

Abstract
Efficient clearance of apoptotic cells (AC) by professional phagocytes is crucial for tissue homeostasis and resolution of inflammation. Macrophages respond to AC with an increase in anti-inflammatory cytokine production, but a diminished release of pro-inflammatory mediators. Mechanisms to explain attenuated pro-inflammatory cytokine formation remain elusive. We provide evidence that peroxisome proliferator-activated receptor γ (PPARγ) coordinates anti-inflammatory responses following its activation by AC. Exposing murine RAW264.7 macrophages to AC prior to LPS-stimulation, reduced NFκB transactivation and lowered target gene expression of e.g. TNFα and IL-6 compared to controls. In macrophages overexpressing a dominant negative (d/n) mutant of PPARγ, NFκB transactivation in response to LPS was restored, while macrophages from myeloid lineage-specific conditional PPARγ knockout mice proved that PPARγ transmitted an anti-inflammatory response, delivered by AC. Expressing a PPARγ-Δaa32-250 deletion mutant, we observed no inhibition of NFκB. Analyzing the PPARγ domain structures within aa32-250, we anticipated PPARγ sumoylation in mediating the anti-inflammatory effect in response to AC. Interfering with sumoylation of PPARγ by mutating the predicted sumoylation site (K77R), or knockdown of the SUMO E3 ligase PIAS1, eliminated the ability of AC to suppress NFκB. ChIP analysis demonstrated that AC prevented the LPS-induced removal of nuclear receptor co-repressor (NCoR) from the κB site within the TNFα promoter. We conclude that AC induce PPARγ sumoylation to attenuate the removal of NCoR, thereby blocking transactivation of NFκB. This contributes to an anti-inflammatory phenotype shift in macrophages responding to AC, by lowering pro-inflammatory cytokine production.
Keywords: monocytes/macrophages, inflammation, phagocytosis, molecular biology
Introduction
Recognition of apoptotic cells (AC3) elicits immunological consequences that received considerable attention over the last years. Professional phagocytes such as dendritic cells and macrophages recognize AC via so called ‘eat me’-signals, with concomitant phagocytosis (1). The uptake of AC avoids secondary necrosis and thus, the release of harmful cell contents. Moreover, ingestion of apoptotic material actively provokes a macrophage phenotype shift, which helps to terminate perpetuating inflammatory responses. The altered macrophage phenotype is characterized by the release of anti-inflammatory mediators such as transforming growth factor β or prostaglandin E2 (2). In addition, these polarized macrophages suppress the production of reactive oxygen species (3), nitric oxide (4) and pro-inflammatory cytokines such as TNFα, IL-1β and IL-6 (1). AC block NFκB activation, which contributes to the diminished production of pro-inflammatory cytokines, although mechanisms how NFκB is inhibited remain unclear (5). Cvetanovic et al. demonstrated that an attenuated NFκB transactivation response and an AC-elicited reduction in target gene expression is cell-cell-contact dependent, but phosphatidylserine-independent (6). Furthermore, it was noticed that NFκB binding to DNA as well as IκB degradation were not affected by AC. As an alternative explanation it was proposed that a limited amount of p300, an established co-factor of NFκB-dependent pro-inflammatory gene expression (7), decreases its activity, although underlying mechanisms remain obscure (5). A potential candidate known to interact with p300 and thereby attenuating an inflammatory response is peroxisome proliferator-activated receptor γ (PPARγ) (8).
PPARγ belongs to the nuclear hormone receptor superfamily of ligand-activated transcription factors and originally has been characterized to be important for adipogenesis and glucose metabolism (9). Induction of PPARγ target genes requires ligand-binding, heterodimerization with the retinoid X receptor (RXR) and subsequent binding to specific peroxisome proliferator response elements. Besides transcriptional activation, PPARγ also suppresses gene induction. In macrophages active PPARγ attenuates the production of various inflammatory mediators such as nitric oxide, TNFα, IL-1β, IL-12 and MMP-9 (10). Several mechanisms are proposed to explain the suppressive role of PPARγ. It is assumed that PPARγ competes for limiting amounts of pro-inflammatory transcriptional co-activators, directly binds transcription factors, interferes with the MAPK cascade (11) and/or prevents the removal of co-repressors from promoter regions of pro-inflammatory target genes (12). Co-activator/co-repressor exchange is a common mechanism controlling the switch from gene repression to gene activation and vice versa. This mechanism is regulated by the removal of co-repressors, their degradation by the ubiquitination/19S proteasome machinery or recruitment of co-activators. Sumoylated PPARγ was shown to prevent NCoR removal, thereby attenuating LPS-induced gene expression. Sumoylation is mediated by the E2 ligase Ubc9 and the SUMO E3 ligase protein inhibitor of activated STAT1 (PIAS1) (12).
Given the interest in macrophage polarization in response to AC, we were intrigued to define a potential link between activation of PPARγ and inhibition of NFκB transactivation. We provide evidence that AC attenuate transactivation of NFκB and associated target gene activation. In macrophages overexpressing a dominant/negative (d/n) mutant of PPARγ inhibition of NFκB no longer occurred, with the further notion that sumoylation of PPARγ at K77 prevents the LPS-induced removal of the NCoR-HDAC3 complex from the NFκB site of pro-inflammatory target gene promoters, i.e. TNFα.
Materials and Methods
Materials
Staurosporine and LPS (from Escherichia coli, serotype 0127:B8) were purchased from Sigma-Aldrich (Deisenhofen, Germany). GW9662 and trichostatin A were bought from Alexis Biochemicals (Lausen, Switzerland). Murine macrophage colony-stimulating factor (M-CSF) was purchased from Peprotech EC (London, UK). Oligonucleotides were obtained from Biomers (Ulm, Germany). Cell culture supplements, FCS and medium came from PAA (Cölbe, Germany).
