

Abstract
Studies have suggested a correlation between the decline in infectious diseases and increase in the incidence of T1D in developed countries. Pathogens influence the disease outcome through innate immune receptors such as toll-like receptors (TLRs). Here, we report the effect of ligation of TLR2 and dectin 1 on APCs, and the influence of innate immune response induced through these receptors on T1D. Exposure of APCs of non-obese diabetic (NOD) mice to zymosan, a fungal cell-wall component that is known to interact with TLR2 and dectin 1, resulted in the release of significant amounts of IL-10, TGF-β1, IL-2, and TNF-α. Treatment of pre- and early-hyperglycemic mice with zymosan resulted in suppression of insulitis leading to a significant delay in hyperglycemia. Importantly, T cells from zymosan treated mice showed reduced ability to induce diabetes in NOD-Scid mice compared to control T cells. Zymosan treatment induced suppression of T1D was associated with an increase in the CD62Lhigh T cell frequencies and enhanced suppressor function of CD4+CD25+ Tregs. Further, activation by anti-CD3-Ab induced larger amounts of TGF-β1 and/or IL-10 production by CD4+CD25+ and CD4+CD25- T cells from zymosan treated mice. These results show that innate immune response through TLR2 and dectin 1 results in suppressor cytokine production by APCs, and promotes regulatory function of T cells. Our study demonstrates the possible involvement of signaling through innate immune receptors such as TLR2 and dectin 1 in reduced T1D incidence under the conditions of low hygiene, and the potential of targeting them for treating T1D.
Keywords: Toll-like receptor 2 (TLR2), Dectin 1, Innate immunity, Type 1 diabetes, Zymosan, antigen presenting cells, T cells, regulatory T cells
Introduction
Autoimmune type 1 diabetes (T1D) is induced by environmental factors affecting the genetic susceptibility of individuals ultimately leading to T cell mediated destruction of insulin-secreting β cells in the pancreatic islets (1). Innate immunity is known to play a key role in initiating an effector T cell response as well as in maintaining tolerance to pancreatic β-cell antigens. The correlation observed between the decline in infectious diseases due to improved hygiene and increase in the incidence of T1D in developed countries suggests a role for innate immunity in preventing autoimmunity against pancreatic β cells (2,3). Innate immune response is mediated through a number of pattern recognition receptors such as Toll-like receptors (TLRs) and c-type lectins that are stimulated primarily by microbial agents (4). Upon interaction with appropriate ligands, these receptors on antigen presenting cells (APCs) and other innate immune cells activate signaling pathways that lead to the production of a wide-array of pro-inflammatory cytokines and/or up-regulation of costimulatory ligands such as CD80 and CD86 (5). Due to the ability of signaling through innate immune receptors to trigger the expression and/or up-regulation of these molecules on APCs, they have been considered important in linking innate and adaptive immune responses (6,7).
Although the role of innate immune receptors such as TLRs in initiating T1D is not clearly understood, inflammatory effects of TLR3, TLR2, and TLR9 mediated signals have been implicated in virus-triggered autoimmune diabetes, sensitizing pancreatic β-cells for apoptosis, and/or activating APCs for efficient self-antigen presentation (8-10). Poly (I:C), a TLR3 ligand, and CpG, a TLR9 ligand, and viral infections in the presence of self-peptides could trigger autoimmune responses leading to T1D in mouse and rat models (8,10-13). Recently, it has been suggested that TLR2 ligation on APCs by necrotic/late apoptotic pancreatic β-cells contributes to the initiation of autoimmune diabetes through the activation of antigen-captured APCs (9).
TLR2 recognizes lipoproteins/lipopeptides (LPs) and peptidoglycan of microorganisms (14,15). This receptor is unique in its ability to form heteromers with TLR1 or TLR6 to mediate intracellular signaling, and bacterial LPs signal through TLR2 in a TLR1- or TLR6-dependent manner (17-19). However, TLR2 is also known to signal in a TLR1 and TLR6 independent manner depending on the chain length of the O-acylated fatty acids as well as the assembling of the polypeptide tail (20-21). Interestingly, TLR1 and TLR6 dependent and independent signals lead to identical pattern of gene activation indicating that heteromerization of TLR2 with TLR1 or TLR6 is evolutionarily developed to expand the ligand spectrum to enable the innate immune system to recognize different structures of LPs present in various pathogens (16).
A significant number of studies have shown that TLR2 ligation by its ligands can trigger a balanced innate immune response that includes both pro- and anti-inflammatory components (22-25). Further, TLR2 agonists have shown the potential to induce not only significant amounts of suppressor cytokines, but also to enhance CD4+CD25+ regulatory T cell (Treg) function (26). On the other hand, recent studies have shown that TLR2 regulate the expansion and function of natural Tregs (27,28). These reports have shown that TLR2 signaling enhanced proliferation of Tregs in vitro and in vivo, resulted in a transient suppression of Foxp3 expression, and loss of their suppressor function. However, these expanded Tregs regained suppressor function and showed the ability to prevent inflammatory condition such as spontaneous colitis (27).
Dectin-1, a c-type lectin that plays a crucial role in the recognition of fungi such as Candida and Aspergillus through β-glucans, is now known to collaborate with TLR2 in inducing a balanced pro- and anti-inflammatory innate immune response by APCs (29,30). This indicates that dectin 1 signaling might also contribute significantly to regulatory T cell expansion and function, and approaches to engage both TLR2 and dectin 1 concurrently would have therapeutic values in preventing and treating autoimmune disorders such as T1D. Recent studies using zymosan, a fungal cell wall component, have demonstrated its ability to interact with both TLR2 and dectin 1 simultaneously and induce large amounts of suppressor cytokines along with TNF-α (31,32). Therefore, we explored the potential use of this agent in therapeutically modulating autoimmunity in T1D by triggering a regulatory innate immune response in diabetes-prone female NOD mice at pre- and early- diabetic stages. Our results demonstrate that zymosan treatment protects NOD mice from T1D even at an early-hyperglycemic stage. The disease protection was associated with a profound increase in IL-10, TGF-β1, IL-2, and TNF-α responses by APCs, enhanced natural Treg function, and changes in the effector T cell phenotype and cytokine profile.
Materials and Methods
Mice
Wild-type female NOD/LtJ and NOD-Scid mice were purchased from the Jackson Laboratory. All animal studies were approved by the animal care and use committee of the University of Illinois at Chicago. Non-fasting glucose levels in the tail vein blood samples of wild-type and NOD-Scid mice were monitored with Ascensia Contour blood glucose test strips and glucose meter (Bayer USA).
Reagents
Purified Sacharomyces cerevisiae zymosan was purchased from Sigma-Aldrich and was boiled for 30 minutes, washed extensively and suspended in PBS as described earlier (31). Purified anti-mouse-TGF-β1 (clone: A75-2), anti-CD16/CD32 (FC block) Abs; FITC-conjugated anti-mouse CD11c, CD4, CD25 Abs and streptavidin; PE-labeled anti-mouse, F4/80, CD80, CD86, CD40, I-Ak (which cross-reacts with I-Ag7), TLR2, dectin 1, CD4, CD25, GITR, PD-1, CTLA-4, CD8, CD62L, CD44, CD127, CCR7, Foxp3, and streptavidin; biotin labeled anti-mouse/human TGF-β1 (clone A-75-3) and anti-LAP (TGF-β1) affinity purified Ab; PE-cy5 labeled anti-mouse CD62L Ab and streptavidin; PE-TR labeled anti-mouse CD4 Ab; and different fluorochrome labeled isotype control Abs (Caltag Laboratories, BD Biosciences, eBiosciences, R&D Systems, Serotec Ltd, and Biolegend Laboratories) were used for FACS analysis. Magnetic bead-conjugated total T cell, CD4+CD25+ T cell, and CD4+ T cell, CD11c+ dendritic cell (DC) isolation kits (Miltenyi Biotec) were used for enriching or depleting T cell sub-populations and DCs. PE-labeled anti-F4/80 or CD11c Ab and anti-PE Ab linked magnetic beads were used for enriching F4/80+ macrophages and CD11c+ DCs. Multiplex reagents, and paired antibodies and standards for ELISA to detect TGF-β1, TNF-α, IL-10, IL-2, IL-4, IL-17, IFN-γ, IL-12, IL-1β, and IL-6 were purchased from R&D system, Millipore, and eBiosciences.
Treating mice with zymosan
Short-term treatment of NOD/Ltj mice of different age groups were carried out by injecting with zymosan (i.p.; 25, 100 or 200 μg/mouse) in PBS on days 1, 3, and 5. 100 μg zymosan per mouse was found to be the adequate and optimum dose based on our preliminary observation that lower dose failed produce a significant delay in hyperglycemia when the treatment was initiated at 8 or 12 weeks of age. Further, early hypgerglycemic mice failed to show significant delay in the disease when treated intermittently even with higher doses. Therefore, two different treatment approaches were undertaken. 1) Intermittent long-term treatment of 8 and 12 week old pre-diabetic mice. These mice were first injected with zymosan (100 μg/mouse) on days 1, 3, 5 (3 injections per week) and again injected 3 times/week on weeks 3, 5, 7, 9, and 11 post-treatment initiation and monitored for hyperglycemia for at least 30 weeks. 2) Continuous treatment of early hyperglycemic mice. Early hyperglycemic mice (glucose levels: 140-200 mg/ml) were injected with zymosan (100 μg/mouse/day) for 30 consecutive days and monitored for hyperglycemia for up to 27-week post-treatment initiation. Control mice were injected with PBS. Glucose levels of test and control mice were tested weekly. Mice with glucose levels >250 mg/dl for two consecutive weeks were considered diabetic.
Bone marrow (BM) derived dendritic cells (BMDCs), peripheral DCs, macrophages, and T cells
BM cells were cultured in complete RPMI 1640 medium containing 10% heat-inactivated FBS in the presence of 20 ng/ml GM-CSF for 2 days and for an additional 3 days in fresh medium containing 20 ng/ml GM-CSF and 5 ng/ml IL-4 (Invitrogen). Cells from 6-day cultures were used for this study.
Peripheral CD11c+ DCs and F4/80+ macrophages were enriched from spleen and pancreatic LN (PnLN) and pancreatic cells using PE-labeled anti- CD11c and F4/80 Abs and anti-PE Ab magnetic beads. Pancreatic tissues and islets were digested with collagenase (Roche, USA) followed by trypsin-EDTA to generate single cell suspension. CD11c+ and F4/80+ cell populations were enriched to >93% using magnetic separation reagents.
