

Abstract
Identification of the cells in the liver that produce alpha-fetoprotein (AFP) during development, in response to liver injury, and during the early stages of chemical hepatocarcinogenesis led to the conclusion that maturation arrest of liver-determined tissue stem cells was the cellular process that gives rise to hepatocellular carcinomas (HCC). When the cellular changes in these processes were compared that of the formation of teratocarcinomas, the hypothesis arose that all cancers arise from maturation arrest of tissue determined stem cells. This was essentially a reinterpretation of the embryonal rest theory of cancer whereby tissue stem cells take the role of embryonal rests. A corollary of the stem cell theory of the origin of cancer is that cancers contain the same functional cell populations as do normal tissues: stem cells, transit-amplifying cells, and mature cells. Cancer stem cells retain the essential feature of normal stem cells: the ability to self-renew. Growth of cancers is due to continued proliferation of cancer transit-amplifying cells that do not differentiate to mature cells (maturation arrest). On the other hand, cancer stem cells generally divide very rarely and contribute little to tumor growth. However, the presence of cancer stem cells in tumors is believed to be responsible for the properties of immortalization, transplantability and resistance to therapy characteristic of cancers. Current therapies for cancer (chemotherapy, radiotherapy, anti-angiogenesis and differentiation therapy) are directed against the cancer transit amplifying cells. When these therapies are discontinued, the cancer re-forms from the cancer stem cells. Therapy directed toward interruption of the cell-signaling pathways that maintain cancer stem cells could lead to new modalities to the prevention of re-growth of the cancer.
Introduction
The goal of this review is to show how analysis of the production of alpha-fetoprotein (AFP) during normal development, after liver injury, during growth of hepatocellular carcinoma (HCC), and during the premalignant stages of chemical hepatocarcinogenesis led to re-establishment of the stem cell theory of cancer. The story begins with the discovery of AFP as a protein present not only in fetal serum (1–3), but also in the serum of both experimental animals (4) and humans (5) with HCC. Although a fetal serum protein, consistent with the properties of AFP, had been reported earlier (1–3), the papers by Abelev et al (4) and Tatarinov (5) were the first to show the commonality of the protein produced by developing fetuses and adult tumors. The discovery of this protein, along with that of carcinoembryonic antigen (6), initiated the field of investigation that became known as “Oncodevelopmental Biology” (7), a term linking the process of carcinogenesis to developmental biology. This conceptualization had been previously formulated by Pierce and co-workers as: “Cancer is a problem of developmental biology” (8).
The studies to be reviewed here began when the author joined the Department of Pathology at the University of Pittsburgh in 1965. The cellular and biochemical changes that preceded the development of HCC during experimental chemical hepatocarcinogenesis were under active investigation by the Chairman of the Department, Prof. Emmanuel Farber, and his collaborators. These studies were directed toward the examination of focal changes in small groups of liver cells (foci), and the development of nodules of increasing size that became know as “neoplastic nodules” that appear in the livers of rats exposed to chemical hepatcarcinogens months before the eventual development of HCC (9,10). These changes implied that HCC arose by “de-differentiation” of adult hepatocytes. Since it had been shown (see above) that AFP was produced by HCC, the question that required answering was: Can the nature of the cells in the liver that give rise to HCC be clarified by analysis of the production of AFP during the carcinogenic process?
Radioimmunoassay for AFP
To address this question, the first objective was to develop a more sensitive and reproducible assay for AFP. I decided that a radioimmunoassay (RIA) for AFP would be the method of choice. RIA is up to 100 times more sensitive than the immunodiffusion assays that were then being used. Fortunately, I was not only taught the principles of RIA, but also had been required to develop an RIA for albumin by Richard S. Farr, as part of the sophomore microbiology laboratories in medical school in 1958. Farr published his extensive studies on antigen-antibody binding and RIA at about the same time (11). This was 2 years before the paper on RIA for insulin was published (12), for which the principle author, Rosalyn Yalow, received the Nobel Prize.
Using the methods recorded in my microbiology laboratory notebook, I developed an RIA for rat AFP (13). To accomplish this, it was first necessary to identify and isolate AFP. From the previous studies of Gitlin et al. (14) it was known that amniotic fluid was a rich source of AFP. An antiserum that would specifically identify rat AFP was made by first immunizing rabbits with rat amniotic fluid, and then absorbing this antiserum with normal rat serum (15, Figure 1). Then AFP was isolated by isoelectric focusing, but this method was adopted only after other methods had proven ineffective (15). Because of difficulties in isolating pure AFP, it took over 5 years to establish the RIA for AFP. During this time period, several others published RIAs for both the human (16,17) and rat (18,19) isoforms of AFP. Before RIA was available, it was not known whether AFP was present in normal adult serum. With the application of the highly sensitive RIA, it became possible to measure normal levels of AFP accurately and reproducibly both in humans (16) and rats (20, Figure 2).
Figure 1. Immunoelectrophoretic identification of AFP.
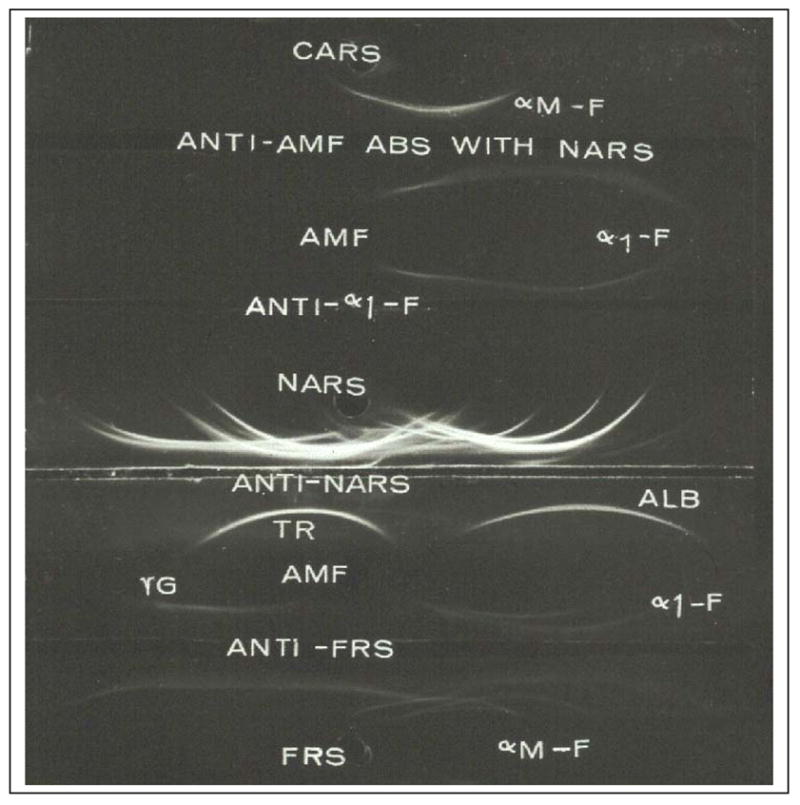
To produce antiserum to AFP rabbits were immunized with rat amniotic fluid (AMF) and the anti-AMF serum was absorbed with normal adult rat serum (NARS). Later AFP was isolated by isoelectric focusing and a specific antiserum was obtained by immunization of a goat with purified AFP (anti-α1-F). CARS- Croton oil induced acute phase rat serum (contains α1- macrofetoglobulin, αM-F); abs – absorbed; NARS-normal adult rat serum; TR-transferrin; ALB-albumin; γG-gamma-globulin; FRS-fetal rat serum. Modified from Sell, et al. [15]
Figure 2. Radioimmunoassay (RIA) for rat AFP.

A. Standard curves for the “high” and the “low” RIA. The low assay was performed by pre-incubation of the unknown or the known standard with the anti-AFP before addition of the radio-labeled AFP. B. The top figure shows the range of AFP levels in normal rat serum. The bottom figure shows results of replicated assays for selected samples for normal rat serum. Each symbol represents the assays for a given serum.
Modified from Sell and Gord [20].
Once our RIA for AFP was available, it was applied to measure the serum concentrations of AFP of the rat in normal development, after liver injury, following growth of transplantable HCC, and during the hepatocarcinogenic process. The results are summarized in Figure 3 (21). During normal pregnancy in the rat the level of AFP is two logs higher in fetal serum at term than in the mother. After birth, the serum level of AFP falls exponentially in the mother, but remains high for 3–4 weeks in the neonatal pup. There is a rapid but temporary elevation after liver injury. Following partial hepatectomy (PH), the serum level is higher in young rats than in older rats. The serum level is also higher after chemical injury than after partial hepatectomy (PH). There is a steady, essentially logarithmic, increase during growth of a transplanted HCC, and a logarithmic decrease after surgical removal of the AFP producing HCC, but re-elevation is seen if the tumor is not fully removed or recurs as metastases. There is a prolonged elevation after treatment with a non-carcinogenic dose, for example, a single cycle of the carcinogen N-2-actylaminofluorine (AAF). These data illustrate the complexity of the production of AFP associated with various physiological and pathological conditions. The examples of elevation of serum AFP during normal development, after acute liver injury, and growth of HCC, and after exposure to chemical hepatocarcinogens, and the nature of the cells that produce AFP, will now be presented more in more detail.
Figure 3. Changes in serum levels of AFP during normal development, after liver injury, during growth of a transplantable hepatoma and during chemical hepatocarcinogenesis.

During pregnancy the serum AFP of the fetus rises exponentially to reach levels 2 logs higher that in the maternal serum. The serum level remains elevated for about 3 weeks after birth and then rapidly falls to the adult normal level. The maternal serum AFP falls immediately after delivery. After liver injury, there is a short elevation of AFP associated with proliferation of liver cells. This is higher after chemical injury than after partial hepatectomy. During growth of a transplantable AFP-producing hepatoma, there is a steady elevation related to growth of the tumor. When the tumor is removed surgically, the AFP level drops, but the lever rises again when there is growth of lung matastases. During chemical hepatocarcinogenesis there may be a prolonged elevation of AFP, even with sub-carcinogenic doses.
Modified from Sell and Becker [21].
AFP in Normal Development
During normal development, AFP is synthesized in the fetal yolk sac endoderm and liver, and then diffuses rapidly into the maternal circulation. Using incorporation of radioactive arginine into cells of various organs, we found that the major sites of synthesis are the yolk sac and liver, with a much lower production in the intestines (22). This pattern was confirmed by Rot analysis for messenger RNA for AFP (mRNAAFP) (23) and by in situ hybridization in collaboration with the late Jose Sala-Trepat (Figure 4). Using radiolabeled proteins injected into various fetal and maternal compartments, we found there to be a rapid equilibration of the labeled proteins in the feto-maternal unit (ref. 24, Figure 5A). Both mRNAAFP and mRNAalbumin are present in high amounts in the liver during late fetal life, but mRNAalbumin is not found in the yolk sac (ref. 25, Figure 5B) After birth, the AFP level drops rapidly in the maternal serum, but remains high in the newborn for about 3–4 weeks (Figure 3). The high AFP level in the newborn rat is associated with continued proliferation of hepatocytes in the liver until about 3 weeks after birth, when liver-cell proliferation ceases. Then the AFP level declines rapidly (26).
Figure 4. Production and distribution of AFP in the Fetal-maternal unit. A. Fetal-maternal distribution of AFP.

Radiolabeled purified AFP was injected into various fetal and maternal fluid comartments and the presence of radioactivity in other fetal and maternal after different times determined. AFP synthesis was determined by incorporation of radiolabel arginine into cultures of tissues and the location of mRNAAFP in fetal and maternal tissues determined. AFP is synthesized in the fetal yolk sac and liver and rapidly enters fetal blood and amniotic fluid, and then the maternal blood. Modified from Sell and Alexander [24]. B, C. In situ hybridization of cDNA for AFP to mRNAAFP in yolk sac endoderm and C. fetal hepatocytes. The black color in b shows mRNA for AFP in yolk sac endoderm; in c in fetal hepatocytes (courtesy of Zoran Ilic).
Figure 5. Aging changes in rat liver and yolk sac mRNAAFP and mRNAALBUMIN.