Cell culture and generation of apoptotic Jurkat cells
RAW264.7 mouse macrophages, RAW264.7 d/n PPARγ macrophages and Jurkat cells were cultured at 37°C, 5% CO2 in RPMI 1640 supplemented with 10% FCS, 100 μg/ml streptomycin and 100 U/ml penicillin. To generate apoptotic Jurkat cells, they were cultured in FCS free medium and stimulated with 0.5 μg/ml staurosporine for 3 h provoking roughly 80% apoptotic cell death as previously described (13). Afterwards cells were washed twice with medium to remove staurosporine. For co-culture, AC were resuspended in medium containing FCS and added to macrophages at a ratio of 5:1. Following experiments, but prior to sample preparation non-ingested AC were removed and macrophages washed twice with PBS excluding variations on results by AC as described earlier (3).
Conditional PPARγ knockout mice, isolation of CD11b+ splenocytes, differentiation and culture of primary murine macrophages
C57BL/6 LysMCre mice (LysMCre+/+) were bred with C57BL/6 PPARγ floxed/floxed (PPARγfl/fl) mice (14) as previously described (15). Genotypes were determined by PCR of tail DNA and deletion of PPARγ was confirmed by analysis of PPARγ-exon 2 mRNA levels in macrophages (14, 16, 17).
Spleens from PPARγfl/fl and LysMCre+/+/PPARγfl/fl mice were removed to prepare single cell suspensions. CD11b positive splenocytes were isolated by MACS seperation using an autoMACS system (Miltenyi Biotec, Bergisch-Gladbach, Germany) following the distributor's instructions. For differentiation of CD11b positive splenocytes, cells were cultured in RPMI supplemented with 10% FCS, 100 μg/ml streptomycin, 100 U/ml penicillin, non-essential amino acids, sodium pyruvate and 25 ng/ml M-CSF for 5 d. Animal experiments followed the guidelines of the Hessian animal care and use committee.
Western blot analysis
3*106 RAW264.7 macrophages were cultured in 10 cm dishes one day prior to experiments. The following day cells were treated with AC for 90 min, followed by stimulation with 1 μg/ml LPS for 1 h. Cell lysis and Western blot analysis was performed as described (18). An anti-PPARγ antibody (1:2000, H100-X from Santa Cruz Biotechnology, Inc., Heidelberg, Germany) and anti-β-tubulin (1:1000, Sigma-Aldrich, Deisenhofen, Germany) were used. Detection and densitometric analysis were performed using the Odyssey infrared imaging system (Li-COR biosciences GmbH, Bad Homburg, Germany).
Vector construction, transient transfection and reporter assay
To restore PPARγ signaling, a PPARγ1 overexpression vector (pcDNA3-PPARγ1 wild-type kindly provided by V.K.K. Chatterjee, University of Cambridge, Cambridge, UK) was used. Overexpression vector pDsRed-Monomer-C1-PPARγ1 wild-type and deletion constructs of DsRed-Monomer-C1-PPARγ1 were previously generated in our lab (13). pDsRed-Monomer-C1-PPARγ1 wild-type was also used for mutation of the sumoylation site (K77R). Mutation was performed with the QuikChange XLII site directed mutagenesis kit (Stratagene, Amsterdam, Netherlands) using the following primer (changed nucleotides are underlined): 5′-GAGTACCAAA GTGCAATCAGAGTGGAGCCTGC-3′.
For reporter analysis 5*104/well RAW264.7 and RAW264.7 d/n PPARγ macrophages were seeded in 24-well plates. The next day, cells were transfected with 1 μg/well pNFκB-Luc or co-transfected with 0.5 μg pNFκB-Luc and 0.5 μg of one of the overexpressing vectors/well respectively, using JetPEI™ transfection reagent (Polyplus transfection, Illkirch, France) as described by the manufacturer. The reporter plasmid contains three κB-sites and was described earlier (19). After transfection, cells were cultured in fresh medium for another 24 h before treatments. All reporter assays were performed in duplicate. Cell extracts were prepared after co-incubation with AC for 90 min and following LPS (1 μg/ml) stimulation for 5 h. Luciferase activity was normalized to protein concentration of each sample. To control transfection efficiency concerning overexpression of the different pDsRed-PPARγ1 constructs, mRNA levels of DsRed were determined by quantitative PCR.
For siRNA experiments, 2*106 RAW264.7 macrophages were transfected with 3 μg siRNA (ON-TARGETplus SMARTpool siRNA against NCoR or PIAS1 from ThermoScientific, Lafayette, CO, USA) using the Nucleofector technology from Amaxa biosystems (Cologne, Germany) according to the manufacturer's protocol. For controls, cells were transfected with Allstars negative control siRNA (Qiagen GmbH, Hilden, Germany). Knockdown was analyzed on mRNA level by quantitative PCR.
Quantitative PCR
Cells were pre-treated with AC for 90 min, followed by stimulation with 1 μg/ml LPS for 3 h. Total RNA was isolated using PeqGold RNAPure Kit (PeqLab Biotechnologie GmbH, Erlangen, Germany) as described by the manufacturer. Reverse Transcription was done with 1 μg of RNA using iScript™ cDNA Synthesis Kit (Bio-Rad GmbH, Munich, Germany). Quantitative PCR was performed with Absolute QPCR SYBRGreen Fluorescein Mix (Abgene, Hamburg, Germany) according to the manufacturer's protocol. Amplification and data analysis were performed using MyiQ iCylcer system from Bio-Rad. The following primer pairs were selected for quantitative PCR: TNFα forward: 5′-CCATTCCTGAGTTCT GCAAAGG-3′, TNFα reverse: 5′-AAGTAGGAAGGCCTGAGATCTTATC-3′; IL-6 forward: 5′-GAACAACGATGATGCACTTGC-3′, IL-6 reverse: 5′-TCTCTGAAGGACTCT GGCTTTG-3′, PPARγ-exon 2 forward: 5′-CACAGAGATGCCATTCTGGC-3′, PPARγ-exon 2 reverse: 5′-GGCCTGTTGTAGAGCTGGGT-3′, DsRed forward: 5′-GAGGTGCAGCAG GACTCCTC-3′, DsRed reverse: 5′-TGGCCTTGTACACGTCTTG-3′, GAPDH forward: 5′-CTCATGACCA CAGTCCATGC-3′, GAPDH reverse: 5′-TTCAGCTCTGGGATGACCTT-3′. For determination of actin, NCoR1 and PIAS1 mRNA levels we used QuantiTect® Primer Assays (Qiagen GmbH, Hilden, Germany). Values were normalized to GAPDH or actin expression.
