

Abstract
Nitric oxide-donating aspirin (NO-ASA) is a promising agent for the control of cancer, whose mechanism of action remains unclear. NF-κB is an important signaling molecule in the pathogenesis of cancer. We studied in several human colon (HT-29, HCT-15, LoVo, HCT116 and SW-480), pancreatic (BxPC-3, MIA PaCa-2) and breast (MDA-MB-231 and MCF-7) cancer cell lines, the effect of NO-ASA on NF-κB activation, determined by electrophoretic mobility shift assays, immunofluorescence and western blot analyses of nuclear proteins. NO-ASA inhibited NF-κB activation, as early as 30 min and with IC50s ranging between 0.83 and 64 μM. Such inhibition was also observed at NO-ASA concentrations that had an insignificant or marginal effect on cell growth. The effect of NO-ASA on NF-κB binding to DNA was significantly correlated with its effect on cell growth (P < 0.05) indicating that the growth inhibitory effect of NO-ASA may be mediated by its effect on NF-κB. Compared with control, NO-ASA decreased NF-κB activation in intestinal epithelial cells of APCmin+/− mice by 38.4% (P < 0.01). Western blot and immunofluorescence analyses revealed that the nuclear levels of the p50 and p65 NF-κB subunits were virtually unaffected, suggesting an inhibitory mechanism different from suppressed subunit translocation into the nucleus. Inhibition of NF-κB activation by NO-ASA may account, at least in part, for its chemopreventive efficacy.
Introduction
Cancer prevention emerges as a promising approach to the control of cancer, a major medical challenge of our times. The feasibility and promise of chemoprevention are exemplified by the case of colon cancer. Despite efforts to develop effective therapies, colon cancer, one of the most prevalent cancers in the western world, is the third leading cause of cancer-related deaths in the USA. The limited efficacy of current treatments for advanced colon cancer has provided the impetus for a concerted effort to identify suitable chemopreventive agents. A (partial) list of such agents includes non-steroidal antiinflammatory drugs (NSAIDs), calcium, antioxidants, polyamine inhibitors, dithiolthiones, polyphenols, vitamin E and selenium (1,2). Epidemiological studies suggested that NSAIDs reduce by about half the incidence of and mortality from colon cancer (3), but recent interventional studies showed the effect of aspirin (ASA) to be much lower, generally between 20 and 30% (3). Compared with non-users, long-term users of NSAIDs have about three times greater relative risk for serious adverse gastrointestinal events (4). The need for a safer and more effective alternative to NSAIDs has led to the synthesis of nitric oxide-donating NSAIDs, currently under intense evaluation as chemopreventive agents against colon and other cancers (5,6).
Nitric oxide-donating NSAIDs are conventional NSAID molecules to which a moiety that releases NO (-ONO2) is covalently attached via a spacer molecule (Figure 1F). The most effective nitric oxide-donating NSAIDs is nitric oxide-donating aspirin (NO-ASA), which is several hundred fold more potent than traditional ASA in inhibiting the growth of colon cancer cells in culture (7). This is also true for cell lines from a variety of tissues such as pancreas, lung, prostate and tongue (8). NO-ASA inhibits the growth of colon cancer cells through a favorable cell kinetic effect that includes decreased proliferation, increased cell death and a block in cell-cycle transitions (9). The mechanism responsible for the enhanced potency of ASA when it is converted to NO-ASA is unknown, but is believed to include effects on cell signaling mediated by cyclooxygenase-2, β-catenin/T-cell factor, nitric oxide synthase-2 and NF-κB (10). Our data on the effect of NO-ASA on NF-κB were of a preliminary nature. Thus, we undertook a detailed study of this potentially significant interaction.
Fig. 1.
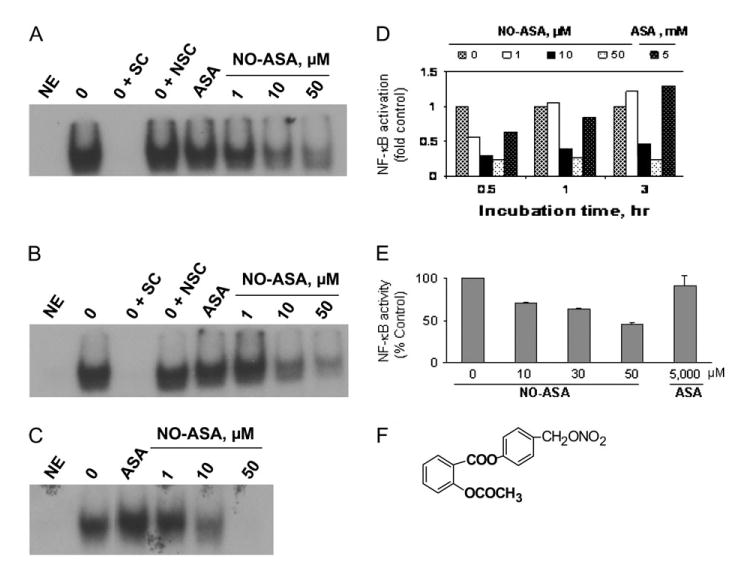
The structure and inhibitory effect of NO-ASA. (A), (B) and (C) Inhibition of NF-κB–DNA binding by NO-ASA. EMSA was performed on nuclear extracts from HT-29 cells treated with NO-ASA for (B) 0.5, (C) 1 and (D) 3 h, as in Materials and methods. (D) Band intensity was determined by densitometry. (E) Inhibition of NF-κB activity by NO-ASA. Nuclear extracts of HT-29 colon cancer cells treated for 3 h with NO-ASA were isolated and the activity of NF-κB was measured by ELISA as in Materials and methods. The IC50 for NF-κB activity as determined under these conditions was 45 μM. This study was repeated twice in duplicate and the results were within 15%. (F) Depiction of the three structural components of NO-ASA (the conventional ASA, -ONO2, the NO-releasing group, and the spacer molecule linking the two). NE, no extract; SC, specific competitor (unlabeled NF-κB oligonucleotide); NSC, non-specific competitor (unlabeled AP-1 oligonucleotide).