The number of moleclules per cell of messenger RNA for AFP and albumin was determined in cultures cells and compared to transplantable Morris hepatoma 7777 cells. mRNAAFP is high in yolk sac and fetal liver until birth. There is no mRNAALBUMIN in the yolk sac, but the number of molecules increased from fetal day 16 until about 2 weeks of age when it reaches adult levels. mRNAAFP falls rapidly after birth and reaches adult levels at 4 weeks of age. The mRNAAFP is higher in hepatoma 7777 than in fetal hepatocytes, but the level of mRNAALBUMIN in hepatoma 7777 is lower than in normal adult hepatocytes. From Sell et al. [25].
A possible function for AFP during development has intrigued investigators since the protein was discovered half a century ago (for reviews see 27,28). Three major hypotheses have been tested. 1. AFP is immunosuppressive serving to protect the fetus from immune rejection by the mother (29–31), 2. AFP carries vital fatty-acid building blocks to the fetal neurons in the developing brain (32), 3. AFP binds to estrogen and prevents masculinization of the developing female brain (33–35). In regard to first hypothesis, immunosuppression, we obtained mixed results (36,37). In most instances, AFP increased the immune responses; for example, AFP increased the induction of cytotoxic T-cells by 9 fold in mice. Of critical importance is to include controls. Both albumin and AFP inhibited mitogen responses in vitro. In support of the second hypothesis is the fact that AFP protein is present in developing neurons (32,38–40), but mRNAAFP is not (41), suggesting that AFP could be a carrier protein to deliver building blocks required for normal brain development. Since it binds fatty acids required for myelination (32), AFP was proposed to be essential for normal myelination. This remained a viable hypothesis until the demonstration that AFP knockout mice, developed by Szpirer and co-workers (33–35), are born and develop normally; that finding essentially eliminated immunosuppression or a carrier function as AFP’s role. However, adult female AFP knockout mice are infertile, due to an inability of the pituitary to produce gonadotropins (34). This “defeminization” of the female brain in the AFP knockout mice can be blocked by administration of estrogen inhibitors (aromatase inhibitors). Thus, if neither AFP nor estrogen is present, defeminization of the female brain does not occur. Szpirer and co-workers concluded that AFP serves to sequester estrogen during development of the mouse brain, thus protecting the developing female mice from defeminization. However, the rodent findings are not necessarily applicable to humans, as human AFP does not bind estrogen. Thus, although various functions have been postulated, the fascinating problem of why there is a specific serum protein that is associated with fetal life, conserved in so many species, remains to be determined convincingly for all species. The next topic of this paper will be the effect of liver injury on AFP production.
AFP and liver injury
Various cells in the hepatocyte lineage respond to injury or loss of liver tissue by proliferation. During this proliferation, AFP is produced and released into the circulation. The ability of mature hepatocytes to restore the lost liver cells by rapid proliferation following partial hepatectomy (PH) or carbon tetrachloride (CCl4) injury led to the conclusion that the liver did not contain liver tissue determined stem cells.
Partial hepatectomy (PH)
The classic model of liver regeneration is the proliferation of mature hepatocytes following two-thirds partial hepatectomy (PH, 42). In this model the lost liver tissue is restored by proliferation of mature hepatocytes in the remaining liver cords (43). Hepatocyte proliferation occurs within the first 48 hr after PH and is followed by an acute elevation of AFP for about 5 days (44). During active proliferation of the hepatocytes, AFP can sometime be detected in hepatocytes undergoing mitosis (45). Thus, whereas AFP is clearly a marker for liver progenitor cells in the yolk sac and fetal liver, there is re-expression of this stem cell marker when adult liver cells proliferate. Repair of centrolobular necrosis of the liver after CCl4 injury is also accomplished by proliferation of the remaining hepatocytes. In fact, Engelhart et al (46) reported that the surviving hepatocytes next to the necrotic central zone expressed AFP prior to division. Thus, liver stem cells are not involved in these repair reactions. However, a different result is obtained from other models of liver injury.
Chemical injury
Treatment of the liver by toxic chemical results in injury at different levels of the liver lobule (47,48). For example, CCl4 produces centrolobular injury, galactosamine and lead nitrate act on mid-zone hepatocytes, allyl alcohol acts on periportal hepatocytes and a number of chemicals (α-naphtyl-isothiocyante, 4,4′-diaminophenylmethane, furan) are toxic to bile duct cells (49). Earlier studies using autoradiography led to the conclusion that the precursors of bile-duct cells and hepatocytes belong to non-overlapping populations (50,51). It was stated that a duct cell or a liver cell “reproduces only its own kind” (51). On the other hand, our studies on liver injury indicated that there is a linage of cells in the liver, and that cells at various differentiation levels in this lineage may respond to injury. Mature hepatocytes proliferate in response to PH and to CCl4 induced centrolobular injury (42–46); midzonal hepatocytes respond to galactosamine and lead nitrate injury (52,53); bipolar ductal cells respond when hepatocyte injury is accompanied by treatments that inhibit hepatocyte proliferation (54,55); and ductular stem cells respond to duct injury induced by chemicals or bile duct ligation (56,57, Figure 6). The presence of small periportal stem cells or hepatocyte progenitor cells is supported by the observation that liver injury by retrorsine is repaired by a population of small periductal hepatocyte-like cells even after the bile ducts have been destroyed by 4,4′-diaminodiphenylmethane (DAPM) (58).
Figure 6. Cellular localization of AFP after various models of liver injury.

A. Immunofluorescence labeling of AFP in mitotic hepatocyte after partial hepatectomy [45]. B. Immunofluorescence labeling of AFP in pre-mitotic hepatocyte after CCl4 induced central injury [46]. C. Immunoperoxidase labeling of AFP in transitional oval cells after periportal injury induced by cocaine [59]. D. Tritiatiate thymidine labeling of oval duct cells and periductular cells after CCl4 injury and treatment with N-2-actylaminofluorene [55].
The possible involvement of small periportal hepatocytes or putative periportal liver stem cells is best illustrated by the proliferative response of small intraportal oval cells to periportal necrosis induced by allyl alcohol in the rat (59–61), or induced by cocaine in the mouse (62). These studies were originally designed to determine whether periportal liver cord cell injury would be repaired by proliferation of mid-zonal hepatocytes that replaced the damaged periportal cells, or by proliferation of putative liver stem cells or biopolar duct cells that then moved into the necrotic periportal zone (59). The cellular response to periportal injury was a marked increase in small periportal cells, which filled the necrotic zone and then appeared to differentiate into hepatocytes (59–62, but see below for an alternative explanation). The finding of a marked increase in small periportal cells after periportal injury raised the possibility that blood-borne stem cells entering the liver in the portal triad contribute to liver regeneration, perhaps through a small-cell intermediate.
Can bone marrow cells become hepatocytes?
To determine whether blood-borne stem cells could contribute to liver regeneration, the contribution of bone marrow derived cells to the liver was assessed after transplantation of donor marked bone marrow (BM) cells. For example, bone marrow cells from male donors are transplanted into female recipients. In this case the donor cells are detected by the presence of the Y-chromosome (for reviews see 63,64). Although there is considerable variation in results, in general very few hepatocytes contain a donor marker (less than 1%). However, in mice with extensive and long-term loss of liver cells due to an inborn error of metabolism, over 50% of the liver cells are derived from BM-cells after transplantation of donor cells without the metabolic defect (65,66). In this case, the liver is repopulated with liver cells that fuse with the BM-derived cells (64,67,68). Because the fused cells are reprogrammed by the DNA of the BM-derived cells to express the gene that is defective in the recipient’s liver, proliferation of these rescued cells leads to a selective survival advantage over the recipient’s cells and restoration of the liver by fused cells. Many fewer donor derived hepatocytes are seen in other models of liver injury, such as in transgenic hepatitis B mice after treatment with retrorsine. However, it is not known how BM derived cells become liver cells in models with less injury.. Published studies on the ability of BM-derived cells to become liver progenitor cells (oval cells or small hepatocytes) have shown inconsistent results (69–77).
To determine whether BM-derived cells could be contributing to the small oval cells seen after chemically induce liver injury, we examined the livers of female mice transplanted with BM from male donors expressing the marker green fluorescent protein (GFP) followed by injury at various levels of the liver lobule: carbon tetrachloride (CCl4), central injury; 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) ductal injury (78); and phenobarbial/cocaine (PC) to produce periportal injury (unpublished data). Hepatocytes proliferate after CCl4-treatment; biliary ducts and cholangioles after DDC, and periportal oval cells after PC. None of the proliferating liver lineage cells in the repair response to injury contained the donor marker. However, the livers of the recipient mice contained large numbers of donor derived Kupffer, stellate, and endothelial cells, as well as an infiltration of small round cells (monocytes) derived from the BM. Months after repair of the injury, the number of hepatocytes containing GFP or Y-chromosome was not significantly different from the numbers in uninjured mice. Thus, it does not appear that BM-derived cells contribute to liver regeneration after injury through a putative stem cell or small hepatocyte intermediate (78). Extensive analysis of the response to PC has not been published but our results show that after this periportal injury there is a massive infiltration of bone marrow derived cells, but that these cells do not become liver cells. In contrast to our earlier interpretation that reconstitution of this injury was accomplished by periductular oval cells, it now appears that the early reconstituting cells are small hepatoctyes (unpublished data).
In conclusion, the general belief that the adult liver does not have stem cells is challenged by the findings on the expression of AFP during the cellular proliferative response to liver injury at various levels of the hepatic lobule. The liver gland consists of the same cellular lineage as in other tissues, i.e., stem cells, transit-amplifying cells (ductule bipolar cells), ductal stem cells and mature hepatocytes. The unusual feature of the liver is not that it lacks stem cells (it does possess them), but rather that hepatocytes, the most differentiated cells of the liver, even those next to the central vein, are able to proliferate in response to injury. Somewhat unexpected was the finding that AFP is produced by all of the cells of the liver lineage when they proliferate, including the mature adult hepatocytes. This indicates that AFP is not only a developmental protein, but also is produced in adult liver in the proliferative response to injury. In the next section, we will see that AFP is also produced by hepatocellular carcinomas, most prominently by proliferating cancer cells. Thus, AFP is not necessarily a marker for liver cancer stem cells because the transit amplifying cells of the cancer also produce AFP.
AFP production by hepatocellular carcinomas
The maintenance of a library of transplantable rat HCCs induced by chemicals by Harold Morris in the 1960s (79) provided pre-clinical models with which the relationship of the serum concentrations of AFP to growth rate, histolologic grade and chromosome composition of HCCs in tumor bearing rats could be determined (80). The author knows of no evidence that the tumor microenvironment contributes to the production of AFP by tumors, other than through support for the proliferation of the cancer cells. Serum AFP levels in rats bearing faster-growing tumors, tumors with poor differentiation, and tumors with aneuploidy were higher than levels in rats with more slowly growing, better-differentiated diploid tumors (80). Clearly, the serum level of AFP correlates closely with the growth rate (number of dividing cells) and size of the tumor. Later clinical studies revealed similar correlations in human patients with HCC. Also, as in humans, serum AFP levels in rats bearing transplantable HCC could be used to follow the growth of HCC and response to therapy (Figure 7). For the preclinical study, we used a fast-growing, transplantable rat HCC that produced high levels of AFP (Morris hepatoma 7777). Following transplantation of hepatoma 7777 cells into the thigh of histocompatible Buffalo rats, the serum concentration of AFP rose exponentially as the tumor enlarged, until about 40 days, when the size of the tumor required that the animals be euthanized (81). At this time-point the serum concentration ranged between 7,000 and 10,000 μg/ml. The kinetics of the elevation of serum AFP paralleled the growth of the tumor. If the transplanted tumors were removed surgically at 17 days, the serum AFP levels fell, with a half-life of about 24 hr. However, rapid elevations occurred with growth of metastatic lesions, mainly in the lung (81). In another experiment, we found that irradiation of the lung at the time of surgical removal of the tumor, could delay or prevent growth of pulmonary metastases. The effect of the irradiation on tumor growth was reflected by serum AFP levels (82). This preclinical model proved to be a good one for the determination of how to use AFP in the diagnosis and management of human patients with AFP producing tumors.
Figure 7. Effect of growth and amputation of transplanted Morris hepatoma 7777 on serum levels of AFP in Buffalo rats.

