

Abstract
Over 100 mutations in Cu/Zn-superoxide dismutase (SOD1) result in familial amyotrophic lateral sclerosis. Dimer dissociation is the first step in SOD1 aggregation, and studies suggest nearly every amino acid residue in SOD1 is dynamically connected to the dimer interface. Post-translational modifications of SOD1 residues might be expected to have similar effects to mutations, but few modifications have been identified. Here we show, using SOD1 isolated from human erythrocytes, that human SOD1 is phosphorylated at threonine 2 and glutathionylated at cysteine 111. A second SOD1 phosphorylation was observed and mapped to either Thr-58 or Ser-59. Cysteine 111 glutathionylation promotes SOD1 monomer formation, a necessary initiating step in SOD1 aggregation, by causing a 2-fold increase in the Kd. This change in the dimer stability is expected to result in a 67% increase in monomer concentration, 315 nm rather than 212 nm at physiological SOD1 concentrations. Because protein glutathionylation is associated with redox regulation, our finding that glutathionylation promotes SOD1 monomer formation supports a model in which increased oxidative stress promotes SOD1 aggregation.
Familial amyotrophic lateral sclerosis (FALS)4 is the hereditary form of amyotrophic lateral sclerosis, a fatal disease characterized by progressive motor neuron loss (1). A subset of FALS is caused by mutations in the gene encoding homodimeric Cu/Zn-superoxide dismutase (SOD1), which forms intraneuronal aggregates (2). Although SOD1 aggregation is involved in SOD1-mediated FALS, it is generally believed that the functional properties of the enzyme are not related to the toxic gain of function imparted by mutations in SOD1 (3). However, the discovery of roles for SOD1 in the regulation of the cellular phosphorylation balance (4) and redox state (5) provides additional avenues for connecting the cellular role of SOD1 to FALS. The classical studies of SOD1 were generally performed using bovine erythrocyte SOD1 or recombinant human SOD1. Although recombinant methods are widely used to produce SOD1 mutants, a disadvantage of studying recombinant SOD1 is the absence of potentially important post-translational modifications present in human tissues. The initial SOD1 crystal structure was solved using bovine erythrocyte SOD1 (6) and no structure of human erythrocyte SOD1 is available. Here we report results using human erythrocyte SOD1 rather than the recombinant enzyme and find that the native enzyme features a consistent pattern of post-translational modifications. Using a combination of “bottom-up” and “top-down” mass spectrometry (MS) approaches, we show that SOD1 isolated from human erythrocytes is post-translationally phosphorylated and glutathionylated. These modifications occur near the SOD1 dimer interface. Because monomer formation is thought to be the first intermediate leading to SOD1 aggregation (7, 8), we tested the dimer stability of modified SOD1 found, as expected, that glutathionylation promotes the formation of SOD1 monomer.
EXPERIMENTAL PROCEDURES
Isolation of hSOD1 from Erythrocytes—Expired human erythrocytes were obtained from the University of North Carolin-Chapel Hill Hospital blood bank. Human erythrocytes were preserved using one of several anticoagulants: AS-1, AS-3, or AS-5 (stored as long as 42 days before expiration). Bovine erythrocytes preserved with 0.38% sodium citrate were purchased from Pel-Freez Biologicals, Rogers, AR. SOD1 was isolated from human erythrocytes using a modification of the protocol originally used by McCord and Fridovich (9). Following acetone precipitation of SOD1, we dialyzed SOD1 against 20 mm Tris, pH 7.8, and performed hydrophobic interaction chromatography and anion exchange chromatography using an AKTA-FPLC to remove trace impurities. For hydrophobic interaction chromatography, dry (NH4)2SO4 was added to bring the final concentration to 55%. The protein was loaded onto a HiTrap phenyl-Sepharose column (GE Healthcare) and eluted with a gradient from 2 to 0 m (NH4)2SO4 in 20 mm Tris, pH 7.8. Fractions containing SOD1 were combined and dialyzed against salt-free 20 mm Tris, pH 7.8, and anion exchange chromatography was performed using a MonoQ column (GE Healthcare) with a gradient from 0 to 1 m NaCl in 20 mm Tris, pH 7.8. The fractions containing SOD1 were dialyzed against 20 mm Tris, 150 mm NaCl, pH 7.8, and concentrated.
Isolation of hSOD1 from Saccharomyces cerevisiae—Human wild type SOD1 was expressed using plasmid yEP351-hwtSOD1 in the EG118 SOD1-knock out yeast strain (both kindly provided by J. S. Valentine). Growth was carried out at 30 °C for 72 h in YPD media. We uses a modified isolation protocol adapted from Goscin and Fridovich (10) where we replaced all steps, after stirring the lysate at room temperature, with high speed centrifugation and dialysis of the supernatant against 20 mm Tris, pH 7.8, to remove the chloroform and ethanol. Trace impurities were removed by MonoQ and phenyl-Sepharose chromatography as described above.
Remetallation of hSOD1 from yeast was performed by successive dialysis at 4 °C against: (a) 50 mm acetate, pH 3.5, 150 mm NaCl, 10 mm EDTA; (b) 50 mm acetate, pH 3.5, 150 mm NaCl; (c) 50 mm acetate, pH 3.5, 150 mm NaCl + CuSO4 (5-fold molar excess compared with [SOD1]); (d) 20 mm Tris, pH 7.8, 150 mm NaCl + ZnCl2 (5-fold molar excess compared with [SOD1]); and (e) 20 mm Tris, pH 7.8, 150 mm NaCl. Upon concentration with a YM-10 ultrafiltration membrane (Millipore, Bedford, MA), a blue-green coloration was observed, indicating incorporation of copper into the enzyme.