NF-κB is a dimer most commonly consisting of the subunits p50 and p65; homodimers of p50, p52 or p65 have also been detected (11). NF-κB is sequestered in the cytoplasm bound to the inhibitory factor IκBα. Phosphorylation of IκBα in response to inducers such as cytokines, reactive oxygen species or infection results in its degradation, unmasking of the nuclear localization signal and activation of NF-κB. As a result, NF-κB translocates to the nucleus and binds to the NF-κB binding site (κB site) in the regulatory region of target genes, thereby promoting the transcription of several regulatory proteins. NF-κB, a strategic signaling molecule in inflammation, promotes cell growth by increasing cell proliferation and inhibiting apoptosis, an effect that directly contributes to carcinogenesis. The nuclear transcriptional factor NF-κB is constitutively expressed in many human cancers, with pancreatic cancer representing a characteristic example (12,13). Over two-thirds of pancreatic cancer display activation of NF-κB, an event considered central to inflammation, a recently appreciated major contributor to carcinogenesis. Of note, NO-ASA is the most effective chemopreventive agent against pancreatic cancer in a preclinical model of pancreatic cancer (14).
Herein, we report our results documenting a profound inhibitory effect of NO-ASA on NF-κB activation, which occurs both in vitro and in vivo and is significantly correlated with NO-ASA's cell growth inhibitory effect. These findings may explain in part the chemopreventive effect of NO-ASA.
Materials and methods
Reagents
We prepared 100-mM stock solutions of para NO-ASA [2-(acetyloxy)benzoic acid 4-(nitrooxymethyl) phenyl ester] synthesized by us (15) and conventional ASA (Sigma, St Louis, MO) in DMSO; the final DMSO concentration in all media was adjusted to 1%. All general solvents and reagents were of high-performance liquid chromatography grade or the highest grade commercially available.
Cell culture
Human colon (HT-29, LoVo, HCT116, HCT-15 and SW-480), breast (MCF-7 and MDA-MB-231) and pancreatic (BxPC-3 and MIA PaCa-2) cell lines (American Type Culture Collection, Manassas, VA) were grown as monolayers in the medium suggested by American Type Culture Collection and supplemented with 10% fetal calf serum (Mediatech, Herndon, VA), penicillin (50 U/ml) and streptomycin (50 μg/ml; Life Technologies, Grand Island, NY). Cells were incubated at 37°C in 5% CO2.
Cell growth
Cell viability/growth was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay according to the manufacturer's instructions (Roche Diagnostics, Nutley, NJ). In brief, cells were seeded in 96-well tissue culture plates at a density of 1 × 104 cells per well and allowed to adhere overnight. On the following day, cells were treated with NO-ASA at concentrations ranging from 1 to 1000 μM for up to 24 h. At the designated time, 10 μl of MTT reagent I was added to the culture media and the plate returned to the 37°C incubator for 4 h, at which time, 100 μl of the solubilization solution (MTT reagent II) was added and incubation continued overnight. Cellular proliferation was determined spectrophotometrically using an enzyme-linked immunosorbent assay (ELISA) plate reader at 590 nm. The MTT assay was validated in our system by the direct trypan blue method.
Nuclear extracts
Nuclear extracts were prepared following standard protocols: trypsinized cells were suspended in lysis buffer to which Nonidet P-40 was added at a subsequent step; nuclei were washed and centrifuged, followed by resuspension in extraction buffer and pelleting (16). Nuclear extracts were stored at −80°C until further analysis.
Electrophoretic mobility shift assays
The NF-κB-DNA binding activity was assessed by reacting extracts from control and cells treated with NO-ASA with 33 nmol of a 32P-end labeled 22-mer double-stranded NF-κB consensus oligonucleotide (AGTTGAGGGGACTTTCCCAGGC; Promega Corp., Madison WI). The specificity of the binding was tested by competition reactions, using 50-fold excess of unlabeled NF-κB (specific) or AP-1 consensus (non-specific) oligonucleotides. The complexes were separated on a 6% gel following standard protocols.
Enzyme-linked immunosorbent assay
TransBinding NF-κB assay was performed using an ELISA plate method in which an oligonucleotide harboring a NF-κB consensus binding site was immobilized on a 96-well plate (Panomics, Fremont, CA). Nuclear extracts isolated from colon cancer cells (10 μg) were added to wells of the ELISA plate. The activated NF-κB p50 from the nuclear extract was allowed to bind specifically to the immobilized oligonucleotide. The bound complex was detected by antibody directed against the p50 subunit. A secondary HRP-conjugated antibody was added, followed by addition of the substrate 3,3′,5,5′-tetramethylbenzidine, and the complete complex was quantified by spectrophotometry.
Western blot analysis
Proteins were extracted, fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes (17). Whole cell lysates were obtained using Radio-Immunoprecipitation Assay buffer (18). Probing with antibodies against the NF-κB subunits p65, p50, IκBα, pIκB-α, Sam68 or β-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA) was done for 1 h at room temperature or overnight at 4°C. The membranes were developed by the enhanced chemiluminescence system (Amersham Biosciences, Piscataway, NJ). Bands were quantified using UVP LABWORKS Imaging Analysis software (UVP LABWORKS, Upland, CA) and normalized to a housekeeping protein.
Immunofluorescence
Cells were washed with phosphate-buffered saline and fixed with methanol at 20°C for 10 min. Nuclei were premeabilized by treatment with 0.1% Triton X-100/phosphate-buffered saline for 5 min. Cells were blocked in 2% BSA/phosphate-buffered saline for 15 min and incubated with rabbit p65, p50 or IgG control antibody (Santa Cruz/1:200) for 1 h at room temperature. Subsequently, cells were incubated with Alexa Fluor® 488 anti-rabbit secondary antibody for 1 h at 4°C (1:2000). The Zeiss LSM 510 META NLO Two-Photon Laser Scanning Confocal Microscope System was used for detection of staining. The intensity of nuclear staining was determined by using the area measurement function within the histogram tool bar of the Zeiss LSM Image Browser.
Animal study
Apcmin/+ mice (Jackson laboratory, Bar Harbor, ME) were treated with NO-ASA (100 mg/kg body weight intrarectally daily) or vehicle for 3 weeks. Following euthanasia, the colon tissue was immediately preserved in 10% buffered formalin and processed for histopathology.