Thirteen Buffalo rats were inoculated in the hind led with 1 X 107 cells from transplantable Morris hepatoma 7777. The animals were then bled 2 times per week and the serum concentration of AFP measure by RIA. At 17 days the tumor bearing legs of 8 of the transplanted mice was amputated. The shaded area shows the serum levels of the 5 control mice that were not treated by surgery. All of the control rats died by 40 days after tumor transplantation. The symbols show the levels of AFP in individual rats following amputation of the tumor. The serum levels fell rapidly after amputation, but elevations were then seen in 7/8 surgically treated rats. Elevation of AFP, in each was associated with growth of metastases in the lung. The serum level of AFP of one of the 8 treated rats fell rapidly to below normal and remained there. This rat showed no evidence of regrowth or metastases of the tumor [81].
Further characterization of the cellular composition and biological behavior of Morris hepatomas in the 1960s revealed that these cancers contained cells with stem cell-like characteristics as recently publicized for other cancers (83,84). Notable among these characteristics were that cancer stem cells are able to grow in vitro (usually on soft agar), are able to initiate tumor growth upon transplantation (tumor initiating cells), and can resist therapy. The fact that cells from these HCCs could be cultured in vitro argues for the presence of cells with stem-like properties (i.e., immortality) in the hepatomas (85). Transplantation of the Morris hepatomas revealed heterogeneity in growth properties. Some of the Morris hepatomas could be transplanted by means of just a few cells and grew very quickly, whereas others could only be transplanted by means of large numbers of cells and grew very slowly (86,87). These findings demonstrated the second property of cancer stem cells noted above, i.e., tumor initiation. Finally, extensive studies on the response to irradiation treatment by William Looney (88) demonstrated that after high-dose radiation, the tumors were able to re-grow, indicating the presence of a therapy-resistant cancer stem cell. Thus, these studies published in the 1960s and 70s demonstrated that Morris hepatomas contained cancer stem cells (see below for more recent studies on cancer stem cells in human hepatomas).
The expression of fetus-associated proteins by Morris hepatomas was interpreted by Van Rensselaer Potter to mean that these hepatomas arose by processes related to normal embryogenesis, i.e., “…oncology is blocked ontogeny” (89–91). Morris HCCs expressed variable levels of adult and fetal liver enzymes (92–95), they secreted variable levels of AFP (80), and they displayed extents of hepatocellular differentiation ranging from well differentiated (‘minimal deviation hepatomas‘) to poorly differentiated (‘maximally deviated HCC’) (96). Potter suggested that the expression of fetal liver-cell markers in HCCs was not due to dedifferentiation, but rather to a block in the maturation of the immature liver cells that gave rise to the cancer. This conception (89) antedated the findings on the production of AFP during chemical hepatocarcinogenesis, which will be covered in the next section of this paper.
Because of the potential for the clinical application of AFP to the diagnosis and monitoring therapy of cancers associated with production of AFP, the author, two post-doctoral fellows and a laboratory technician at the University of Pittsburgh developed a test kit (FETO-TECT) for the detection of human AFP that became used throughout the world from 1969 thorough 1974 (97). The test was based on a simple double diffusion in agar plate, and could be applied in the field without requirement of a laboratory (Figure 8). It was especially useful in high HCC incidence areas of the World, such as South Africa and China. However, the Feto-Tect test was removed from interstate commerce in 1974 by the United States Food and Drug Administration because over-interpretation of CEA measurements had led to unnecessary surgery for colon cancer (97). Even though assays for AFP were not a problem, they were linked to CEA as oncodevelopmental markers and also removed. Even when the test for CEA was reinstated, that for AFP was not. We were unable to obtain an explanation. When AFP testing finally gained approval, the tests for certification were so expensive to conduct that only large companies could afford to obtain the data required. Now AFP testing for HCC is a standard approved procedure.
Figure 8. FETO-TECT double-diffusion-in-agar for detection of human AFP.

The test kit included a control positive AFP serum (left well), a control negative serum (bottom well), and anti-AFP (right well). The patient’s serum to be tested was placed in the top well. The reaction of the control positive with the anti-AFP resulted in a precipitin-in-agar line that ran between the top and bottom wells. If the patient’s serum contained AFP, the line would deviate from the top well. The sensitivity of the test was about 100ng/ml. This test kit was sold internationally by Fernwood Laboratory, Pittsburgh Pa. from 1969 until 1974. In 1974 the test was taken off the market by the United States Food and Drug Administration because of it’s association with carcinoembryonic antigen (CEA) as an oncocancer antigen. The company was formed by two fellows, Richard Baney and James Storey, and a technician, Art Park, in the author’s laboratory in 1968 [97].
In conclusion, AFP production by HCC is related to growth rate, size, and chromosome composition of the tumor. Slowly growing, well-differentiated HCC generally are not associated with elevated serum AFP, whereas fast-growing, poorly differentiated HCCs produce high serum AFP concentrations. However, significant exceptions are found: some fast-growing, near-diploid HCC do not cause elevations, whereas some slow growing euploid HCCs do. The production of AFP by HCC identifies the protein as an “onco” protein and represents the re-expression of a developmental product during oncogenesis. Characterization of the growth properties, transplantability and response to therapy of the experimental Morris rat hepatomas supports the conclusion that these tumors contain cancer stem cells. The next section reviews the data on the production of AFP during chemical hepatocarcinogensis, that led to the hypothesis that HCCs arise from stem cells (98).
AFP and chemical hepatocarcinogenesis
De-differentiation: foci to nodules to cancer
Detailed histologic and histochemical analyses by many investigators studying chemical hepatocarcinogenesis, led to the hypothesis that liver cancer arises from de-differentiation of hepatocytes. For a detailed review of this subject, with more complete references than listed in this paper, the reader is referred to a recent review by the author (98). The induction of human liver cancer by chemicals began to be seen in Japan in the 1930s and in other places in the 1940s due to development of the chemical dye industry. Pathologists clearly described the major histologic findings in the liver that preceded the appearance of liver cancer in experimental models of chemical hepatocarcinogenesis (99–102). The two major observations were the proliferation of small round cells and the development of nodules in the liver that came to be called neoplastic nodules. After World War II, hepatocarcinogenesis became a major area of investigation in the United States, as exemplified by the extensive work of Emmanuel Farber (103,104) and Henry Pitot (105), as well as in Germany (106–110). The extensive proliferation of small round cells, designated “oval cells” by Farber (9,10,103,104) and the development of foci of altered staining of hepatocytes and increasing nodularity in the liver, were described. However, the striking sequence of foci → nodule → HCC was interpreted to mean that HCC developed by dedifferentiation of hepatocytes (98,110). This interpretation became the accepted dogma for the cellular origin of HCC.
AFP production during chemical hepatocarcinogenesis
Analysis of the production of AFP during chemical hepatocarcinogenesis led to a major change in the paradigm for the cellular origin of cancer. As noted above, the author joined the department of Emmanuel Farber in 1965, at the height of the investigation of the biochemical and cellular changes in the liver during chemical hepatocarcinogenesis. It was generally thought that the determination of the biochemical changes taking place during development of neoplastic nodules would reveal how and why cells become cancerous (98–110). With the discovery in 1963 that AFP was associated with cancer (4), it was postulated that the decipherment of where AFP was produced during the development of HCC, would further our knowledge of the carcinogenic process. After the sensitive RIA for AFP was developed (20), we undertook an extensive study of the kinetics of the elevation of serum AFP during the hepatocarcinogenic process (see review, 21). Unexpectedly, we found that the kinetics of elevation varied, depending upon which of the chemical carcinogenic models was used (21,111); this finding forced the realization that the cellular localization of AFP is regimen-dependent (47). The regimen-specificity of AFP localization will be illustrated for four regimens: cyclic N-2 fluorenylacteamide (AAF), choline–deficiency AAF (CD-AAF), the Solt-Farber model (SF), and diethyl-nitrosamine (DEN).
Cyclic AAF (112)
The cyclic AAF regimen of George Teebor and Fred Becker was chosen first for study, because of the consistent progression of liver lesions from foci to nodules to cancer. In this model, AAF is fed for 3-week on, 1-week off cycles. Since the AAF is in itself toxic if fed continuously, the week-off periods allow the rats to recover. At the end of the first cycle, small cellular foci of 8 to 10 altered cells are seen. If the feeding is discontinued, the foci disappear. At the end of the second cycle, small nodules are present in the liver cords. Again, if feeding is discontinued, the nodules disappear. After the third cycle, larger nodules are grossly visible. Again, if feeding is discontinued, the nodules disappear. After the fourth cycle, the nodules are much larger and distort the liver grossly. If feeding AAF is discontinued at this point, liver cancer will develop about 21 weeks after the 4th cycle. After the fourth cycle, hundreds of nodules of different sizes may be evident in the liver. Most of these regress, but a few do not (persistent nodules); of the latter, a small number were believed to become HCC. When the serum AFP levels were measured by RIA, in contrast to what was expected, there was an immediate elevation during the first cycle (111). The serum AFP levels fell during the 3rd and 4th cycles when the nodules were the predominant feature, and then rose later when HCCs developed. When the liver was examined histologically, the early AFP rise correlated with a marked increase in the number of small oval cells in between the nodules (113). Thus, the elevation of serum AFP correlated not with the development of nodules, but rather with the presence of oval cells. Immunofluorescent cellular localization of AFP revealed that the AFP was present in the oval cells and not in the foci or nodules. This unexpected localization indicated that the origin of HCC is not through the foci to nodule pathway, but instead through the small, AFP+ oval cells. Since the HCCs that develop in this model produce AFP, it is more likely that the HCCs arise from oval cells than from hepatocytes. Based on these data and the results of the some of the injury experiments described above, we postulated that tissue determined stem cells exist in the liver, either in the terminal ducts or the intraportal zone, and that these “stem cells” proliferated in response to carcinogens and gave rise to HCC (25,114).
CD-AAF
To determine the origin of the oval cells, we chose the CD-AAF model of Hishi Shinozuka and Benito Lombardi. In this model, there is very rapid appearance of oval cells within days of the start the regimen (115,116). The rapid proliferation of the oval cells allowed us to label the proliferating cells by pulse labeling using tritiated thymidine (117,118). Light microscopy revealed that there is within 1–2 days, an increase in labeled small round cells next to the bile ducts in the intraportal zone, and then an increase in labeled cells within the bile ducts. During the next 2–3 weeks, labeled oval cells extend across the liver lobule to the central vein (118). By electron microscopy, the first identifiable labeled cells were cells containing very few organelles, and located in the intraportal tissue outside the basement membrane of the bile ducts (118). The results of these experiments confirmed our previous speculation that putative liver stem cells are located in the intraportal zone as well as within the bile ducts (25,114).
Solt-Farber (SF) model
The complex SF regimen was designed to activate proliferation of hepatocytes initiated following exposure to carcinogen (DEN). The supposition is that when proliferation of non-initiated hepatocytes is inhibited (AAF treatment), there will be selective stimulation of proliferation of the initiated hepatocytes following PH (119). Thus, the regimen consists of administration of DEN (initiation), followed 2-weeks later by a 2-week treatment with AAF (inhibits hepatocyte proliferation), with a PH done during the middle of the AAF exposure (stimulates proliferation of initiated cells). Using this model, we found a rapid increase in AFP occurring shortly after the PH (120). The level AFP increased until about 2 months after PH and then dropped to essentially normal. Another elevation of AFP occurred after 10 months when HCC began to form. However, in contrast to the cyclic-AAF model, the AFP in this model, was not found in periportal cells, but rather in newly formed duct cells (120). Thus, in this model, the cells giving rise to HCC appear to be the bipolar bile ductular progenitor cells.
DEN
After continuous exposure of rats to DEN, serum AFP levels do not rise until 6–8 weeks after exposure, but then steadily increase as HCCs grow (120). Cellular localization is first seen not in oval cells or duct cells, but in small atypical foci of hepatocytes. These foci enlarge to form microcarcinomas and later HCC. Thus, in this model HCCs appear to arise directly from hepatocytes (120).
Summary
A summary of the cellular changes in the liver arising from various carcinogen regimens is shown in Figure 9. Depending on the regimen, HCC can arise from intraportal non-ductal stem cells, from ductal bipolar transit amplifying progenitor cells, or from hepatocytes. This variability indicates that HCC can arise from a number of cell types in the hepatocyte lineage: stem cells, transit-amplifying cells, and mature hepatocytes (121). If cancer is a problem of maturation arrest, we would expect that the differentiation phenotype for HCC arising from stem cells would differ from that of HCC arising from ductal progenitor cells or from mature hepatocytes. However, all of these protocols result in HCC. The missing link in the cellular hierarchy of the liver and liver cancer appears to be the liver cancer of early childhood, hepatoblastoma (122).
