

Abstract
Retinol dehydrogenase 12 (RDH12) is a novel member of the short-chain dehydrogenase/reductase superfamily of proteins that was recently linked to Leber’s congenital amaurosis 3 (LCA). We report the first biochemical characterization of purified human RDH12 and analysis of its expression in human tissues. RDH12 exhibits ~2000-fold lower Km values for NADP+ and NADPH than for NAD+ and NADH and recognizes both retinoids and lipid peroxidation products (C9 aldehydes) as substrates. The kcat values of RDH12 for retinaldehydes and C9 aldehydes are similar, but the Km values are, in general, lower for retinoids. The enzyme exhibits the highest catalytic efficiency for all-trans-retinal (kcat/Km ~900 min−1 μM−1), followed by 11-cis-retinal (450 min−1 mM−1) and 9-cis-retinal (100 min−1 mM−1). Analysis of RDH12 activity toward retinoids in the presence of cellular retinol-binding protein (CRBP) type I or cellular retinaldehyde-binding protein (CRALBP) suggests that RDH12 utilizes the unbound forms of all-trans- and 11-cis-retinoids. As a result, the widely expressed CRBPI, which binds all-trans-retinol with much higher affinity than all-trans-retinaldehyde, restricts the oxidation of all-trans-retinol by RDH12, but has little effect on the reduction of all-trans-retinaldehyde, and CRALBP inhibits the reduction of 11-cis-retinal stronger than the oxidation of 11-cis-retinol, in accord with its higher affinity for 11-cis-retinal. Together, the tissue distribution of RDH12 and its catalytic properties suggest that, in most tissues, RDH12 primarily contributes to the reduction of all-trans-retinaldehyde; however, at saturating concentrations of peroxidic aldehydes in the cells undergoing oxidative stress, for example, photoreceptors, RDH12 might also play a role in detoxification of lipid peroxidation products.
Retinol dehydrogenase 12 (RDH12)1 is a novel member of the short-chain dehydrogenase/reductase superfamily of proteins that is highly expressed in photoreceptors (1) and was recently associated with Leber’s congenital amaurosis 3 (LCA) (2, 3). LCA is a genetically heterogeneous group of disorders characterized by retinal dystrophy affecting both rods and cones with onset of symptoms in early childhood and progression to legal blindness in early adulthood (www.sph.uth.tmc.edu/Retnet). Although RDH12 is obviously important for vision, its exact physiological role in the eye and other tissues is not yet clear.
Initial analysis of the biochemical properties of RDH12 showed that this enzyme catalyzes the oxidation and reduction of both all-trans- and cis-retinoids in the presence of NADP+ or NADPH as cofactors (1). In the cells, all-trans-and 11-cis-retinoids bind to their respective binding proteins. All-trans isomers of retinol and retinaldehyde bind to the ~15 kDa cellular retinol-binding proteins (CRBPs) of four different types (4–7). CRBP type I (CRBPI) exhibits the most widespread tissue distribution and has a greater affinity for all-trans-retinol than for all-trans-retinaldehyde (8–10). 11-cis-Retinoids bind to the 36 kDa cellular retinaldehyde-binding protein (CRALBP), which exhibits a greater affinity for 11-cis-retinaldehyde than for 11-cis-retinol (11). CRALBP is abundant in the retinal pigment epithelium and Müller glial cells (12, 13). It has been suggested that the physiologically relevant retinol dehydrogenases should be able to utilize retinoids bound to their respective carrier proteins (14). Whether RDH12 can metabolize CRBPI- or CRALBP-bound retinoids is not yet known.
RDH12 shares the highest sequence similarity (~79%) with RDH11 (also known as PSDR1 (15) and RalR1 (16, 17)). The mouse ortholog of RDH11 named short-chain aldehyde reductase (SCALD) exhibits high reductive activity toward toxic lipid peroxidation products such as medium-chain (>C6) aldehydes trans-2-nonenal, nonanal, and cis-6-nonenal (18). Lipid peroxidation has been associated with a number of retinal diseases, for example, age-related maculopathy and diabetic retinopathy. The abundance of poly-unsaturated fatty acids in the membrane-rich photoreceptors renders the retina extremely vulnerable to oxidative attack (19–23). If RDH12 is active toward medium-chain fatty aldehydes, it could play a role in the detoxification of lipid peroxidation products in photoreceptors.