Microcapillary (μ) ESI-FT-ICR-MS Analysis (Top-down Approach)—MS spectra are acquired using a hybrid Qe-Fourier Transform Ion Cyclotron Resonance (FT-ICR)-Mass Spectrometer, equipped with a 12.0-tesla actively shielded magnet (Apex Qe-FTICR-MS, 12.0 T AS, Bruker Daltonics, Billerica, MA), and an Apollo II microelectrospray source. The voltages on the μESI sprayer, interface plate, heated capillary exit, deflector, ion funnel, and skimmer were set at 4.3 kV, 3.9 kV, 300 V, 250 V, 175 V, and 80 V, respectively. The temperature of the μESI source was maintained at 180 °C. Desolvation was carried out using a nebulization gas flow (2.0 bar) and a countercurrent drying gas flow (4.0 liters/s). The electron capture dissociation (ECD) hollow dispenser cathode was heated to increase the temperature inside the ICR cell above 180 °C without application of ECD bias, eliminating non-covalent adducts. SOD1 sample solutions were directly infused using a syringe pump (Harvard Apparatus, Holliston, MA) and a 250-μl syringe (Hamilton, Reno, NV), and electrosprayed at an infusion flow rate of 90 μl/h. Before transfer, ion packets are accumulated inside the collision cell for a duration of 0.2 s. 50 MS scans per spectrum were acquired in the ICR cell with a resolution of 580,000 at m/z 400.
MS/MS Fragmentation for Sequencing Full-length Proteins—FT-MS/MS spectra were obtained from collision-induced dissociation (CID) performed with in-source collision-activated dissociation followed by collision cell-activated dissociation, or from ECD. In CID experiments, argon was used as the collision gas (PAr ≈ 1.6 × 10-6 torr). Precursor ions were isolated with Q1 (Mass Selective Quadrupole) and subjected to collision cell-activated dissociation using an isolation window width of 5 Da. 50 MS/MS scans were acquired in the ICR cell with a resolution of 580,000 at m/z 400. In ECD experiments, precursor ions were isolated with Q1 and subjected to ICR cell. Isolation window width was 10 Da. Low energy electrons were generated by a heated hollow dispenser cathode with a bias voltage of -1.5 V. ECD lens voltage was set at 15.0 V. The electrons produced by the hollow cathode were pulsed into the ICR cell with a pulse length of 0.0050 s, causing fragmentation of the ions that were already trapped in the ICR cell. To maximize the ion population before irradiation, the cell was filled with 5 iterations of ion accumulation from the external collision cell of the isolated precursor as suggested (11). In both CID and ECD experiments, 50 MS/MS scans per spectrum were acquired in the ICR cell with a resolution of 580,000 at m/z 400.
Tempo LC MALDI Fractionation and MALDI-TOF-TOF Analysis (Bottom-up Approach)—SOD1 protein samples were digested with trypsin (Promega, Madison, WI) overnight at 37 °C in an enzyme to substrate ratio of 1:40 by mass. After desalting with ZipTipC18 (Millipore), 0.5 μl of the tryptic digests were directly spotted on a 384-well stainless steel matrix-assisted laser desorption/ionization (MALDI) target plate for pre-scan using a MALDI time-of-flight/time-of-flight (TOF/TOF) tandem mass spectrometer (ABI 4800 Proteomics Analyzer, Applied Biosystems, Foster City, CA). In an analytical run the peptide digests were further separated using a reverse phase liquid chromatography (LC) system (Tempo LC MALDI, Applied Biosystems, Foster City, CA). 10 μl of tryptic peptides were injected and directly loaded on a C18 capillary column (Monolithic silica RP C18 endcapped, 100 μm inner diameter, 15 cm length, Chromolith ® CapRod ®, Merck KGaA, Darmstadt, Germany) with a loading buffer (H2O/acetonitrile/trifluoroacetic acid, 98/2/0.1, v/v/v), and separated using a gradient from 100% A (H2O/acetonitrile/trifluoroacetic acid, 98/2/0.1, v/v/v) to 40% B (H2O/acetonitrile/trifluoroacetic acid, 2/98/0.1, v/v/v) over 70 min, and 40-80% B in 10 min, at a flow rate of 1.0 μl/min. The column eluates are mixed with a matrix solution of ∼7.0 mg/ml re-crystallized α-cyano-4-hydroxycinnamic acid (Sigma) in H2O/acetonitrile, 30/70 (v/v), and 5 mm ammonium citrate, deposited on a 2000-well stainless steel MALDI target plate at a frequency of 0.15 Hz, and analyzed by ABI 4800 MALDI TOF/TOF (200 Hz Nd:YAG laser, OptiBeam™). Each spot was analyzed in MS mode in the mass range of 800-4000 Da, by accumulating ion signals over 1200 laser shots. Up to 15 ions with a signal-to-noise ratio less than 30 were selected for MS/MS analysis, which was performed with a +1.0 kV collision energy and 1.0 × 10-6 torr collision gas (air) pressure. Acquisition of an MS/MS spectrum was interrupted when 2000 laser shots (40 sub-spectra from 50 laser shots each) were accumulated. Mass calibration was applied by spotting the standard peptide mixture at six places around the target plate (12). Acquired MS and MS/MS spectra were searched using Mascot 2.2 (Matrix Science, London, UK) embedded in GPS 3.5 (Applied Biosystems).
Phosphatase Treatment of SOD1—To prepare dephosphorylated. SOD1 we use agarose-linked alkaline phosphatase (Sigma). The beads were washed using 20 mm Tris, 150 mm NaCl, pH 7.8, buffer and added to a solution of SOD1 at a 1:200 (v/v) ratio. The reaction was performed overnight at 37 °C.