Figure 9. Cellular origin of proliferating liver cells during various models of chemical hepatocarcinogenesis in the rat.

The top of the figure shows the location of the cellular reaction to cyclic AAF. A. AFP positive intrahepatic oval cells by immunofluorescence after cyclic AFP [113]; B. New duct cells staining for OV-6, a monoclonal antibody to oval cells, during the cellular reaction to the Solt-Farber model of hepatocarcinogenesis [121]; C. AFP postitive microcarcinomas by immunoperoxidase after DEN exposure [120].
Hepatoblastomas
Hepatoblastomas are tumors of childhood with approximately two-thirds occurring within the first 2 years of life, and 90% within the first 5 years of life (122). Hepatoblastomas are made up of cells at various stages of differentiation, from small round embryonal cells to fetal-like cells that form primitive liver cords (123,124). The small cells grow in sheets and are similar to neuroblastoma and other primitive tumors of childhood. This embryonal phenotype suggests that there is a loss of potential of liver-determined stem cells with aging, such that HCCs arising in the livers of adults are the more highly differentiated HCC, whereas those arising in the livers of young children reflect the less differentiated stage of the liver-determined stem cell (98). A relationship of the cellular origin of liver cancer to the stage of maturation arrest of liver lineage cells is shown in Figure 10.
Figure 10. Diagram of cellular lineage of hepatocarcinogenesis.

Cancers of the liver may arise from fetal hepatocytes (hepatoblastomas) or from adult cells at different stages of the hepatocyte lineage, stem cells, bipolar ductal progenitor cells or mature hepatocytes. This model differs from most tissue lineages because even the most mature cells in the hepatocyte lineage, the central hepatocytes, may proliferate in response to injury. This implies that there are no terminally differentiated cells in the liver, and thus HCCs may arise from any cell in the liver lineage. The lineage of teratocarcinomas is illustrated in Fig. 11 [98].
From this model, we conclude that HCCs arise from maturation arrest of differentiation and not by dedifferentiation (47,114,120,121). Thus, even though some HCCs arise from hepatocytes, liver cancer is not an exception to the general rule that cancers arise from stem cells, since hepatocytes represent one stage of liver-cell differentiation. Mature hepatocytes are not terminally differentiated and can be activated to proliferate. The conclusion that liver cancer - one of the least likely cancers to arise from stem cells - actually do so, led to a general hypothesis that essentially all cancers arise from maturation arrest of tissue-determined stem cells (125).
Is there a carcinogenic role for AFP?
The association of AFP production by pre-malignant cells and early carcinomas suggested a possible role of AFP in tumorigenesis or tumor progression (27). There is no evidence known to this author that the presence of production of AFP has a role in induction of cancer, but appears to be re-expression of a developmental protein. However, it is possible that AFP contributes to controlling progression. For example, some tumors have a receptor for AFP and the serum level of this receptor may be more accurate than other tumor markers (126,127). Reaction of AFP with these receptors may affect nuclear transcription, apoptosis or signal transduction, but these activities are demonstrated in vitro and as yet have no treatment applications have even approached study (27). On the other hand, AFP, especially of murine species, is known to bind estrogen, and AFP (128,129) or peptides derived from AFP (130) may inhibit growth of estrogen dependent tumors. Again, since human AFP binds estrogen poorly, it is unlikely that human AFP has this effect. However, the possibility of treating estrogen dependent tumors with AFP peptides has been proposed (130).
The Stem Cell Theory of the Origin of Cancer
The theory that cancers arise from stem cells was the first rational theory of cancer. It was first propounded by Joseph Claude Anselme Recamier in 1829 (131), and later by Robert Remark (132). Prior to this theory, cancers had been considered to arise though an imbalance of black bile, an extension of the humoral theory of disease. A variation of the humoral theory was favored by Sir James Paget, and even Rudolph Virchow, the father of pathology, believed that cancers arose from connective tissue during chronic inflammation (133). The concept that cancers arise from stem cells became known as the embryonal rest theory of cancer (134,135). This theory postulated that cancers arose in adult tissues from small collections of embryonal cells that did not differentiate. Although this idea became popular, other ideas gradually superseded it. For example, Hugo Rippert thought that cancers came from displaced tissue that lost the restraining influence of surrounding tissue, and Amedee Borrel favored parasitic infection as a cause. By 1914, Theodor Boveri had found chromosomal abnormalities in cancers, and he concluded that cancers arose from mature cells that mutated. Such findings led William Seaman Bainbridge, in his monumental book “The Cancer Problem”, to reach the following conclusion: “The congenital or embryonic theory of the origin of cancer has received no support whatever from the experimental and comparative investigations of recent times.” (136). However, studies on teratocarcinoma pointed the way back to the stem cell theory of the origin of cancer and cancer stem cells.
Teratocarcinoma
Rudolf Virchow, although noting the resemblance between developing embryonic tissues and the tissues that made up teratocarcinomas, concluded that cancers arose from inflammatory granulation tissue (133). The contribution of the study of teratocarcinoma to reconsideration of the theory of the stem cell origin of cancer took place after World War II. Teratocarcinomas are cancers arising usually in the testes or ovaries, which contain a mixture of normal, mature, well-differentiated cells representing essentially every mature tissue of the body. Thus, most of the cells of a teratocarcinoma are benign mature cells. The malignant stem cells of a teratocarcinoma form a structure that resembles an early embryo (embryoid body; 137). Only the cells in the embryoid body are actively proliferating and only they give rise to the other differentiated tissues in the tumor, including brain, muscle, bone, bone marrow, eyes, secretary glands, and skin. Teratocarcinomas usually also contain yolk sac elements that produce AFP, and choriocarcinoma elements that produce human chorionic gonadotropin HCG. These markers can be used for diagnosis and for tracking the results of therapy of those teratocarcinomas which contain yolk sac or chorionic elements (138–140). In 1954, LeRoy Stevens, a veteran fighter pilot from WWII, published his studies on the production of transplantable teratocarcinomas through injection of the normal male germinal cells from an SIJ/129 mouse, that has a high rate of spontaneously appearing teratocarcinoma, into the normal testes of a 129 mouse (141). The transplantation of these cells resulted in growth of teratocarcinomas arising from the normal germinal cells, proving that cancers can arise from normal germinal stem cells placed into an abnormal tissue environment (142,143). The concept that environmental signals can be critical in the development of cancer was predicted by Cohnheim in 1875 (135), and restated by Rippert in 1911 (144). Incidentally, transplantation of extra-embryonic yolk sac cells of 9 day old embryos to the rat kidney capsule results in growth of yolk sac tumors that produce AFP, whereas transplantation of the embryonic portion of the embryo gives rise to teratomas that do not produce AFP (145).
Extensive study of the growth and differentiation properties of experimental teratocarcinomas by Barry Pierce and co-workers established that many of the progeny of the malignant cells of teratocarcinomas differentiate into mature benign tissue (8,146). This was also shown to be the case for other cancers specifically, squamous cell carcinoma (147). The difference between cancer and normal tissue renewal is that in normal tissue renewal, the number of cells that are proliferating is essentially equal to the number of cell terminally differentiating (undergoing apoptosis), whereas in cancer the number of cells that are proliferating (transit-amplifying cells) is greater than the number of cells that are entering terminal differentiation, because of maturation arrest of the cancer cells in the transit amplifying population (125).
The most impressive demonstration of the role of the environment, in inducing differentiation of cancer stem cells, is the blastocyst transplantation experiments. Using the method developed by Brinster (148) for injection of cells into developing blastocysts, Mintz and Illmense (149), and Papaioannou et al. (150), reported in 1975 that teratocarcinoma stem cells could be controlled by the environment of the developing blastocyst. When teratocarcinoma cells were injected into the inner cell mass of the developing blastocyst and the embryos allowed to mature, the carcinoma-derived cells differentiated into fully functioning mature tissue in chimeras made up of mixtures of carcinoma-derived cells and blastocyst-derived cells. Pierce et al. (151) then demonstrated that the germinal layers of the blastocysts determine the types of cells into which the transplanted embryonal carcinoma cells develop. Thus, the actual carcinoma stem cells undergo complete differentiation to benign cells; leading to the conclusion that “tumors are caricatures of normal tissue renewal” (152). A diagram illustrating the differentiation pathway of cancers arising from germinal cells, and the possible stages of maturation arrests is shown in Figure 11. Embryonal carcinomas can be treated with agents such as retinoic acid (vitamin A), which induces differentiation of the malignant cells to benign cells (153). However, human teratocarcinomas are more effectively treated by surgery and chemotherapy. The ability to induce differentiation in teratocarcinomas from malignant to benign cells establishes the precedent for differentiation therapy of cancer (154–156).
Figure 11. Diagram of cellular lineage of germinal cell tumors.

The lineage of germinal cell cancers begins with the toti-potential neoplastic germinal cells and includes embryonal carcinoma, seminoma and spermatocytic seminoma. Embryonal carcinoma cells may give rise to chroiocarcinomas, yolk sac carcinomas, teratomas and mixed cell tumors.
Differentiation therapy of leukemias
The success of differentiation therapy is arguably best exemplified by the treatment of myeloid leukemias (157,158), where appropriate differentiation therapy forces differentiation of malignant cells. Examples of the various forms of myeloid leukemia are chronic myeloid, subacute promyeloid and acute myeloid (blastic). Each of these is caused by gene translocations that result in constitutive activation of signaling cascades or blocking of apoptotic pathways. Differentiation therapy is directed toward disruption of these signaling pathways, or activation of apoptotic pathways. For example, Gleevec binds to and inactivates the bcr/abl tyrosine kinase formed in chronic myeloid leukemia. This inactivation blocks the signaling pathway and allows the cells to differentiate (159). In subacute promyeloid leukemia, fusion of the promyeloid leukemia region (PML) with the retinoic acid receptor (RARα) gene results in inhibition of the formation of cytoplasmic granules required for differentiation (160,161). In this way the maturation of the leukemic cells is blocked at the promyelocyte stage. Treatment with retinoic acid induces breakdown of the PML/RARα gene fusion product and allows the promyelocytic leukemia cells to differentiate. In acute myeloblastic lukemias (AML) there is usually both activation of signaling pathways for proliferation and inhibition of an apoptotic pathway (162). These provide a one-two punch, so that the number of proliferating cells is increased and the cells produced do not die. Treatments with agents that inhibit the activation pathway and activate apoptosis in AML are now being tested clinically (163,164).
Cancer stem cells
As discussed above, there are two components to cancer stem cell models: cancers arise from tissue stem cells and cancers contain stem cells. The preceding sections of this paper have largely covered the hypothesis that cancers arise from stem cells. In the last section of this paper, the concept of cancers containing stem cells and therapy directed to these cancer stem cells will be presented. First, there are three major biological properties of cancer stem cells: the ability to grow in soft agar, the capacity to transplant the tumor (tumor initiating cells) and the ability to resist radio-and chemotherapy.
Growth in agar
Growth of cancer cells on soft agar was first described for HeLa cells in 1955 (165) and later for cells from primary human tumors (166). Salman (167) found that 1:1,000 to 1:100,000 cells from a primary human adenocarcinoma would form colonies in soft agar (called “tumor colony forming units”) and proposed to use these cultured cells to study the effects of various drug treatments. This range of proportions of tumor colony forming units is similar to the proportions recently found for the tumor initiating cells in acute myelogenous leukemia (see below).
Tumor transplantation and tumor initiating cells
The second property proposed for a cancer stem cell is that it can initiate tumor growth when transplanted to syngeneic or immuno-compromised recipients (168–173, see review 83). The frequency of tumor-initiating cells was later found to be of the same order of magnitude as the frequency of cells that survive after chemo- or radio-therapy and grow in soft agar. The evidence for transplantable liver cancer stem cells was discussed above under the properties of Morris hepatomas. Briefly, some of the Morris hepatomas could be transplanted by means of just a few cells and grew very quickly, whereas others could only be transplanted by means of large numbers of cells and grew very slowly. These observations again suggest cellular heterogeneity in hepatomas, but neither systematic dilution nor fluctuation analysis experiments were performed. Consequently, there is as yet no clear identification, in this model system, of the percentage of cells with tumor initiating properties for HCC.