Immunohistochemistry
Tissue samples were isolated, fixed in 10% formalin, embedded in paraffin and sections were placed on slides. The tissue sections were dewaxed and rehydrated according to standard protocols. Tissue sections were pre-treated for antigen retrieval by boiling in a microwave oven for 15 min in a 0.01 M solution of citric acid solution (pH 6.0). Staining was performed according to the instructions for rapid immunohistochemical staining using the VECTASTAIN kit (Vector Laboratories, Burlingame, CA). The primary antibody (Chemicon International, Temecula, CA) detected the activated form of NF-κB, as it specifically recognized the epitope overlapping the nuclear location signal of the NF-κB heterodimer (19). An isotypic antibody control was included with each tissue section.
Scoring
It was performed using Adobe PhotoShop. By using the ‘sampled colors’ function, the positive colors were converted to white and the background was deleted. The histogram function was used to generate the luminosity, which was proportional to the intensity of staining. The gradient luminosities were calculated and used to obtain the ‘mean value’ generating the ‘immunohistochemistry (IHC) luminosity index’. In an independent experiment, we examined the activation of p65 in different tissue sections derived from the same tissue blocks, as described above. The ‘extent of expression’ of activated NF-κB in the epithelial cells of each sample was scored based on the proportion of cells staining positive (0 = no staining, 1+ = up to 1/3 of cells, 2+ = 1/3–2/3, 3+ = 2/3 to 90% and 4+ = 90–100% of cells). We also scored the ‘intensity of staining’ using the following scale: 0 = no staining, 1+ = light brown staining, 2+ = between 2+ and 3+ and 3+ = maximal staining.
Results
NO-ASA inhibits the growth of adenocarcinoma cell lines
In agreement with previous findings, NO-ASA profoundly inhibited cell growth (9). The IC50 for cell growth inhibition in response to NO-ASA, shown in Table I, varied among cell lines originating from a given tissue as well as between cancer types. The IC50 for colon cancer cell lines treated with NO-ASA for 24 h ranged between 10 ± 0.9 and 57 ± 6.3 μM. The IC50s for the pancreatic (range 9.1–22.0 μM) and breast (range 5.2–14.4 μM) cancer cell lines were, in general, smaller than those of colon. These results underscore the broad spectrum of the in vitro cell growth inhibitory effect of NO-ASA.
Table I.
NO-ASA inhibits the growth of human adenocarcinoma cell lines
| Cell line | Apc | p53 | COX-2 | ER | IC50 (μM) |
|---|---|---|---|---|---|
| Colon | |||||
| SW-480 | Mutant | Mutant | − | 33.0 ± 5.0 | |
| HT-29 | Mutant | Mutant | + | 16.0 ± 2.2 | |
| HCT-15 | Mutant | Wild-type | − | 10.0 ± 0.9 | |
| LoVo | Mutant | Wild-type | +a | 20.6 ± 1.4 | |
| HCT116 | Wild-type | Wild-type | − | 57.0 ± 6.3 | |
| Pancreas | |||||
| BxPC-3 | Mutant | Mutant | + | 9.1 ± 0.12 | |
| MIA PaCa-2 | Mutant | − | 22.0 ± 2.0 | ||
| Breast | |||||
| MCF-7 | Wild-type | Wild-type | −b | + | 14.4 ± 1.3 |
| MDA-MB-231 | Wild-type | Mutant | + | − | 5.2 ± 2.1 |
NO-ASA inhibits the activation of NF-κB in various colon cancer cell lines
Given the modulating effect of NF-κB on cell growth, we investigated whether NO-ASA affects the activation of NF-κB in these cell lines. We have observed increase in apoptosis and necrosis in cells treated with NO-ASA for 24–72 h (9). It was, however, important to examine these cells for the effect of NO-ASA on NF-κB prior to the induction of overt cellular damage. Study of NO-ASA induced cell death during incubation periods <24 h revealed that at 3 h there was no apoptosis. Therefore, in most experiments presented here cells were treated with NO-ASA for up to 3 h.
As shown in Figure 1, NO-ASA inhibited NF-κB activation evaluated using electrophoretic mobility shift assays (EMSAs). Such inhibition occurred as early as 30 min and at as low a NO-ASA concentration as 10 μM; at 3 h, 50 μM of NO-ASA blocked essentially all NF-κB binding to DNA. The IC50 for this effect was time dependent, being 5.6, 7.8 and 8.6 μM at 0.5, 1 and 3 h, respectively. In contrast, conventional ASA 5 mM had only a marginal effect on the DNA binding of NF-κB.
NF-κB activity in response to NO-ASA was also analyzed using an ELISA method. Nuclear extracts of HT-29 colon cancer cells treated with NO-ASA for 3 h were quantified for NF-κB activity. Similar to what was observed by EMSA, NO-ASA inhibited NF-κB activity in cells treated for 3 h in a concentration-dependent manner (Figure 1E); the IC50 for this effect was 33.5 μM.
To determine whether the inhibitory effect of NO-ASA on NF-κB binding was unique to HT-29 cells, we examined four additional human colon cancer cell lines differing with respect to their Apc and p53 status and COX-2 expression (Table I). As seen in Figure 2, at 3 h NO-ASA's IC50 for the binding of NF-κB to DNA was 5 μM for LoVo colon cancer cells, 0.83 μM for HCT-15, 46.3 μM for HCT116 and 64 μM for SW-480. Similar to HT-29 cells, the effect of ASA on NF-κB binding was marginal on all these cell lines.
Fig. 2.