Size Exclusion Chromatography—Size exclusion chromatography was carried out using a Superdex 200 10/300 column (GE) and a 200-μl injection loop. The void volume of the column was ∼8 ml and a flow rate of 0.5 ml/min was used at 4 °C. For measurement of the dimer dissociation constant, Kd, samples of SOD1 at pH 7.8 were diluted to 1.25 nm to facilitate dimer dissociation using the column running buffer (20 mm Tris, pH 7.8, 150 mm NaCl, and 10 μg/liter bovine serum albumin to prevent SOD1 adherence to the column and the glass collection tubes). Samples are incubated at 37 °C for up to 5 h to ensure proper equilibration. 250-μl fractions were collected throughout the chromatography experiment for use in immunoblotting. Fractions were acetone precipitated by the addition of 4 volumes of cold acetone to the sample and a 1-h incubation at -20 °C. The samples are pelleted for 30 min at 20,000 rpm. The precipitated SOD1 was resuspended in protein solvent containing sodium dodecyl sulfate and β-mercaptoethanol and resolved by polyacrylamide gel electrophoresis. Following transfer to a polyvinylidene fluoride membrane and blocking with 5% bovine serum albumin, blots were probed with a sheep anti-SOD1 primary antibody (Calbiochem number 574597) and a rabbit anti-sheep alkaline phosphatase-conjugated secondary antibody (Thermo Scientific number 31360). The blots are incubated in ECF reagent (Amersham Biosciences) and visualized using a GE Healthcare STORM 840 Phosphor-Imager. Quantification of band intensities is performed using the ImageQuant software (GE Healthcare).
Kd Determination Using SOD1 Activity—We tested for a reduction in the rate of 6-hydroxydopamine autoxidation as previously described (13). The assay was sufficiently sensitive to allow measurement of SOD1 concentrations as low as 100 pm. The activity of glutathionylated and non-glutathionylated 2.5 nm SOD1 was measured before and after a 3-h equilibration at 37 °C. Activity assays performed after 3 and 5 h incubation at 37 °C show no additional loss in activity, indicating that the monomer-dimer equilibrium was reached at 3 h. The percent dimer dissociation at equilibrium was determined using the following equation,
% Dimer dissociation
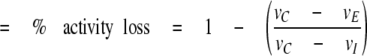 |
where vc is the initial rate of the control reaction, vE is the initial rate of the reaction containing equilibrated SOD1, and vI is the initial rate of the reaction measured immediately upon dilution of SOD1 from a concentrated stock solution. Assuming a physiological SOD1 concentration of 40 μm (14), we calculated the monomer concentration based on the Kd. For a Kd of 2.25 nm, we expect the monomer concentration to be 212 nm. For a Kd of 5 nm, we expect a monomer concentration of 315 nm, a 67% increase.
RESULTS
SOD1 Is Modified in Human Erythrocytes—Using a 12-tesla FT-ICR-MS with ultrahigh mass accuracy and resolution, full-length wild-type SOD1 from human erythrocytes was found to have a monoisotopic mass of 15837.8902 Da (Fig. 1A), corresponding to the theoretical mass of 15837.8812 Da for N-terminal acetylated wild-type human SOD1. In addition to the acetylated form, two strong peaks with masses of 15935.8702 and 16033.8462 Da corresponding to the respective addition of one (+98 Da) or two (+196 Da) hydrated phosphate groups were observed, as well as a third peak corresponding to the modification of SOD1 by glutathione (+305 Da). The protein was treated with alkaline phosphatase, which resulted in the loss of the +98 and +196 Da peaks (Fig. 1c), suggesting both sites are related to phosphorylation.
FIGURE 1.

Post-translational modification of SOD1. μESI-FTICR-MS spectra of: A, human SOD1 isolated from erythrocytes; B, high temperature spectrum of human erythrocyte SOD1 causing phosphorylation peaks to shift from +98 to +80 Da; and C, alkaline phosphatase-treated human SOD1 isolated from erythrocytes. D, a UV trace from anion exchange separation of three populations of human erythrocyte SOD1, the unlabeled peak is composed of impurities. E, a comparison of the μESI-FTICR-MS spectra for populations 1-3 from panel D showing SOD1 enrichment of glutathionylation in population 3. Phosphorylation is indicated by solid vertical arrows. Glutathionylation is indicated by the dashed arrow.
To differentiate between the non-covalent association of H3PO4 or H2SO4 and a hydrated phosphate ester, both expected to result in the addition of ∼98 Da, we heated the ICR cell to above 180 °C. Heating results in the conversion of +98 and +196 Da peaks to +80 and +178 Da, respectively, but has no effect on the +305-Da shift (Fig. 1B). The CID/ECD spectra of the +80-Da peak (supplemental Fig. S1), which imply phosphorylation at threonine 2 (Thr-2) (Fig. 2B), are similar to those of the +98-Da peak (supplemental Fig. S2), indicating that the +98-Da peak belongs to single phosphorylation plus one water molecule. Similarly, the CID/ECD spectra of the +178-Da peak indicates that it possibly arises from double phosphorylation plus one water molecule (supplemental Fig. S4). A +160-Da peak corresponding to double phosphorylation was only observed in SOD1 extracted from freshly drawn human erythrocytes (supplemental Fig. S9). However, the signal intensities of the +160, +178, and +196 Da peaks were insufficient for FT-ECD fragmentation experiments in mapping the second modification site.
FIGURE 2.

Location of SOD1 modifications in human erythrocytes. A, the SOD1 homodimer (Protein Data bank code 2V0A) with glutathionylated Cys-111 and phosphorylated Thr-2 modeled using PYMOL. B, the SOD1 dimer interface showing the relative positions of the phosphate and glutathione moieties. C, a μESI-FTICR-MS-CID MS/MS spectrum with the phosphorylation at Thr-2 for a representative b-ion illustrated. A complete MS/MS spectrum is shown in supplemental Fig. S1. D, a Tempo LC-MALDI-TOF-TOF MS/MS spectrum with several fragment ions from the parent peptide 92DGVADVSIEDSVISLSGDHCIIGR115 highlighted, which are shifted by glutathionylation. Complete MS/MS spectra are shown in supplemental Figs. S3 and S4.