With the relatively recent recognition that the cells with stem cell properties in a cancer might be identified by “stem cell” markers, a number of attempts to identify such cells in hepatocellular carcinoma have been reported. By gene expression analysis, Lee et al (174) were able to identify at least three types of human liver cancers and poor prognosis is associated with HCC with less differentiated expression. Specific markers that appear to identify stem-like cells in human hepatocellular carcinoma include CD 133 (175–177), CD90 (178,179), ABCG2 (180), epithelial cell adhesion molecule (EpCAM, 181), and Met (182). CD133 and CD90 expression are associated with fetal liver cell marker expression, tumor initiation, culture in vitro and chemoresistance (175–179), properties attributed to cancer stem cells. Further study of the properties of these cells may lead to modalities of therapy for hepatocellular carcinoma directed at the cancer stem cells (see below)
The transplantable tumor cells that are resistant to therapy may differ from other cells in the cancer (cancer stem cells as distinct from the cancer transit-amplifying cells), or transplantable cells may merely be quiescent cells that respond poorly to therapy, and survive the challenges of transplantability. These alternatives were presented in commentaries 1994. Trott (183) supported the concept that the cells from transplantable mouse tumors meet the criteria of a tumor stem cell; i.e., “re-growth of the tumour preceded by clonal expansion from a single cell with unlimited proliferative potential”. He concluded that tumors contain the same populations of cells as normal tissue, consistent with the proposal of Pierce et al., derived from studies on teratocarcinoma (8). On the other hand, in a companion paper Denekamp (184), argued that the putative cancer stem cells might be considered the least differentiated cells of a cancer cell population, functionally and kinetically different from the remaining mass of tumor cells, but not necessarily stem cells.
Cancer stem cells and therapy
The recognition that cancers contain the same lineages of cells as normal tissues, i.e. stem cells, transit amplifying cells, and differentiated cells (125,152), suggests that a cure for cancer should be directed against the cancer stem cells. Therefore, even if all of the proliferating transit-amplifying tumor cells were killed therapeutically, tumorous tissues would re-emerge from therapy-resistant cancer stem cells. In support of this possibility, highly tumorigenic subpopulations of putative cancer initiating ‘stem’ cells derived from human glioblastomas have been found to display resistance to radiation because of increased protection against DNA damage (185). Provocative qualitative and quantitative theoretical discussions of immortal DNA strands, an obligate property of normal stem cells; and certain predicted DNA-repair deficiencies in somatic cancer cells have also been reported (186,187). However, little is known about the role of putative liver cancer stem cells in resistance to therapy, although it may be presumed that re-growth of HCCs after effective treatment may be mediated by putative liver cancer stem cells. Although this cannot be proven, it is the most likely explanation.
The models of therapy now in use are directed against the transit-amplifying cells. As depicted in Figure 12, radiation therapy, chemotherapy, and anti-angiogenic therapy inhibit proliferation of actively dividing cells, whereas differentiation therapy inactivates signals that cause maturation arrest thereby allowing terminal differentiation of transit-amplifying cells (154–158). Even if these approaches are successful, the cancer stem cell remains, and the tumor retains the potential to re-grow from the cancer stem cells.
Figure 12. Levels of therapy for cancer.

Radiation therapy and chemotherapy kill proliferating transit amplifying cells of a cancer. Anti-angiogenic therapy inhibits the blood supply to the proliferating cells. Differentiation therapy reverses maturation arrest and allows proliferating cancer cells to terminally differentiate. Even if these approaches are successful, cancers may re-grow from cancer stem cells that are not affected by these therapies. It is hypothesized that cancer stem cell inhibition therapy would block the proliferation of cancer stem cells and prevent re-growth [188].
New experimental therapy approaches are directed toward signaling pathways active in maintaining cancer stem cells through use of inhibitory RNA or small blocking molecules. Some of these blocking agents are listed in Table 1 (156). If stem cell therapy is to be applied successfully, then the re-growth of the cancer after successful treatment of the cancer transit amplifying cells can be prevented.
Footnotes
The Abbot Award Lecture; presented at the 35th Meeting of the International Society for Oncodevelopmental Biology and Medicine, September 17, 2007, Prague, Czech Republic. This paper reviews the data from Dr Sell’s laboratory testing the hypothesis that liver cancer is due to dedifferentiation of mature cells. These data did not support this hypothesis. For detained historical reference citations to alpha-fetoprotein and carcinogenesis see: Sell S. Stem cells in hepatocarcinogenesis. Cell Science Reviews, ISN NO. 1742-8130, 2006, and for extensive citations on the biological properties of AFP see: Mizejewski GJ. Biological roles of alpha-fetoprotein during pregnancy and perinatal development. Exp Biol Med 229:430-463, 2004
Dedicated to Garri Israelevich Abelev on the occasion of his 80th birthday, January 10, 2008.
References
- 1.Bergstrand CF, Czar B. Demonstration of a new protein fraction in serum from the human fetus. Scand J Cllin Lab Invest. 1961;8:174. doi: 10.3109/00365515609049266. [DOI] [PubMed] [Google Scholar]
- 2.De Muralt G, Roulet DLA. Etude immunologique des proteines seriques foetales humaines. Helv Paediat Acta. 1961:517–533. [Google Scholar]
- 3.Masopust J. Development of serum protein spectrum during childhood. Rev Czech Med. 1962;8:214–225. [PubMed] [Google Scholar]
- 4.Abelev GI, Perova SD, Khramkova NI, Postnikova AS, Irlin IS. Production of embryonal α-globulin by transplantable mouse hematomas. Transplantation. 1963;1:174–180. doi: 10.1097/00007890-196301020-00004. [DOI] [PubMed] [Google Scholar]
- 5.Tatarinov YS. Presence of embryonal α-globulin in the serum of patients with primary hepatocellular carcinomas. Vopr Med khimii. 1964:90–91. [Google Scholar]
- 6.Gold P, Freedman SL. Specific carcinoembryonic antigens of the human digestive system. J Exp Med. 1965;122:467–481. doi: 10.1084/jem.122.3.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fishman WH, Sell S. Onco-developmental gene expression. New York: Academic Press, Inc.; 1976. pp. 1–788. [Google Scholar]
- 8.Pierce GB, Shikes R, Fink LM. Cancer: a problem of developmental biology. Engelweed Cliffs, NJ.: Prentice Hall Inc.; 1978. pp. 1–242. [Google Scholar]
- 9.Farber E. Ethionine Carcinogenesis. Adv Cancer Res. 1963;7:383–474. doi: 10.1016/s0065-230x(08)60986-0. [DOI] [PubMed] [Google Scholar]
- 10.Farber E, Cameron R. The sequential analysis of liver cancer induction. Adv Cancer Res. 1979;31:125–226. doi: 10.1016/s0065-230x(08)60658-2. [DOI] [PubMed] [Google Scholar]
- 11.Farr RSA. Quantitative Immunochemical measure of the primary interaction between I*BSA and antibody. J Infectious Dis. 1958;103:237–262. doi: 10.1093/infdis/103.3.239. [DOI] [PubMed] [Google Scholar]
- 12.Yalow RS, Berson SA. Immunoassay of endogenous insulin in man. J Clin Invest. 1960;39:1157–1175. doi: 10.1172/JCI104130. (NOBEL PRIZE) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sell S. Radioimmunoassay of rat α1-fetoprotein. Cancer Res. 1973;33:1010–1015. [PubMed] [Google Scholar]
- 14.Gitlin D, Boesman M. Sites of α-fetoprotein synthesis in the human and in the rat. J Cllin Invest. 1967;46:1010–1016. doi: 10.1172/JCI105590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sell S, Jalowayski I, Bellone C, Wepsic HT. Isolation and characterization of rat α1-fetoprotein. Cancer Research. 1972;32:1184–1189. [PubMed] [Google Scholar]
- 16.Ruoslahti D, Seppala M. Studies on carcino-fetal proteins III. Development of a radioimmunoassay for α-fetoprotein. Demonstration of α-fetoprotein in healthy human adults. Intern J Cancer. 1971;8:374–383. doi: 10.1002/ijc.2910080304. [DOI] [PubMed] [Google Scholar]
- 17.Purves LR, Purves M. Serum AFP VI. The radioimmunoassay evidence for the presence of AFP in the serum of normal people and during pregnancy. S Afr Med J. 1972;46:1290–1298. [PubMed] [Google Scholar]
- 18.Oakes DD, Shuster J, Gold P. Radioimmunoassay for α1-fetoprotein in the serum of rats. Cancer Res. 1972;32:2753–2760. [PubMed] [Google Scholar]
- 19.Hirai H, Nishi S, Watabe H. Radioimmunoassay for α-fetoprotein. In: Peeters H, editor. Protides of the Biological Fluids. Pergamon Press; Oxford and New York: 1973. pp. 579–587. [Google Scholar]
- 20.Sell S, Gord D. Refinement of radioimmunoassay for detection of 1 ng rat alpha-1 fetoprotein. Immunochemistry. 1973;10:439–442. doi: 10.1016/0019-2791(73)90013-x. [DOI] [PubMed] [Google Scholar]
- 21.Sell S, Becker FF, Sell S, Becker FF. Guest Editorial: Alpha-fetoprotein. J Natl Cancer Inst. 1978;60:19–26. doi: 10.1093/jnci/60.1.19. [DOI] [PubMed] [Google Scholar]
- 22.Sell S, Skelly H. Tissue sites of alpha1-fetorpotein synthesis by the rat during pregnancy and hepatoma growth. J Natl Cancer Inst. 1976;56:645–648. doi: 10.1093/jnci/56.3.645. [DOI] [PubMed] [Google Scholar]
- 23.Sala-Trepat JM, Dever J, Sargent TD, Thomas K, Sell S, Bonner J. Changes in expression of albumin and α-fetoprotein genes during rat liver development and neoplasia. Biochemistry. 1979;18:2167–2178. doi: 10.1021/bi00578a006. [DOI] [PubMed] [Google Scholar]
- 24.Sell S, Alexander D. Rat alpha-1-fetoprotein. V. Catabolism and fetal-maternal distribution. J Nat Cancer Inst. 1974;52:1483–1489. doi: 10.1093/jnci/52.5.1483. [DOI] [PubMed] [Google Scholar]
- 25.Sell S, Sala-Trepat J, Sargent T, Thomas K, Nahon J-L, Goodman T, Bonner J. Molecular mechanisms of control of albumin and alphafetoprotein production: A system to study the early effects of chemical hepatocarcinogens. Cell Biology Intl Reports. 1980;4:235–254. doi: 10.1016/0309-1651(80)90056-9. [DOI] [PubMed] [Google Scholar]
- 26.Sell S, Nichols M, Becker FF, Leffert JL. Hepatocyte proliferation and alpha1-fetoprotein in pregnant, neonatal and partially hepatectomized rets. Cancer Res. 1974;34:865–871. [PubMed] [Google Scholar]
- 27.Mizejewski GJ. Alpha-fetoprotein structure and function: relevance to isofoms, epitopes, and conformational variants. Exp “Biol Med. 2001;226:377–408. doi: 10.1177/153537020122600503. [DOI] [PubMed] [Google Scholar]
- 28.Mizejewski GJ. Biological roles of alpha-fetoprotein during pregnancy and perinatal development. Exp Biol Med. 2004;229:439–463. doi: 10.1177/153537020422900602. [DOI] [PubMed] [Google Scholar]
- 29.Parmely MJ, Thompson JS. Inhibition of rat lymphocyte cultures by serum fractions rich in alpha-fetoprotein. In: Masseyeff R, editor. Alpha-fetoprotein. Inserm; Paris: 1974. p. 467. [Google Scholar]
