26, and Ig 91-98 (thin black line from Bullard et al. (6) and red line from Watanabe et al. (18)). The UF is the average of all domains contained in the fragments, and the stretch speed (in nm/s) is the imposed speed corrected for cantilever bending (except for data from Bullard et al. (6)). Results of Ig 24-26 are shown in the absence and presence of αB-crystallin. B, effect of αB-crystallin on UF of Ig 1-8, Ig 8-15, and Ig 24-26. The mean UF of all domains contained in the fragments is shown. The increase in UF of Ig 24-26 is significantly larger than that of Ig 1-8 (p < 0.001) and Ig 8-15 (p < 0.001).
We also measured the CL of the eight unfolding peaks in Ig 1-8 and Ig 8-15; results of the two fragments at three stretch velocities largely overlap and are indistinguishable (see Fig. 6B). The slope of the line fit to CL versus peak number indicates the average CL gain per unfolding event; the average of the three velocities is 30.7 ± 0.6 nm for Ig 1-8 and 30.8 ± 0.5 for Ig 8-15. These values are significantly smaller (p = 0.02) than those of the Ig 24-26 domains, where the average CL gain is 32.8 ± 0.6 nm (see also the inset of Fig. 6B). The CL of the first unfolding peak indicates the CL of the fragments with folded Ig domains with a value (average of the three velocities) of 43.1 ± 3.4 nm for Ig 1-8 and 42.9 ± 2.3 nm for Ig 8-15. This amounts to ∼5.4 nm/folded domain.
Examples of Ig 1-8 and Ig 8-15 force-extension curves in the presence of αB-crystallin are shown in Fig. 5, C and D. The curves are similar in appearance to those in the absence of αB-crystallin. WLC analysis revealed that the average domain-induced CL gain (slope of CL versus peak number) of the two fragments is 30.7 nm, which is identical to the value in the absence of αB-crystallin. UF levels are shown in Fig. 6, C and D (to keep the work manageable, we only studied a single stretch speed (1000 nm/s)). Results indicate that UF of both fragments is significantly increased by αB-crystallin; the average increase of all domains is 33 ± 2.4 for Ig 1-8 and is 23 ± 6.4 pN for Ig 8-15. Fig. 7B compares the increase in UF in the presence of αB-crystallin of Ig 1-8, Ig 8-15, and Ig 24-26. The UF increase of Ig 24-26 is significantly higher than that of Ig 1-8 and Ig 8-15. Finally, we also measured the effect of the two αB-crystallin mutants, R120G and R157H, on UF of Ig 1-8 and Ig 8-15 and found that R120G did not affect UF, whereas R157H significantly increased UF of domains in both Ig 1-8 and Ig 8-15 but less than WT αB-crystallin (see Table 4).
DISCUSSION
αB-crystallin is a member of the small heat shock protein family of molecular chaperones that binds to proteins in the early stages of denaturation, holding them in a folding-competent state (25). In addition to being present at high levels in the lens, small heat shock proteins are found in many tissues with high levels of αB-crystallin expressed in cardiac and skeletal muscle. Small heat shock proteins have subunit masses in the range 12-43 kDa and typically exist as large oligomeric species with a size distribution in the case of αB-crystallin of 200-800 kDa (25). Immunoelectron microscopy on heart muscle has shown that the N2B-Us, one of the important spring elements in titin, is a main site of association between αB-crystallin and titin (6), and we therefore focused in this study on the mechanical changes in the N2B-Us that result from αB-crystallin binding. Because in vitro binding studies indicate binding of αB-crystallin to Ig domains, we also studied the Ig domains that flank the N2B-Us as well as all Ig domains that make up the proximal tandem Ig segment, selected because these domains are thought to be relatively unstable and to unfold at low forces (16, 20). Single molecule force spectroscopy revealed significant αB-crystallin effects, a reduction in the PL of the N2B-Us and an increase in the UF of the Ig domains, with the largest effects on the Ig domains that flank the N2B-Us. These effects were attenuated by the R157H mutation in αB-crystallin and abolished by the R120G mutation. Below we discuss these findings in detail.