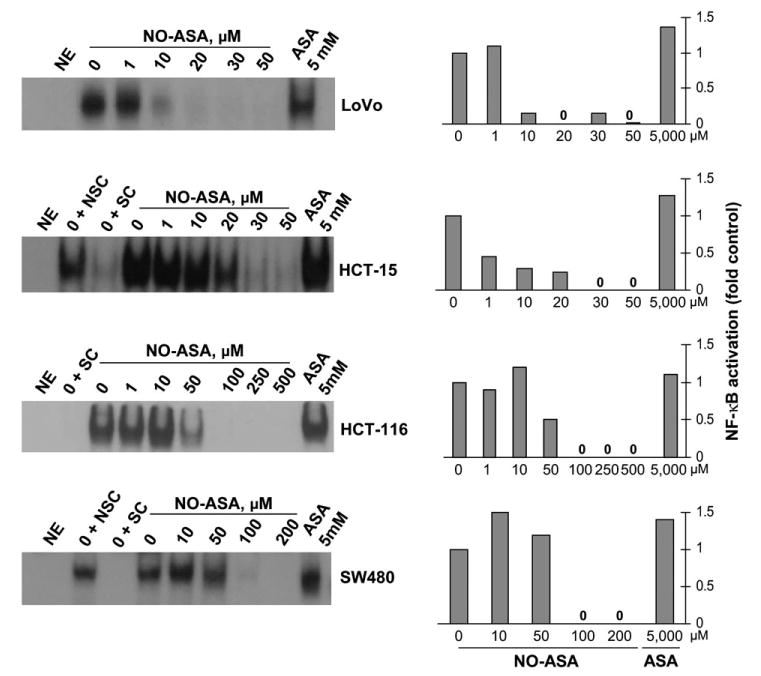
NO-ASA inhibits NF-κB–DNA binding in human colon cancer cell lines. LoVo, HCT-15, HCT116 and SW-480 cells were treated with NO-ASA or ASA for 3 h at the indicated concentrations. EMSAs were performed as described in Materials and methods. Abbreviations are as defined in Figure 1. The results of the densitometric analysis of each band are shown on the right. Each study was repeated at least three times giving results within 15%.
Overall, NO-ASA decreased the binding of NF-κB in a concentration-dependent manner. However, we did observe a slight increase in most cell lines treated with low concentrations of NO-ASA. This increase was consistent between experiments and was not due to unequal sample loading; equal sample loading was confirmed in parallel control samples immunoblotted for the nuclear protein Sam68 (data not shown).
NO-ASA inhibits the activation of NF-κB in breast and pancreatic cancer cell lines
In much the same fashion as seen with colon cancer cell lines, NO-ASA inhibited nuclear NF-κB binding in breast and pancreatic cells. As determined by EMSA, NO-ASA decreased the baseline binding of NF-κB to DNA in breast and pancreatic cancer cell lines in a concentration-dependent manner (Figure 3A). In MCF-7 and MDA-MB-231 breast cancer cells, at 3 h NO-ASA decreased NF-κB–DNA binding starting at 10 μM and this inhibition was virtually complete at 50 μM. The IC50s for this effect were 8.9 and 30 μM for MCF-7 and MDA-MB-231, respectively. Similarly, NO-ASA inhibited the DNA binding of NF-κB in the pancreatic cancer cell lines BxPC-3 and MIA PaCa-2 with an IC50 of 11.5 and 7 μM, respectively. These effects mirror NO-ASA's effect on HT-29 colon cancer cells, shown in Figure 1, suggesting that most probably this effect is independent of the tissue origin of any given cell line.
Fig. 3.
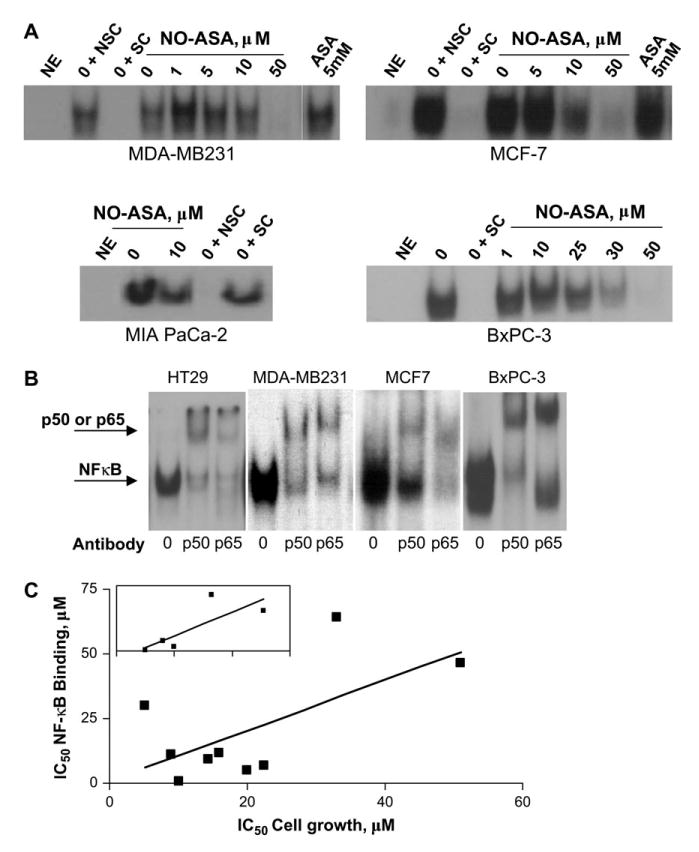
NO-ASA inhibits NF-κB–DNA binding in cancer cell lines of varying origins. (A) The ability of nuclear NF-κB to bind its cognate binding site after treatment with varying concentrations of NO-ASAwas determined using EMSA performed as in Materials and methods. The cancer cell lines indicated here were treated with NO-ASA or ASA for 3 h. Abbreviations are as defined in Figure 1. (B) Supershift analysis using antibodies to p50 or p65. Each study was repeated at least three times giving similar results. (C) The association between the inhibition of growth and NF-κB binding by NO-ASA. The IC50 for NF-κB binding is graphed against that for cell growth in all nine cell lines (colon, breast and pancreas) that we studied. The correlation between the two is statistically significant (P < 0.05). Inset: a similar graph for the five colon cancer cell lines; the association between these two parameters is statistically significant (P < 0.05).
The role of NF-κB inhibition in cancer cell growth
As shown in Figure 3C, we explored whether the effect of NO-ASA on NF-κB binding to DNA correlated with its effect on cell growth. When we combined all nine cell lines (colon, pancreatic and breast) into one group, the IC50s for NF-κB activation and cell growth were significantly associated (P < 0.05). A similar result was obtained when we restricted our analysis to the five colon cell lines (P < 0.05). These findings suggest that NF-κB probably mediates the cell growth inhibitory effect of NO-ASA on cancer cell lines, regardless of their tissue of origin.