Total T cells were enriched to >98% using T cell negative selection kit (Miltenyi). CD4+, CD4+CD25-, and CD4+CD25+ T cell sub-populations were enriched to >95% using CD25+ T cell isolation kit from Miltenyi Biotec according to the manufacturer's directions. Isolated cells were washed, stained with FITC-, PE- and APC labeled appropriate Abs, and tested for purity by FACS before use. Enriched CD4+CD25+ cells were also analyzed for Foxp3 expression by FACS.
FACS analysis
Freshly isolated and ex vivo cultured cells were washed using PBS supplemented with 2% FBS and 10 mM EDTA (pH 7.4) and blocked with anti-CD16/CD32 Fc block Ab or 5% rat serum on ice for 15 min. For surface staining, cells were incubated with FITC-, PE-, and PECy5 or PE-TR-labeled appropriate antibodies in different combinations on ice for 30 min, and washed thrice before analysis. For intracellular staining, surface stained cells were fixed and permeablized using reagents from eBiosciences, incubated with fluorochrome labeled appropriate Abs, and washed thrice prior to analysis. Stained cells were acquired using FACS-Calibur or LSR (BD Biosciences) flow-cytometer, and the data were analyzed using WinMDI or Weasel application. Cells were also stained using isotype-matched control Abs and considered background controls. Specific regions were marked, and the gates and quadrants were set while analyzing the data based on these isotype control background staining. At least 10,000 cells were analyzed in each experiment.
Cytokine analysis
Cell-free supernatants collected from DC, macrophage and T cell cultures were tested for cytokine levels by ELISA or Luminex multiplex assays. Assays were carried out for detecting IL-2, IL-4, IL-10, IFN-γ, TGF-β1, IL-17, TNF-α, IL-10, IL-12, IL-1β, and IL-6. ELISA was carried out using paired Abs and standards (Ebiosciences and R&D systems). Multiplex assay was carried out as per the manufacturer's (Millipore) directions and the data were acquired and analyzed using a luminex-100 instrument and application from Biorad. The amount of a particular cytokine was determined using an appropriate cytokine-specific standard curve.
Adoptive T cell transfer experiment
Total T cells were enriched from spleen cells of control and zymosan treated mice using magnetic bead based pan T cell isolation (negative selection) kit (Miltenyi). Enriched cells, which contained both CD4+ and CD8+ populations, were transferred to 6-wk old NOD-Scid mice (i.v.; 2×106 cells/mouse), glucose levels were tested every third day for up to 75 days.
In vitro assays
All in vitro assays with mouse primary cells were done in RPMI medium supplemented with 10% FBS, L-glutamine (2 mM), HEPES (15 mM), sodium pyruvate (1 mM), 2-ME (5 × 10-5 M), penicillin (100 U/ml), streptomycin (0.1 mg/ml), and fungizone (1 μg/ml). BMDCs, and CD11c+ and F4/80+ cell fractions enriched from spleen were cultured with or without zymosan (10 or 50 μg/ml), for different time-points and the supernatants obtained from these cultures were used for cytokine analysis. Cells were examined for surface marker expression by FACS.
In some assays, spleen and pancreatic LN (PnLN) cells (2×105 cells/well) from zymosan treated and control mice were stimulated with anti-CD3 Ab (2 μg/ml) for 48 h. Enriched CD4+CD25+ and CD4+CD25- T cells from treated and untreated mice were stimulated with anti-CD3 and CD28 Abs (2 μg/ml each) with or without zymosan (50 μg/ml). Spent media from stimulated and non-stimulated wells were tested for cytokines. Cells harvested from these cultures were tested for the levels of expression of surface markers by FACS.
For co-culture assay, CD4+CD25- T cells from untreated control mice were labeled with CFSE, cultured with CD4+CD25+ T cells from zymosan treated or control mice in the presence of anti-CD3 and CD28 Abs. Effector and regulatory T cells were co-cultured at varying ratios, but the total number of cells was maintained at 5×105/well to avoid discrepancy in the proliferative response due to the difference in the cell density. On day 5, to test for CFSE dilution, cells from these cultures were collected, stained with PE labeled Abs to CD4, and analyzed by FACS.
Histochemical analysis of pancreatic tissues
Pancreata were fixed in 10% formaldehyde, 5-μm paraffin sections were made, and stained with hematoxylin and eosin (H&E). Stained sections were analyzed in a blinded fashion, using a grading system, in which 0 = no evidence of infiltration, 1 = peri-islet infiltration, 2 = <25%, 3 = 25–50% infiltration, 4 = 50-100% infiltration of each islet, and 5 = complete loss or only remnants of islets seen as described earlier (33). About 100 islets were examined for each group.
Statistical analysis
Mean, SD, and statistical significance (p-value) were calculated using Microsoft Excel, SPSS, or GraphPad statistical application. Since same number of test (zymosan treated or exposed) and control values (data points) were compared, paired two tailed t-test was employed unless specified. Log-rank analysis was performed to compare TID incidence (hyperglycemia) of test group with that of the control group. Fisher's exact test was employed for comparing the total number of infiltrated islets in the test groups versus the control group. A p-value of ≤0.05 was considered significant.
Results
Zymosan exposure induces IL-10, TGF-β1, IL-2, and TNF-α production by APCs
It has been shown that zymosan can interact with both TLR2 and dectin 1(23,23). To examine the effect of engagement of TLR2 and dectin 1 on APCs from NOD mice, BMDCs, splenic DCs, and peritoneal and splenic macrophages were incubated with zymosan for different time-points, and examined for surface activation markers and secreted cytokines. BMDCs and peritoneal macrophages, but not splenic DCs and macrophages, showed a significant difference in the levels of expression of costimulatory molecules on the surface upon exposure to zymosan compared to untreated control APCs (Fig. 1A). While BMDCs showed considerable upregulation of CD80, CD86, and MHC II, peritoneal macrophages showed up-regulation of CD86 and CD40, but a marked down regulation of CD80 upon in vitro exposure to zymosan. Although all macrophage and DC preparations showed considerable levels of TLR2 and dectin 1 on the surface, exposure to zymosan had no significant effect on splenic DCs and macrophages in terms of the expression levels of activation markers, CD80, CD86, CD40 and MHC II. These results suggest that zymosan can influence different populations of APCs differently and, upon treatment, may modulate T cell function in vivo.
FIGURE 1.
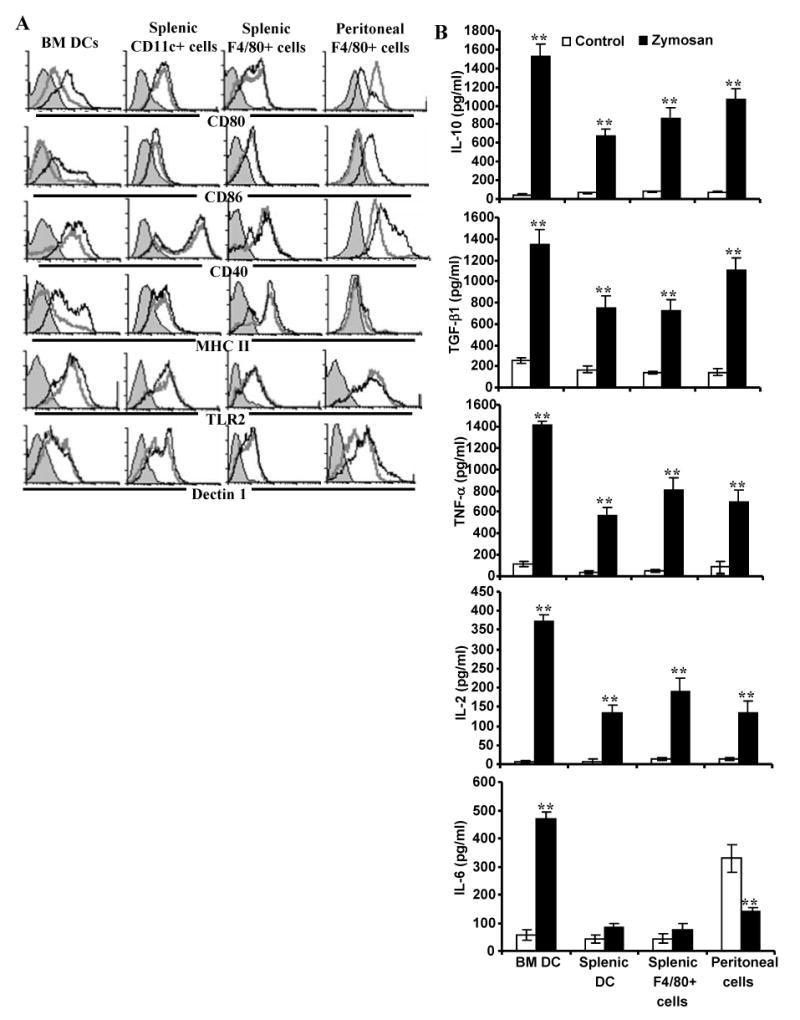
Zymosan modulates APC phenotype and cytokine profiles. Immature DCs generated in vitro from BM cells (BMDCs), freshly isolated splenic CD11c+ DCs and F4/80+ macrophages, and peritoneal exudate cells (1×106 cells/ml) were left untreated or exposed to zymosan (50 μg/ml) for 48 h. Cells were obtained from eight week old female NOD mice. A) Cells were stained using fluorochrome labeled anti-mouse CD11c or F4/80 Ab and anti-mouse CD80, CD86 CD40, I-Ag7, TLR2, or dectin 1 Ab. CD11c+ population for DCs and F4/80+ population for macrophages were gated for the graphs shown. Representative isotype control Ab staining histogram (gray filled) that is overlaid with marker specific staining histograms of untreated (gray open) and zymosan treated (black open) cells are shown. B) Cytokine levels in supernatants obtained from the above cultures at 48 h time-point were tested by ELISA. Mean±SD of values from three to four individual experiments carried out in triplicate are shown. A minimum of 9 data points of zymosan group were compared to that of respective control group by 2-tailed paired t-test to obtain p-values. *, p <0.01; **, p <0.001.