Chromatin immunoprecipitation (ChIP) assay
3*106 RAW264.7 cells were seeded in 10 cm plates and cultured over night. Before crosslinking, cells were pretreated with AC (90 min) followed by 1 μg/ml LPS (1 h) afterwards. ChIP assays were performed as described by Nelson et al. (20). For each sample we crosslinked cells combined from three 10 cm plates. NCoR was precipitated using 1 μg of anti-NCoR from Affinity Bioreagents (Golden, CO, USA), an established ChIP assay antibody (12). A 211 bp fragment of the TNFα promoter spanning an established κB response element (21), was amplified. For Mock-IP we used 1 μg of normal rabbit IgG from Millipore/Upstate (Billerica, MA, USA). 15% DNA of each probe was used for input controls. The following primers were used: ChIP-TNFα forward: 5′-GGCTTGTGAGGTCCGTGAAT-3′, ChIP-TNFα reverse: 5′-GAAAGCTGGGTGCATAAGGG-3′.
Statistical analysis
Each experiment was performed at least three times and statistical analysis was done with One- or Two-way-ANOVA modified with Bonferroni's multiple comparison test, respectively. In the case of ChIP assay and Western blot analysis representative data of at least three independently performed experiments are shown. *p < 0.05, **p < 0.01, ***p < 0.001
Results
PPARγ attenuates NFκB transactivation and target gene expression in response to AC
To investigate whether AC activate PPARγ thereby blocking NFκB transactivation, we performed NFκB reporter assays in RAW264.7, compared to RAW264.7 macrophages expressing a d/n PPARγ mutant (3). The mutant is characterized by two amino acid substitutions (L468A/E471A), which impair ligand-dependent PPARγ transactivation and the interaction with co-activators such as p300 (22). Macrophages were co-incubated with AC at a ratio of 1:5 for 90 min and then stimulated with 1 μg/ml LPS for 5 h. LPS stimulation caused an approximately 3-fold induction of reporter activity compared to controls, i.e. resting cells (data not shown). Following the interaction with AC, NFκB-dependent transactivation in RAW264.7 macrophages was reduced by roughly 50% compared to LPS stimulation (Fig. 1A). Inhibition was completely reversed in RAW264.7 d/n PPARγ expressing cells, suggesting a causative role of PPARγ in reducing NFκB activation. To prove the importance of PPARγ, we overexpressed PPARγ1 wild-type in RAW264.7 d/n PPARγ macrophages to restore its functionality. This was achieved by transfecting RAW264.7 d/n PPARγ macrophages with the NFκB reporter plasmid in combination with a PPARγ1 wild-type encoding vector. As expected, overexpression of PPARγ1 wild-type in d/n PPARγ macrophages restored the inhibitory potency of AC on NFκB transactivation, with the notion that inhibition was comparable to control cells (Fig. 1A). To further strengthen the role of PPARγ, RAW264.7 cells were prestimulated for 3 h with 1 μM GW9662, a specific PPARγ antagonist (23). Thereafter, macrophages were exposed to AC followed by LPS stimulation as described above. GW9662 completely abrogated the ability of AC to block NFκB reporter activity (Fig. 1B), but did not alter the LPS response (data not shown). These data suggest a role of PPARγ in blocking NFκB transactivation in response to AC. To exclude variations in PPARγ expression accounting for alterations seen under our conditions, we checked PPARγ expression by Western blot analysis. Neither LPS nor AC changed the expression of PPARγ in RAW264.7 macrophages (Fig. 1C).
Figure 1. Attenuated NFκB reporter activity is restored in RAW264.7 d/n PPARγ macrophages.

Cells were incubated for 90 min with AC (ratio 1:5), followed by the treatment with LPS (1 μg/ml, 5 h) in (A) RAW264.7, RAW264.7 d/n PPARγ macrophages and RAW264.7 d/n PPARγ cells overexpressing PPARγ1 wild-type and in (B) RAW264.7 macrophages pretreated with 1 μM GW9662 for 3 h. NFκB reporter activity in control macrophages, stimulated with LPS alone was set to 1. Statistics in (A) were analyzed with two-way-ANOVA modified with Bonferroni's multiple comparison test. In (B) statistical analysis was done with one-way-ANOVA modified with Bonferroni's multiple comparison test. Each column represents the mean value of duplicate determinations of a minimum of four independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001. (C) PPARγ expression in RAW264.7 was determined by Western analysis. Macrophages were co-incubated with AC for 90 min and treated with 1 μg/ml LPS for 1 h afterwards. Data are representative of three independent experiments. Statistics were analyzed with one-way-ANOVA modified with Bonferroni's multiple comparison test.