We found that human erythrocyte SOD1 is phosphorylated at Thr-2 (Fig. 2B, supplemental Figs. S1 and S2), but due to difficulty in sequencing doubly phosphorylated SOD1, even after the enrichment of phosphorylated protein, we can only report that a second phosphorylation is possibly located at either Thr-58 or Ser-59 (supplemental Fig. S4). Residues 58 and 59 are located in the zinc loop in SOD1, which both chelates the zinc atom at the active site and accounts for several dimer interface contacts.
The site of glutathionylation in human SOD1 was determined by analyzing tryptic digestions of human erythrocyte SOD1 with Tempo LC-MALDI fractionation and MALDI-TOF-TOF. The acquired MS and MS/MS spectra were searched using Mascot 2.2 embedded in GPS 3.5. In 13 unique SOD1 peptides, we found mass shifts of +305 Da from C-terminal ions y5 to y20 relative to the MS/MS spectra of the unmodified peptides (supplemental Fig. S3), which indicates that cysteine 111 (Cys-111) is susceptible to glutathionylation. Peptides containing cysteines 6, 57, and 146 were found not to be glutathionylated (supplemental Fig. S5).
Human erythrocyte SOD1 was separated into multiple charge populations by anion exchange chromatography (Fig. 1D). Each population shows distinct modification patterns in its mass spectrum (Fig. 1E). The SOD1 that elutes at the highest ionic strength, population 3 (Fig. 1, D and E), has a dominant peak at m/z = 16142.9600 Da, corresponding to the acetylated protein derivatized with glutathione. The extent of phosphorylation varies in SOD1 populations 1 and 2 that elute at low and medium salt concentrations (Fig. 1, D and E). A pool of SOD1 that is simultaneously phosphorylated and glutathionylated was also observed (Fig. 1E).
Distribution of Modifications in Individual Erythrocyte Samples—The modifications described above were observed in SOD1 isolated from erythrocytes pooled from multiple donors and as such, any modifications could have arisen due primarily to contributions from a single donor. To investigate the consistency of glutathionylation and phosphorylation levels between individuals, we next compared SOD1 from individual samples of human erythrocytes. Of six individual samples, five contained both phosphorylation and glutathionylation (Table 1). One sample lacked glutathione but was significantly phosphorylated. Assuming independent probabilities of phosphorylation and glutathionylation of SOD1, we expect the probability of observing both modifications to be the product of the individual frequencies. In erythrocyte samples 1-3, the calculated frequency of combined phosphorylation and glutathionylation is significantly smaller than the measured frequency, suggesting that phosphorylation and glutathionylation are co-dependent in the case of SOD1. However, the observed frequency of combined modification in samples 4 and 5 is only marginally higher than the expected frequency and is within the experimental error. This behavior may arise from differences in phosphatase activity among individuals, or other variables not controlled for in this study.
TABLE 1.
Comparison of modifications in independent erythrocyte samples
SOD1 isolated from individual samples of human erythrocytes are analyzed by μESI-FTICR-MS without separation of modification states. We calculate the frequencies of phosphorylation (fP), glutathionylation (fG), and both (fPG) from the percentages of each modified SOD1 species in the sample as follows: fP = (%P + %PG)/100; fG = (%G + PG)/100; and fPG = %PG/100. Assuming that phosphorylation and glutathionylation are independent processes, we expect the frequency of simultaneous phosphorylation and glutathionylation to be fP · fG = fPG. The sample blood types are as follows: 1 is AB−, 2 is AB+, 3-5 are O+, and 6 is O−.
| Sample No. | Unmodified SOD1 | Phosphorylated SOD1 | Glutathionylated SOD1 | Both |
|---|---|---|---|---|
| 1 | 51.1 ± 11% | 8.5 ± 1.8% fP = 0.30 ± 0.05 | 18.8 ± 3.9% fG = 0.41 ± 0.06 | 21.7 ± 4.6% Expected = 12 ± 4% |
| 2 | 36.9 ± 2.9% | 6.6 ± 1.3% fP = 0.29 ± 0.01 | 34.0 ± 3.6% fG = 0.57 ± 0.05 | 22.6 ± 0.3% Expected = 16.5 ± 1.6% |
| 3 | 41.1 ± 5.0% | 14.4 ± 4.4% fP = 0.33 ± 0.05 | 25.8 ± 1.5% fG = 0.45 ± 0.02 | 18.7 ± 1.6% Expected = 14.7 ± 2.3% |
| 4 | 53.6 ± 0.7% | 22.5 ± 4% fP = 0.35 ± 0.04 | 17.1 ± 3.6% fG = 0.29 ± 0.04 | 12.2 ± 1.0% Expected = 10.2 ± 1.8% |
| 5 | 45.2 ± 3.0% | 14.6 ± 0.5% fP = 0.27 ± 0.01 | 31.5 ± 3.1% fG = 0.44 ± 0.03 | 12.0 ± 0.3% Expected = 11.6 ± 0.9% |
| 6 | 54.4 ± 11.4% | 45.6 ± 9.6% | None | None |
Modifications in Freshly Drawn Human Erythrocytes—The phosphorylation and glutathionylation of human erythrocyte SOD1 does not occur as a result of long-term blood storage. We isolated SOD1 from human erythrocytes immediately following their collection from two donors. The results are summarized in Table 2. Mass spectra from each donor (ages 37 and 73 years) included major peaks related to both SOD1 phosphorylation and glutathionylation. In addition, we observed peaks related to oxidized SOD1. These peaks were shifted +16 and +32 Da from the unmodified mass of SOD1 (supplemental Fig. S9). Because there are no +16 or +32 Da shifted peaks relative to either phosphorylated or glutathionylated SOD1, we postulate that Cys-111 is the site of the oxidation, as has been observed in vitro (15). Alternatively, histidine residues in SOD1 might be oxidized because SOD1 lacks methionine and the oxidation of histidine residues in SOD1 has been observed previously (16). It is interesting to note the abundance of oxidized SOD1 species in sample B compared with sample A given that the more highly oxidized sample was drawn from a 73-year-old donor, whereas sample A was drawn from a 37-year-old donor. However, a comparison based on only two samples is insufficient to support a hypothesis that the difference is related to age.