Culture supernatants from control and zymosan-exposed APCs were examined for secreted pro- and anti- inflammatory cytokines. BM and splenic DCs, and peritoneal and splenic macrophages produced profound amounts of IL-10, TGF-β1, TNF-α, and IL-2 compared to control DCs (Fig. 1B). While zymosan induced a significantly higher amount of IL-6 in BM DCs, the IL-6 response by peritoneal macrophages upon zymosan exposure was significantly lower compared to the un-exposed control. Importantly, splenic DCs and macrophages did not produce significant amounts of IL-6. Similarly, exposure to zymosan did not trigger secretion of detectable amounts of IL-12(p70), and IL-1β by DCs and macrophages (not shown). These observations show that zymosan can trigger both anti- and pro- inflammatory innate-immune responses by APCs.
In vivo effect of TLR2 and dectin 1 engagement by zymosan on APCs
To examine the in vivo effect of TLR2 and dectin 1 engagement on APCs, zymosan treated mice were tested for their DC and macrophage phenotypes and cytokine profiles. Fig. 2A demonstrates that macrophages and DCs from spleen, pancreas and PnLN of zymosan treated mice are not significantly different from cells of control mice in terms of the levels of expression of antigen presentation related activation markers, TLR2, and dectin 1 on the surface. This observation was similar to that observed in vitro in Fig. 1A. However, peritoneal macrophages from zymosan treated mice expressed relatively lower levels of CD86, but high MHC II compared to controls, which were different from that observed upon in vitro exposure of these cells to zymosan. This difference could be due to prolonged exposure of macrophages to zymosan and various cytokines in vivo.
FIGURE 2.
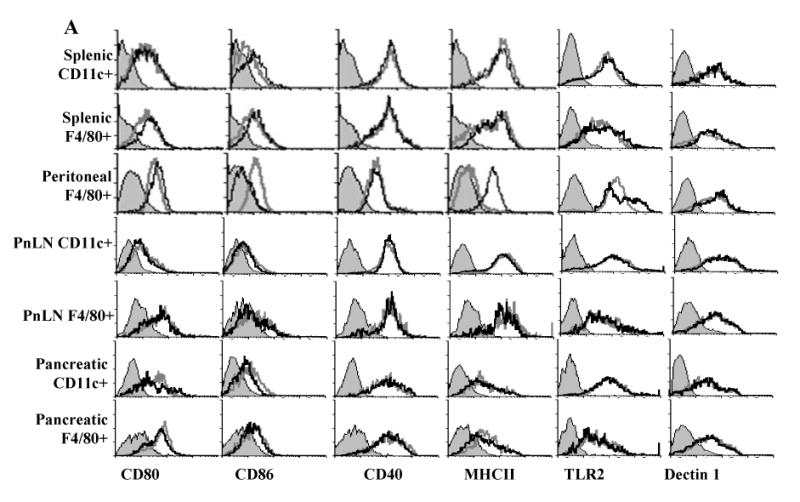

Zymosan triggers TNF-α and anti- inflammatory cytokine production by APCs in vivo. Eight-week old female NOD mice were treated i.p. with zymosan (100 μg/mouse/day) or PBS (control) on days 1, 3, and 5. Splenic, pancreatic (Pn), and pancreatic LN (PnLN) CD11c+ DCs and F4/80+ macrophages, and peritoneal cells obtained from these mice on day 7 were tested for surface markers and ex vivo spontaneous cytokine response. A) Cells were stained using fluorochrome labeled anti-mouse CD11c or F4/80 Ab and anti-mouse CD80, CD86, CD40, I-Ag7, TLR2 or Dectin 1 Ab. CD11c+ population for DCs and F4/80+ population for macrophages were gated for the graphs shown. Representative isotype control Ab staining histogram (gray, filled) that is overlaid with marker specific staining histograms of cells from untreated (gray; open) and zymosan treated (black; open) mice are shown. B) CD11c+ and/or F4/80+ cells isolated from spleen or peritoneum (1×106 cells/ml), and pancreas and PnLNs (5×105 cells/ml) were cultured for 48 h. Cell-free supernatants from these cultures were tested for cytokine levels by ELISA or luminex multiplex assay. Mean±SD of values from three individual experiments carried out in triplicate are shown. Cells from at least 5 mice were pooled to obtain sufficient pancreatic and PnLN DCs and macrophages for every individual experiment. A minimum of 9 data points of zymosan group were compared to same number of data points of respective control group by 2-tailed paired t-test to obtain p-values. *, p <0.01; **, p <0.001.
As shown in Fig. 2B, CD11c+ DCs and F4/80+ macrophages from spleen, pancreas and PnLNs produced significant amounts of IL-10, TGF-β1, IL-2, and TNF-α as observed for BM derived, splenic and peritoneal APCs in vitro. Similar to in vitro effects, while zymosan exposed splenic APCs did not produce significant amounts of IL-6, peritoneal APCs and PnLN DCs produced significantly lower levels of IL-6 compared to control cells (Fig. 2B; bottom panels). In addition, these APCs did not produce significant levels of IL-1β and IL-12 upon exposure to zymosan (not shown). These results demonstrate that zymosan induces production of anti- and pro- inflammatory cytokines that are known to influence natural Treg function and adoptive immune response.
Zymosan treatment prevents hyperglycemia in pre-diabetic NOD mice
Considering the effect of TLR2 and Dectin 1 ligation on APCs in terms of suppressor cytokine production, we examined whether treatment using zymosan can influence the T1D outcome in NOD mice. Younger (4-week old) mice, but not pre-diabetic mice, treated for a short-term (on days 1, 3, and 5) with zymosan produced a significant delay in hyperglycemia. These 4-week old mice showed delay of hyperglycemia up to 7 weeks upon short-term treatment with a lower (25 μg/mouse) or higher (100 μg/mouse) doses of zymosan as compared to controls (not shown). However, 8 and 12 week old mice failed to show significant delay in hyperglycemia when doses lower than 100 μg/mouse were used for short-term or intermittent treatment. Therefore, pre-diabetic NOD mice of 8 weeks and 12 weeks of age were treated intermittently with zymosan for a prolonged period (thrice on alternate weeks for 6 weeks) and monitored weekly for hyperglycemia at least until 40 weeks of age (30 weeks post-treatment initiation). As observed in Fig. 3A, treatment of 8- and 12-week old mice with zymosan prevented hyperglycemia in a majority of mice for at least 30 weeks post-treatment initiation and for about 20 weeks after the termination of treatment. Pancreatic tissues of mice from parallel experiments were examined for insulitis 1 week after the termination of treatment. Zymosan treated mice showed significantly suppressed immune cell infiltration and destruction of pancreatic islets (Fig. 3B and C). Importantly, significant numbers of islets were free of infiltration (about 50% islets), or with minimum infiltration (about 40% islet with grade 1 and 2 insulitis), in zymosan treated mice compared to control mice (more than 70% islets with grade 3 and 4 insulitis).
FIGURE 3.
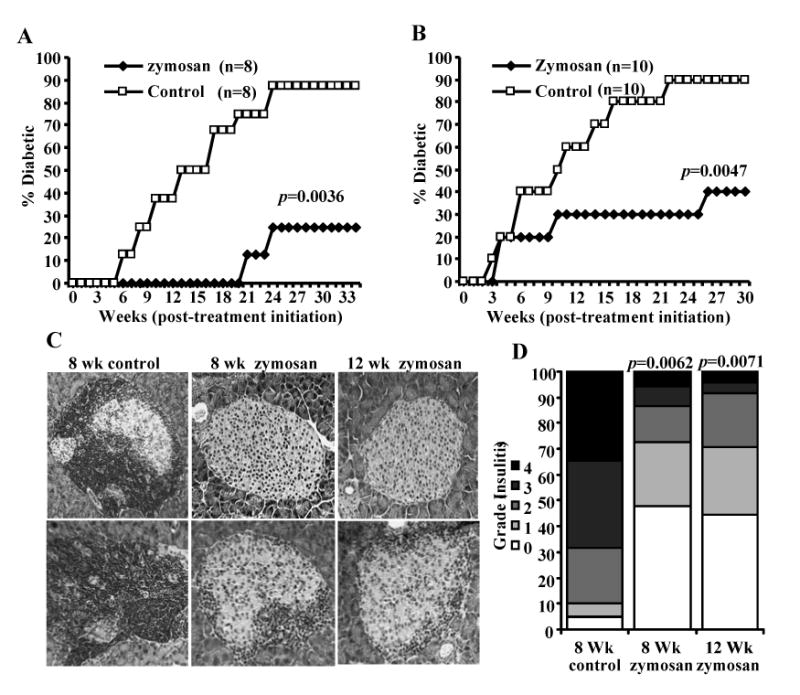
Activation of innate immune response using zymosan prevents hyperglycemia in NOD mice. Eight week old (A) and 12 week old (B) euglycemic female NOD mice were treated with zymosan or PBS (control) starting week 1 (on days 1, 3 and 5 with 100 μg/mouse/day; repeated similar three injections on weeks 3, 5, 7, 9 and 11). Mice were checked every week for hyperglycemia and glucose level of 250 mg/dl for two consecutive weeks was considered diabetic. Eight and 10 mice/group were included for A and B respectively. This experiment was repeated and obtained similar results. Log-rank test was performed to compare the hyperglycemia incidence in zymosan treated and control groups of mice and the p-value is shown on each graph. C) Euglycemic zymosan-treated and control mice from parallel experiments were euthanized two weeks after the last injection with zymosan, pancreatic tissues were processed for H&E staining to evaluate insulitis. Two representative islet areas (left panel) for each group and the percentages of islets with different levels of lymphocyte infiltration plotted as bar diagram (right panel) are shown. D) Sections of pancreatic tissues from at least 6 mice/group were examined for immune cell infiltration, and the insulitis severity was scored as described in materials and methods. Insulitis score of a total of 100 islets/group were plotted as bar diagram. Since 90% of the 12-week control group from parallel experiments turned hyperglycemic at the time of testing, insulitis could not be examined in sufficient number of mice from this group. Therefore, both 8 and 12 week zymosan groups are compared with 8 week control group. Fisher's exact test was employed for comparing the infiltrated islets in the test groups versus the control group. p-values are shown over test group bars.