To elucidate a functional consequence of NFκB inhibition, we analyzed the expression of proinflammatory, established NFκB target genes such as TNFα (21) and IL-6 (24) in macrophages treated with AC and LPS. To this end we determined IL-6 and TNFα mRNA amount by quantitative PCR in RAW264.7 and RAW264.7 d/n PPARγ macrophages. Cells were co-incubated with AC for 90 min and treated with 1 μg/ml LPS for 3 h afterwards. In response to LPS, mRNA expression of TNFα and IL-6 was at least 50-fold increased compared to unstimulated cells and this response was set to a relative mRNA increase of 1 (Fig. 2A and 2B). Recognition of AC by RAW264.7 macrophages prior to stimulation with LPS reduced TNFα expression by roughly 60% (Fig. 2A), while IL-6 mRNA expression was diminished by 90% (Fig. 2B). To substantiate a role of PPARγ, experiments were also performed in RAW264.7 d/n PPARγ macrophages. Pretreating cells with AC, followed by LPS stimulation restored TNFα expression (Fig. 2A) and largely reversed suppressed formation of IL-6 (Fig. 2B) thus, underscoring the impact of PPARγ on NFκB target gene expression. To verify the physiological significance of PPARγ for the anti-inflammatory response, we analyzed TNFα mRNA levels in primary murine macrophages from PPARγfl/fl (Control) and myeloid lineage-specific conditional PPARγ knockout mice (Mac-PPARγ KO). CD11b positive splenocytes were differentiated with 25 ng M-CSF for 5 d. Knockout of PPARγ was proven by quantifiying PPARγ-exon 2 mRNA amount, which was reduced in PPARγ deficient macrophages by 90% (Fig. 2C). Following their differentiation, macrophages were treated with AC and LPS as described. Recognition of AC diminished LPS-induced TNFα expression by roughly 80%. This reduction was significantly mitigated in PPARγ knockout macrophages (Fig. 2D), supporting the notion of a PPARγ-dependent anti-inflammatory phenotype switch in response to AC.
Figure 2. PPARγ accounts for attenuated TNFα mRNA expression in response to AC.
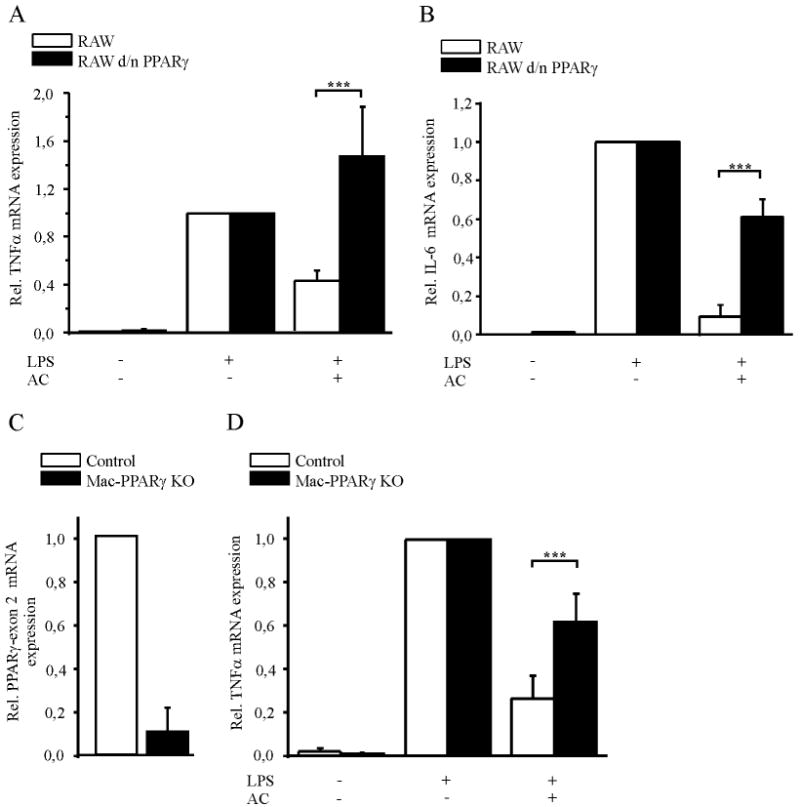
(A) TNFα and (B) IL-6 mRNA expression were measured by quantitative PCR in RAW264.7 and RAW264.7 d/n PPARγ cells. Macrophages were co-incubated with AC for 90 min (ratio 1:5) and afterwards treated with 1 μg/ml LPS for 3 h. As a control, macrophages were stimulated with LPS alone and relative mRNA expression was set to 1. Data were normalized to GAPDH mRNA levels. (C) PPARγ-exon 2 mRNA expression and (D) TNFα mRNA expression in primary murine macrophages from PPARγfl/fl (Control) and conditional PPARγ knockout mice (Mac-PPARγ KO) were measured by quantitative PCR. Cells were stimulated as described in (A) and (B). Statistics were analyzed with two-way-ANOVA modified with Bonferroni's multiple comparison test. n ≥ 5, ***p < 0.001
Identification of PPARγ domains required for NFκB inhibition
With the following experiments we explored mechanisms of PPARγ-mediated transrepression of NFκB activity. This was accomplished by NFκB reporter assays in RAW264.7 d/n PPARγ macrophages overexpressing various deletion constructs of DsRed-tagged PPARγ1, that were previously verified for their expression by Western blot analysis (18). The DsRed-PPARγ1 wild-type encoding vector was included as a control. As expected, overexpression of DsRed-PPARγ1 wild-type restored NFκB inhibition in response to AC in comparison to RAW264.7 d/n PPARγ macrophages (Fig. 3A). Overexpression of DsRed-PPARγ1-Δaa32-250 failed to restore the ability of AC to inhibit NFκB transactivation, comparable to the situation seen in RAW264.7 d/n PPARγ cells (Fig. 3A). Amino acids 32-250 of PPARγ span a region of the ligand-independent activation domain AF1, the DNA binding domain and a part of the hinge domain. From the results we concluded that besides the AF2 domain, responsible for ligand-binding, amino acids within the region 32-250 are needed to inhibit NFκB. Next we showed that overexpression of DsRed-PPARγ1-Δaa309-319, a PPARγ deletion construct lacking a region that appears important for co-factor binding (25, 26), restored NFκB inhibition (Fig. 3A). These data suggest that amino acids 309-319, within the ligand-binding domain, are dispensable for blocking NFκB transactivation in response to AC, whereas amino acids 32-250 seem to play a role. Considering that sumoylation of PPARγ and concomitant prevention of NCoR removal is a postulated mechanism for transrepression, we analyzed protein motives and noticed a possible sumoylation site at K77 within the AF1 domain. Therefore, we reasoned that sumoylation of PPARγ may contribute to block NFκB transactivation.