The effect of NO-ASA on the nuclear levels of NF-κB subunits p50 and p65
Supershift analyses demonstrated that in these cell lines the NF-κB heterodimer consists of p50 and p65 (Figure 3B). A non-specific antibody, used as a control, failed to generate the supershift (data not shown). To determine whether the inhibition of NF-κB activation by NO-ASA was due to decreased nuclear amounts of the NF-κB subunits, we monitored the levels of p50 and p65. At NO-ASA concentrations that inhibited the binding of NF-κB to DNA, the p50 and p65 subunits were present at amounts equal to or modestly below control levels (Figure 4A). Therefore, the inhibition of NF-κB binding appears to occur without the loss or appreciable reduction of the NF-κB subunits (p50 and p65) in the nucleus. Results from immunofluorescence analyses of HT-29 and BxPC-3 cells treated with NO-ASA were consistent with those of the immunoblot analyses. As seen in Figure 4B, the fluorescence from p50 and p65 in the nuclei of HT-29 colon cancer cells treated with various concentrations of NO-ASA remained virtually unchanged compared with untreated controls, with the exception of p50 levels that were increased at the highest NO-ASA concentration (100 μM). In contrast, the levels of both p50 and p65 increased significantly over controls following treatment with TNF-α. Results for the BxPC-3 cell line (data not shown) were similar.
Fig. 4.
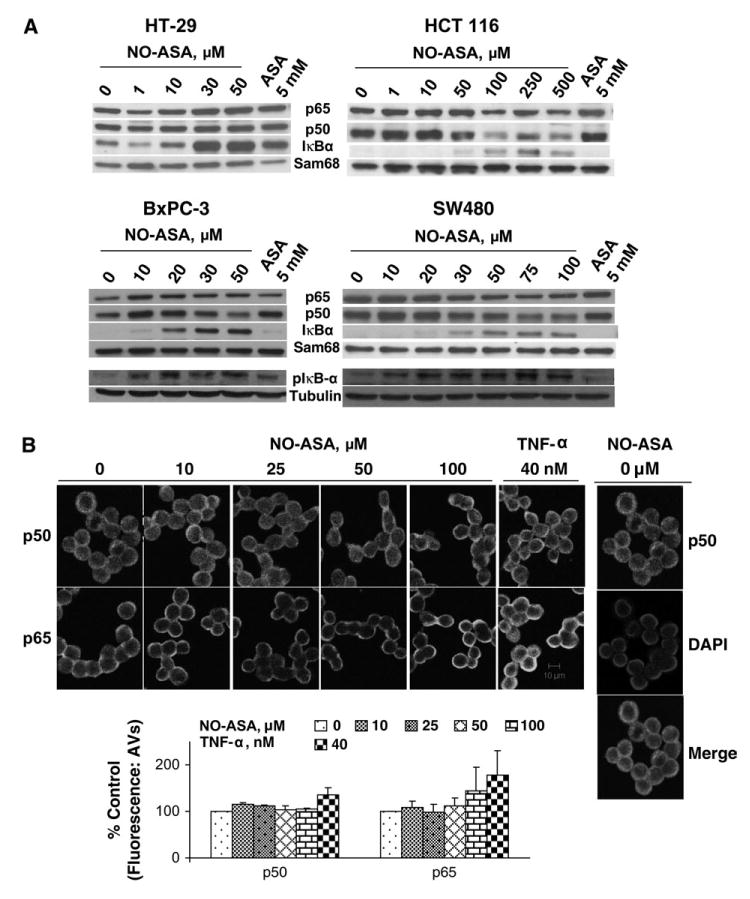
Nuclear levels of NF-κB subunits in cancer cells treated with NO-ASA. (A) Protein nuclear extracts from three colon (HT-29, HCT116 and SW-480) and one pancreatic (BxPC-3) human cancer cell line treated with NO-ASA or ASA for 3 h were subjected to western blot analysis using antibody against p50, p65, IκBα and Sam68 (loading control). Although the levels of p50 and p65 are in some of them decreased, they are higher than expected considering their level of binding to the labeled DNA probe (compare with corresponding bands in Figures 1, 2 and 3). Bottom of western blot: pIκB levels in whole cell lysates from human colon (SW480) and pancreatic (BxpC-3) cancer cell lines. (B) Immunofluorescence analysis of NF-κB subunits in HT-29. HT-29 colon cancer cells treated with NO-ASA or TNF-α for 3 h were reacted with anti-p50 (top panel) and anti-p65 (lower panel) antibodies or an isotypic control (data not shown). Immunofluorescence was detected using a Two-Photon Laser Scanning Confocal Microscope as in Materials and methods. In all instances, 4′,6-diamidino-2-phenylindole staining was used to identify the nucleus (representative figure on side). Fluorescence was quantified and plotted; TNF-α is the positive control for NF-κB activation. Horizontal bar: 10 μm.
To clarify this unusual situation, we also assayed nuclear levels of IκBα, a protein known to export NF-κB subunits from the nucleus to the cytoplasm by physically associating with them (22,23). As shown in Figure 4A, the nuclear levels of IκBα were increased in response to NO-ASA. In fact, there appears to be roughly an inverse relationship between the inhibition of NF-κB by NO-ASA and the nuclear levels of IκBα. That the nuclear levels of IκBα that we observed are indeed nuclear and not cytoplasmic contaminates generated during its isolation procedure was documented by examining the Sam68 (nuclear) and tubulin (cytoplasmic) control proteins. Thus, it is unlikely that lack of IκBα accounts for maintaining p50 and p65 at around baseline levels. In addition, we examined cellular levels of pIκB. As shown in Figure 4A, the phosphorylation of IκB increased in response to NO-ASA treatment as compared with control levels. Thus, the inhibitory effect of NO-ASA on NF-κB binding cannot be solely attributed to sequestering of NF-κB in the cytoplasm.
We considered that IκBα might engage in a futile physical interaction with the NF-κB subunits such that it prevented their binding to the NF-κB recognition sequence, while also abrogating their translocation into the cytoplasm. Thus, we attempted to coimmunoprecipitate IκBα and p50 or p65 and also to assess their potential colocalization by confocal microscopy. Both efforts proved negative (data not shown).
NO-ASA decreases the activation of NF-κB in Min (Apcmin/+) mice
We assessed whether NO-ASA modulates the NF-κB signaling system in vivo as it does in vitro. Thus, we examined the activation of NF-κB in the colon of 6-week-old female Min (Apcmin/+) mice treated with NO-ASA (100 mg/kg body weight intrarectally daily for 3 weeks). These mice represent an excellent model system of intestinal cancer, as they recapitulate many of its molecular features (24).