Treatment of early-hyperglycemia mice with zymosan prevents diabetes
To examine whether prolonged treatment with zymosan can suppress autoimmunity and prevent hyperglycemia in mice that show early signs of hyperglycemia, mice with glucose levels 140-200 mg/dl were treated for 30 consecutive days with zymosan. Fig. 4 shows that while 100% control mice turned hyperglycemic within 9 week of treatment initiation, more than 70% of zymosan treated early-hyperglycemic mice remained non-diabetic for at least 20 weeks after the treatment was terminated. Further, examination of the pancreatic tissue in non-diabetic mice 20 weeks after the termination of treatment (aged between weeks 36-45) demonstrated that zymosan treated mice have a significant number of infiltration free islets even when compared to control mice that were at early hyperglycemic stages (glucose: 140-200 mg/dl; aged between weeks 12-20). These results indicate that lymphocyte infiltration into pancreatic islets is abrogated by innate immune response induced through TLR2 and dectin 1 by zymosan. Considering the fact that a majority of the remaining islets in the control mice had severe insulitis, it appears that anti-inflammatory cytokines induced through TLR2 and dectin 1 engagement have a role in suppressing immune cell infiltration into islets and reducing the insulitis severity.
FIGURE 4.
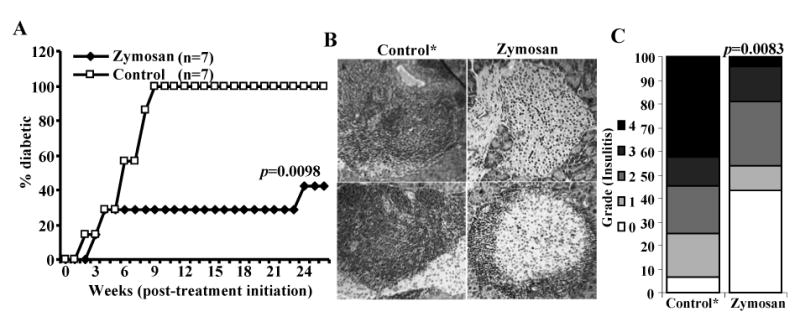
Activation of innate immune response using zymosan suppresses hyperglycemia in NOD mice. A) Early hyperglycemic mice (glucose levels between 140 and 200 mg/dl; screened from 12-20-week-old mice, pooled, and randomly picked) were treated i.p. with PBS or zymosan (100 μg/mouse/day) for 30 consecutive days. Mice were bled every week to check for glucose levels. Mice with glucose levels 250 mg/dl for two consecutive weeks were considered diabetic. Seven mice were included in each group and the experiment was repeated with similar results. Log-rank test was performed to compare the hyperglycemia incidence in zymosan treated and control groups of mice and the p-value is shown on the graph. B) Pancreatic tissue obtained from sets of 5 zymosan treated euglycemic treated and control mice* 20 weeks after the final injection with zymosan were processed for H&E staining to evaluate insulitis. Two representative islet areas (left panel) for each group and the percentages of islets with different levels of lymphocyte infiltration plotted as bar diagram (right panel) are shown. At least 100 islets were examined for each group. * Since 100% control mice developed overt hyperglycemia within 9 weeks post-treatment initiation, fresh batch of mice with glucose levels between 140 and 200 mg/dl at the time of testing were included as control. Fisher's exact test was employed for comparing the infiltrated islets in the test groups versus the control group. p-value is shown over the test group bar.
T cells from zymosan treated mice are less efficient in inducing diabetes in NOD-Scid mice
Since zymosan treatment suppressed autoimmunity and prevented hyperglycemia in female NOD mice and the immune cell infiltration into pancreatic islets was significantly affected, we assumed that T cell function in zymosan treated mice might also have been altered along with the APC function as observed in Figs 1 and 2. Earlier studies have shown that splenocytes or purified splenic T cells of diabetic and pre-diabetic NOD mice can induce hyperglycemia in immune deficient NOD-Scid mice upon adoptive transfer (34,35). Therefore, diabetogenicity of purified total T cells that include both CD4+ and CD8+ populations from zymosan treated mice was compared to that of T cells from untreated mice in immune deficient NOD-Scid mice. Fig. 5 demonstrates that while T cells from control mice induced hyperglycemia in 100% of NOD-Scid mice within 55 days, T cells from zymosan treated mice failed to induce diabetes in 80% of the mice for at least 75 days. These results indicated that T cell function is altered as a result of innate immune response induced by zymosan.
FIGURE 5.

Zymosan treatment suppresses the diabetogenicity of T cells. Purified T cells from euglycemic PBS (control) or zymosan treated mice (8-week old NOD mice that were treated with 100 μg/mouse/day as described above for Fig. 3 and splenic T cells isolated 15 days after the final dose of zymosan) were adoptively transferred into 8-week old NOD-Scid mice (i.v.; 2×106 T cells/mouse) and monitored for hyperglycemia every third day. Mice with glucose levels 250 mg/dl for two consecutive weeks were considered diabetic. Five mice were included in each group and the experiment was repeated with similar results. Log-rank test was performed to compare the hyperglycemia incidence in zymosan treated and control groups of mice to obtain p-value.
T cells from zymosan treated mice produce IL-10 and TGF-β1 upon activation
To examine the difference between T cells from control and zymosan treated mice, pancreatic LN (PnLN) and spleen cells were stimulated using anti-CD3 Ab and spent medium was tested for various cytokines. As observed in Fig. 6, both PnLN and spleen cells secreted large amounts of IL-10 and TGF-β1 compared to cells from control mice upon activation. On the other hand, IFN-γ response by T cells from zymosan treated mice was significantly lower compared to control cells. Further, T cells from both control and zymosan treated mice produced comparable levels of IL-4 and IL-17 (not shown). These results indicated that T cell function in zymosan treated mice is also modulated, perhaps, by IL-10, TGF-β1, and IL-2 secreted by APCs upon signaling through TLR2 and dectin 1.
FIGURE 6.

T cells from zymosan treated mice produce profound amounts of TGF-β1 and IL-10. Spleen and pancreatic LN (PnLN) cells (1×106cells/ml) obtained from PBS or zymosan treated 8-week old NOD mice (treated as described above for Fig. 3; cells obtained 15 days after the final dose of zymosan) were left non-stimulated or stimulated using anti-CD3 Ab (2μg/ml) for 48 h, supernatants were tested for IL-10, TGF-β1, and IFN-γ by ELISA or luminex multiplex assay. Background cytokine values of supernatants from non-stimulated cultures were subtracted from respective anti-CD3 Ab stimulated cultures to exclude cytokine released by APCs spontaneously, and plotted as bar-diagrams. Mean±SD of values from three separate experiments carried out in triplicate are shown. PnLN cells from three mice were pooled for each individual experiment to obtain sufficient number of T cells. A minimum of 9 data points of zymosan group were compared to same number of data points of respective control group by 2-tailed paired t-test to obtain p-values. **, p <0.001.
Zymosan treated mice have increased CD62Lhigh T cells
CD62L+ T cells are considered less diabetogenic and have regulatory properties in NOD mice (36,37). Therefore, we examined the frequency of CD62L+ T cells in zymosan treated mice compared to untreated controls. Higher frequency of CD62Lhigh CD4+ T cells was detected in both spleen and PnLNs in zymosan treated mice compared to controls (Fig. 7A). Importantly, while majority of T cells in PnLNs of pre-diabetic NOD mice appeared to be CD62Llow, more than 50% of these cells in zymosan treated mice were found to be CD62Lhigh. Further, the CD62Lhigh and CD62Llow T cell distribution appeared to be similar in both PnLNs and spleens of zymosan treated mice. Similar CD62L expression pattern was observed on CD8+ T cells from the spleen and PnLN of zymosan treated mice compared to control mice (not shown). T cells from zymosan treated and control mice were also examined for other memory markers such as CD44, CD127, and CCR7. As observed in Fig. 7B, significantly higher number of spleen and PnLN cells from zymosan treated mice appeared to have naive T cell phenotype (CD44lowCD62Lhigh) compared to these cells from control mice. While majority of CD62Llow cells from PnLNs of control mice were CD44low, a significant number of these cells in zymosan treated mice were CD44high. Further analysis showed that significantly lower proportions of PnLN and spleen T cells from zymosan treated mice were CD127+ and CCR7+ compared to T cells from control mice. These observations indicate that the T cell phenotype is altered and/or T cells with memory phenotype are replaced by T cells with naïve and/or regulatory phenotype in zymosan treated mice. This result, in conjunction with the observation from Fig. 6, suggests that altered T cell function and distribution may have contributed to reduced immune cell infiltration into pancreatic islets, and day in hyperglycemia.
FIGURE 7.
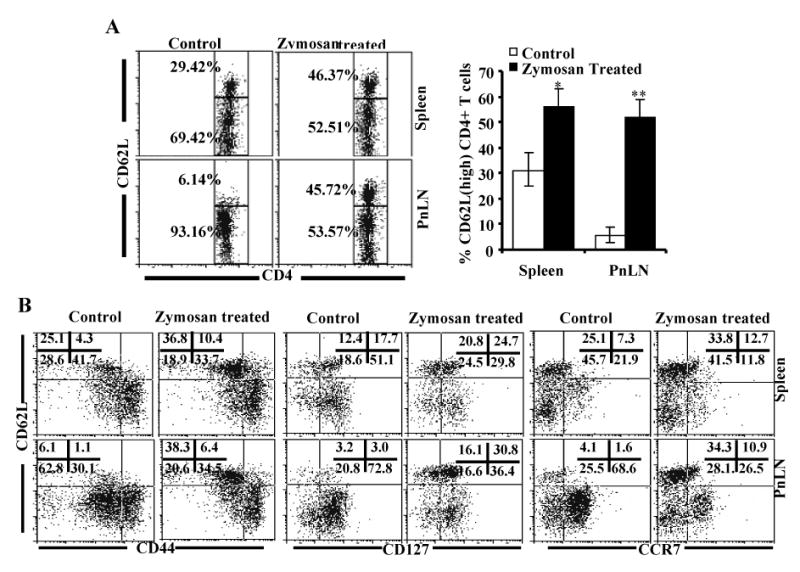
Zymosan treated mice carry large numbers of CD62Lhigh T cells. Female NOD mice (8-week old) were treated with PBS (control) or zymosan as described above for Fig. 3. A) Spleen and pancreatic LN (PnLN) cells obtained from these mice 15 days after the final dose of zymosan were stained using fluorochrome labeled anti-mouse CD4 and CD62L Abs, and analyzed by FACS. CD4+ population was gated for the panels shown here. Representative plots of spleen and PnLN cells and the percentage values of CD62Lhigh and CD62Llow CD4+ populations are shown (left panel). Mean ± values of CD62Lhigh CD4+ T cell frequencies from five mice tested in triplicate in each group are shown (right panel). Two-tailed paired t-test was performed to compare the frequency of CD4+CD62Lhigh T cells from control and zymosan treated mice. *, p<0.01; **, p<0.001. B) Splenic and PnLN CD4+ T cells were also examined for additional memory markers, CD44, CD127, and CCR7 by FACS. CD4+ population was gated for the panels shown here. Percentage values for quadrants are shown in each graph.