- 30.Murgita RA, Tomasi TB. Suppression of the immune response by α-fetoprotein. I the effect of mouse α-fetoprotein on the primary and secondary antibody response. J Exp Med. 1975;141:269–286. doi: 10.1084/jem.141.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murgita RA, Tomasi TB. Suppression of the immune response by α-fetoprotein II. The effect of mouse α-fetoprotein on mixed lymphocyte reactivity and mitogen0induced lymphocyte transformation. J Exp Med. 1975;141:440–452. doi: 10.1084/jem.141.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uriel J, Trojan J, Moro R, Pineiro A. Intracellular uptake of α-fetoprotein: a maker of neuronal differentiation. Ann NY Acad Sci. 1983;417:321–329. doi: 10.1111/j.1749-6632.1983.tb32875.x. [DOI] [PubMed] [Google Scholar]
- 33.Gabant P, Forrester L, Nichols J, Van Reeth T, De Mees C, Pajack B, Watt A, Smitz J, Alexandre H, Szpirer C, Szpirer J. Alpha-fetoprotein, the major fetal serum protein, is not essential for embryonic development but is required for female fertility. PNAS. 2002;99:12865–12890. doi: 10.1073/pnas.202215399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeMees C, Laes J-F, Bakker J, Smitz J, Hennuy B, Van Vorren P, Gabant P, Szpierer J. Szpirer Alpha-fetoportein controls female fertility and prenatal development of the gonadotropin-realsing hormone pathway through an antiestrogenic action. Mole Cell Biology. 2006;26:2012–2018. doi: 10.1128/MCB.26.5.2012-2018.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bakker J, DeMees C, Souhard Q, Balthazart J, Gabant P, Szpirer J, Szpirer C. Alpha-fetoprotein protects the developing female mouse brain from masculinization and defeminization by estrogens. Nature Neuroscience. 2006;9:220–226. doi: 10.1038/nn1624. [DOI] [PubMed] [Google Scholar]
- 36.Sheppard HW, Sell S, Threfts P, Bahu R. Effects of α-fetoprotein on murine immune responses. I Studies on mice. J Immunol. 1977;119:91–97. [PubMed] [Google Scholar]
- 37.Sheppard HW, Sell S, Threfts P, Bahu R. Effects of α-fetoprotein on murine immune responses. II Studies on rats. J Immunol. 1977;119:98–103. [PubMed] [Google Scholar]
- 38.Attardi B, Ruoslathi E. Foetoneonatal oestrodiol-binding protein in mouse brain cytosol is α-fetoprotein. Nature. 1976;263:685–687. doi: 10.1038/263685a0. [DOI] [PubMed] [Google Scholar]
- 39.Benno RW, Williams TH. Evidence for intracellular localizatiion of alpha-fetoprotein in the developing rat brain. Brain Res. 1978;142:182–186. doi: 10.1016/0006-8993(78)90189-0. [DOI] [PubMed] [Google Scholar]
- 40.Trojan J, Uriel J. Intracellullar locallization of AFP and serum albumin in the central nervous system of the developing rat. CR Acad Sci (Paris) 1979;289:1157–1160. [PubMed] [Google Scholar]
- 41.Sell S, Longley MA, Boulter J. α-fetoprotein and albumin gene expression in brain and other tissues of fetal and adult rats. Developmental Brain Res. 1985;22:49–53. doi: 10.1016/0165-3806(85)90067-7. [DOI] [PubMed] [Google Scholar]
- 42.Higgins GM, Anderson RM. Experimental pathology of the liver. Restoration of the liver of the white rat following partial surgical removal. AMA Arch Pathol. 1991;139:1352–1368. [Google Scholar]
- 43.Steiner JW, Pera ZM, Taichman LB. Cell population dynamics in the liver. A review of quantitative morphological techniques applies to the study of physiological and pathological growth. Exp Mole Pathol. 1966;5:146–181. doi: 10.1016/0014-4800(66)90012-8. [DOI] [PubMed] [Google Scholar]
- 44.Sell S, Nichols M, Becker FF, Leffert JL. Hepatocyte proliferation and alpha1-fetoprotein in pregnant, neonatal and partially hepatectomized rets. Cancer Res. 1974;34:865–871. [PubMed] [Google Scholar]
- 45.Sell S. Heterogeneity of Alpha-fetoprotein (AFP) and albumin containing cells in normal and pathological permissive states for AFP production: AFP containing cells induced in adult rats recapitulate the appearance of AFP containing hepatocytes in fetal rats. Oncodevelopmental Biol & Med. 1980;1:93–105. [PubMed] [Google Scholar]
- 46.Engelhardt NV, Lazareva MN, Abelev GI, Uryaeva IV, Factor VM, Brodsky VY. Detection of α-fetoprotein in mouse liver differentiated hepatocytes before their progression through S phase. Nature. 1976;263:146–148. doi: 10.1038/263146a0. [DOI] [PubMed] [Google Scholar]
- 47.Sell S. The role of liver progenitor cells in repair of liver injury and in liver transplantation. Wound Repair Regener. 2001;9:467–482. doi: 10.1046/j.1524-475x.2001.00467.x. [DOI] [PubMed] [Google Scholar]
- 48.Sell S. Heterogeneity and plasticity of hepatocyte lineage cells. Hepatol. 2001;33:738–750. doi: 10.1053/jhep.2001.21900. [DOI] [PubMed] [Google Scholar]
- 49.Sell S. Comparison of liver progenitor cells in human atypical ductular reactions with those seen in experimental models of liver injury. Hepatology. 1998;27:317–331. doi: 10.1002/hep.510270202. [DOI] [PubMed] [Google Scholar]
- 50.Grisham JW, Porta EA. Origin and fate of proliferated hepatic ductal cells in the rat: electron microscopic and autoradiographic studies. Exp Mole Pathol. 1964;3:242–261. doi: 10.1016/0014-4800(64)90057-7. [DOI] [PubMed] [Google Scholar]
- 51.Ruben E. The origin and fate of proliferated bile ductular cells. Exp Mole Pathol. 1964;3:279–286. doi: 10.1016/0014-4800(64)90059-0. [DOI] [PubMed] [Google Scholar]
- 52.Sell S, Reynolds RD, Reutter WJ. Rat alpha1-fetoprotein: appearance after galactosamine-induced liver injury. J natl. Cancer Inst. 53:289–291, 1974. Natl Cancer Inst. 1974;53:289–291. doi: 10.1093/jnci/53.1.289. [DOI] [PubMed] [Google Scholar]
- 53.Tournier I, Legres L, Schoevaert D, Feldman G, Bernuau D. Cellular ahalysis of α-fetoprotein gene acitivation during carbon tetrachloride and D-galactosamine-induced acute liver injury in rats. Lab Invest. 1998;59:657–665. [PubMed] [Google Scholar]
- 54.Petersen BE, Zajec AF, Michalopoulos GK. Hepatic oval cell activation in response to injury following chemically induced periportal of pericentral damage in rats. Hepatol. 1998;27:1030–1038. doi: 10.1002/hep.510270419. [DOI] [PubMed] [Google Scholar]
- 55.Yin L, Lynch D, Ilic Z, Sell S. Proliferation and differentiation of ductular liver precursor cells and non-parenchymal cells during the restitutive response of the rat liver to carbon tetrachloride injury after inhibition of hepatocyte proliferation by n-2-acetylaminofluorine. Histol Histopathol. 2002;17:65–81. doi: 10.14670/HH-17.65. [DOI] [PubMed] [Google Scholar]
- 56.Lopez M, Mazzanti L. Experimental investigation of alpha-naphthylisothiocyanate as a hyperplastic agent of the biliary ducts in the rat. J Pathol Bacteriol. 1955;69:243–250. doi: 10.1002/path.1700690132. [DOI] [PubMed] [Google Scholar]
- 57.Sell S. Comparison of oval cells induced in rat liver by feeding N-2-fluorenylacetamide in a choline devoid diet and bile ducts induced by feeding 4,4-diaminodiphenylmethane. Cancer Res. 1983;43:1761–1767. [PubMed] [Google Scholar]
- 58.Best DH, Coleman WB. Bile duct destruction by 4,4′-diaminodiphenylmethane does not block the small hepatocyte-like progenitor cell response in retrorsine-exposed rats. Hepatology. 2007;46:1611–1619. doi: 10.1002/hep.21876. [DOI] [PubMed] [Google Scholar]
- 59.Yavorkovsky L, Lai E, Ilic Z, Sell S. Participation of small intra-portal stem cells in the restitutive response to periportal injury induced by allyl alcohol. Hepatology. 1995;21:1702–1712. [PubMed] [Google Scholar]
- 60.Sell S. Electron microscopic identification of putative liver stem cells and intermediate hepatocytes following periportal necrosis induced in rats by allyl alcohol. Stem Cells. 1997;15:378–385. doi: 10.1002/stem.150378. [DOI] [PubMed] [Google Scholar]
- 61.Yin L, Lynch D, Sell S. Participation of different cell types in the restitutive response of the rat liver to periportal injury induced by allyl alcohol. J Hepatol. 1999;31:492–507. doi: 10.1016/s0168-8278(99)80043-9. [DOI] [PubMed] [Google Scholar]
- 62.Rosenberg D, Ilic Z, Sell S. Proliferation of hepatic lineage cells of normal C57BL and IL-6 knockout mice after cocaine-induced periportal injury. Hepatology. 2000;31:948–955. doi: 10.1053/he.2000.5410. [DOI] [PubMed] [Google Scholar]
- 63.Sell S. Adult stem cell plasticity. Stem Cell Reviews. 2005;1:1–8. doi: 10.1385/SCR:1:1:001. [DOI] [PubMed] [Google Scholar]
- 64.Thorgeirsson SS, Grisham JW. Hematopoietic cells as hepatocyte stem cells: a critical review of the evidence. Hepatology. 2006;43:2–8. doi: 10.1002/hep.21015. [DOI] [PubMed] [Google Scholar]
- 65.Grompe M, al-Dhalimy M, Finegold M, et al. Loss of fumarylacetoacetate hydrolase is responsible for the neonatal hepatic dysfunction phenotype of lethal albino mice. Genes Dev. 1993;7:2298–2307. doi: 10.1101/gad.7.12a.2298. [DOI] [PubMed] [Google Scholar]
- 66.Lagasse E, Connors H, Al-Dhalimy M, et al. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nature Med. 2000;6:1229–1234. doi: 10.1038/81326. [DOI] [PubMed] [Google Scholar]
- 67.Vassilopoulos G, Wang PR, Russell DW. Transplanted bone marrow regenerates liver by cell fusion. Nature. 2003;422:901–904. doi: 10.1038/nature01539. [DOI] [PubMed] [Google Scholar]
- 68.Wang X, Willenbring H, Akkari Y, Torimaru Y, Foster M, Al-Dhalimy M, Lagasse E, Finegold M, Olson S, Grompe M. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature. 2003;422:897–901. doi: 10.1038/nature01531. [DOI] [PubMed] [Google Scholar]
- 69.Vig P, Russo FP, Edwards RJ, Tadrous PJ, Wright NA, Thomas HC, Alison MR, Forbes SJ. The sources of parenchymal regeneration after chronic hepatocellular liver injury in mice. Hepatology. 2006;43:316–324. doi: 10.1002/hep.21018. [DOI] [PubMed] [Google Scholar]
- 70.Petersen BE, Bowen WC, Patrene KD, et al. Bone marrow as a potential source of hepatic oval cells. Science. 1999;284:1168–1170. doi: 10.1126/science.284.5417.1168. [DOI] [PubMed] [Google Scholar]
- 71.Oh SH, Witek RP, Bae SH, et al. Bone marrow-derived hepatic oval cells differentiate into hepatocytes in 2-acetylaminofluorene/partial hepatectomy-induced liver regeneration. Gastroenterology. 2007;132:1077–1087. doi: 10.1053/j.gastro.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 72.Menthena A, Deb N, Oertel M, et al. Bone marrow progenitors are not the source of expanding oval cells in injured liver. Stem cells. 2004;22:1049–1061. doi: 10.1634/stemcells.22-6-1049. [DOI] [PubMed] [Google Scholar]
- 73.Moritoki Y, Ueno Y, Kanno N, et al. Lack of evidence that bone marrow cells contribute to cholangiocytic repopulation during experimental cholestatic ductal hyperplasia. Liver International. 2006;26:457–466. doi: 10.1111/j.1478-3231.2006.01250.x. [DOI] [PubMed] [Google Scholar]
- 74.Wulf GG, Luo K-L, Jackson KA, Brenner MK, Goodell MA. Cells of the hepatic side population contribute to liver regeneration and can be replenished by bone marrow stem cells. J of Hematol. 2003;88:368–378. [PubMed] [Google Scholar]
- 75.Rountree CB, Barsky L, Ge S, Senadheera S, Crooks GM. A CD133 expressing murine liver oval cell population with bi-lineage potential. Stem Cells. 2007;25:2419–2429. doi: 10.1634/stemcells.2007-0176. [DOI] [PubMed] [Google Scholar]