In the present study, we expressed and purified RDH12 to determine its catalytic efficiency for the oxidation and reduction of retinoids and medium-chain aldehydes, examined the effects of CRBPI and CRALBP on RDH12 activity toward all-trans- and 11-cis-retinoids, and determined whether RDH12 mRNA is expressed in extraocular tissues.
EXPERIMENTAL PROCEDURES
Construction of Vectors for Expression of Wild-Type and His6-Tagged RDH12 in Sf9 Cells
The full-length cDNA for human RDH12 was PCR-amplified from the retina cDNA using primers 5′-ATGCTGGTCACCTTGGGACTGCTCACC-3′ and 5′-CTACTCCCACCGGATTCCTAGAAGC-3′ and TA-cloned into pCR2.1-TOPO vector. To prepare a vector for expression of RDH12 in insect Sf9 cells, the RDH12 cDNA was excised from pCR2.1-TOPO using EcoRI restriction endonuclease, gel-purified using the QIAQuick gel extraction kit (Qiagen Inc., Valencia, CA), and cloned into the EcoRI site of the baculovirus transfer vector pVL1393. The clone with the correct orientation of the insert was identified based on the restriction digest and verified by sequencing. Recombinant baculovirus was produced by cotransfection of Sf9 cells with the transfer vector and the linearized Sapphire Baculovirus DNA (Orbigen Inc., San Diego, CA) according to the manufacturer’s instructions.
For purification of RDH12, a construct encoding RDH12 protein fused with a His6-tag on the C-terminus was first prepared in the pET28a vector (Novagen, Madison, WI). To create this construct, the RDH12 cDNA in pCR2.1-TOPO vector was PCR-amplified using the forward primer 5′-CTCCCATGGTGGTCACCTTGGGACTGCT-3′, containing an NcoI restriction site (site underlined), and the reverse primer 5′-GCCAAGCTTCTCCCACCGGATTCCTAGAAG-3′, containing a HindIII restriction site (site underlined). PCR amplification was performed for 35 cycles, with denaturation for 1 min at 94 °C, annealing for 1 min at 53 °C, and elongation for 1.5 min at 72 °C. The PCR product was gel-purified, digested with NcoI and HindIII restriction endonucleases, and cloned into the respective sites of pET28a vector (Novagen, Madison, WI) in-frame with the C-terminal His6-tag provided by pET28a.
Then, the cassette encoding RDH12–His6 fusion protein was transferred from pET28a to pVL1393 vector for expression in Sf9 cells, because RDH12 was expressed in E. coli at a very low level. The RDH12–His6 fusion cassette was excised from the pET28a vector using DraIII (flanking the 3′ end of the cassette) and XbaI (flanking the 5′ end of the cassette) restriction endonucleases and gel-purified. The cleaved DraIII site was blunt-ended using T4 DNA polymerase (New England Biolabs, Inc., Beverly, MA) before cleaving the RDH12-containing pET28a with XbaI, because pVL1393 did not have a matching DraIII site. The excised XbaI-blunt RDH12–His6 cDNA construct was gel-purified and ligated into pVL1393 baculovirus transfer vector. pVL1393 (BD Biosciences Pharmingen, San Diego, CA) was prepared by cleaving it, first, with PstI endonuclease, followed by treatment with T4 DNA polymerase to blunt-end the PstI site, and then cleaving the pVL1393 vector with XbaI to provide a 5′ cohesive end for the ligation of the XbaI-cleaved end of the RDH12–His6 cDNA. The ligation was carried out using T4 DNA ligase (New England Biolabs, Inc., Beverly, MA). The final construct was verified by DNA sequencing. Recombinant baculovirus was produced as described above.