αB-crystallin Effect on N2B-Us—The recombinant N2B protein that we stretched consists of the 572-residue N2B-Us, flanked at one end by Ig24 and Ig25 and at the other by Ig26. Thus, when three unfolding peaks are detected in the force-extension trace, the WLC fit to the trace leading up to the first unfolding peak provides a PL and CL estimate of the N2B-Us. The PL under control conditions was 0.65-0.71 nm (Table 1), which is close to the published N2B-Us PL values of 0.66 nm (16), 0.65 nm (19), and 0.4-0.7 nm (17). The measured CL was ∼195 nm, which is slightly less than that expected from a fully unfolded polypeptide (572 residues × 0.38 nm/residue, or ∼215 nm). Low N2B-Us CL values have been reported by Leake et al. (∼100 and ∼180 nm), but this was probably due to the absence of reducing agent in their experimental buffer, resulting in disulfide cross-links that lowered CL (see Ref. 17). Our experiments were carried out in the presence of 5 mm dithiothreitol, and disulfide cross-links are unlikely to have existed. A possible explanation for the shorter CL in our work is the presence of structural elements in the N2B-Us with stability greater than that of the first to unfold the Ig domain. The contribution to the CL of a fully unfolded polypeptide is ∼0.38 nm/residue, but for structured elements, this value is much less (e.g. 0.15 nm/residue for an α-helix). Thus, the CL that we measured suggests that the N2B-Us is not completely random coil when the first Ig domain unfolds but instead is likely to contain regions with secondary structures.
The presence of secondary structures in the N2B-Us is also suggested by the significantly reduced PL of the N2B-Us in the presence of αB-crystallin (∼0.2 versus ∼0.7 nm). Such reduction is hard to explain if the N2B-Us were to be a completely unfolded polypeptide (the unfolded state has an extremely low affinity for αB-crystallin (25)); instead, it suggests the presence of structural elements in the N2B-Us that are stabilized by αB-crystallin and that resist therefore unfolding during stretch. Based on work with mutant lysozymes of varying stability, it has been suggested that αB-crystallins have a low capacity, high affinity binding mode to the more nearly native substrates and a high capacity, low affinity binding mode to more destabilized substrates (25). The low affinity binding to less native substrates prevents the chaperones from promoting protein unfolding, and the high affinity mode to nearly native substrates protects them from further unfolding. Thus, our findings suggest that the N2B-Us contains secondary structure elements that can be stabilized by αB-crystallins.
The N2B-Us structure has not been solved, and there is currently no experimental evidence for the existence of secondary structure. However, all available computer algorithms that predict secondary structure (available on the World Wide Web) indicate with high confidence that the N2B-Us sequence is a largely unstructured protein (random coil) with multiple pockets of α-helix (comprising 10-15% of the sequences) and β-sheet (5-10%). For example, Profile network Prediction Heidelberg (26) predicts 11 α-helical segments (each 5-20 residues in size) and 11 β-sheet segments (each 5-10 residues in size). Thus, the tertiary structure of the N2B-Us is likely to consist of random coil and α-helical and β-sheet structural elements. Stretching of the N2B-Us is likely to be initially dominated by extension of random coil sequences, with at higher extensions unfolding of secondary structures. When an α-helix unfolds, the CL of a single residue increases from 0.15 to 0.38 nm, giving rise to a reduction in force (an abrupt CL gain also explains the force reduction seen when Ig domains unfold). This force reduction is expected to be low if the number of residues that unfold at one time is small; we estimate, using the computational approach explained in our previous work (19), that when the average α-helical segment (10 residues in size) unfolds, the ensuing force reduction is at most several pN, which is below the detection limit of the atomic force microscope. Although the individual unfolding events within the N2B-Us might not be resolved, cumulatively they are expected to have the effect of depressing force. It follows that when the probability of unfolding is reduced by αB-crystallin, force during stretch will be higher, and the WLC fits will reveal lower PL values (force and PL are inversely related; see Equation 4). Thus, the existence of multiple structural elements in the N2B-Us that unfold during stretch and that can be stabilized by αB-crystallin might explain the lower PL. An alternative interpretation for the effect of αB-crystallin on the N2B-Us force and PL is that αB-crystallin binding stabilizes the tertiary structure of the N2B-Us and that this increases the resistance to extension of the N2B-Us, including of random coil sequences. As a result, force would be elevated (and PL would be reduced).