- 76.Roundtree CB, Wang X, Ge S, Barsky L, Zhu J, Gonzales I, Crooks GM. Bone marrow fails to differentiate into liver epithelium during murine development and regeneration. Hepatol. 2007;45:1250–1260. doi: 10.1002/hep.21600. [DOI] [PubMed] [Google Scholar]
- 77.Ochsner SA, Strick-Marchand H, Qiu Q, Venable S, Dean A, Wilde M, Weiss MC, Darlington GJ. Transcriptional profiling of bipotential embryonic liver cells to identify liver progenitor cell surface markers. Stem Cells. 2007;25:2476–2487. doi: 10.1634/stemcells.2007-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Swenson ES, Guest I, Ilic Z, Mazzeo M, Lizardi P, Hardiman C, Sell S, Krause DS. Hepatocyte nuclear factor-1 as a marker of epithelial phenotype reveals marrow–derived hepatocytes, but not duct cells, after liver injury in mice. Stem Cells. 7658;7:1768–1777. doi: 10.1634/stemcells.2008-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morris HP, Wagner BP. Induction and transplantation of rat hepatomas with different growth rate (including “minimal deviation” hepatomas) In: Busch H, editor. Methods in Cancer Res. Vol. 4. New York: Academic Press, Inc; 1968. pp. 125–152. [Google Scholar]
- 80.Sell S, Morris HP. Relationship of rat α1-fetoprotein to growth rate and chromosome composition of Morris hepatomas. Cancer Res. 1974;34:1413–1417. [PubMed] [Google Scholar]
- 81.Sell S, Wepsic HT, Nickel R, Nichols M. Rat alpha1-fetoprotein IV. the effect of growth and surgical removal of Morris hepatoma 7777 upon the serum alpha1-fetoprotein concentrations of Buffalo rats. J Natl Cancer Inst. 1974;52:133–137. doi: 10.1093/jnci/52.1.133. [DOI] [PubMed] [Google Scholar]
- 82.Sell S, Stillman D, Michaelsen M, Alaimo J, VonEssen C. Prevention of pulmonary metastases by irradiation of the lung: A model system employing a transplantable hepatoma and alpha1-fetoprotein as an index of tumor growth. Rad Res. 1977;69:54–64. [PubMed] [Google Scholar]
- 83.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 84.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea—a paradigm shift. Cancer Res. 2006;66:1883–1890. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 85.Van Wijk T. Regulation of DNA synthesis in cultured rat hepatoma cells. Int Rev Cytol. 1983;85:63–107. doi: 10.1016/s0074-7696(08)62370-0. [DOI] [PubMed] [Google Scholar]
- 86.Looney WB, Mayo AA, Janners MY, et al. Cell proliferation and tumor growth in hepatoma 3924A. Cancer Res. 1971;31:821–825. [PubMed] [Google Scholar]
- 87.Morris HP, Meranze DR. Induction and some characteristics of “minimal deviation” and other transplantable rat hepatomas. Recent Results Cancer Res. 1974;44:103–111. [Google Scholar]
- 88.Kovacs CJ, Evans MJ, Wakefield JA, Looney WB. A comparative study of the response to radiation by experimental tumors with markedly different growth characteristics. Rad Res. 1977;72:455–468. [PubMed] [Google Scholar]
- 89.Potter VR. Recent trends in cancer biochemistry: the importance of studies on fetal tissue. Canad Cancer Conf. 1968;8:9–30. [PubMed] [Google Scholar]
- 90.Potter VR. Phenotypic diversity in experimental hepatomas; the concept of partially blocked ontogeny. Br J Cancer. 1978;38:1–23. doi: 10.1038/bjc.1978.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Potter VR. The present status of the blocked ontogeny hypothesis of neoplasia: the Thalassemia connection. Oncodevel Biol Med. 1981;2:243–266. [PubMed] [Google Scholar]
- 92.Ono T. Enzyme patterns and malignancy of experimental hepatomas. Bann Monograph No. 1966;1:189–206. [Google Scholar]
- 93.Sugimura T, Matsushima T, Kawachi T, Hirata Y, Kawabe S. Molecular species of aldolases and hexokinases in experimental hepatomas. Gann Monograph No. 1966;1:143–150. [Google Scholar]
- 94.Schapira F. Aldolase isozymes in cancer. Eur J Cancer. 1966;2:131–134. doi: 10.1016/0014-2964(66)90005-3. [DOI] [PubMed] [Google Scholar]
- 95.Matsushima T, Kawabe S, Shibuya M, Sugimura T. Aldolase isozyme in rat tumor cells. Biochem Biophys Res Comm. 1968;30:565–570. doi: 10.1016/0006-291x(68)90090-9. [DOI] [PubMed] [Google Scholar]
- 96.Farber E. On the concept of minimal deviation in the study of the biochemistry of cancer. Cancer Res. 1968;28:1210–1211. [PubMed] [Google Scholar]
- 97.Sell S. Diagnostic applications of alpha-fetoprotein. Government regulations prevent full application of a clinically useful test. Human Pathol. 1981;12:959–963. doi: 10.1016/s0046-8177(81)80252-3. [DOI] [PubMed] [Google Scholar]
- 98.Sell S. Stem cells in hepatocarcinogenesis. Cell Science Reviews. 2006 ISN NO. 1742–8130. [Google Scholar]
- 99.Yoshida T. o-amidoazotoluol; uber die expermentelle annulare lebercirrhose des kaninchems. Trans Japan Pathol Soc. 1935;25:409–411. [Google Scholar]
- 100.Kinosita R. Special report: studies on the cancerogenic chemical substances. Trans Japan Pathol Soc. 1937;27:655–725. [Google Scholar]
- 101.Orr JW. The histology of the rat’s liver during the course of carcinogenesis by butter-yellow (p-dimethylaminoazobenzene) J Pathol & Bacteriol. 1940;50:393–408. [Google Scholar]
- 102.Opie EL. The pathogenesis of tumors of the liver produced by butter yellow. J Exp Med. 1944;80:231–246. doi: 10.1084/jem.80.3.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Farber E. Similarities in the sequence of early histologic changes induced in the liver by ethionine, 2-acetylaminofluorene, and 3′-methyl -4-dimethylamionazobenzene. Cancer Res. 1956;16:142–148. [PubMed] [Google Scholar]
- 104.Farber E, Sarma DSR. Hepatocarcinogenesis: a dynamic cellular perspective. Lab Invest. 1987;56:4–22. [PubMed] [Google Scholar]
- 105.Pitot HC. The natural history of neoplastic development: the relation of experimental models to liver cancer. Cancer. 1982;49:1206–1211. doi: 10.1002/1097-0142(19820315)49:6<1206::aid-cncr2820490623>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 106.Scherer E. Neoplastic progression in experimental hepatocarcinogenesis. Biochim Biophys Acta. 1984;738:219–236. doi: 10.1016/0304-419x(83)90005-7. [DOI] [PubMed] [Google Scholar]
- 107.Rabes HM. Development and growth of early preneoplastic lesions induced in the liver by chemical carcinogens. J Cancer Res Clin Oncol. 1983;106:85–91. doi: 10.1007/BF00395384. [DOI] [PubMed] [Google Scholar]
- 108.Bannasch P. Sequential cellular changes during chemical carcinogenesis. J Cancer Res Clin Oncol. 1984;108:11–22. doi: 10.1007/BF00390968. [DOI] [PubMed] [Google Scholar]
- 109.Bannasch P, Hacker HJ, Klinek F, Mayer D. Hepatocellular glycogenosis and related pattern of enzymatic changes during hepatocarcinogenesis. Adv Enzyme Regul. 1984;22:97–121. doi: 10.1016/0065-2571(84)90010-4. [DOI] [PubMed] [Google Scholar]
- 110.Aterman KJ. The stem cells of the liver – a selective review. Cancer Clin Oncol. 1992;118:87–115. doi: 10.1007/BF01187498. [DOI] [PubMed] [Google Scholar]
- 111.Becker FF, Sell S. Differences in serum alphafetoprotein concentrations during the carcinogenic sequences resulting from exposure to diethylnitrosamine or acetylaminofluorene. Cancer Res. 1979;39:1437–1442. [PubMed] [Google Scholar]
- 112.Teebor GW, Becker FF. Regression and persistence of hyperplastic hepatic nodules induced by N-2-fluorenylacetamide and their relationsip to hepatocarcinogenesis. Cancer Res. 1971;31:1–3. [PubMed] [Google Scholar]
- 113.Sell S. Distribution of alpha-fetoprotein and albumin containing cells in the livers of Fisher rats fed four cycles of N-2-fluorenylacetamide. Cancer Research. 1978;75:181–186. [PubMed] [Google Scholar]
- 114.Sell S, Leffert HL. An evaluation of cellular lineages in the pathogenesis of experimental heatocellular carcinoma. Hepatology. 1982;2:77–86. doi: 10.1002/hep.1840020113. [DOI] [PubMed] [Google Scholar]
- 115.Shinozuka H, Lombardi B, Sell S, Iammarino RM. Modification of ethionine liver carcinogenesis by choline deficiency: Early histological and functional alterations. Cancer Res. 1978;38:1092–1098. [PubMed] [Google Scholar]
- 116.Shinozuka H, Lombardi B, Sell S, Iammarino RM. Enhancement of DL-ethionine-induced liver carcinogenesis in rats fed a choline devoid diet. J Natl Cancer Inst. 1978;61:813–818. [PubMed] [Google Scholar]
- 117.Sell S, Osborn K, Leffert HL. Autoradiography of “oval cells” appearing early in the livers of rats fed N-2-fluorenylacetamide in choline devoid diet. Carcinogenesis. 1981;2:7–14. doi: 10.1093/carcin/2.1.7. [DOI] [PubMed] [Google Scholar]
- 118.Sell S, Salman J. Light and electronmicroscopic autoradiographic analysis of proliferating cells during the early stages of chemical hepatocarcinogenesis in the rat induced by feeding N-2-fluorenylacetamide in a choline deficient diet. Am J Pathol. 1984;114:287–300. [PMC free article] [PubMed] [Google Scholar]
- 119.Solt DB, Farber E. New principle for the analysis of chemical carcinogenesis. Nature. 1976;263:702–703. [Google Scholar]
- 120.Sell S, Dunsford H. Evidence for the stem cell origin of hepatocellular carcinoma and cholangiocarcinoma. Am J Pathol. 1989;134:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 121.Sell S. The role of determined stem-cells in the cellular lineage of hepatocellular carcinoma. Int J Dev Biol. 1993;33:189–201. [PubMed] [Google Scholar]
- 122.Schmidt D, Harms D, Lang W. Primary hepatic tumours in childhood. Virchows Archiv (A) 1985;407:387–405. doi: 10.1007/BF00709986. [DOI] [PubMed] [Google Scholar]
- 123.Kasai M, Watanabe I. Histologic classification of liver cell carcinoma in infancy and childhood and its clinical evaluation. Cancer. 1970;25:551–563. doi: 10.1002/1097-0142(197003)25:3<551::aid-cncr2820250309>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 124.Haas R, Muczynski KA, Krailo M, et al. Histopathology and prognosis in childhood hepatoblastona and hepatocarcinoma. Cancer. 1989;64:1082–1095. doi: 10.1002/1097-0142(19890901)64:5<1082::aid-cncr2820640520>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 125.Sell S, Pierce GB. Biology of Disease: Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab Invest. 1994;70:6–21. An AACR LANDMARK PAPER. [PubMed] [Google Scholar]
- 126.Moro R, Tanaoki T, Wegmann TG, Longnecker BM, Laderoute MP. Monoclonal antibodies directed against a sidespread oncofetal antigen: the alpha-fetoprotein receptor. Rumour Biol. 1993;14:116–130. doi: 10.1159/000217864. [DOI] [PubMed] [Google Scholar]
- 127.Moro R, Tcherkassova H, Song E, Shen G, Moro R, Schmid T, Hu X, Kummer A, Chen C. RECAF, a new broad-spectrum cancer marker. Personal communication. [Google Scholar]
- 128.Sonnenschein C, Soto AM. Growth inhibition of estrogen-sensitive tumor cells in newborn rats. Probable role of alpha-fetoprotein. N Natl Cancer Inst. 1979;63:835–841. doi: 10.1093/jnci/63.3.835. [DOI] [PubMed] [Google Scholar]
- 129.Sonnenschein D, Ucci A, Soto AAM. Growth inhibition of estrogen-sensitive rat mammary tumors. Effect of an alpha-fetoprotein-secreting hepatoma. J Natl Cancer Inst. 1980;64:1147–1152. [PubMed] [Google Scholar]