Figure 3. Domain analysis and sumoylation of PPARγ.
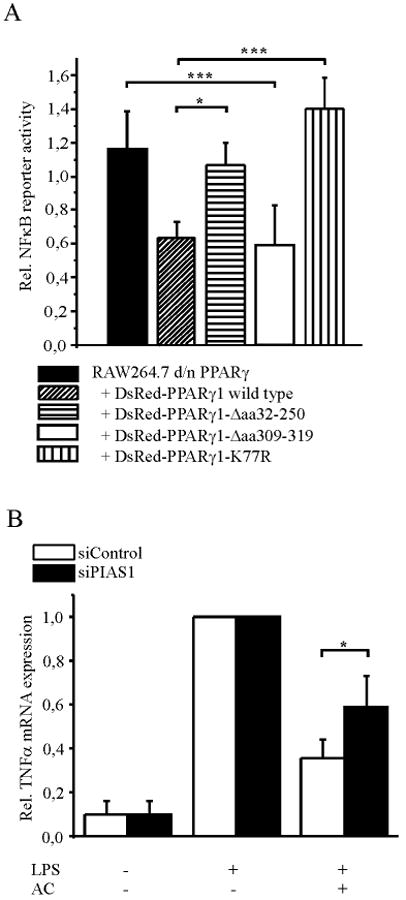
(A) NFκB reporter activity was measured in RAW264.7 d/n PPARγ cells overexpressing deletion constructs of pDsRed-PPARγ1 or pDsRed-PPARγ1-K77R as indicated. Cells were co-transfected with pNFκB-Luc. Reporter activity was measured after co-incubation with AC for 90 min followed by stimulation with 1 μg/ml LPS for 5 h. As a control, cells were stimulated with LPS alone and values set to 1 (data not shown). (B) The impact of PIAS1 on TNFα expression was analyzed by siRNA knockdown of PIAS1. Two days after transfection of RAW macrophages with PIAS1 siRNA or siControl, cells were co-incubated with AC for 90 min, followed by LPS stimulation (1 μg/ml LPS, 3 h). TNFα mRNA levels were measured by quantitative PCR, the LPS response was set to one. Statstics were analyzed with two-way-ANOVA modified with Bonferroni's multiple comparison test. *p < 0.05, ***p < 0.001
Sumoylation of PPARγ prevents the removal of NCoR from NFκB binding sites
Interfering with the removal of NCoR from promoter regions of different pro-inflammatory genes such as inducible NO synthase (iNOS) has been described as a mechanism for PPARγ-mediated transrepression that occurs after sumoylation of PPARγ (12). Among other proteins, HDAC3 is associated to the co-repressor complex mediating transcriptional repression (27). In a first approach we inhibited HDAC3 to see whether the NCoR/HDAC3 complex might be involved in blocking NFκB activity. RAW264.7 macrophages were pretreated with 10 nM of the histone deacetylase inhibitor trichostatin A (TSA) 1 h prior to AC addition, followed by LPS stimulation and subsequent determination of NFκB reporter activity. In the presence of TSA, NFκB was not any longer inhibited by AC, whereas TSA alone did not alter the LPS response (Supplemental Fig. S1). Although this experiment may suggest HDAC-mediated NFκB inhibition, it remains unclear whether PPARγ is sumoylated and concomitantly retains NCoR bound at the promoter. Taking into consideration that amino acids 32-250 are involved (Fig. 3A), we reasoned K77 to be sumoylated. To approach this possibility, we mutated the sumoylation site (K77R) in the pDsRed-PPARγ1 wild-type encoding vector. Experimentally, we performed NFκB reporter assays in the RAW264.7 d/n PPARγ macrophages and overexpressed DsRed-PPARγ1-K77R and DsRed-PPARγ1 wild-type. Along with our expectations, the K77R-mutated protein was unable to restore NFκB inhibition compared to the DsRed-PPARγ1 wild-type protein (Fig. 3A). As PIAS1 mediates PPARγ sumoylation (12), we knocked down PIAS1 by siRNA to abrogate PPARγ sumoylation and analyzed its relevance in attenuating TNFα expression in response to AC. Two days after transfection of siRNA, RAW264.7 cells were exposed to AC and LPS as described above and TNFα as well as PIAS1 mRNA levels were determined by quantitative PCR. AC reduced LPS-induced TNFα formation in RAW264.7 macrophages transfected with control siRNA. In comparison PIAS1 knockdown by approximately 50% at the mRNA level did not affect LPS-induced TNFα expression, but significantly reduced the ability of AC to attenuate TNFα mRNA expression (Fig. 3B). Therefore, lowering PPARγ sumoylation attenuates an AC-provoked anti-inflammatory phenotype shift. To verify that sumoylated PPARγ prevents NCoR removal to inhibit NFκB, we analyzed the relevance of NCoR for AC to block NFκB by siRNA knockdown of NCoR. As a result of transfection the extent of TNFα induction by LPS was reduced, while NCoR expression remained unchanged by transfection with control siRNA compared to non-transfected cells. In control siRNA transfected cells recognition of AC reduced LPS-induced TNFα expression by approximately 50%, whereas knockdown of NCoR by roughly 50%, as determined by quantitative PCR (data not shown), significantly reverted TNFα expression (Fig. 4A). To verify that sumoylated PPARγ affects the occupancy of NFκB sites by NCoR, we examined the association of NCoR within the TNFα promoter by ChIP analysis. RAW264.7 and RAW264.7 d/n PPARγ macrophages were pretreated with AC for 90 min and stimulated with 1 μg/ml LPS for 1 h afterwards or remained as controls. Under control conditions NCoR associated with the NFκB site of the TNFα promoter and it was cleared from that promoter region in response to LPS in the wild-type as well as d/n PPARγ overexpressing macrophages (Fig. 4B and C). In RAW264.7 macrophages, NCoR remained bound to the promoter after recognition of AC despite LPS-stimulation, whereas in RAW264.7 d/n PPARγ macrophages NCoR was cleared from the promoter in response to AC, followed by LPS stimulation. These effects were statistically significant (Fig. 4D).