TABLE 2.
Comparison of modifications in freshly drawn single erythrocyte samples
SOD1 isolated immediately after removal from donors and analyzed by μESI-FTICR-MS. Donor age is listed under the sampe I.D. All frequencies calculated as described in Table I. The percentage of unmodified SOD1 is calculated using the sum of the intensities of SOD1 peaks, which are neither phosphorylated nor glutathionylated (i.e. oxidized SOD1 species are considered unmodified).
| Sample I.D. | Unmodified SOD1 | Phosphorylated SOD1 | Glutathionylated SOD1 | Both |
|---|---|---|---|---|
| A: age 37 | 38.6 ± 5.8% | 13.3 ± 2.0% fP = 0.23 ± 0.03 | 38 ± 5.7% fG = 0.48 ± 0.06 | 10.1 ± 1.5% Expected = 11.3 ± 2.0% |
| B: age 73 | 37.4 ± 5.6% | 8.4 ± 1.3% fP = 0.22 ± 0.02 | 40.8 ± 6.1% fG = 0.54 ± 0.06 | 13.4 ± 2.0% Expected = 11.% ± 1.7% |
Species/System Effects on Modifications—Because S. cerevisiae contain a copper chaperone for SOD1, it is widely used for expressing recombinant human SOD1, but do phosphorylation and glutathionylation of human SOD1 occur in yeast or other systems used to isolate SOD1? To answer this question, we purified and analyzed recombinant SOD1 from yeast, as well as endogenous SOD1 from both yeast and bovine erythrocytes. Mass spectra of human SOD1 isolated from S. cerevisiae indicate an exact match with the acetylated, unmodified SOD1 found in human erythrocytes (supplemental Fig. S8). We separated a sample of yeast-expressed human SOD1 into subpopulations of high and low negative net charge and found similar mass shifts in the deconvoluted mass spectra indicating SOD1 glutathionylation but not phosphorylation (supplemental Fig. S8). The mass spectrum of SOD1 isolated from bovine erythrocytes using the identical protocol used for human erythrocytes indicates bovine SOD is neither phosphorylated nor glutathionylated, nor is endogenous yeast SOD1 (supplemental Figs. S6 and S7, respectively), an expected result given that neither species has a cysteine at position 111 in SOD1. Cysteine 6 is conserved between bovine and human SOD1 but we find no glutathionylation of this residue in either species. We hypothesize that SOD1 phosphorylation was not previously observed due to the common use of bovine erythrocyte SOD1 or recombinant human SOD1.
Effect of Modifications on SOD1 Dimer Dissociation—Thr-2 appears to be at a sufficient distance so as not to participate directly in the SOD1 dimer interface. However, phosphorylation can change both the structure and dynamics of proteins (17) and thus, the distance from the interface and the active site does not preclude Thr-2 phosphorylation from altering the stability or activity of SOD1. Cys-111 residues in each SOD1 monomer are directly apposed in a cleft at the dimer interface. We postulated that the addition of the glutathione tripeptide to Cys-111 will result in a measurable decrease in SOD1 dimer stability. Because of the proximity of both Thr-2 phosphorylation and Cys-111 glutathionylation to the dimer interface and evidence that dimer stability is important for preventing SOD1 aggregation (18, 19), we measured the effect of glutathionylation and phosphorylation on the in vitro stability of dimeric SOD1.
Using size exclusion chromatography, we compared SOD1 species separated by anion exchange as described above (peaks 1 and 3 in Fig. 1E). Altered dimer stability of SOD1 in peak 3 relative to peak 1 is expected to be the result of Cys-111 glutathionylation because substoichiometric amounts of phosphorylation are present in both SOD1 species. Due to the unusually high stability of the SOD1 dimer (the dissociation constant of human SOD1 isolated from yeast is less than 10 nm (20)), it was necessary to perform the experiment at low concentrations. To observe dissociation into monomers, we performed size exclusion chromatography using 1.25 nm SOD1. We compared SOD1 with and without glutathionylation and found that glutathionylation has a marked destabilizing effect on the SOD1 dimer. Progressive SOD1 dimer dissociation was observed upon incubation at 37 °C for up to 5 h. We observed a 3-fold increase in monomer formation in glutathionylated SOD1 relative to SOD1 unmodified by glutathione (Fig. 3).
FIGURE 3.

SOD1 dimer is destabilized by glutathionylation. Non-glutathionylated (SOD1) and glutathionylated (GS-SOD1) SOD1 diluted to 1.25 nm is allowed to equilibrate at 37 °C and resolved by size exclusion chromatography to monitor dimer dissociation. Glutathionylated SOD1 dissociates into monomer upon incubation at low concentrations, whereas non-glutathionylated SOD1 begins to dissociate only slightly after 5 h at 37 °C. Inset, Western blots of 1.25 nm size exclusion chromatography fractions are aligned above a size exclusion chromatograph of 30 μm GS-SOD1 dimer to illustrate the dissociation of GS-SOD at low concentration.