Preparation of Purified RDH12–His6
Expression of recombinant proteins in insect Sf9 cells was carried out as described previously for RDH11 and other microsomal SDRs (16, 24–26). Briefly, Sf9 cells were infected with recombinant virus at a virus/cell ratio of 10:1 and incubated at 28 °C for 3–4 days. Sf9 cells were harvested by centrifugation at 5000g for 10 min and resuspended in 10 mM Hepes (pH 7.4), 250 mM sucrose, 0.2 mM EDTA, 5 mM β-mercaptoethanol, 1.5 μg/mL aprotinin, 1.5 μg/mL leupeptin, and 1.0 μg/mL pepstatin A. The cell suspension was homogenized using a French press mini-cell at 800 psi. Phenylmethylsulfonyl fluoride was added to 5 μM immediately after homogenization. The unbroken cells, cellular debris, nuclei, and mitochondria were removed by centrifugation at 12000g for 20 min. Microsomes containing RDH12 were pelleted by centrifugation at 105000g for 2.5 h and resuspended in 90 mM potassium phosphate, pH 7.4, 40 mM KCl (Buffer A), plus 0.1 mM EDTA, 1 mM dithiothreitol, and 20% glycerol. Protein concentration was determined by the method of Lowry et al. (27) using bovine serum albumin as a standard.
For purification of RDH12–His6, the 105000g pellet of RDH12–His6 microsomes was resuspended in a buffer containing 50 mM KH2PO4 (pH 8.0), 500 mM KCl, 5 mM β-mercaptoethanol, and 10% glycerol (Buffer B) and supplemented with inhibitors of proteases, 20 mM imidazole, and 15 mM detergent 1,2-diheptanoyl-sn-glycero-3-phosphocholine (DHPC) (Avanti Polar Lipids, Alabaster, AL) (28). To extract RDH12–His6, the microsomal membranes were incubated in the extraction buffer with DHPC for 1 h under vortexing on ice followed by centrifugation at 105000g for 2.5 h to remove the insoluble material. Binding of solubilized RDH12–His6 to Ni2+–NTA resin was carried out in a batch mode according to the manufacturer’s instructions (Qiagen Inc.). Following a 2 h-incubation with DHPC extract on ice, the resin was washed with 65 bed volumes of buffer B supplemented with inhibitors of proteases, 50 mM imidazole, and 2 mM DHPC. RDH12–His6 was eluted with a stepwise gradient of 100, 150, 200, 250, and 300 mM imidazole in buffer B plus 2 mM DHPC. Fractions were analyzed by electrophoresis in a 12% denaturing polyacrylamide gel in the presence of sodium dodecyl sulfate (SDS–PAGE). The actual molecular mass of RDH12–His6 (36.5 kDa) correlated well with the experimentally predicted molecular mass (36 kDa). Those fractions that contained homogeneous RDH12–His6 were combined and concentrated. To remove imidazole, the buffer in the eluate was exchanged for buffer A plus 2 mM DHPC, 2 mM β-mercaptoethanol, and 10% glycerol using Centriplus concentrators (Millipore Corp., Billerica, MA). Purified RDH12–His6 preparations were stored at −80 °C for several months without loss of activity.
Preparation of Sf9 Microsomes Containing RDH5 (11-cis-Retinol Dehydrogenase)
EST clone C152778 encoding RDH5 was obtained from Research Genetics (Huntigton, AL). The full-length cDNA was amplified using primers GCT GGA TCC ATG TGG CTG CCT CTT CTG CT (forward primer, BamHI site underlined) and AGG GAA TTC TCA GTA GAC TGC TTG GGC AG (reverse primer, EcoRI site underlined) and cloned into the baculovirus vector pVL1393. Preparation of the recombinant baculovirus, expression of the protein in Sf9 cells, and isolation of the microsomal fraction were carried out as described for RoDH-4 (24).
The activity of RDH5 was determined using 0.4 μg of RDH5-expressing Sf9-microsomes, 1 mM NAD+, and 5 μM 11-cis-retinol alone or in the presence of 5 μM CRALBP. The 0.5 mL-reactions were carried out for 10 min at 37 °C.