Similar to total T cells, significantly higher proportion of CD4+CD25+ T cells in zymosan treated mice are CD62Lhigh
Ineffective natural Treg (CD4+CD25+ T cells) function has been implicated in the progression of T1D in NOD mice. Although the Treg frequency appears to be significantly not different at different stages of the disease and in different age groups, their ability to exert suppressive effect on effector T cells is diminished at later stages in NOD mice and in T1D patients (38,39). Therefore, we examined whether naturally existing Tregs are affected by zymosan treatment. Although there was no selective increase in the frequency of CD62Lhigh CD4+CD25+ population was observed in zymosan treated mice, a higher proportion of CD4+CD25+ T cell population in these mice was CD62Lhigh compared to control mice (Fig. 8A). In other wards, increase in the CD62Lhigh Treg frequency in zymosan treated mice was comparable to that of total CD4+ T cells (right panels of Figs 7A and 8A). Importantly, as observed in Fig. 8B, Foxp3+ Treg frequencies in spleen and PnLNs of zymosan treated mice were not significantly different from that of untreated control mice. These results indicate that CD62Lhigh sub-populations may have a role in the reduced diabetogenicity of splenic T cells from zymosan treated mice.
FIGURE 8.
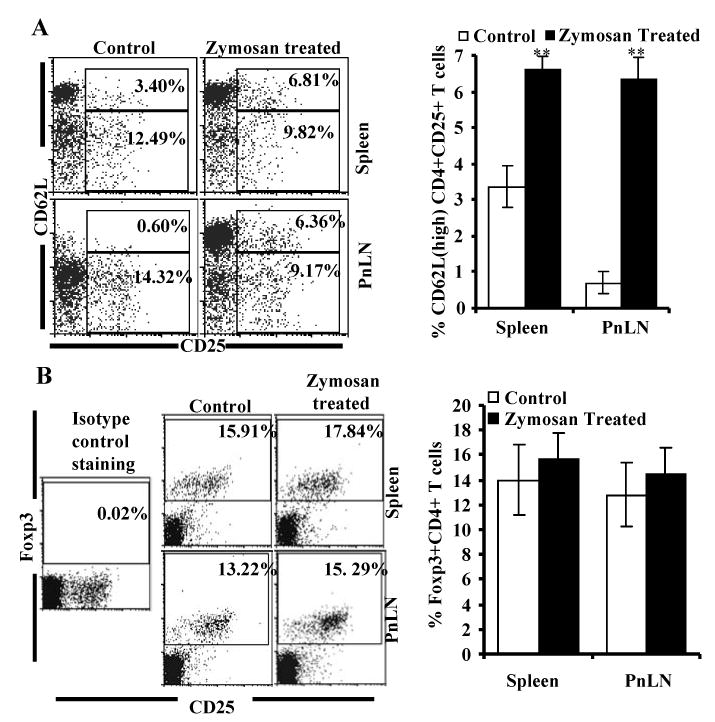
Zymosan treated mice carry significantly higher frequency of CD25+CD62Lhigh but not Foxp3+ CD4+ T cells compared to controls. Eight-week-old female NOD mice were treated with PBS (control) or zymosan as described above. A) Spleen and PnLN cells obtained from these mice 15 days post-treatment were stained using anti-mouse CD4-PE-TR, CD62L-PEcy5 and CD25+ PE reagents, and analyzed by FACS. B) Cells were also stained using anti-mouse CD4-PE-TR and CD25-alexa-488 reagents, permeablized and further stained with anti-mouse Foxp3-PE, and analyzed. CD4+ positive population was gated for the panels and the representative plots and percentage values are shown (left panels). Mean ± SD values of CD4+CD25+C62Lhigh or CD4+Foxp3+ T cell frequencies of samples from five individual mice tested in triplicate are also shown (right panels). Two-tailed paired t-test was performed to compare the frequency of these populations from zymosan treated and control mice. **, p<0.001.
CD4+CD25+ Tregs from zymosan treated mice show enhanced suppressor function
The ability of CD4+CD25+ T cells from zymosan treated mice in suppressing the effector T cell (CD4+CD25-) response was tested in comparison with cells from control mice in a co-culture assay. CD4+CD25+ T cells enriched from zymosan treated and control mice were tested for Treg frequency by examining for Foxp3 expression before using in T cell suppression assays. As observed in Fig. 9A, about 94% of Tregs enriched from both zymosan treated and control mice were Foxp3+. While CD4+CD25+ T cells from both control and zymosan treated NOD mice demonstrated a significant ability to suppress CD3/CD28 ligation induced proliferation of effector T cells, the suppressive ability per cell appeared to be significantly higher in CD4+CD25+ cells from zymosan treated mice compared to controls (Fig. 9B). This shows that natural Treg function is enhanced by innate immune response through TLR2 and dectin 1 and it could partially explain the disease protective effect of zymosan therapy.
FIGURE 9.
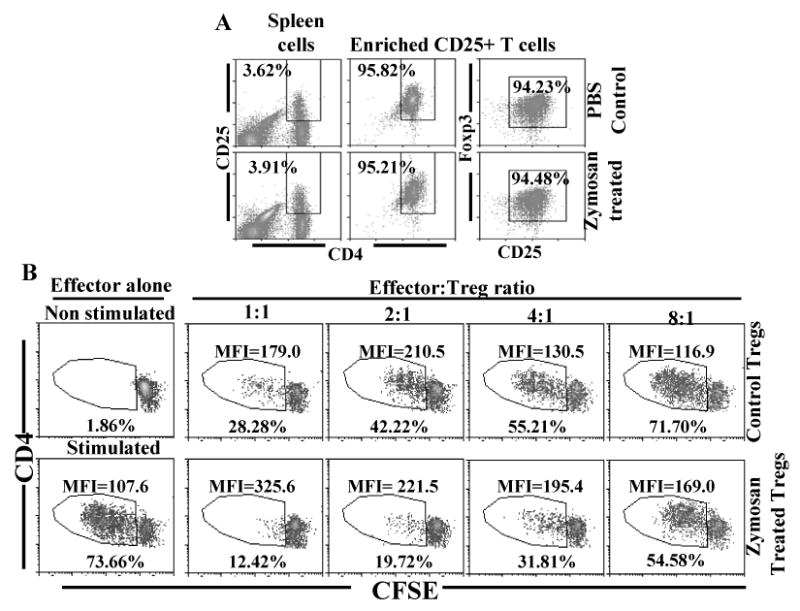
Innate immune response induced by zymosan enhances the suppressor efficiency of CD4+CD25+ Tregs. A) CD4+CD25+ T cells were enriched from control and zymosan treated mice spleens by magnetic sorting reagents, and tested for purity by FACS. B) Purified splenic CD4+CD25+ T cells were co-cultured with CFSE labeled CD4+CD25- T cells from control mice at varying effector : Treg ratios in the presence of anti-CD3 Ab and APCs. CFSE dilution in CD4+CFSE+ gated population was examined by FACS on day 4. For effectors alone, total number of cells was equalized to that of co-culture wells using unlabeled CD4+CD25- T cells from control mice. Percentage and mean fluorescence intensity (MFI) values of cells with CFSE dilution among CFSE+ CD4+ T cells are shown in each plot. This assay was repeated thrice in triplicate with similar results.
CD4+CD25+ T cells from zymosan treated mice produce TGF-β1 and IL-10
Since CD4+CD25+ T cells from zymosan treated mice demonstrated enhanced ability to suppress effector T cell proliferation, levels of surface molecules and secreted cytokines of these cells were compared to that of control CD4+CD25+ T cells. The frequency of CD4+CD25+ T cells with surface bound active and latent TGF-β1 (LAP), expression levels of GITR, PD-1, and CTLA-4 on these cells were comparable to that of cells from control mice (not shown). However, activation using anti-CD3 and CD28 Abs revealed that CD4+CD25+ T cells from zymosan treated mice can produce higher amounts of TGF-β1 and IL-10 compared to control CD4+CD25+ T cells (Fig. 9). On the other hand, CD4+CD25- T cells from zymosan treated mice produced significantly higher amounts of IL-10, but not TGF-β1, compared to corresponding cells from control mice. These results show that IL-10 and TGF-β1 could be major mediators of enhanced suppressor function of CD4+CD25+ T cells from zymosan treated mice.
Discussion
With the exception of a recent study that suggested a role for TLR2 engagement by dying pancreatic β cells in initiating T1D (9), the effect of TLR2 and dectin 1 signaling in T1D is largely unknown. Therefore, to understand the effect of innate immune response induced through TLR2 and Dectin 1 in modulating T1D, pre-diabetic and early- hyperglycemic NOD mice were treated with zymosan. We found that induction of innate immune response using zymosan in these mice leads to their protection from T1D. This disease protective effect appears to be contributed by copious amounts of anti-inflammatory cytokines produced by APCs and enhanced function of the regulatory wing of the immune system.
Consistent with earlier reports (31,32), exposure of DCs and macrophages to zymosan resulted in the secretion of significant amounts of IL-10, TNF-α and IL-2. Zymosan also triggered the production of large amounts of TGF-β1 by DCs and macrophages in vivo and in vitro suggesting that treatment using this agent will produce suppressive effects on autoimmunity in T1D. Unlike signaling through inflammatory TLRs such as TLR3, TLR4, and TLR9 that induces profound amounts of IL-6, IL-1β, and/or IL-12 (40,41), zymosan did not trigger these inflammatory cytokines by peripheral APCs such as macrophages and DCs indicating that signaling through these receptors produces a regulatory innate immune response. Importantly, TNF-α has contradictory effects in NOD mice depending on the insulitis stage (42). It acts destructive at early stages, but protective at later stages of insulitis. TNF-α is also known to indirectly promote natural Treg function through aiding effective APC function (43,44). This cytokine, in the presence of large amounts of IL-10, TGF-β1 and IL-2, may produce a protective effect at all stages of T1D. IL-10 and TGF-β1 have proven their protective effects in T1D and other inflammatory diseases by promoting Treg function and by suppressing pathogenic T cell response (45-47). IL-2, on the other hand, is essential for maintaining peripheral tolerance through promoting the survival and propagation of natural Tregs (48,49). Therefore, we assumed that induction of regulatory innate immune response through TLR2 and dectin 1 using zymosan will suppress the ongoing autoimmunity in NOD mice and protect them from T1D.