Treatment of Min (Apcmin/+) mice with NO-ASA decreased the number of tumors in the small intestine by 59% (P < 0.01), compared with vehicle-treated controls (25). Small intestinal tissue from these animals was examined by IHC, using an antibody that recognizes the activated form of NF-κB. The results are summarized in Figure 5. Compared with controls, NO-ASA decreased the activation of NF-κB by 38.4% (P < 0.01), as determined by the IHC luminosity index. Wild-type control mice treated with vehicle or NO-ASA showed little or no positive staining with this antibody, demonstrating essentially no NF-κB activation. This finding is consistent with the notion that NF-κB is activated during carcinogenesis (26).
Fig. 5.
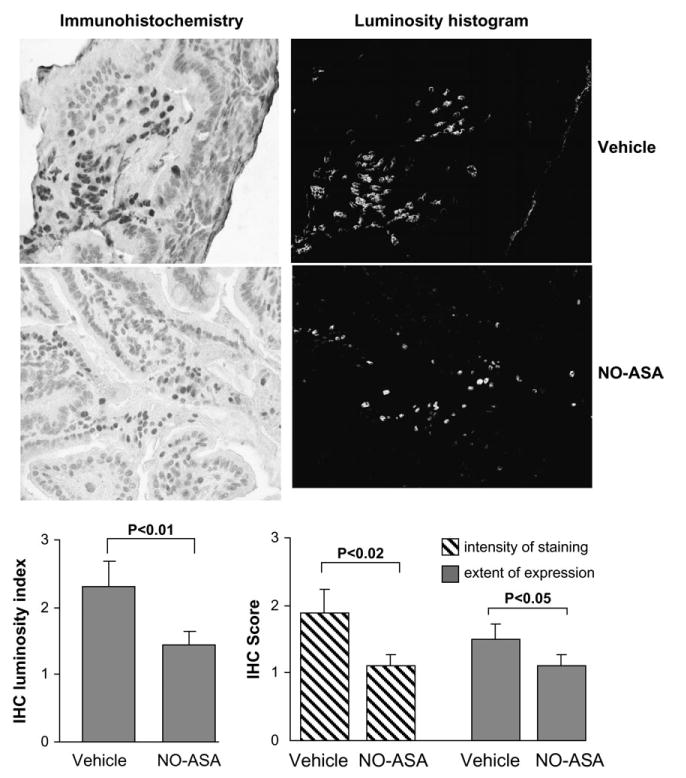
NO-ASA inhibits NF-κB activation in the intestine of Min mice. Immunohistochemical analysis of small intestinal tissue from Min mice treated with NO-ASA or vehicle as in Materials and methods. Representative images (magnification, ×400) of immunohistochemically stained tissues from NO-ASA (upper left) or vehicle (lower left) mice are shown along with the corresponding images of the histogram. The luminosity index for each group is shown in the left graph; NO-ASA decreased it by 38.4% (P < 0.01). In an independent experiment, immunohistochemically stained tissue sections were scored for extent of expression of activated NF-κB in the epithelial cells (decreased 47% by NO-ASA) as well as for its intensity of staining (decreased 33% by NO-ASA) as defined in Materials and methods. Vertical bars: standard error of the mean; n = 10 for each group.
In an independent experiment, different sections of the same tissue samples were stained by IHC and scored for NF-κB expression in terms of its intensity and extent, as defined in Materials and methods. NO-ASA decreased the extent of expression of activated NF-κB in murine intestinal epithelial cells by 47% (P < 0.02) and its intensity by 33% (P < 0.05). Both sets of data are in close agreement.
Discussion
Our data indicate that NO-ASA inhibited the activation of N-FκB in a variety of human cancer cell lines through an apparently novel mechanism. Inhibition of NF-κB activation was also noted in an animal model of intestinal cancer.
Our study included human cancer cell lines derived from three clinically important cancers, colon, pancreas and breast, each represented by more than one cell line. In agreement with our previous findings, NO-ASA inhibited potently the growth of all these cell lines (8,10). The presence or absence of COX-2 had no apparent effect on drug potency, whereas the estrogen receptor positive breast cancer cell line appeared less susceptible (by a factor of three) to the inhibitory effect of NO-ASA compared with the one that was estrogen receptor negative. Conventional ASA, even at millimolar concentrations, had no appreciable effect on cancer cell growth, as noted previously (8,10).
NO-ASA inhibited NF-κB activation as determined either by the EMSA or by an ELISA method assessing its biological activity. Inhibition of NF-κB activation was noted as early as 30 min after exposure to NO-ASA, even before any signs of cell death associated with exposure to NO-ASA. The effect of NO-ASAwas clearly concentration dependent as assessed by both EMSA and ELISA. It is of interest, however, that in some cell lines at relatively low concentrations of NO-ASA, NF-κB activation was modestly increased (up to 10–30%) by EMSA. No such stimulation was observed by the ELISA method.
We explored the mechanism of the inhibitory effect of NO-ASA on NF-κB by determining the nuclear levels of its two subunits. Supershift assays revealed that in all cell lines that we tested, the NF-κB dimer was made up of p50 and p65. In the one pancreatic and three colon cell lines that we studied, we demonstrated that over a wide range of NO-ASA concentrations associated with changes in NF-κB activation, the levels of p65 were either unchanged or very modestly decreased compared with controls. A similar observation was made for p50, although at the highest NO-ASA concentrations there was a clear and consistent reduction in its levels. Immunofluorescence analysis of these levels gave similar results, while the positive control (treated with TNF-α) generated the expected rise in p50 and p65 levels. It is clear that for any given concentration of NO-ASA, the NF-κB protein levels in the nucleus cannot account for its reduced binding to the κB DNA recognition sequence. This observation establishes that the reduced NF-κB activity in response to NO-ASA is brought about through a mechanism that does not involve quantitative changes of the NF-κB proteins. In addition, the induction of cytoplasmic levels of pIκB is further evidence that NO-ASA does not reduce the nuclear levels of NF-κB. Thus, the inhibitory binding effect of NO-ASA was not a result of sequestering of the protein in the cytoplasm by IκB. Our conclusion differs from the numerous examples of decreased NF-κB activation owing to diminished nuclear levels of NF-κB subunits [e.g. (27–29)], a mechanism clearly consistent with current understanding of NF-κB regulation.