Effect of αB-crystallin on Ig Domains—We studied the Ig 24-26 domains that flank the N2B-Us and the Ig 1-15 domains that make up the proximal tandem segment. It is noteworthy that the 32.8 nm average contour length gain per unfolding event in Ig24 is 2.1 nm larger than that of Ig domains in Ig 1-15 (Fig. 6B, inset) and 4.5 nm larger than previously measured for the Ig 91-98 domains (18). Some differences in CL gain can be expected due to the different number of residues contained in the Ig domains of titin (18, 27). It is unlikely, however, that the large CL gain of the Ig 24-26 domain can be explained this way, because the domains have an average of 85 residues (predicted by the sequence of Ref. 11 and using SMART (Simple Modular Architecture Research Tool) on the World Wide Web), which is less than the 92 residues on average contained in the Ig 1-15 and 89 residues in Ig 91-98 (18, 27). The contour length gain upon unfolding can be calculated from the number of residues in the domain times 0.38 nm/residue minus the size of a folded domain (5.3 nm; see “Results”). The predicted values are 28.5 for Ig 91-98 domains and 29.8 nm for Ig 1-15 domains, both of which are very close to the measured values (28.7 and 30.7 nm). However, the calculated value for I24-26 domains is 27.0 nm, or ∼6 nm less than measured. A possible explanation for the much larger measured CL gain of Ig 24-26 is that these values are not solely due to Ig domain unfolding but have contributions from the short unique sequences that flank Ig24 (shown in Fig. 5; top schematic). It is also possible that some of the structured N2B-Us sequences discussed above are involved; if they unfold after unfolding of the first Ig domain, the obtained CL gain will exceed that of the Ig domain unfolding event. Thus, the longer than expected CL gain of the unfolding peaks, the lower than expected CL of the N2B-Us (see above), and the effect of αB-crystallin on the PL of the N2B-Us (above) can all be explained by secondary structures in the N2B-Us.
Another unique feature of the Ig 24-26 domains is that their UF values are the lowest of the so far characterized Ig domains; the average UF at the stretch velocities that we used is ∼40 pN less than that of Ig 1-15 and ∼60 pN less than that of Ig 91-98 (see Fig. 7B). UF of Ig domains is likely to be a function of the number of hydrogen bonds between the A′ and the G′ strand that are located at the force-bearing C and N termini (28), as well as other noncovalent interactions (hydrophobic contacts) and differences in the angles between strands of their force-bearing C and N termini (18). Thus, our work suggests sequence differences between Ig 24-26 domains and those of the proximal and distal tandem Ig segments that result in a lower number of force-bearing interactions at the termini of the Ig 24-26 domains.
We found that the UF of Ig 24-26 domains is highly sensitive to αB-crystallin, and their UF is increased on average by ∼50 pN. In vitro binding assays previously showed that binding of αB-crystallin to Ig domains that flank the N2B-Us is differential with no detectable binding to Ig24/25 but binding to Ig26/27 (6). Based on this, we expected that only some of the Ig 24-26 force peaks would be increased. Instead, we observed that all of them are higher (Table 3) and by a similar amount (the increase relative to the control value of the first peak tends to be somewhat larger than that of the second peak, but this difference does not reach statistical significance (p = 0.07)). Our work suggests that in vitro binding assays are not ideal for determining which domains are stabilized by αB-crystallin (possibly, domains need to be mechanically stressed before they bind αB-crystallin). This view is supported by the previous work on Ig 91-98, which showed very weak binding to αB-crystallin in dot blot assays and no binding in pull-down assays but a clear stabilizing αB-crystallin effect in atomic force microscope assays, with a ∼30 pN UF increase (6). Hence, we propose that αB-crystallin can interact with a broad range of Ig domains, including Ig 24-26 and Ig 1-15. It is interesting that the UF increase of Ig 24-26 (51 pN) is nearly double that of Ig 1-15 (28 pN), suggesting that those domains that intrinsically are least stable (Ig 24-26) gain most from the stabilizing effect of αB-crystallin. In summary, our work shows that αB-crystallin reduces the unfolding probability of Ig 24-26 and Ig 1-15 and that this effect is most pronounced for the Ig 24-26 domains.