A recent report indicated that TLR2 signaling by necrotic pancreatic β-cells contributes to the activation of APCs and initiation of the β cell specific autoimmune response in NOD mice (9). However, in contrast to inflammatory responses induced by ligands for other TLRs, stimulation of TLR2 induced large amounts of pro-inflammatory cytokine, TNF-α as well as anti-inflammatory cytokines (30-32). Recent studies have also shown that TLR2 engagement enhances natural Treg function (26), and helps expand Treg numbers (27,28). These reports, and our observation that zymosan can trigger significant amounts of TGF-β1, IL-10, and IL-2 secretion by APCs from NOD mice prompted us to examine the potential of zymosan, which is known to bind to TLR2 and the synergistic receptor, dectin 1 in modulating T1D. Importantly, TLR2 and Dectin 1 receptors are also known to collaborate in activating NF-κB and NFAT leading to the production of IL-2 and IL-10 (32,50) indicating that profound disease modulation could be achieved using zymosan.
Treatment of pre-diabetic and early hyperglycemic NOD mice using zymosan clearly demonstrated that the innate immune response induced using zymosan can result in profound protection of these mice from diabetes. Studies have shown that insulitis can be detected in NOD mice as early as 3-4 weeks of age in NOD mice and therapeutic approaches have been widely effective at this stage. Short-term treatment of NOD mice at 4 weeks of age with zymosan was sufficient to delay the disease for about 7 weeks. However, since an effective treatment at, or close to, hyperglycemic stage is one of the major challenges in treating T1D, the efficacy of prolonged treatment in preventing the disease was tested. Our observations show that disease in older age groups (8 and 12 wk) and in early-hyperglycemic mice (glucose: 140-200 mg/dl) could be prevented for a significantly long-time by intermittent and/or prolonged treatment using zymosan suggesting that innate immune response through receptors such as TLR2 and Dectin 1 can have a protective effect on pre- and early- hyperglycemic stages of the disease.
T1D can be induced in immune-deficient NOD-Scid mice by adoptively transferring T cells from diabetic and pre-diabetic NOD mice (34,35,51). We assumed that an immunotherapy that suppresses the disease outcome could negatively affect the pathogenicity of peripheral T cells and, therefore, tested the diabetogenic ability of T cells from zymosan treated mice in comparison with control T cells using NOD-Scid mice. In fact, our results show that T cells from zymosan treated mice failed to induce hyperglycemia in majority of NOD-Scid mice indicating that innate immune response induced through TLR2 and dectin 1 has a profound suppressive effect on the T cell function.
Lowered ability of T cells from zymosan treated mice to induce diabetes is associated with a reduction in the frequency of CD62Llow T cells in lymphoid organs including PnLNs. In NOD mice, diabetogenicity of CD62Llow cells is known to be higher compared to CD62Lhigh T cells (36,37). In addition, CD62Lhigh CD4+ T cells have been found to have regulatory properties, and the CD4+CD25+CD62Lhigh population has been considered a more efficient Treg population than CD4+CD25+CD62Llow cells (36,52,53). Importantly, a significantly higher number of PnLNs T cells are CD62Llow compared to spleen and other peripheral lymphoid organs at any given stage and are indicative of the inflammatory nature of pancreatic microenvironment in NOD mice (33). However, unlike controls, zymosan treated mice had similar CD62L high and low T cells ratios in both spleen and PnLNs.
Memory T cells are highly heterogeneous in terms of expression of surface markers including chemokine and cytokine receptors. Mouse memory T cell populations with varying levels of expression of CD62L, CD44, CD127, CD122, and CCR7 have been reported (54-57). Reduced frequency of CD62Llow cells in zymosan treated mice compared to controls indicated that memory T cells may be affected by zymosan treatment. Our observation that relatively higher proportions of T cells from the PnLNs and spleens of zymosan treated mice have naïve T cell phenotype (CD62LhighCD44lowCCR7-) compared to controls supported this notion. In addition, while a significant portion of CD62Llow T cells from the PnLNs of control mice expressed CD127, CCR7, and low levels of CD44, majority of these cells from zymosan treated mice expressed CD127, and high levels of CD44 indicating that innate immune response induced upon TLR2 and dectin 1 engagement has a significant effect on T cell phenotype. Although further studies are needed to understand the functional significance of these phenotypic alterations, our observations suggest that innate immune response induced through TLR2 and dectin 1 affect the pathogenicity of T cells, and zymosan treatment leads to repopulation of non-pathogenic T cells in the pancreatic microenvironment. Our observations that PnLN and spleen T cells from zymosan treated mice produce higher amounts of IL-10 and TGF-β1, but significantly lower levels of IFN-γ upon activation further indicate that the T cell function is, in fact, altered in zymosan treated mice.
TGF-β1 and IL-2 play a critical role in the maintenance of effective natural Treg function (45,48,49). On the other hand, IL-10 and TGF-β1 can negatively affect the production of pro-inflammatory cytokines (such as IL-1β and IL-6 by APCs) that have an abrogative effect on Foxp3+ natural Tregs (58-60). Interestingly, although zymosan treatment did not result in a significant increase in the natural Foxp3+ Treg numbers in vivo, Tregs from treated mice secreted profoundly higher amounts of TGF-β1 and IL-10 upon CD3 ligation, and demonstrated an enhanced ability to suppress effector T cells compared to Tregs from untreated mice. These observations stress not only on a positive role of innate immune response through receptors such as TLR2 and dectin 1 on natural Treg function, but also on the importance of innate immunity through these receptors in maintaining peripheral tolerance.
It is now known that mutually dependent steady state functions of APCs and Tregs are important to maintain peripheral T cell tolerance (61). Two major defects in the immune system that have widely been reported in T1D are antigen presenting function and Treg properties (62,63). Our observation that DCs and macrophages from zymosan treated mice produce significant amounts of IL2, IL-10, and TGF-β1 indicate that these factors could be responsible, at least in part, for the alteration of T cell phenotype and function.
TLRs and other innate immune receptors are widely expressed on APCs, and cytokines produced by them upon ligation of innate immune receptors bridge innate and adaptive arms of the immune system (7). However, recent reports indicated that TLRs are also expressed on various other immune cell populations including T and B cells, and they might have a direct role in modulating adaptive immune responses upon antigen specific activation (64,65). Therefore, it is likely that T cell function in zymosan treated mice is affected, in part, also by direct ligation of TLR2 and/or dectin 1.
A number of studies have demonstrated the ability of microbial agents in preventing T1D in NOD mice (66-69). Injection of mycobacteria or complete freund's adjuvant (CFA) could prevent T1D for a long duration in NOD mice (67,68,70). Although the mechanism is not known, a role of TNF-α produced upon microbial triggering of innate immune response in the disease prevention has been suggested (70). Interestingly, recent studies have demonstrated that dectin-1 functions together with TLR2 to mediate macrophage activation by mycobacteria (71,72). Our observations suggest that these synergistically acting receptors, TLR2 and dectin 1 have the unique ability to induce cytokines such as IL-10, TGF-β1, and IL-2 by APCs upon ligation by zmosan and prevent T1D in NOD mice. These observations also indicate that protection from T1D achieved by others (67,68,70) through treating with mycobacteria or CFA could involve innate immune response induced through innate immune receptors such as TLR2 and dectin 1.
Based on the observation that zymosan can trigger the secretion of large amounts of suppressor cytokines by APCs, it can be assumed that adaptive self-antigen specific T cell response could be skewed towards protective regulatory type. Further, considering the fact that innate immune receptors are expressed not only on APCs, but also on T cells (64,65), direct effects of ligation of these receptors in the alteration of T cell phenotype and function can not be ruled out. Therefore, additional studies are needed to understand the effect of ligation of these receptors on APCs and T cells in influencing adaptive self-antigen specific T cell response. Studies are also needed to understand the effect of innate immune response triggered through receptors such as TLR2 and dectin 1 on target tissue and insulitis. Nevertheless, our study clearly demonstrates the protective role of innate immune response through TLR2 and dectin 1 by zymosan in T1D and agonistic compounds for these receptors could prove effective in preventing/treating T1D in humans. These observations also suggest that environmental factor induced prevention of T1D in NOD mice may be dependent on innate immune response induced through receptors such as TLR2 and dectin 1.
FIGURE 10.

CD4+CD25+ T cells from zymosan treated mice secrete large amounts of IL-10 and TGF-β1. CD4+CD25+ and CD4+CD25- T cells from control and zymosan treated mice were examined for cytokine profiles. Enriched T cell populations were cultured in the presence 10 U/ml recombinant IL-2 with and without anti-CD3 and anti-CD28 Abs (2 μg/ml each), spent medium was tested for TGF-β1 and IL-10. Each bar represents mean ± SD of three separate assays carried out in triplicate. Three to four mice/group were used for each experiment. Two-tailed paired t-test was performed to compare cytokine secretion by T cells from control and zymosan treated mice. *, p<0.01; **, p<0.001.
Abbreviations
- TLR
toll-like receptor
- DCs
dendritic cells
- Tregs
regulatory T cells
- Foxp3
foxhead box p3 transcription factor
- TGF-β1
transforming growth factor 1 beta
- T1D
Type 1 diabetes
- LP
lipoprotein/lipopeptide
Footnotes
This work was supported in part by the Department of Surgery, UIC, National Institutes of Health grants R21A1059745 and R21AI069848 and the Juvenile Diabetes Research Foundation awards 1-2005-27 and 32-2008-343.