The possibility that changes in the nuclear levels of IκBα, a protein known to export NF-κB subunits from the nucleus to the cytoplasm (30,31), may account for the observed unusual levels of p50 and p65 in the nucleus is highly unlikely. IκBα levels increased in an apparently inverse proportion to the inhibition of NF-κB by NO-ASA, indicating that p50 and p65 levels were not influenced by quantitative changes in their transport protein IκBα. The possibility that IκBα might prevent NF-κB binding to its recognition sequence while also abrogating the translocation of its subunits into the cytoplasm is also unlikely, as we have failed to demonstrate any such interaction.
We can speculate, for example, that the subunits of NF-κB are modified by NO-ASA in a way that prevents them from binding to DNA. A prime candidate for such a modification is S-nitrosylation of cysteine residues, similar to what we speculated regarding β-catenin (32). Our preliminary data indicate that both p50 and p65 are nitrosylated in response to NO-ASA. In such a case, one could invoke steric hindrance from cysteine S-NOs affecting the κB-binding domain of the protein. Alternatively, conformational changes of one or both NF-κB subunits as a result of S-nitrosylation could explain the observed results.
NF-κB is known to modulate cell proliferation and cell death, especially by apoptosis, depending on biological context (e.g. cell or tissue type) (26). Our previous work with these and other cell lines (33) has demonstrated that NO-ASA inhibits cell growth through its combined effects on cell renewal and cell death. Our demonstration that the IC50 for inhibition of NF-κB is significantly correlated with the IC50 for the inhibition of cell growth is in keeping with such a role of NF-κB in cell kinetics. This association was statistically significant when we combined all nine cell lines (colon, pancreatic and breast) into one group as well as when we examined the five colon cell lines as a separate group. This observation indicates that the association between NF-κB and cancer cell growth in response to NO-ASA is probably a generalized property.
Based on our data, it is difficult to conclude that the effect of NO-ASA on NF-κB is responsible, alone or in combination with other effects, for its pharmacological effects against cancer. Nevertheless, such a possibility is plausible. NF-κB is an important factor in carcinogenesis through both its modulation of cell kinetics and its central role in inflammation; NF-κB is currently being appreciated as an important link between inflammation and cancer (34). For example, NF-κB is activated in over two-thirds of human pancreatic cancers, participates in early events of pancreatic carcinogenesis through its interactions with signaling pathways and suppression of its activation restores pancreatic cell kinetics, mainly normalizing the suppressed apoptosis of pancreatic cancer (reviewed in ref. 35).
In conclusion, our findings document a quantitatively significant in vitro inhibitory effect of NO-ASA on the activation of NF-κB in a variety of cell lines from three important human cancers, colon, pancreas and breast. This effect was also observed in vivo in a model of intestinal cancer. Although the mechanism of this inhibition is not yet deciphered, it is clearly different from that involving reduced nuclear levels of the constituent proteins of NF-κB. Since NF-κB inhibition by NO-ASA correlates with its cell growth inhibitory effect, this effect may have direct relevance to the chemopreventive effect of NO-ASA and, at least in part, may explain it.
Acknowledgments
Funding: National Institutes of Health (K01 CA10660, R01 CA92423, R01 101019).
Abbreviations
- ASA
aspirin
- ELISA
enzyme-linked immunosorbent assay
- EMSA
electrophoretic mobility shift assay
- IHC
immunohistochemistry
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NO-ASA
nitric oxide-donating aspirin
- NSAID
non-steroidal anti-inflammatory drug
Footnotes
Conflict of Interest Statement: None declared.
References
- 1.Gustin DM, et al. Chemoprevention of colon cancer: current status and future prospects. Cancer Metastasis Rev. 2002;21:323–348. doi: 10.1023/a:1021271229476. [DOI] [PubMed] [Google Scholar]
- 2.Brenner DE, et al. Cancer chemoprevention: lessons learned and future directions. Br J Cancer. 2005;93:735–739. doi: 10.1038/sj.bjc.6602765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baron JA. Epidemiology of non-steroidal anti-inflammatory drugs and cancer. Prog Exp Tumor Res. 2003;37:1–24. doi: 10.1159/000071364. [DOI] [PubMed] [Google Scholar]
- 4.Bjorkman DJ. Current status of nonsteroidal anti-inflammatory drug (NSAID) use in the United States: risk factors and frequency of complications. Am J Med. 1999;107:3S–8S. doi: 10.1016/s0002-9343(99)00362-9. discussion 8S–10S. [DOI] [PubMed] [Google Scholar]
- 5.Konturek PC, et al. NO-releasing aspirin exerts stronger growth inhibitory effect on Barrett's adenocarcinoma cells than traditional aspirin. J Physiol Pharmacol. 2006;57(suppl 12):15–24. [PubMed] [Google Scholar]
- 6.Rao CV, et al. Nitric oxide-releasing aspirin and indomethacin are potent inhibitors against colon cancer in azoxymethane-treated rats: effects on molecular targets. Mol Cancer Ther. 2006;5:1530–1538. doi: 10.1158/1535-7163.MCT-06-0061. [DOI] [PubMed] [Google Scholar]
- 7.Williams JL, et al. Nitric oxide-releasing nonsteroidal anti-inflammatory drugs (NSAIDs) alter the kinetics of human colon cancer cell lines more effectively than traditional NSAIDs: implications for colon cancer chemoprevention. Cancer Res. 2001;61:3285–3289. [PubMed] [Google Scholar]
- 8.Kashfi K, et al. Nitric oxide-donating nonsteroidal anti-inflammatory drugs inhibit the growth of various cultured human cancer cells: evidence of a tissue type-independent effect. J Pharmacol Exp Ther. 2002;303:1273–1282. doi: 10.1124/jpet.102.042754. [DOI] [PubMed] [Google Scholar]