Disease-causing αB-crystallin Mutations—We studied two αB-crystallin mutations, R120G and R157H. The missense mutation R120G is located within the conserved α-crystallin domain (14). This mutation has been linked to a number of diseases, including cataract and desmin-related myopathy, an inherited muscle disorder in humans characterized by intracellular accumulation of R120G αB-crystallin and desmin (29, 30). Desmin filaments play an important role in cardiomyocytes, where they maintain the structural integrity of the cell by linking adjacent myofibrils to each other. Extensive studies have shown that R120G αB-crystallin has altered secondary, tertiary, and quaternary structures compared with the wild type protein, which presumably diminish its chaperone ability (30). Our work is the first to directly assay for chaperone function of the R120G mutant by measuring its effect on single titin molecule mechanics. A consistent finding of our work was that the mechanical properties of titin were unaltered by the addition of R120G αB-crystallin; no effect was seen on the N2B-Us PL or on UF of the Ig 24-26 and Ig 1-15 domains (Tables 1, 2, 3, 4). Thus, the chaperone effect of αB-crystallin on titin appears to be completely lost by the R120G mutation, and this is likely to be a major part of the pathophysiological significance of the R120G mutation.
The R157H αB-crystallin mutation was recently found in a familial DCM patient; the mutation affects an evolutionarily conserved amino acid residue among α-crystallins (13). Mammalian two-hybrid assays revealed that the mutation decreases the binding to the N2B-Us with no significant reduction in binding to the flanking Ig domains Ig26/27 (13). Our work shows that, unlike R120G, the R157H mutant has a significant effect on the single molecule mechanics of titin, since it lowers the N2B-Us PL and increases the unfolding force of its flanking Ig domains as well as that of Ig 1-15 domains. The effect, however, is significantly less than that of WT αB-crystallin. If this reduced chaperone activity of the R157H mutant is a primary cause of DCM in patients carrying this mutation, it follows that having the full chaperone effect of WT αB-crystallin is important for maintaining the biological functions of the N2B-Us. These functions include providing elasticity toward the upper limit of the physiological sarcomere length range (12) and binding two members of the FHL (four-and-a-half LIM domain) protein family, FHL1 (31) and FHL2 (32, 33). Recent evidence indicates that FHL1 functions in the hypertrophic biomechanical stress response (31) and that FHL2 functions in localizing various metabolic enzymes (32). In addition, the N2B-Us contains sequences that can be phosphorylated by PKA (34) and PKG (35) and that thereby tune the compliance of the N2B-Us spring. It is likely that these important N2B-Us functions (binding FHL1/2, responding to phoshorylation) depend on local secondary structures and that αB-crystallin is required to maintain these structures, especially when the heart is stressed, such as during ischemia (29). Importantly, our findings show that if the chaperone function of αB-crystallin is fully (R120G) or partially (R157H) abolished, structured elements in the N2B-Us and Ig domains become more vulnerable to unfolding, and this is likely to contribute to the pathophysiology of patients carrying these mutations.
Acknowledgments
We thank Joe Popper, Joshua Nedrud, and Brian Anderson for outstanding technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant HL62881 (to H. G.). This work was also supported by Deutsche Forschungsgemeinschaft Grant La668/12-1 (to S. L.).