Kd Determination Using an Assay for SOD1 Activity—Monomeric SOD1 was postulated to have reduced activity based on findings that mutations forcing monomeric SOD1 show an 80% activity loss (21). We therefore postulated that activity loss can be used as an indicator of dimer dissociation upon dilution from high concentrations in measuring the Kd of SOD1. The Kd is important considering that concentration of the SOD1 species nucleating aggregation depends upon the nth power of the monomer concentration, where n is the number of monomers in the nucleating species. Small increases in the Kd, therefore, result in large increases in the number of nuclei, especially as the number of monomers in the nucleus increases. Using a spectrophotometric assay sufficiently sensitive to assay sub-nanomolar concentrations SOD1, we measured the reduction in the reaction rate in the presence of 2.5 nm SOD1 at equilibrium (3 h), normalized to the activity immediately after dilution (Fig. 4). We observed a loss in SOD1 activity concomitant with dimer dissociation, evident as a reduction in the initial reaction rate relative to the control reaction. We tested the effect of glutathionylation on dimer stability by comparing SOD1 species separated by anion exchange as described above (peaks 1 and 3 in Fig. 1E). Although the initial reaction rate was similar (2 × 10-4 absorbance units s-1), the glutathionylated SOD1 showed a higher rate at equilibrium, indicating less SOD1 activity and therefore, more dimer dissociation. Based on the percent reduction in activity, we estimated the Kd to be 2.25 nm for unmodified SOD1 and 5 nm for modified SOD1.
FIGURE 4.

Measurement of dimer dissociation using an assay for SOD1 activity. An activity assay comparing glutathionylated SOD1 (GS-SOD1) to unmodified SOD1 (SOD1) at 2.5 nm. The % activity change due to dimer dissociation was estimated by assaying samples of SOD1 after a 3-h equilibration at 37 °C (dashed lines) and normalizing against non-equilibrated samples (solid lines). A control reaction containing only 6-hydroxydopamine in sample buffer is shown in solid blue.
DISCUSSION
Post-translational Modifications of Native Human SOD1—Here we report that SOD1 can be phosphorylated at Thr-2 and at either Thr-58 or Ser-59. We also confirm previous findings of SOD1 glutathionylation and map that modification to Cys-111. Previously in our laboratory, a series of molecular dynamics simulations was carried out that showed that residues in every region of SOD1 show high connectivity to the dimer interface (22) and that residues closely associated with its stability reside on the surface of SOD1 (23). Other recent findings suggest that loss of copper and zinc at the metal binding site results in structural fluctuation at the dimer interface (24), providing further rationale for diverse perturbations resulting in SOD1 aggregation via dimer destabilization. In light of these findings, we postulated that post-translational modifications, like point mutations, would have a similar effect on the structure and dynamics of SOD1. To test the effect of post-translational modifications on the enzyme, we separated a sample of SOD1 into a population that contained a low percentage of phosphorylation and a population that was very highly glutathionylated while also phosphorylated to a similar degree as the non-glutathionylated enzyme. We found that these two forms of SOD1 differ in their propensity toward forming monomers, the glutathionylated SOD1 forming monomer more readily. Our previous finding that monomeric SOD1 is the first intermediate formed during SOD1 aggregation suggests that glutathionylation, which has a destabilizing effect on the SOD1 dimer, may affect the initiation of SOD1 aggregation in FALS.
Glutathionylation has been reported several times in erythrocyte SOD1, including erythrocytes collected from FALS patients (25, 26), and treatment of apo-SOD1 with GSSG causes aggregation in vitro (27). We show here that only Cys-111 glutathionylation is present in vivo. However, glutathionylation of SOD1 has also been performed in vitro and shown to modify each of the four cysteines (27). The involvement of Cys-111 disulfide bonding during SOD1 aggregation has been suggested (28). However, bovine SOD1 lacks Cys-111, yet is capable of aggregating (7). Therefore, Cys-111 glutathionylation need not interfere with SOD1 aggregation by blocking a specific disulfide bond in the aggregated state.
Oxidation of SOD1 in Freshly Drawn Human Erythrocytes—We observe SOD1 oxidation, in addition to phosphorylation and glutathionylation, in freshly drawn human erythrocytes. Although the sample number is not sufficient to show a relationship between age and the degree of SOD1 oxidation, the two samples show differences in this property. Erythrocytes are short-lived cells and have an average lifespan of 120 days in humans. Therefore, the SOD1 used in our experiments was isolated from both young and old cells. Because virtually all of the unmodified SOD1 in one of the samples was oxidized, we conclude that differences in SOD1 oxidation levels are not the result of a cumulative process wherein a population of SOD1 is slowly modified as each individual erythrocyte “ages,” but rather an instantaneous process in which the population of SOD1 responds to differences in the individuals that may or may not be age-related, such as oxidizing conditions in erythrocytes or the surrounding plasma.
Modification Differences in Different Systems—When studying the effects of mutations on SOD1, it is important to have a proper benchmark for SOD1 as it exists inside human tissues. It is clear from our comparison of human SOD1 isolated from both human erythrocytes and yeast that the latter model for native SOD1 lacks at least one feature of native SOD1, phosphorylation. Because we studied wild type SOD1 isolated from human tissue, we believe that phosphorylation and glutathionylation are features of the SOD1 native state and not a result of a disease-related process. However, analysis of spinal cord tissue from sporadic amyotrophic lateral sclerosis patients revealed many kinases that are up-regulated and found dramatic differences in the phosphorylation of multiple proteins (29), suggesting that changes in SOD1 phosphorylation are possible in FALS.
Possible Regulatory Role of SOD1—Phosphorylation and glutathionylation may impact the function of SOD1 through the mediation of interactions with other proteins. SOD1 is involved in cellular redox regulation through a direct interaction with the Rac1-NOX2 enzyme complex (5). This example of a physical interaction between SOD1 and a functional enzyme complex illustrates one mechanism through which phosphorylation and/or glutathionylation of SOD1 could play a significant role without altering its traditionally recognized activity as a free radical scavenger.