- 9.Kashfi K, et al. Positional isomerism markedly affects the growth inhibition of colon cancer cells by nitric oxide-donating aspirin in vitro and in vivo. J Pharmacol Exp Ther. 2005;312:978–988. doi: 10.1124/jpet.104.075994. [DOI] [PubMed] [Google Scholar]
- 10.Williams JL, et al. Growth inhibition of human colon cancer cells by nitric oxide (NO)-donating aspirin is associated with cyclooxygenase-2 induction and beta-catenin/T-cell factor signaling, nuclear factor-kappaB, and NO synthase 2 inhibition: implications for chemoprevention. Cancer Res. 2003;63:7613–7618. [PubMed] [Google Scholar]
- 11.Pande V, et al. NF-kappaB in human disease: current inhibitors and prospects for de novo structure based design of inhibitors. Curr Med Chem. 2005;12:357–374. doi: 10.2174/0929867053363180. [DOI] [PubMed] [Google Scholar]
- 12.Fujioka S, et al. Function of nuclear factor kappaB in pancreatic cancer metastasis. Clin Cancer Res. 2003;9:346–354. [PubMed] [Google Scholar]
- 13.Wang W, et al. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999;5:119–127. [PubMed] [Google Scholar]
- 14.Ouyang N, et al. Nitric oxide-donating aspirin prevents pancreatic cancer in a hamster tumor model. Cancer Res. 2006;66:4503–4511. doi: 10.1158/0008-5472.CAN-05-3118. [DOI] [PubMed] [Google Scholar]
- 15.Penning TD, et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide (SC-58635, celecoxib) J Med Chem. 1997;40:1347–1365. doi: 10.1021/jm960803q. [DOI] [PubMed] [Google Scholar]
- 16.Natarajan K, et al. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc Natl Acad Sci USA. 1996;93:9090–9095. doi: 10.1073/pnas.93.17.9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirano H, et al. Microsequence analysis of the N-terminally blocked proteins immobilized on polyvinylidene difluoride membrane by western blotting. Electrophoresis. 1993;14:839–846. doi: 10.1002/elps.11501401134. [DOI] [PubMed] [Google Scholar]
- 18.Sefton BM, editor. Labeling Cultured Cells with 32Pi and Preparing Cell Lysates for Immunoprecipitation. John Wiley & Sons, Inc.; New York, NY: 2005. [DOI] [PubMed] [Google Scholar]
- 19.Zabel U, et al. Nuclear uptake control of NF-kappa B by MAD-3, an I kappa B protein present in the nucleus. EMBO J. 1993;12:201–211. doi: 10.1002/j.1460-2075.1993.tb05646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen WS, et al. Tumor invasiveness and liver metastasis of colon cancer cells correlated with cyclooxygenase-2 (COX-2) expression and inhibited by a COX-2-selective inhibitor, etodolac. Int J Cancer. 2001;91:894–899. doi: 10.1002/1097-0215(200102)9999:9999<894::aid-ijc1146>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 21.Liu XH, et al. Differential expression and regulation of cyclooxygenase-1 and -2 in two human breast cancer cell lines. Cancer Res. 1996;56:5125–5127. [PubMed] [Google Scholar]
- 22.Tam WF, et al. Cell-specific association and shuttling of IkappaBalpha provides a mechanism for nuclear NF-kappaB in B lymphocytes. Mol Cell Biol. 2001;21:4837–4846. doi: 10.1128/MCB.21.14.4837-4846.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aguilera C, et al. Efficient nuclear export of p65-IkappaBalpha complexes requires 14-3-3 proteins. J Cell Sci. 2006;119:3695–3704. doi: 10.1242/jcs.03086. [DOI] [PubMed] [Google Scholar]
- 24.Lipkin M, et al. Preclinical mouse models for cancer chemoprevention studies. Ann N Y Acad Sci. 1999;889:14–19. doi: 10.1111/j.1749-6632.1999.tb08719.x. [DOI] [PubMed] [Google Scholar]
- 25.Williams JL, et al. NO-donating aspirin inhibits intestinal carcinogenesis in Min (APC(Min/+)) mice. Biochem Biophys Res Commun. 2004;313:784–788. doi: 10.1016/j.bbrc.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 26.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 27.Ichikawa H, et al. Isodeoxyelephantopin, a novel sesquiterpene lactone, potentiates apoptosis, inhibits invasion, and abolishes osteoclastogenesis through suppression of nuclear factor-kappaB (nf-kappaB) activation and nf-kappaB-regulated gene expression. Clin Cancer Res. 2006;12:5910–5918. doi: 10.1158/1078-0432.CCR-06-0916. [DOI] [PubMed] [Google Scholar]
- 28.Nair AS, et al. Deguelin, an Akt inhibitor, suppresses IkappaBalpha kinase activation leading to suppression of NF-kappaB-regulated gene expression, potentiation of apoptosis, and inhibition of cellular invasion. J Immunol. 2006;177:5612–5622. doi: 10.4049/jimmunol.177.8.5612. [DOI] [PubMed] [Google Scholar]
- 29.Rishi L, et al. Nitric oxide induces apoptosis in cutaneous T cell lymphoma (HuT-78) by downregulating constitutive NF-kappaB. Biochim Biophys Acta. 2007;1770:1230–1239. doi: 10.1016/j.bbagen.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 30.Tam WF, et al. IkappaB family members function by different mechanisms. J Biol Chem. 2001;276:7701–7704. doi: 10.1074/jbc.C000916200. [DOI] [PubMed] [Google Scholar]
- 31.Huang TT, et al. Postrepression activation of NF-kappaB requires the amino-terminal nuclear export signal specific to IkappaBalpha. Mol Cell Biol. 2001;21:4737–4747. doi: 10.1128/MCB.21.14.4737-4747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nath N, et al. Nitric oxide-donating aspirin inhibits beta-catenin/T cell factor (TCF) signaling in SW480 colon cancer cells by disrupting the nuclear beta-catenin-TCF association. Proc Natl Acad Sci USA. 2003;100:12584–12589. doi: 10.1073/pnas.2134840100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeh RK, et al. NO-donating nonsteroidal antiinflammatory drugs (NSAIDs) inhibit colon cancer cell growth more potently than traditional NSAIDs: a general pharmacological property? Biochem Pharmacol. 2004;67:2197–2205. doi: 10.1016/j.bcp.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 34.Karin M, et al. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Z, et al. NF-kappaB, inflammation and pancreatic carcinogenesis: NF-kappaB as a chemoprevention target (review) Int J Oncol. 2006;29:185–192. [PubMed] [Google Scholar]