Preparation of Apo- and Holo- Forms of CRBPI
CRBPI was expressed in E. coli as a C-terminal fusion to a bifunctional tag, consisting of the chitin binding domain (CBD) and the intein (CBD-intein). For purification of CRBPI using the C-terminal CBD-intein tag, the corresponding cDNA was cloned between the NdeI and XhoI restriction sites of pKYB1 vector (New England Biolabs Inc., Beverly, MA). Expression and purification of CRBPI fused to CBD-intein were carried out using the IMPACT-CN protein purification system (New England Biolabs Inc.). Specifically, M9ZB medium supplemented with 15 μg/mL of kanamycin was inoculated with freshly grown culture of BL21(DE3) cells transfected with CRBPI-pKYB1 vector. Expression of the protein was induced by the addition of 1 mM IPTG at OD600 of 0.8. Cells were incubated in a shaker at room temperature overnight. Harvested cells were resuspended in an ice-cold 20 mM Tris-HCl (pH 8.0), 500 mM NaCl, 1 mM EDTA (buffer C) supplemented with 5 mM benzamidine and 4 mM PMSF. The cells were broken by French press followed by sonication on ice to reduce viscosity. The cell extract was clarified by centrifugation at 20000g for 30 min and then slowly loaded onto the chitin column. The column was washed with 25–30 bed volumes of buffer C followed by a quick flushing with 3 bed volumes of buffer C supplemented with 50 mM DTT. The flow in the column was stopped, and the column was left at room temperature for 16 h. After the induction of the cleavage reaction, CRBPI was released from the intein tag and was eluted from the column using 4 bed volumes of buffer C. As judged by SDS–PAGE analysis, the preparation of CRBPI was nearly homogeneous. The yield of CRBPI was ~15–20 mg/L of culture.
To prepare holo-CRBPI, an aliquot of purified apo-CRBPI was saturated with a 2-fold molar excess of all-trans-retinol at room temperature for 1 h. Unbound retinol was removed by gel-filtration on G50 Sephadex column as described previously (28). The A350/A280 ratio of the holo-CRBPI preparation was 1.76. Holo-CRBPI was stored in small aliquots at −80 °C.
All-trans-retinal complexed with CRBPI was prepared by incubating 20 μM apo-CRBPI with 25 μM all-trans-retinal for 20 min on ice, followed by purification of the complex from unbound retinal by gel-filtration through Sephadex G-50 spin column.
Preparation of Apo- and Holo- Forms of CRALBP
Full-length coding sequence of CRALBP cDNA was PCR-amplified using the forward primer 5′-CCATAGGCCATATGTCAGAAGGGGTG -3′, containing an NdeI restriction site (site underlined), and the reverse primer 5′-GAGGGATCCTCAGAAGGCTGTGTTCTCAGCTTG -3′, containing a BamHI restriction site (site underlined). The PCR product was gel-purified and cloned between the respective sites of the pET19 vector. CRALBP protein fused to the N-terminal His10-tag was expressed and purified as described previously (11).
To prepare holo-CRALBP, purified CRALBP was desalted and incubated for 20 min on ice with a 1.2-fold molar excess of 11-cis-retinal, which was prepared in ethanol. To remove the unbound retinal, holo-CRALBP was applied onto Ni–NTA affinity resin; the resin was washed, and holo-CRALBP was eluted with 250 mM imidazole in 40 mM potassium chloride and 90 mM potassium phosphate (pH 7.4) buffer. Residual unbound 11-cis-retinal was removed by Sephadex G-50 spin chromatography. The ratio of the extinction coefficients for holo-CRALBP with bound 11-cis-retinal (ε280/ε425 = 2.8) (11) was used to estimate CRALBP-binding stoichiometry from observed A280/A425 absorbance spectral ratios obtained by UV–visible spectrophotometry.
HPLC Analysis of RDH12 Activity
Catalytic activity of the membrane-bound RDH12 or purified RDH12–His6 was assayed in buffer A at 37 °C in siliconized glass tubes (24). The 500-μL reactions were terminated by the addition of 1 mL cold methanol. Retinoids were extracted twice with 2 mL of hexane, separated in hexane/tert-butyl-methyl ether (96:4) mobile phase at 2 mL/min flow rate, and analyzed using a Waters 2996 Photodiode Array Detector. The stationary phase was a Waters Spherisorb S3W column (4.6 mm × 100 mm). Elution of retinoids was monitored at 350 nm. On a typical chromatogram, elution times were as follows: 3.17 min for 9-cis-retinal, 4.38 min for all-trans-retinal, 14.41 min for 9-cis-retinol, and 15.59 min for all-trans-retinol. 11-cis-Retinoids were separated at a 1.5 mL/min flow rate with typical elution times of 3.58 min for 11-cis-retinal and 14.95 min for 11-cis-retinol. Retinoids were quantified by comparing their peak areas to a calibration curve constructed from peak areas of a series of standards. Using the known molar extinction coefficients of 9-cis-retinol and 9-cis-retinal in ethanol (42300 and 36100, respectively), we calculated the extinction coefficients in hexane as 43765 for 9-cis-retinol with λmax at 320 nm and 38737 for 9-cis-retinal with λmax at 364 nm.