References
- 1.Jahromi MM, Eisenbarth GS. Cellular and molecular pathogenesis of type 1A diabetes. Cell Mol Life Sci. 2007;64:865–872. doi: 10.1007/s00018-007-6469-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gale EAM. The rise of childhood type 1 diabetes in the 20th century. Diabetes. 2002;51:3353–3361. doi: 10.2337/diabetes.51.12.3353. [DOI] [PubMed] [Google Scholar]
- 3.Bodansky HJ, Staines A, Stephenson C, Haigh D, Cartwright R. Evidence for an environmental effect in the aetiology of insulin dependent diabetes in a transmigratory population. Br Med J. 1992;304:1020–1022. doi: 10.1136/bmj.304.6833.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van den Berg TK, Yoder JA, Litman GW. On the origins of adaptive immunity: innate immune receptors join the tale. Trends Immunol. 2004;25:11–16. doi: 10.1016/j.it.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 5.Roy CR, Mocarski ES. Pathogen subversion of cell-intrinsic innate immunity. Nat Immunol. 2007;8:1179–1187. doi: 10.1038/ni1528. [DOI] [PubMed] [Google Scholar]
- 6.Albiger B, Dahlberg S, Henriques-Normark B, Normark S. Role of the innate immune system in host defense against bacterial infections: focus on the Toll-like receptors. J Intern Med. 2007;261:511–528. doi: 10.1111/j.1365-2796.2007.01821.x. [DOI] [PubMed] [Google Scholar]
- 7.Belardelli F, Ferrantini M. Cytokines as a link between innate and adaptive antitumor immunity. Trends Immunol. 2002;23:201–208. doi: 10.1016/s1471-4906(02)02195-6. [DOI] [PubMed] [Google Scholar]
- 8.Moriyama H, Wen L, Abiru N, Liu E, Yu L, Miao D, Gianani R, Wong FS, Eisenbarth GS. Induction and acceleration of insulitis/diabetes in mice with a viral mimic (polyinosinic-polycytidylic acid) and an insulin self-peptide. Proc Natl Acad Sci USA. 2002;99:5539–5544. doi: 10.1073/pnas.082120099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim HS, Han MS, Chung KW, Kim S, Kim E, Kim MJ, Jang E, Lee HA, Youn J, Akira S, Lee MS. Toll-like receptor 2 senses beta-cell death and contributes to the initiation of autoimmune diabetes. Immunity. 2007;27:321–333. doi: 10.1016/j.immuni.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 10.Lang KS, Recher M, Junt T, Navarini AA, Harris NL, Freigang S, Odermatt B, Conrad C, Ittner LM, Bauer S, Luther SA, Uematsu S, Akira S, Hengartner H, Zinkernagel RM. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat Med. 2005;11:138–145. doi: 10.1038/nm1176. [DOI] [PubMed] [Google Scholar]
- 11.Rasschaert J, Ladrière L, Urbain M, Dogusan Z, Katabua B, Sato S, Akira S, Gysemans C, Mathieu C, Eizirik DL. Toll-like receptor 3 and STAT-1 contribute to double-stranded RNA+ interferon-gamma-induced apoptosis in primary pancreatic beta-cells. J Biol Chem. 2005;280:33984–33991. doi: 10.1074/jbc.M502213200. [DOI] [PubMed] [Google Scholar]
- 12.Dogusan Z, García M, Flamez D, Alexopoulou L, Goldman M, Gysemans C, Mathieu C, Libert C, Eizirik DL, Rasschaert J. Double-stranded RNA induces pancreatic beta cell apoptosis by activation of the TLR3 and IRF-3 pathways. Diabetes. 2008;57:1236–1245. doi: 10.2337/db07-0844. [DOI] [PubMed] [Google Scholar]
- 13.Fukushima K, Abiru N, Nagayama Y, Kobayashi M, Satoh T, Nakahara M, Kawasaki E, Yamasaki H, Ueha S, Matsushima K, Liu E, Eguchi K. Combined insulin B: 9-23 self-peptide and polyinosinic-polycytidylic acid accelerate insulitis but inhibit development of diabetes by increasing the proportion of CD4+Foxp3+ regulatory T cells in the islets in non-obese diabetic mice. Biochem Biophys Res Commun. 2008;367:719–724. doi: 10.1016/j.bbrc.2007.12.191. [DOI] [PubMed] [Google Scholar]
- 14.Lien E, Sellati TJ, Yoshimura A, Flo TH, Rawadi G, Finberg RW, Carroll JD, Espevik T, Ingalls RR, Radolf JD, Golenbock DT. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J Biol Chem. 1999;274:33419–33425. doi: 10.1074/jbc.274.47.33419. 1999. [DOI] [PubMed] [Google Scholar]
- 15.Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between Toll-like receptors. Proc Natl Acad Sci USA. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farhat K, Riekenberg S, Heine H, Debarry J, Lang R, Mages J, Buwitt-Beckmann U, Roschmann K, Jung G, Wiesmuller KH, Ulmer AJ. Heterodimerization of TLR2 with TLR1 or TLR6 expands the ligand spectrum but does not lead to differential signaling. J Leukoc Biol. 2008;83:692–701. doi: 10.1189/jlb.0807586. [DOI] [PubMed] [Google Scholar]
- 17.Lee JY, Zhao L, Youn HS, Weatherill AR, Tapping R, Feng L, Lee WH, Fitzgerald KA, Hwang DH. Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J Biol Chem. 2004;279:16971–16979. doi: 10.1074/jbc.M312990200. [DOI] [PubMed] [Google Scholar]
- 18.Takeda K, Takeuchi O, Akira S. Recognition of lipopeptides by Toll-like receptors. J Endotoxin Res. 2002;8:459–463. doi: 10.1179/096805102125001073. [DOI] [PubMed] [Google Scholar]
- 19.Omueti KO, Beyer JM, Johnson CM, Lyle EA, Tapping RI. Domain exchange between human toll-like receptors 1 and 6 reveals a region required for lipopeptide discrimination. J Biol Chem. 2005;280:36616–36625. doi: 10.1074/jbc.M504320200. [DOI] [PubMed] [Google Scholar]
- 20.Buwitt-Beckmann U, Heine H, Wiesmuller KH, Jung G, Brock R, Akira S, Ulmer AJ. TLR1- and TLR6-independent recognition of bacterial lipopeptides. J Biol Chem. 2005;281:9049–9057. doi: 10.1074/jbc.M512525200. [DOI] [PubMed] [Google Scholar]
- 21.Buwitt-Beckmann U, Heine H, Wiesmuller KH, Jung G, Brock R, Akira S, Ulmer AJ. Toll-like receptor 6-independent signaling by diacylated lipopeptides. Eur J Immunol. 2005;35:282–289. doi: 10.1002/eji.200424955. [DOI] [PubMed] [Google Scholar]
- 22.Sing A, Reithmeier-Rost D, Granfors K, Hill J, Roggenkamp A, Heesemann J. A hypervariable N-terminal region of Yersinia LcrV determines Toll-like receptor 2-mediated IL-10 induction and mouse virulence. Proc Natl Acad Sci USA. 2005;102:16049–16054. doi: 10.1073/pnas.0504728102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiura K, Kataoka H, Yasuda M, Inoue N, Shibata K. The diacylated lipopeptide FSL-1 induces TLR2-mediated Th2 responses. FEMS Immunol Med Microbiol. 2006;48:44–55. doi: 10.1111/j.1574-695X.2006.00119.x. [DOI] [PubMed] [Google Scholar]
- 24.Martin M, Schifferle RE, Cuesta N, Vogel SN, Katz J, Michalek SM. Role of the phosphatidylinositol 3 kinase-Akt pathway in the regulation of IL-10 and IL-12 by Porphyromonas gingivalis lipopolysaccharide. J Immunol. 2003;171:717–725. doi: 10.4049/jimmunol.171.2.717. [DOI] [PubMed] [Google Scholar]
- 25.Polumuri SK, Toshchakov VY, Vogel SN. Role of phosphatidylinositol-3 kinase in transcriptional regulation of TLR-induced IL-12 and IL-10 by Fc gamma receptor ligation in murine macrophages. J Immunol. 2007;179:236–246. doi: 10.4049/jimmunol.179.1.236. [DOI] [PubMed] [Google Scholar]
- 26.Zanin-Zhorov A, Cahalon L, Tal G, Margalit R, Lider O, Cohen IR. Heat shock protein 60 enhances CD4+ CD25+ regulatory T cell function via innate TLR2 signaling. J Clin Invest. 2006;116:2022–2032. doi: 10.1172/JCI28423. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc Natl Acad Sci USA. 2006;103:7048–7053. doi: 10.1073/pnas.0601554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sutmuller RP, Den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ, Joosten LA, Akira S, Netea MG, Adema GJ. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485–494. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goodridge HS, Underhill DM. Fungal Recognition by TLR2 and Dectin-1. Handb Exp Pharmacol. 2008;183:87–109. doi: 10.1007/978-3-540-72167-3_5. [DOI] [PubMed] [Google Scholar]
- 30.Underhill DM. Collaboration between the innate immune receptors dectin-1, TLRs, and Nods. Immunol Rev. 2007;219:75–87. doi: 10.1111/j.1600-065X.2007.00548.x. [DOI] [PubMed] [Google Scholar]
- 31.Dillon S, Agrawal S, Banerjee K, Letterio J, Denning TL, Oswald-Richter K, Kasprowicz DJ, Kellar K, Pare J, Dyke Tvan, Ziegler S, Unutmaz D, Pulendran B. Yeast zymosan, a stimulus for TLR2 and dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. J Clin Invest. 2006;116:916–928. doi: 10.1172/JCI27203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Slack EC, Robinson MJ, Hernanz-Falcón P, Brown GD, Williams DL, Schweighoffer E, Tybulewicz VL, Sousa CReise. Syk-dependent ERK activation regulates IL-2 and IL-10 production by DC stimulated with zymosan. Eur J Immunol. 2007;37:1600–1612. doi: 10.1002/eji.200636830. [DOI] [PubMed] [Google Scholar]
- 33.Li R, Perez N, Karumuthil-Melethil S, Vasu C. Bone marrow is a preferential homing site for autoreactive T-cells in type 1 diabetes. Diabetes. 2007;56:2251–2259. doi: 10.2337/db07-0502. [DOI] [PubMed] [Google Scholar]
- 34.Savinov AY, Wong FS, Chervonsky AV. IFN-gamma affects homing of diabetogenic T cells. J Immunol. 2001;167:6637–6643. doi: 10.4049/jimmunol.167.11.6637. [DOI] [PubMed] [Google Scholar]
- 35.Yadav D, Judkowski V, Flodstrom-Tullberg M, Sterling L, Redmond WL, Sherman L, Sarvetnick N. B7-2 (CD86) controls the priming of autoreactive CD4 T cell response against pancreatic islets. J Immunol. 2004;173:3631–3639. doi: 10.4049/jimmunol.173.6.3631. [DOI] [PubMed] [Google Scholar]