Figure 4. PPARγ antagonizes the removal of NCoR.
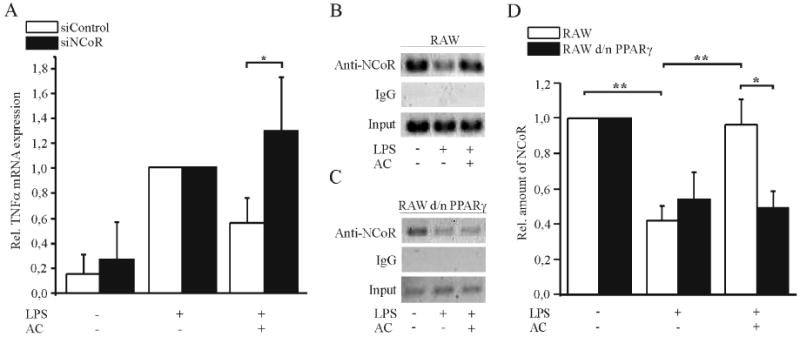
(A) The role of NCoR for TNFα expression was analyzed by siRNA knockdown of NCoR1. Two days after transfection of RAW264.7 macrophages with NCoR siRNA or siControl, cells were co-incubated with AC for 90 min, followed by stimulation with 1 μg/ml LPS for 3 h. TNFα mRNA levels were measured by quantitative PCR, the LPS response was set to one. (B. C) Association of NCoR with the κB response element within the TNFα promoter was analyzed by ChIP analysis. (B) RAW264.7 macrophages and (C) RAW264.7 d/n PPARγ macrophages were incubated with AC for 90 min and 1 μg/ml LPS for 1 h afterwards. Data are representative for three independent experiments. (D) Statistical evaluation of ChIP assays showing the relative amount of NCoR vs. input controls. Statistics were analyzed with two-way-ANOVA modified with Bonferroni's multiple comparison test. *p > 0.05, **p < 0.01, ***p < 0.001
Our data support the assumption that PPARγ sumoylation in response to AC regulates the activity of NFκB via NCoR, which contributes to immune modulation of macrophages.
Discussion
PPARγ was recently implicated in macrophage polarization provoked by IL-4. The reduced production of pro-inflammatory cytokines such as IL-6 and the enhanced expression of the mannose receptor as a differentiation marker was attributed to PPARγ-mediated suppression or gene induction (28). Besides IL-4, AC also induce a switch of macrophages towards an alternatively activated phenotype, although underlying mechanisms are insufficiently clear. We provide evidence that PPARγ gets activated, most likely sumoylated in response to AC, which is essential for blocking NFκB transactivation. Our basic observation that AC attenuate LPS-induced NFκB transactivation is in line with the work of Cvetanovic et al., demonstrating a diminished NFκB activity in response to AC (5). The notion that PPARγ not only inhibits NFκB (29-31) but also is activated by AC (3), stimulated our interest in identifying underlying molecular mechanisms. We followed a molecular and pharmacological approach to establish the contributing role of PPARγ by using cells that express a dominant negative (d/n) mutant of PPARγ. This mutant carries two amino acid substitutions in the AF2 domain (L468A/E471A) of the protein, which impair ligand-dependent PPARγ transactivation and the interaction with co-activators, e.g. p300 (22). In RAW264.7 d/n PPARγ macrophages inhibition of NFκB in response to AC was completely relieved. As a prove of concept, functionality in RAW264.7 d/n PPARγ macrophages was restored by overexpressing PPARγ1 wild-type, which again suppressed NFκB reporter activity in response to AC. Pharmacologically, the impact of PPARγ was further corroborated by using GW9662 to antagonize PPARγ, which restored NFκB reporter activity after adding AC to macrophages. During these studies AC were co-cultured with macrophages for 6.5 h, which comprises a 90 min lasting preincubation period with AC, followed by LPS stimulation for 5 h. During the entire incubation period AC remained in the medium, without removing non-ingested cells. However, removing non-phagocytosed cells after 30 min followed by LPS stimulation failed to block NFκB activity (data not shown). Variations in the stimulation regimes of macrophages with AC may affect macrophage plasticity. Majai et al. observed that a treatment of cells with LPS for 30 min, incubations with AC for 25 min, followed by removing non-ingested cells, lowered the amount of TNFα when measured 18-24 h later (32). This response was not antagonized by GW9662. Likely, pro-inflammatory stimuli given to macrophages prior to confronting them with AC might activate distinct pathways, e.g. receptor desensitization, that contribute to the diversity of anti-inflammatory responses, with the further possibility that short vs. long incubation periods differ towards the involvement of PPARγ.
To verify the inhibitory role of PPARγ in our system, we not only followed NFκB reporter activity, but also searched for the expression of NFκB downstream target genes i.e. TNFα and IL-6. Their expression was reduced in response to AC in RAW264.7 and primary murine macrophages, but cytokine formation was partially in the case of IL-6 and fully reversed in the case of TNFα, when exposing d/n PPARγ cells to AC. Supporting evidence came from experiments in PPARγ knockout macrophages, where the inhibitory effect of AC on NFκB activity was restored. These data support conclusions by Odegaard et al. using macrophages from PPARγ knockout mice, showing that activation of PPARγ by IL-4 attenuated IL-6 expression (28). In addition, the role of PPARγ for macrophage polarization is further corroborated by Bouhlel et al., showing that IL-4 switches the macrophage phenotype towards an anti-inflammatory one by activating PPARγ (33). Moreover, IL-13 activates PPARγ to generate an anti-inflammatory macrophage phenotype (28, 34).