Determination of Kinetic Constants
The apparent Km values for the reduction of retinals were determined at 1 mM NADPH and six concentrations of all-trans-retinal (0.01–1.0 μM), 11-cis-retinal (0.125–4 μM), or 9-cis-retinal (0.01–5 μM). The apparent Km values for the oxidation of retinols were determined at 1 mM NADP+ and six concentrations of all-trans-retinol (0.5–10.0 μM), 11-cis-retinol (0.125–2 μM), or 9-cis-retinol (0.2–3.2 μM). Reaction rates were determined based on the percent substrate conversion, which was calculated as follows. Retinol and retinaldehyde obtained from each reaction mixture were quantified based on their peak area values using the respective calibration curves as described above. The amounts of retinol and retinaldehyde were summarized to obtain the total amount of retinoids (equivalent to the initial substrate amount) in the reaction (100%). Then, the amount of product was divided by the total amount of retinoids to determine the percent conversion at each substrate concentration. The extraction efficiencies for retinol and retinaldehyde were similar.
9-cis-Retinol was prepared enzymatically by RDH12-catalyzed reduction of 9-cis-retinal and purified by HPLC. 11-cis-Retinal was obtained from the National Eye Institute. 11-cis-Retinol was prepared by the reduction of 11-cis-retinal catalyzed by RDH5. Specifically, 20 μM 11-cis-retinal solubilized with the equimolar BSA was incubated with RDH5-expressing Sf9 microsomes in the presence of 0.5 mM NADPH. Retinoids were extracted by hexane and separated using HPLC. 11-cis-Retinol was collected; mobile phase was evaporated under nitrogen flow, and the residue was dissolved in ethanol and solubilized with the equimolar BSA to obtain a 10–15 μM stock.
The apparent Km values for cofactors were determined at 5 μM all-trans-retinal or all-trans-retinol and six concentrations of NADPH (0.25–20.0 μM), NADP+ (0.2–10.0 μM), NADH (0.1–20.0 mM), or NAD+ (1.0–10.0 mM). The reaction rate was linear with up to 0.16 μg/mL of RDH12–His6 for 15 min in the presence of 10 μM all-trans-retinal as substrate. The background value without cofactor was determined for each concentration of substrate and was subtracted from each data point.
Kinetic constants for the reduction of nonanal, 6-cis-nonenal, trans-2-nonenal, and 4-hydroxynonenal were determined spectrophotometrically at 37 °C in 1 mL of buffer A with 100 μM NADPH. Aldehyde solutions were prepared in ethanol. The concentrations of aldehydes varied between 1 and 150 μM. The concentration of ethanol was the same in all reaction mixtures and did not exceed 0.75%. Ethanol was not inhibitory with up to 3% final concentration. Control reactions contained all the components with the exception of aldehyde substrates. The amount of purified enzyme was 0.2–0.4 μg/reaction. Six to eight points were obtained in duplicates for each kinetic curve in three to four independent experiments.
Initial velocities (nmol/min of product formed per milligram of protein) were obtained by nonlinear regression analysis. Kinetic constants were calculated using GraFit (Erithacus Software Ltd., U.K.) and expressed as the means ± SD. The results shown are representative of three to four experiments. Goodness of fit was tested using the F test (29). The probability value was calculated from the reduced χ2 values and was considered to be significant if it was lower than 0.1.