- 36.Lepault F, Gagnerault MC. Characterization of peripheral regulatory CD4+ T cells that prevent diabetes onset in non obese diabetic mice. J Immunol. 2000;164:240–247. doi: 10.4049/jimmunol.164.1.240. [DOI] [PubMed] [Google Scholar]
- 37.You S, Slehoffer G, Barriot S, Bach JF, Chatenoud L. Unique role of CD4+CD62L+ regulatory T cells in the control of autoimmune diabetes in T cell receptor transgenic mice. Proc Natl Acad Sci USA. 2004;101 2:14580–14585. doi: 10.1073/pnas.0404870101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brusko T, Wasserfall C, McGrail K, Schatz R, Viener HL, Schatz D, Haller M, Rockell J, Gottlieb P, Clare-Salzler M, Atkinson M. No alterations in the frequency of FOXP3+ regulatory T-cells in type 1 diabetes. Diabetes. 2007;56:604–612. doi: 10.2337/db06-1248. [DOI] [PubMed] [Google Scholar]
- 39.Brusko TM, Wasserfall CH, Clare-Salzler MJ, Schatz DA, Atkinson MA. Functional defects and the influence of age on the frequency of CD4+ CD25+ T-cells in type 1 diabetes. Diabetes. 2005;54:1407–1414. doi: 10.2337/diabetes.54.5.1407. [DOI] [PubMed] [Google Scholar]
- 40.Barr TA, Brown S, Ryan G, Zhao J, Gray D. TLR-mediated stimulation of APC: Distinct cytokine responses of B cells and dendritic cells. Eur J Immunol. 2007;37:3040–3053. doi: 10.1002/eji.200636483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mazzoni A, Segal DM. Controlling the Toll road to dendritic cell polarization. J Leukoc Biol. 2004;75:721–730. doi: 10.1189/jlb.1003482. [DOI] [PubMed] [Google Scholar]
- 42.Christen U, Wolfe T, Möhrle U, Hughes AC, Rodrigo E, Green EA, Flavell RA, von Herrath MG. A dual role for TNF-alpha in type 1 diabetes: islet-specific expression abrogates the ongoing autoimmune process when induced late but not early during pathogenesis. J Immunol. 2001;166:7023–7032. doi: 10.4049/jimmunol.166.12.7023. [DOI] [PubMed] [Google Scholar]
- 43.Wu AJ, Hua H, Munson SH, McDevitt HO. Tumor necrosis factor-alpha regulation of CD4+CD25+ T cell levels in NOD mice. Proc Natl Acad Sci USA. 2002;99:12287–12292. doi: 10.1073/pnas.172382999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee LF, Xu B, Michie SA, Beilhack GF, Warganich T, Turley S, McDevitt HO. The role of TNF-alpha in the pathogenesis of type 1 diabetes in the nonobese diabetic mouse: analysis of dendritic cell maturation. Proc Natl Acad Sci USA. 2005;102:15995–16000. doi: 10.1073/pnas.0508122102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–1067. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng XX, Steele AW, Hancock WW, Stevens AC, Nickerson PW, Roy-Chaudhury P, Tian Y, Strom TB. A noncytolytic IL-10/Fc fusion protein prevents diabetes, blocks autoimmunity, and promotes suppressor phenomena in NOD mice. J Immunol. 1997;158:4507–4513. [PubMed] [Google Scholar]
- 47.Huang X, Zhu J, Yang Y. Protection against autoimmunity in nonlymphopenic hosts by CD4+ CD25+ regulatory T cells is antigen-specific and requires IL-10 and TGF-beta. J Immunol. 2005;175:4283–4291. doi: 10.4049/jimmunol.175.7.4283. [DOI] [PubMed] [Google Scholar]
- 48.Fontenot JD, Rasmussen JP, Gavin M, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 49.Tang Q, Adams JY, Panaranda C, Melli K, Piaggio E, Sgouroudis E, Piccirillo CA, Salomon BL, Bluestone JA. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity. 2008;28:687–697. doi: 10.1016/j.immuni.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med. 2003;197:1107–1117. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Christianson SW, Shultz LD, Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes. 1993;42:44–55. doi: 10.2337/diab.42.1.44. [DOI] [PubMed] [Google Scholar]
- 52.Friedline RH, Wong CP, Steeber DA, Tedder TF, Tisch R. L-selectin is not required for T cell-mediated autoimmune diabetes. J Immunol. 2002;168:2659–2666. doi: 10.4049/jimmunol.168.6.2659. [DOI] [PubMed] [Google Scholar]
- 53.Szanya V, Ermann J, Taylor C, Holness C, Fathman CG. The subpopulation of CD4+CD25+ splenocytes that delays adoptive transfer of diabetes expresses L-selectin and high levels of CCR7. J Immunol. 2002;169:2461–2465. doi: 10.4049/jimmunol.169.5.2461. [DOI] [PubMed] [Google Scholar]
- 54.Bingaman AW, Patke DS, Mane VR, Ahmadzadeh M, Ndejembi M, Bartlett ST, Farber DL. Novel phenotypes and migratory properties distinguish memory CD4 T cell subsets in lymphoid and lung tissue. Eur J Immunol. 2005;35:3173–186. doi: 10.1002/eji.200526004. [DOI] [PubMed] [Google Scholar]
- 55.Unsoeld H, Krautwald S, Voehringer D, Kunzendorf U, Pircher H. CCR7+ and CCR7- memory T cells do not differ in immediate effector cell function. J Immunol. 2002;169:638–641. doi: 10.4049/jimmunol.169.2.638. [DOI] [PubMed] [Google Scholar]
- 56.Dooms H, Wolslegel K, Lin P, Abbas AK. Interleukin-2 enhances CD4+ T cell memory by promoting the generation of IL-7R alpha-expressing cells. J Exp Med. 2007;204:547–557. doi: 10.1084/jem.20062381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huehn J, Siegmund K, Lehmann JC, Siewart C, Haubold U, Feuerer M, Debes GF, Lauber J, Frey O, Przybylski GK, Niesner U, Rosa Mdela, Schmidt CA, Brauer R, Buer J, Scheffold A, Hamann a. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med. 2004;199:303–313. doi: 10.1084/jem.20031562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang S, Xia C, Morel L. IL-6 produced by dendritic cells from lupus-prone mice inhibits CD4+CD25+ T cell regulatory functions. J Immunol. 2007;178:271–279. doi: 10.4049/jimmunol.178.1.271. [DOI] [PubMed] [Google Scholar]
- 59.Bellelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 60.O'Sullivan BJ, Thomas HE, Pai S, Santamaria P, Iwakura Y, Steptoe RJ, Kay TW, Thomas R. IL-1 beta breaks tolerance through expansion of CD25+ effector T cells. J Immunol. 2006;176:7278–7287. doi: 10.4049/jimmunol.176.12.7278. [DOI] [PubMed] [Google Scholar]
- 61.Cools N, Ponsaerts P, Van Tendeloo VF, Berneman ZN. Balancing between immunity and tolerance: an interplay between dendritic cells, regulatory T cells, and effector T cells. J Leukoc Biol. 2007;82:1365–1374. doi: 10.1189/jlb.0307166. [DOI] [PubMed] [Google Scholar]
- 62.Kukreja A, Cost G, Marker J, Zhang C, Sun Z, Lin-Su K, Ten S, Sanz M, Exley M, Wilson B, Porcelli S, Maclaren N. Multiple immuno-regulatory defects in type-1 diabetes. J Clin Invest. 2002;109:131–140. doi: 10.1172/JCI13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alard P, Manirarora JN, Parnell SA, Hudkins JL, Clark SL, Kosiewicz MM. Deficiency in NOD antigen-presenting cell function may be responsible for suboptimal CD4+CD25+ T-cell-mediated regulation and type 1 diabetes development in NOD mice. Diabetes. 2006;55:2098–2105. doi: 10.2337/db05-0810. [DOI] [PubMed] [Google Scholar]
- 64.Kabelitz D. Expression and function of Toll-like receptors in T lymphocytes. Curr Opin Immunol. 2007;19:39–45. doi: 10.1016/j.coi.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 65.Xu D, Liu H, Komai-Koma M. Direct and indirect role of Toll-like receptors in T cell mediated immunity. Cell Mol Immunol. 2004;1:239–246. [PubMed] [Google Scholar]
- 66.Feillet H, Bach JF. On the mechanisms of the protective effect of infections on type 1 diabetes. Clin Dev Immunol. 2004;11:191–194. doi: 10.1080/17402520400004557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harada M, Kishimoto Y, Makino S. Prevention of overt diabetes and insulitis in NOD mice by a single BCG vaccination. Diabetes Res Clin Pract. 1990;8:85–89. doi: 10.1016/0168-8227(90)90017-n. [DOI] [PubMed] [Google Scholar]
- 68.Bras A, Aguas AP. Diabetes-prone NOD mice are resistant to Mycobacterium avium and the infection prevents autoimmune disease. Immunology. 1996;89:20–25. doi: 10.1046/j.1365-2567.1996.d01-717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Toyota T, Satoh J, Oya K, Shintani S, Okano T. Streptococcal preparation (OK-432) inhibits development of type I diabetes in NOD mice. Diabetes. 1986;35:496–499. doi: 10.2337/diab.35.4.496. [DOI] [PubMed] [Google Scholar]
- 70.Ryu S, Kodama S, Ryu K, Schoenfeld DA, Faustman DL. Reversal of established autoimmune diabetes by restoration of endogenous beta cell function. J Clin Invest. 2001;108:63–72. doi: 10.1172/JCI12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yadav M, Schorey JS. The beta-glucan receptor dectin-1 functions together with TLR2 to mediate macrophage activation by mycobacteria. Blood. 2006;108:3168–3175. doi: 10.1182/blood-2006-05-024406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shin DM, Yang CS, Yuk JM, Lee JY, Kim KH, Shin SJ, Takahara K, Lee SJ, Jo EK. Mycobacterium abscessus activates the macrophage innate immune response via a physical and functional interaction between TLR2 and dectin-1. Cell Microbiol. 2008 Apr 16; doi: 10.1111/j.1462-5822.2008.01151.x. 2008. Epub ahead of print. [DOI] [PubMed] [Google Scholar]