Despite mounting evidence for a role of PPARγ in macrophage polarization, molecular mechanisms explaining repression of NFκB, one crucial transcription factor regulating the inflammatory repertoire of macrophages, by AC are ill defined. Proposed strategies how PPARγ represses NFκB, comprises competition with co-activators or inhibition of co-repressor clearance (10). To approach mechanisms, we analyzed domains of PPARγ being involved. DsRed-PPARγ1 wild-type as well as DsRed-PPARγ1-Δaa309-319 attenuated NFκB activity in response to AC. Considering that amino acids 309-319 are required for binding transcriptional co-activators (25, 26) ruled a simple co-activator scavenging of e.g. p300, to explain inhibition of NFκB, out. Interestingly, overexpression of DsRed-PPARγ1-Δaa32-250 restored NFκB transactivation, compared to the action of DsRed-PPARγ1 wild-type. Deleted amino acids in PPARγ1-Δaa32-250 span a part of the AF1 domain, the DNA binding domain, a part of the hinge domain and thus, contain a predicted sumoylation site at K77. Sumoylation of PPARγ attenuates NFκB target gene expression by preventing NCoR removal from NFκB binding sites in various promoter regions of target genes such as iNOS (12). NCoR is a component of a co-repressor complex, containing transducin beta-like protein-1 (TBL1), TBLR1 and HDAC3, the latter one mediating transcriptional repression (27). A potential role for the NCoR-associated HDAC3 was supposed when the HDAC inhibitor TSA reversed PPARγ-dependent repression of iNOS (12). In analogy, TSA reversed inhibition of NFκB by AC, suggesting that a similar mechanism might operate in response to AC. ChIP analysis confirmed that NCoR is cleared from the NFκB site within the TNFα promoter after LPS stimulation, but remained bound when macrophages were prestimulated with AC. Pascual et al. noticed that sumoylated PPARγ suppressed the NFκB target gene iNOS (12). The model predicts that NCoR/HDAC3 associates with κB binding sites along with TBL1 and TBLR1, which are required for ubiquitination of NCoR in response to pro-inflammatory stimuli (35). Following sumoylation, PPARγ binds to NCoR/HDAC3 and prevents the recruitment of the ubiquitination/19S proteasome machinery that normally degrades the co-repressor complex. This scenario requires ligand-dependent PPARγ activation and K365 sumoylation (12). PPARγ contains two possible sumoylation sites at K77 and K365 and our experiments with the PPARγ aa32-250 deletion fragment pointed to the involvement of K77 rather than K365. Indeed, overexpression of DsRed-PPARγ1-K77R in RAW264.7 d/n PPARγ cells failed to restore NFκB respression, indicating that sumoylation of PPARγ at K77 represses NFκB transactivation. Our studies do not rule out the possibility that PPARγ is also sumoylated at K365, but at least this would not to be sufficient for NFκB inhibition under our experimental conditions. Furthermore, knockdown of the SUMO E3 ligase PIAS1, which mediates PPARγ sumoylation (12), reversed the inhibitory ability of AC. In the future, it will require new imaginative experiments to understand how AC sumoylate PPARγ.
It is becoming more and more evident that PPARγ essentially contributes to a macrophage phenotype shift. Our data suggest that this signaling circuit operates under conditions when AC re-program immune functions of macrophages, exemplified by an altered NFκB-mediated target gene expression profile. We propose that sumoylated PPARγ attenuates NFκB transactivation in response to AC by preventing NCoR co-repressor displacement. This helps to understand how AC affect the remarkable plasticity of macrophages associated with decreased pro-inflammatory cytokine production.
Supplementary Material
Footnotes
This work was supported by grants from Deutsche Forschungsgemeinschaft (Br 999, FOG 784, Excellence Cluster Cardiopulmonary System), Deutsche Krebshilfe, Sander Foundation, LiFF and European Community (PROLIGEN).