Glutathione plays a key role in establishing the redox state of the cell (30). The high ratio of reduced (GSH) to oxidized (GSSG) glutathione is tightly controlled by both glutathione reductase, which uses NADPH to reduce GSSG to GSH, and the active removal of GSSG from the cytosol (31). During scenarios of oxidative stress, the concentration of GSSG increases dramatically (32) and reversibly glutathionylates free cysteines in what is thought to be a general protective mechanism against permanent oxidation of proteins by reactive oxygen or nitrogen species (33). In addition to its general role, several examples of specific functional roles for protein glutathionylation are documented (34-37). Recent evidence also suggests that SOD1 may be involved in growth factor signaling through the production of H2O2, which acts as a second messenger in modulating the activity of protein-tyrosine phosphatases and thus, the balance of phosphorylation in the cell (4). Predicated on these findings, we present a model for the impact of SOD1 modification on oxidative protein-tyrosine phosphatase regulation (Fig. 5).
FIGURE 5.

Model for the participation of SOD1 in protein-tyrosine phosphatase
(PTP) redox regulation. SOD1 interacts with redox regulation of
phosphatase activity through its conversion of superoxide
 to hydrogen peroxide
(H2O2). Glutathione reduces the activity of SOD1 (red
dashed line) by facilitating monomer formation. SOD1 monomer
formation is also associated with aggregation, and ribbon structures
are used to show the proposed destabilizing effect on the SOD1 dimer as a
result of Cys-111 glutathionylation.
to hydrogen peroxide
(H2O2). Glutathione reduces the activity of SOD1 (red
dashed line) by facilitating monomer formation. SOD1 monomer
formation is also associated with aggregation, and ribbon structures
are used to show the proposed destabilizing effect on the SOD1 dimer as a
result of Cys-111 glutathionylation.
SOD1 glutathionylation increases dimer dissociation, facilitating aggregation in the presence of contributing factors such as oxidative damage or destabilizing FALS mutations. Even a modest effect of glutathionylation on SOD1 dimer dissociation, combined with the effects of mutations, could affect FALS-related aggregation over time. Furthermore, a SOD1 variant engineered to be monomeric retains only 20% activity relative to the wild type dimer (21), and therefore we postulate that glutathionylation, by inducing dimer dissociation, inhibits SOD1 activity (Fig. 5).
Decreased Dimer Stability: Possible Implications for FALS—The link between SOD1 mutations, protein aggregation, and FALS is not fully understood, but there are multiple reports showing that dimer dissociation is an early event during SOD1 aggregation (7, 8). Our finding that modifications can facilitate SOD1 dimer dissociation suggests a possible link between the normal characteristics of SOD1 and its role in FALS. Although relatively modest, a 2-fold increase in Kd resulting from SOD1 modification translates to nearly a 70% increase in SOD1 monomer concentration. Because nucleation of SOD1 aggregation is dependent on at least the square of the monomer concentration, we expect a 70% increase in monomer concentration to have a marked effect on the nucleation of SOD1 aggregates. For example, the formation of a hypothetical nucleus made up of 3 glutathionylated SOD1 monomers becomes roughly 5 times more likely and a nucleus of 6 monomers 24 times more likely relative to unmodified SOD1.
Supplementary Material
Acknowledgments
We thank Dr. Joan S. Valentine for providing the EG118 yeast strain and yEP-351:hwtSOD1 vector and Dr. Matthew P. Torres for helpful discussions. We also thank the University of North Carolina Hospitals Blood Bank for providing expired human erythrocytes.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM080742 (to N. V. D.) and 1R01AI064806-01A2 (to X. C.). This work was also supported by United States Department of Energy Grant (BER) DE-FG02-07ER64422 (to X. C.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1-S9.
Footnotes
The abbreviations used are: FALS, familial amyotrophic lateral sclerosis; SOD1, Cu/Zn-superoxide dismutase; μ-ESI-FT-ICR-MS, microcapillary electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry; ECD, electron-capture dissociation; CID, collision-induced dissociation; MALDI, matrix-assisted laser desorption/ionization; TOF, time-of-flight; LC, liquid chromatography.
References
- 1.Cleveland, D. W., and Rothstein, J. D. (2001) Nat. Rev. Neurosci. 2 806-819 [DOI] [PubMed] [Google Scholar]
- 2.Bruijn, L. I., Houseweart, M. K., Kato, S., Anderson, K. L., Anderson, S. D., Ohama, E., Reaume, A. G., Scott, R. W., and Cleveland, D. W. (1998) Science 281 1851-1854 [DOI] [PubMed] [Google Scholar]
- 3.Reaume, A. G., Elliott, J. L., Hoffman, E. K., Kowall, N. W., Ferrante, R. J., Siwek, D. F., Wilcox, H. M., Flood, D. G., Beal, M. F., Brown, R. H., Jr., Scott, R. W., and Snider, W. D. (1996) Nat. Genet. 13 43-47 [DOI] [PubMed] [Google Scholar]