- 130.Bennett JA, Mesfin FB, Andersen TT, Gierthy JF, Jacobson HI. A peptide derived from α-fetoprotein prevents the growth of estrogen-dependent human breast cancers sensitive and resistant to tamoxifen. PNAS. 2002;99:2211–2215. doi: 10.1073/pnas.251667098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Recamier JCA. Recherches sur the traitement du cancer: par la compression methodique simple ou combinee, et sur l’histoire general de la meme maladie. Vol. 2. Paris Gabon: 1829. [Google Scholar]
- 132.Remark R. Ein beitrag zur entwickelungsgeschichte der krebshaften geschwulste. Deut Klin. 1854;6:70–174. [Google Scholar]
- 133.Virchow R. Die krankhaften Geschwulste. Berlin: 1863. [Google Scholar]
- 134.Durante F. Nesso fisio-pathologico tra la struttura dei nei materni e la genesi di alcuni tumori maligni. Arch Memori eed Osservazioni di chirugia practica. 1874;11:217–226. [Google Scholar]
- 135.Cohnheim J. Congenitales, quergestreiftes muskelsarkon der nireren. Virchows Arch. 1875;65:64. [Google Scholar]
- 136.Bainbridge WS. The Cancer Problem. The Macmillan Co.; New York: 1914. [Google Scholar]
- 137.Peyron A. Sue la presence des cellules genitales primordiales dans les bouton embryonnaires des embryones parthenogenetiques chez l’homme. CR Acad Sci (Paris) 1938;206:1680–1683. [Google Scholar]
- 138.Braunstein GD, McIntire KR, Waldmann TA. Discordance of human chorionic gonadotropin and alpha fetooprotein in testicular teratocarcinomas. Cancer. 1973;31:1065–1068. doi: 10.1002/1097-0142(197305)31:5<1065::aid-cncr2820310505>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 139.Willemse PHB, Sleijfer DT, Schraffordt Koops H, De Bruijn HWA, Oosterhuis JW, Brouwers TM, Ockhuizen T, Marrink J. Tumor markers in patients with non-seminomatous germ cell tumors of the testis. Oncodevelolpmental Biol & Med. 1981;2:117–128. [PubMed] [Google Scholar]
- 140.Vogelzang NJ, Lange PH, Goldman A, Vessela RH, Fraley EE, Kennedy BJ. Acute changes of α-fetoprotein and human chorionic gonadotropin during induction chemotherapy of gem cell tumors. Cancer Res. 1982;42:4855–4861. [PubMed] [Google Scholar]
- 141.Stevens LE, Little CC. Spontaneous testicular teratomas in inbred strains of mice. Proc Natl Acad Sci USA. 1954;40:1080–1087. doi: 10.1073/pnas.40.11.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Stevens LC. Origin of testicular teratomas from primordial germ cells in mice. J Natl Cancer Inst. 1967;38:549–552. [PubMed] [Google Scholar]
- 143.Stevens LC. Experimental production of testicular teratomas in mice. Proc natl Acad Sci USA. 1964;52:654–661. doi: 10.1073/pnas.52.3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rippert V. Das carcinoma des menschen. Geschwulstlehre; Bonn: 1911. [Google Scholar]
- 145.Damjanov I, Skreb N, Sell S. Origin of embryo-derived yolk sac carcinomas. Int J Cancer. 1977;19:526–530. doi: 10.1002/ijc.2910190413. [DOI] [PubMed] [Google Scholar]
- 146.Pierce GB, Dixon FL, Varney EL. Teratocarcinogenic and tissue forming potentials of the cell types comprising neoplastic embryoid bodies. Lab Invest. 1960;9:583–602. [PubMed] [Google Scholar]
- 147.Pierce GB, Wallace C. Differentiation of malignant cells to benign cells. Cancer Res. 1971;31:127–124. AN AACR SCIENTIFIC LANDMARK. [PubMed] [Google Scholar]
- 148.Brinster RL. Effect of cells transferred into the mouse blastocyst on subsequent development. J Exp med. 1974;140:1049–1056. doi: 10.1084/jem.140.4.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci. 1975;72:3583–3589. doi: 10.1073/pnas.72.9.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Papaioannou VE, McBurndy MW, Gardner RL, Evans MJ. Fate of teratocarcinoma cells injected into early mouse embryos. Nature. 1975;258:70–73. doi: 10.1038/258070a0. [DOI] [PubMed] [Google Scholar]
- 151.Pierce GB, Arechaga J, Jones A, Lewellyn A, Wells RS. The fate of embryonal-carcinoma cells in mouse blastocysts. Differentiation. 1987;33:247–253. doi: 10.1111/j.1432-0436.1987.tb01564.x. [DOI] [PubMed] [Google Scholar]
- 152.Pierce GB, Spears WC. Tumors as caricatures of the process of tissue renewal: prospects for therapy by directing differentiation. Cancer Res. 1988;48:1196–1204. [PubMed] [Google Scholar]
- 153.Yoskitake T, Itoyama S. Treatment of primary mediastinal germ cell tumors. Tinsho Kyobu Geka. 1989;9:29–34. [PubMed] [Google Scholar]
- 154.Sell S. Stem cell origin of cancer and differentiation therapy. CRC Crit Rev Onco/Hematology. 2004;51:1–28. doi: 10.1016/j.critrevonc.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 155.Sell S. Potential gene therapy strategies for cancer stem cells. Current Gene Therapy. 2006;6:579–591. doi: 10.2174/156652306778520674. [DOI] [PubMed] [Google Scholar]
- 156.Sell S. Cancer and stem cell signaling: a guide to preventive and therapeutic strategies for cancer stem cells. Stem Cell Rev. 2007;3:1–6. doi: 10.1007/s12015-007-0015-5. [DOI] [PubMed] [Google Scholar]
- 157.Sell S. Leukemia: Stem cells, maturation arrest, and differentiation therapy. Stem Cell Rev. 2005;1:197–205. doi: 10.1385/SCR:1:3:197. [DOI] [PubMed] [Google Scholar]
- 158.Sell S. Cancer stem cells and differentiation therapy. Tumor Biol. 2006;27:59–70. doi: 10.1159/000092323. [DOI] [PubMed] [Google Scholar]
- 159.Drucker BJ, Guilhot F, O’Brian SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Eng J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 160.Segalla S, Rinaldi L, Kilstrup-Nielsen C, Badaracco G, et al. Retinoic acid receptor alpha fusion to PML affects in transcriptional and chromatin-remodeling properties. Mol Cell Biol. 2003;23:8795–8808. doi: 10.1128/MCB.23.23.8795-8808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pitha-Rowe I, Petty WJ, Kitareewan S, Dmitrovsky E. Retinoid target genes in acute promyelocytic leukemia. Leukemia. 2003;17:1723–1730. doi: 10.1038/sj.leu.2403065. [DOI] [PubMed] [Google Scholar]
- 162.Chalandon Y, Schwaller J. Targeting mutated protein tyrosine kinases and their signaling pathways in hematologic malignancies. Haematologica. 2005;90:949–968. [PubMed] [Google Scholar]
- 163.Gilliand DG, Griffin JD. The role of FLT3 in hematopoiesis and leukemia. Blood. 2002;100:1332–1342. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- 164.Feldman EJ. Faresyltransferase inhibitors in melodysplastic syndrome. Current Hematol Rep. 2004;4:186–190. [PubMed] [Google Scholar]
- 165.Puck TT, Marcus PI. A rapid method of viable cell titration and clone production with HeLa cells in tissue culture: use of x-irradiated cells to supply conditioning factors. PNAS. 1955;41:432–437. doi: 10.1073/pnas.41.7.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Buick RN. In vitro clonogenicty of primary human tumor cells: Quantitation and relationship to tumor stem cells. In: Salmon S, editor. Cloning of Humor Tumor Stem cells. AR Liss; New York: 1980. pp. 15–20. Chapter 2. [PubMed] [Google Scholar]
- 167.Salmon S. In: In vitro effects of drugs on human tumor stem cell assays, in Cloning of Human Tumor Stem Cells. Salmon S, editor. AR Liss; New York: 1980. pp. 197–312. [Google Scholar]
- 168.Greene HSN. The significance of the heterologous translatability of human cancer. Cancer. 1952;5:24–44. doi: 10.1002/1097-0142(195201)5:1<24::aid-cncr2820050106>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 169.Reinhard MC, Goltz HL, Warner SC. Further studies on the quantitative determination of the growth of a transplantable mouse adenocarcinoma. Cancer Res. 1952;5:102–106. [Google Scholar]
- 170.Hewitt HB. Transplantation of mouse sarcomas with small numbers of single cells. Nature. 1952;170:622–623. doi: 10.1038/170622a0. [DOI] [PubMed] [Google Scholar]
- 171.Lapidot T, Sirard C, Bormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 172.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 173.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MR. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lee J-S, Heo J, Libbrecht L, Chu I-N, Kaposi-Novak P, Calvisi DF, Mikaelyan A, Roberts LR, Demetris AJ, Sun A, Nevens F, Roskams T, Thorgerisson SS. A novel prognostic subtype of human hpatocellular carcinoma derived from hepatic progenitor cells. Nature Med. 2006;12:410–416. doi: 10.1038/nm1377. [DOI] [PubMed] [Google Scholar]
- 175.Yin S, Li J, Hu C, Chen X, Yao M, Yan M, Jiang G, Ce C, Xie H, Wan D, Yang S, Zheng S, Gu J. CD133 positive hematocellular carcinoma cells posess high capacity for tumorigenicity. Int J Cancer. 2007;120:1444–1450. doi: 10.1002/ijc.22476. [DOI] [PubMed] [Google Scholar]
- 176.Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO, Zheng BJ, Guan XY. Identification and characterization of tumorigenic liver cancer stem/progeniotro cells. Gastroenterol. 2007;132:2542–2556. doi: 10.1053/j.gastro.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 177.Ma S, Lee TK, Zheng B-J, Chan KW, Guan X-Y. Cd133+ HCC canacer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749–1758. doi: 10.1038/sj.onc.1210811. [DOI] [PubMed] [Google Scholar]
- 178.Yang ZF, Ngai P, Ho DW, Yu WC, Ng MNP, Lau CK, Li MLY, Tam KO, Lam CT, Poon RTP, Fan ST. Identification of local and circulating cancer stem cells in human liver cancer. Hepatology. 2008;47:919–928. doi: 10.1002/hep.22082. [DOI] [PubMed] [Google Scholar]
- 179.Yang ZF, Ho DW Ng MN, Lau CK, Yu WC, Ngal P, Chu PEK, Lan CT, Poon RTP, Fan ST. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153–166. doi: 10.1016/j.ccr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 180.Zen Y, Fujii T, Yoshikawa S, Takamura H, Tani T, Ohtia T, Nakanuma Y. Histological and culture studies with respect to ABCG2 expression support the existence of a cancer cell hierarchy in human hepatocellular carcinoma. Am J Pathol. 2007;170:1750–1762. doi: 10.2353/ajpath.2007.060798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yamashita T, Forgues M, Wang W, Kim JW, Ye Q, Jia H, Budu A, Zanetti KA, Chen Y, Qin L-X, Tang Z-Y, Wang XW. EpCAM and α-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008;68:1451–1461. doi: 10.1158/0008-5472.CAN-07-6013. [DOI] [PubMed] [Google Scholar]
- 182.Kaposi-Novak P, Lee J-S, Gomez-Quiroz L, Coulouarn C, Factor VM, Thorgerisson SS. Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J Clin Invest. 2006;116:1582–1595. doi: 10.1172/JCI27236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Trott KR. Tumour stem cells: the biological concept and its application in cancer treatment. Radiotherapy and Oncology. 1994;30:1–5. doi: 10.1016/0167-8140(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 184.Denekamp J. Tumour stem cells: facts, interpretation and consequences. Radiotherapy and Oncology. 1994;30:6010. doi: 10.1016/0167-8140(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 185.Rich JN. Cancer stem cells in radiation resistance. Cancer Res. 2007;67:8980–8984. doi: 10.1158/0008-5472.CAN-07-0895. [DOI] [PubMed] [Google Scholar]
- 186.Cairns J. Somatic stem cells and the kinetics of mutagenesis and carcinogenesis. Proc Natl Acad Sci U S A. 2002;99:10567–10570. doi: 10.1073/pnas.162369899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cairns J. Cancer and the immortal strand hypothesis. Genetics. 2006;174:1069–1072. doi: 10.1534/genetics.104.66886. [DOI] [PMC free article] [PubMed] [Google Scholar]