Abbreviations used in this paper: AC: apoptotic cells, PPARγ: peroxisome proliferator-activated receptor γ, d/n: dominant negative, NCoR: nuclear receptor co-repressor, HDAC: histone deacetylase, TBL1: transducin beta-like protein-1, PIAS1: protein inhibitor of activated STAT1; TSA: trichostatin A, iNOS: inducible nitric oxide synthase
References
- 1.Krysko DV, D'Herde K, Vandenabeele P. Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis. 2006;11:1709–1726. doi: 10.1007/s10495-006-9527-8. [DOI] [PubMed] [Google Scholar]
- 2.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johann AM, von Knethen A, Lindemann D, Brune B. Recognition of apoptotic cells by macrophages activates the peroxisome proliferator-activated receptor-gamma and attenuates the oxidative burst. Cell Death Differ. 2006;13:1533–1540. doi: 10.1038/sj.cdd.4401832. [DOI] [PubMed] [Google Scholar]
- 4.Johann AM, Barra V, Kuhn AM, Weigert A, von Knethen A, Brune B. Apoptotic cells induce arginase II in macrophages, thereby attenuating NO production. Faseb J. 2007;21:2074–2012. doi: 10.1096/fj.06-7815com. [DOI] [PubMed] [Google Scholar]
- 5.Cvetanovic M, Ucker DS. Innate immune discrimination of apoptotic cells: repression of proinflammatory macrophage transcription is coupled directly to specific recognition. J Immunol. 2004;172:880–889. doi: 10.4049/jimmunol.172.2.880. [DOI] [PubMed] [Google Scholar]
- 6.Cvetanovic M, Mitchell JE, Patel V, Avner BS, Su Y, van der Saag PT, Witte PL, Fiore S, Levine JS, Ucker DS. Specific recognition of apoptotic cells reveals a ubiquitous and unconventional innate immunity. J Biol Chem. 2006;281:20055–20067. doi: 10.1074/jbc.M603920200. [DOI] [PubMed] [Google Scholar]
- 7.Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci U S A. 1997;94:2927–2932. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li M, Pascual G, Glass CK. Peroxisome proliferator-activated receptor gamma-dependent repression of the inducible nitric oxide synthase gene. Mol Cell Biol. 2000;20:4699–4707. doi: 10.1128/mcb.20.13.4699-4707.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes Dev. 2000;14:1293–1307. [PubMed] [Google Scholar]
- 10.Ricote M, Glass CK. PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta. 2007;1771:926–935. doi: 10.1016/j.bbalip.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Knethen A, Brune B. PPARgamma--an important regulator of monocyte/macrophage function. Arch Immunol Ther Exp (Warsz) 2003;51:219–226. [PubMed] [Google Scholar]
- 12.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weigert A, Johann AM, von Knethen A, Schmidt H, Geisslinger G, Brune B. Apoptotic cells promote macrophage survival by releasing the antiapoptotic mediator sphingosine-1-phosphate. Blood. 2006;108:1635–1642. doi: 10.1182/blood-2006-04-014852. [DOI] [PubMed] [Google Scholar]
- 14.Akiyama TE, Sakai S, Lambert G, Nicol CJ, Matsusue K, Pimprale S, Lee YH, Ricote M, Glass CK, Brewer HB, Jr, Gonzalez FJ. Conditional disruption of the peroxisome proliferator-activated receptor gamma gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Mol Cell Biol. 2002;22:2607–2619. doi: 10.1128/MCB.22.8.2607-2619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 16.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 17.Shah YM, Morimura K, Gonzalez FJ. Expression of peroxisome proliferator-activated receptor-gamma in macrophage suppresses experimentally induced colitis. Am J Physiol Gastrointest Liver Physiol. 2007;292:G657–666. doi: 10.1152/ajpgi.00381.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.von Knethen A, Soller M, Tzieply N, Weigert A, Johann AM, Jennewein C, Kohl R, Brune B. PPARgamma1 attenuates cytosol to membrane translocation of PKCalpha to desensitize monocytes/macrophages. J Cell Biol. 2007;176:681–694. doi: 10.1083/jcb.200605038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Knethen A, Callsen D, Brune B. NF-kappaB and AP-1 activation by nitric oxide attenuated apoptotic cell death in RAW 264.7 macrophages. Mol Biol Cell. 1999;10:361–372. doi: 10.1091/mbc.10.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson JD, Denisenko O, Bomsztyk K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc. 2006;1:179–185. doi: 10.1038/nprot.2006.27. [DOI] [PubMed] [Google Scholar]
- 21.Collart MA, Baeuerle P, Vassalli P. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol. 1990;10:1498–1506. doi: 10.1128/mcb.10.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gurnell M, Wentworth JM, Agostini M, Adams M, Collingwood TN, Provenzano C, Browne PO, Rajanayagam O, Burris TP, Schwabe JW, Lazar MA, Chatterjee VK. A dominant-negative peroxisome proliferator-activated receptor gamma (PPARgamma) mutant is a constitutive repressor and inhibits PPARgamma-mediated adipogenesis. J Biol Chem. 2000;275:5754–5759. doi: 10.1074/jbc.275.8.5754. [DOI] [PubMed] [Google Scholar]
- 23.Leesnitzer LM, Parks DJ, Bledsoe RK, Cobb JE, Collins JL, Consler TG, Davis RG, Hull-Ryde EA, Lenhard JM, Patel L, Plunket KD, Shenk JL, Stimmel JB, Therapontos C, Willson TM, Blanchard SG. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry. 2002;41:6640–6650. doi: 10.1021/bi0159581. [DOI] [PubMed] [Google Scholar]
- 24.Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol. 1990;10:2327–2334. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 26.Westin S, Kurokawa R, Nolte RT, Wisely GB, McInerney EM, Rose DW, Milburn MV, Rosenfeld MG, Glass CK. Interactions controlling the assembly of nuclear-receptor heterodimers and co-activators. Nature. 1998;395:199–202. doi: 10.1038/26040. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Wang J, Nawaz Z, Liu JM, Qin J, Wong J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. Embo J. 2000;19:4342–4350. doi: 10.1093/emboj/19.16.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, Chawla A. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 30.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 31.Chung SW, Kang BY, Kim SH, Pak YK, Cho D, Trinchieri G, Kim TS. Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-gamma and nuclear factor-kappa B. J Biol Chem. 2000;275:32681–32687. doi: 10.1074/jbc.M002577200. [DOI] [PubMed] [Google Scholar]
- 32.Majai G, Sarang Z, Csomos K, Zahuczky G, Fesus L. PPARgamma-dependent regulation of human macrophages in phagocytosis of apoptotic cells. Eur J Immunol. 2007;37:1343–1354. doi: 10.1002/eji.200636398. [DOI] [PubMed] [Google Scholar]
- 33.Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, Staels B, Chinetti-Gbaguidi G. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–143. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 34.Coste A, Dubourdeau M, Linas MD, Cassaing S, Lepert JC, Balard P, Chalmeton S, Bernad J, Orfila C, Seguela JP, Pipy B. PPARgamma promotes mannose receptor gene expression in murine macrophages and contributes to the induction of this receptor by IL-13. Immunity. 2003;19:329–339. doi: 10.1016/s1074-7613(03)00229-2. [DOI] [PubMed] [Google Scholar]
- 35.Perissi V, Aggarwal A, Glass CK, Rose DW, Rosenfeld MG. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell. 2004;116:511–526. doi: 10.1016/s0092-8674(04)00133-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.