- 4.Juarez, J. C., Manuia, M., Burnett, M. E., Betancourt, O., Boivin, B., Shaw, D. E., Tonks, N. K., Mazar, A. P., and Donate, F. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 7147-7152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harraz, M. M., Marden, J. J., Zhou, W., Zhang, Y., Williams, A., Sharov, V. S., Nelson, K., Luo, M., Paulson, H., Schoneich, C., and Engelhardt, J. F. (2008) J. Clin. Investig. 118 659-670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richardson, J., Thomas, K. A., Rubin, B. H., and Richardson, D. C. (1975) Proc. Natl. Acad. Sci. U. S. A. 72 1349-1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khare, S. D., Caplow, M., and Dokholyan, N. V. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 15094-15099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rakhit, R., Crow, J. P., Lepock, J. R., Kondejewski, L. H., Cashman, N. R., and Chakrabartty, A. (2004) J. Biol. Chem. 279 15499-15504 [DOI] [PubMed] [Google Scholar]
- 9.McCord, J. M., and Fridovich, I. (1969) J. Biol. Chem. 244 6049-6055 [PubMed] [Google Scholar]
- 10.Goscin, S. A., and Fridovich, I. (1972) Biochim. Biophys. Acta 289 276-283 [DOI] [PubMed] [Google Scholar]
- 11.Borchers, C. H., Thapar, R., Petrotchenko, E. V., Torres, M. P., Speir, J. P., Easterling, M., Dominski, Z., and Marzluff, W. F. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 3094-3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pflieger, D., Junger, M. A., Muller, M., Rinner, O., Lee, H., Gehrig, P. M., Gstaiger, M., and Aebersold, R. (2008) Mol. Cell. Proteomics 7 326-346 [DOI] [PubMed] [Google Scholar]
- 13.Heikkila, R. E., and Cabbat, F. (1976) Anal. Biochem. 75 356-362 [DOI] [PubMed] [Google Scholar]
- 14.Kurobe, N., Suzuki, F., Okajima, K., and Kato, K. (1990) Clin. Chim. Acta 187 11-20 [DOI] [PubMed] [Google Scholar]
- 15.Fujiwara, N., Nakano, M., Kato, S., Yoshihara, D., Ookawara, T., Eguchi, H., Taniguchi, N., and Suzuki, K. (2007) J. Biol. Chem. 282 35933-35944 [DOI] [PubMed] [Google Scholar]
- 16.Rakhit, R., Cunningham, P., Furtos-Matei, A., Dahan, S., Qi, X. F., Crow, J. P., Cashman, N. R., Kondejewski, L. H., and Chakrabartty, A. (2002) J. Biol. Chem. 277 47551-47556 [DOI] [PubMed] [Google Scholar]
- 17.Baker, J. M., Hudson, R. P., Kanelis, V., Choy, W. Y., Thibodeau, P. H., Thomas, P. J., and Forman-Kay, J. D. (2007) Nat. Struct. Mol. Biol. 14 738-745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ray, S. S., Nowak, R. J., Strokovich, K., Brown, R. H., Jr., Walz, T., and Lansbury, P. T., Jr. (2004) Biochemistry 43 4899-4905 [DOI] [PubMed] [Google Scholar]
- 19.Ray, S. S., Nowak, R. J., Brown, R. H., Jr., and Lansbury, P. T., Jr. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 3639-3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doucette, P. A., Whitson, L. J., Cao, X., Schirf, V., Demeler, B., Valentine, J. S., Hansen, J. C., and Hart, P. J. (2004) J. Biol. Chem. 279 54558-54566 [DOI] [PubMed] [Google Scholar]
- 21.Ferraroni, M., Rypniewski, W., Wilson, K. S., Viezzoli, M. S., Banci, L., Bertini, I., and Mangani, S. (1999) J. Mol. Biol. 288 413-426 [DOI] [PubMed] [Google Scholar]
- 22.Khare, S. D., and Dokholyan, N. V. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 3147-3152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khare, S. D., Ding, F., and Dokholyan, N. V. (2003) J. Mol. Biol. 334 515-525 [DOI] [PubMed] [Google Scholar]
- 24.Ding, F., and Dokholyan, N. V. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 19696-19701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakanishi, T., Kishikawa, M., Miyazaki, A., Shimizu, A., Ogawa, Y., Sakoda, S., Ohi, T., and Shoji, H. (1998) J. Neurosci. Methods 81 41-44 [DOI] [PubMed] [Google Scholar]
- 26.Marklund, S. L., Andersen, P. M., Forsgren, L., Nilsson, P., Ohlsson, P. I., Wikander, G., and Oberg, A. (1997) J. Neurochem. 69 675-681 [DOI] [PubMed] [Google Scholar]
- 27.Furukawa, Y., and O'Halloran, T. V. (2005) J. Biol. Chem. 280 17266-17274 [DOI] [PubMed] [Google Scholar]
- 28.Banci, L., Bertini, I., Durazo, A., Girotto, S., Gralla, E. B., Martinelli, M., Valentine, J. S., Vieru, M., and Whitelegge, J. P. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 11263-11267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu, J. H., Zhang, H., Wagey, R., Krieger, C., and Pelech, S. L. (2003) J. Neurochem. 85 432-442 [DOI] [PubMed] [Google Scholar]
- 30.Maher, P. (2006) Antioxid. Redox Signal. 8 1941-1970 [DOI] [PubMed] [Google Scholar]
- 31.Leier, I., Jedlitschky, G., Buchholz, U., Center, M., Cole, S. P., Deeley, R. G., and Keppler, D. (1996) Biochem. J. 314 433-437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schafer, F. Q., and Buettner, G. R. (2001) Free Radic. Biol. Med. 30 1191-1212 [DOI] [PubMed] [Google Scholar]
- 33.Ghezzi, P. (2005) Free Radic. Res 39 573-580 [DOI] [PubMed] [Google Scholar]
- 34.Adachi, T., Pimentel, D. R., Heibeck, T., Hou, X., Lee, Y. J., Jiang, B., Ido, Y., and Cohen, R. A. (2004) J. Biol. Chem. 279 29857-29862 [DOI] [PubMed] [Google Scholar]
- 35.Cross, J. V., and Templeton, D. J. (2004) Biochem. J. 381 675-683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klatt, P., Molina, E. P., De Lacoba, M. G., Padilla, C. A., Martinez-Galesteo, E., Barcena, J. A., and Lamas, S. (1999) FASEB J. 13 1481-1490 [DOI] [PubMed] [Google Scholar]
- 37.Wang, J., Boja, E. S., Tan, W., Tekle, E., Fales, H. M., English, S., Mieyal, J. J., and Chock, P. B. (2001) J. Biol. Chem. 276 47763-47766 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.