Footnotes
The abbreviations used are: DCM, dilated cardiomyopathy; WLC, wormlike chain; PL, persistence length; CL, contour length; UF, unfolding force; AB, assay buffer; pN, piconewtons; WT, wild type.
References
- 1.Horwitz, J. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 10449-10453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhat, S. P., and Nagineni, C. N. (1989) Biochem. Biophys. Res. Commun. 158 319-325 [DOI] [PubMed] [Google Scholar]
- 3.Horwitz, J. (2000) Semin. Cell Dev. Biol. 11 53-60 [DOI] [PubMed] [Google Scholar]
- 4.Wang, X., Osinska, H., Gerdes, A. M., and Robbins, J. (2002) J. Card. Fail. 8 S287-S292 [DOI] [PubMed] [Google Scholar]
- 5.Golenhofen, N., Arbeiter, A., Koob, R., and Drenckhahn, D. (2002) J. Mol. Cell Cardiol. 34 309-319 [DOI] [PubMed] [Google Scholar]
- 6.Bullard, B., Ferguson, C., Minajeva, A., Leake, M. C., Gautel, M., Labeit, D., Ding, L., Labeit, S., Horwitz, J., Leonard, K. R., and Linke, W. A. (2004) J. Biol. Chem. 279 7917-7924 [DOI] [PubMed] [Google Scholar]
- 7.Granzier, H. L., and Labeit, S. (2004) Circ. Res. 94 284-295 [DOI] [PubMed] [Google Scholar]
- 8.Trombitas, K., Jin, J. P., and Granzier, H. (1995) Circ. Res. 77 856-861 [DOI] [PubMed] [Google Scholar]
- 9.Linke, W. A., and Granzier, H. (1998) Biophys. J. 75 2613-2614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu, Y., Cazorla, O., Labeit, D., Labeit, S., and Granzier, H. (2000) J. Mol. Cell Cardiol. 32 2151-2162 [DOI] [PubMed] [Google Scholar]
- 11.Labeit, S., and Kolmerer, B. (1995) Science 270 293-296 [DOI] [PubMed] [Google Scholar]
- 12.Helmes, M., Trombitas, K., Centner, T., Kellermayer, M., Labeit, S., Linke, W. A., and Granzier, H. (1999) Circ. Res. 84 1339-1352 [DOI] [PubMed] [Google Scholar]
- 13.Inagaki, N., Hayashi, T., Arimura, T., Koga, Y., Takahashi, M., Shibata, H., Teraoka, K., Chikamori, T., Yamashina, A., and Kimura, A. (2006) Biochem. Biophys. Res. Commun. 342 379-386 [DOI] [PubMed] [Google Scholar]
- 14.Vicart, P., Caron, A., Guicheney, P., Li, Z., Prevost, M. C., Faure, A., Chateau, D., Chapon, F., Tome, F., Dupret, J. M., Paulin, D., and Fardeau, M. (1998) Nat. Genet. 20 92-95 [DOI] [PubMed] [Google Scholar]
- 15.Muhle-Goll, C., Habeck, M., Cazorla, O., Nilges, M., Labeit, S., and Granzier, H. (2001) J. Mol. Biol. 313 431-447 [DOI] [PubMed] [Google Scholar]
- 16.Li, H., Linke, W. A., Oberhauser, A. F., Carrion-Vazquez, M., Kerkvliet, J. G., Lu, H., Marszalek, P. E., and Fernandez, J. M. (2002) Nature 418 998-1002 [DOI] [PubMed] [Google Scholar]
- 17.Leake, M. C., Grutzner, A., Kruger, M., and Linke, W. A. (2006) J. Struct. Biol. 155 263-272 [DOI] [PubMed] [Google Scholar]
- 18.Watanabe, K., Muhle-Goll, C., Kellermayer, M. S., Labeit, S., and Granzier, H. (2002) J. Struct. Biol. 137 248-258 [DOI] [PubMed] [Google Scholar]
- 19.Watanabe, K., Nair, P., Labeit, D., Kellermayer, M. S., Greaser, M., Labeit, S., and Granzier, H. (2002) J. Biol. Chem. 277 11549-11558 [DOI] [PubMed] [Google Scholar]
- 20.Labeit, D., Watanabe, K., Witt, C., Fujita, H., Wu, Y., Lahmers, S., Funck, T., Labeit, S., and Granzier, H. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 13716-13721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bustamante, C., Marko, J. F., Siggia, E. D., and Smith, S. (1994) Science 265 1599-1600 [DOI] [PubMed] [Google Scholar]
- 22.Granzier, H., Kellermayer, M., Helmes, M., and Trombitas, K. (1997) Biophys. J. 73 2043-2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kellermayer, M. S., Smith, S. B., Granzier, H. L., and Bustamante, C. (1997) Science 276 1112-1116 [DOI] [PubMed] [Google Scholar]
- 24.Vaughan-Jones, R. D., Spitzer, K. W., and Swietach, P. (2009) J. Mol. Cell Cardiol. 46 318-331 [DOI] [PubMed] [Google Scholar]
- 25.Horwitz, J. (2003) Exp. Eye Res. 76 145-153 [DOI] [PubMed] [Google Scholar]
- 26.Rost, B., and Sander, C. (1993) J. Mol. Biol. 232 584-599 [DOI] [PubMed] [Google Scholar]
- 27.Witt, C. C., Olivieri, N., Centner, T., Kolmerer, B., Millevoi, S., Morell, J., Labeit, D., Labeit, S., Jockusch, H., and Pastore, A. (1998) J. Struct. Biol. 122 206-215 [DOI] [PubMed] [Google Scholar]
- 28.Lu, H., Isralewitz, B., Krammer, A., Vogel, V., and Schulten, K. (1998) Biophys. J. 75 662-671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumarapeli, A. R., and Wang, X. (2004) J. Mol. Cell Cardiol. 37 1097-1109 [DOI] [PubMed] [Google Scholar]
- 30.Treweek, T. M., Rekas, A., Lindner, R. A., Walker, M. J., Aquilina, J. A., Robinson, C. V., Horwitz, J., Perng, M. D., Quinlan, R. A., and Carver, J. A. (2005) FEBS J. 272 711-724 [DOI] [PubMed] [Google Scholar]
- 31.Sheikh, F., Raskin, A., Chu, P. H., Lange, S., Domenighetti, A. A., Zheng, M., Liang, X., Zhang, T., Yajima, T., Gu, Y., Dalton, N. D., Mahata, S. K., Dorn, G. W., 2nd, Heller-Brown, J., Peterson, K. L., Omens, J. H., McCulloch, A. D., and Chen, J. (2008) J. Clin. Invest. 118 3870-3880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lange, S., Auerbach, D., McLoughlin, P., Perriard, E., Schafer, B. W., Perriard, J. C., and Ehler, E. (2002) J. Cell Sci. 115 4925-4936 [DOI] [PubMed] [Google Scholar]
- 33.Radke, M. H., Peng, J., Wu, Y., McNabb, M., Nelson, O. L., Granzier, H., and Gotthardt, M. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 3444-3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamasaki, R., Wu, Y., McNabb, M., Greaser, M., Labeit, S., and Granzier, H. (2002) Circ. Res. 90 1181-1188 [DOI] [PubMed] [Google Scholar]
- 35.Kruger, M., Kotter, S., Grutzner, A., Lang, P., Andresen, C., Redfield, M. M., Butt, E., Dos Remedios, C. G., and Linke, W. A. (2009) Circ. Res. 104 87-94 [DOI] [PubMed] [Google Scholar]
- 36.Trombitas, K., Freiburg, A., Centner, T., Labeit, S., and Granzier, H. (1999) Biophys. J. 77 3189-3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bang, M. L., Centner, T., Fornoff, F., Geach, A. J., Gotthardt, M., McNabb, M., Witt, C. C., Labeit, D., Gregorio, C. C., Granzier, H., and Labeit, S. (2001) Circ. Res. 89 1065-1072 [DOI] [PubMed] [Google Scholar]