RDH12 activity toward steroids was assayed using [3H]-labeled 5α-androstan-3α-ol-17-one (androsterone), 5-androsten-3β-ol-17-one (dehydroepiandrosterone), 4-pregnen-3,20-dione (progesterone), 4-pregnen-11β,21-diol-3,20-dione (corticosterone), 5α-pregnan-3α-ol-20-one (allopregnanolo-ne), and 14C-labeled 5α-androstan-17β-ol-3-one (dihydrotestosterone) (NEN Life Science Products, Boston, MA). Aqueous solutions of steroids were prepared as described previously (24). Reactions were carried out for 1 h at 37 °C in 150 μL of reaction buffer in the presence of 1 mM NADPH and 5 μM steroid compound. The amount of the microsomal protein in the reaction mixture was 5 μg. Reaction products were extracted and analyzed by thin-layer chromatography (25). To visualize tritiated steroids, TLC plates were exposed to a tritium phosphorimager screen. [14C]-labeled steroid was detected by exposure to an X-ray film.
Incubations of All-trans-Retinol with CRBPI or BSA
All-trans-retinol or all-trans-retinaldehyde dissolved in ethanol was added to phosphate-buffered saline containing 50 mM DTT and either 20 μM apo-CRBPI or 20 μM bovine serum albumin (BSA) to the final concentration of 20 μM. Tubes containing retinol and equimolar CRBPI or BSA were sonicated for 10 min in an ultrasonic waterbath to disperse retinol. Sonication had no effect on the isomeric composition of retinol. Samples were incubated at 4 °C or at room temperature in the light or covered with foil as indicated. At selected time points, 500 μL-aliquots were extracted with hexane and analyzed by HPLC.
Quantitative Real-Time PCR Analysis of RDH12 Expression in Human Tissues
Real time PCR was carried out in triplicates for each tissue using SYBR green PCR master mix (Applied Biosystems, Foster City, CA), 0.2 μM of each primer, and 12.5 pg of human retina marathon ready cDNA (Clontech Laboratories, Inc., Palo Alto, CA) or 200 pg of other human tissue cDNA in multiple tissue cDNA panel I and II (Clontech). The PCR protocol was 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 30 s. PCR was performed and analyzed using an ABI Prism 7700 sequence detector. PCR without cDNA templates did not produce significant amplification products. Specificity of the primers was verified by the amplification of a single PCR product, which was determined by observing a single dissociation curve from each tissue. Primers for the real-time PCR were as follows: RDH12 forward, 5′-TCT TGA CCC TTC TGG GAA TG-3′; RDH12 reverse, 5′-CCA GAG GGT TGC TGA CTT TC-3′; CRBPI forward, 5′-CCAGTGGATCGAGGGTGATGAG-3′; CRBPI reverse, 5′-TCCTGCTGATTCCTTGGGACAA-3′; glyceraldehyde-3-phosphate dehydrogenase (GAPDH) forward, 5′-ACT TCA ACA GCG ACA CCC ACT C-3′; and GAPDH reverse, 5′-CAC CCT GTT GCT GTA GCC AAA-3′.
Relative expression of RDH12 and CRBPI was calculated according to the ABI prism 7700 sequence detection system user bulletin no. 2. Briefly, RDH12 and CRBPI expression was normalized to GAPDH and then to their respective expression levels in the liver, using the following formula: ΔΔCTgene(tissue) = ΔCT(gene–GAP)tissue − ΔCT(gene–GAP)liver; relative expression = 2−ΔΔCTgene (30). Tissues expressing low amounts of the gene had a standard deviation similar to that observed in the tissues expressing high amounts of the transcript.
RESULTS
Purification and Kinetic Characterization of RDH12–His6
To obtain purified RDH12, the protein was expressed in Sf9 cells as a C-terminal fusion to a six-histidine tag. The His6-tagged RDH12 was associated with the microsomal membranes of Sf9 cells. This subcellular localization was identical to that of the untagged wild-type recombinant RDH12 expressed in Sf9 cells as a control. The level of expression and the all-trans-retinal reductase activity of the His6-tagged RDH12 were also very similar to those of wild-type RDH12 (data not shown).
RDH12–His6 was purified from DHPC-solubilized microsomal membranes using metal affinity chromatography as described under Experimental Procedures. Purified RDH12–His6 was nearly homogeneous (Figure 1) with an average all-trans-retinal reductase activity of ~1000 (nmol/min)/mg, which represented a 22-fold increase over the specific activity of the starting microsomal preparation of RDH12.
Figure 1.
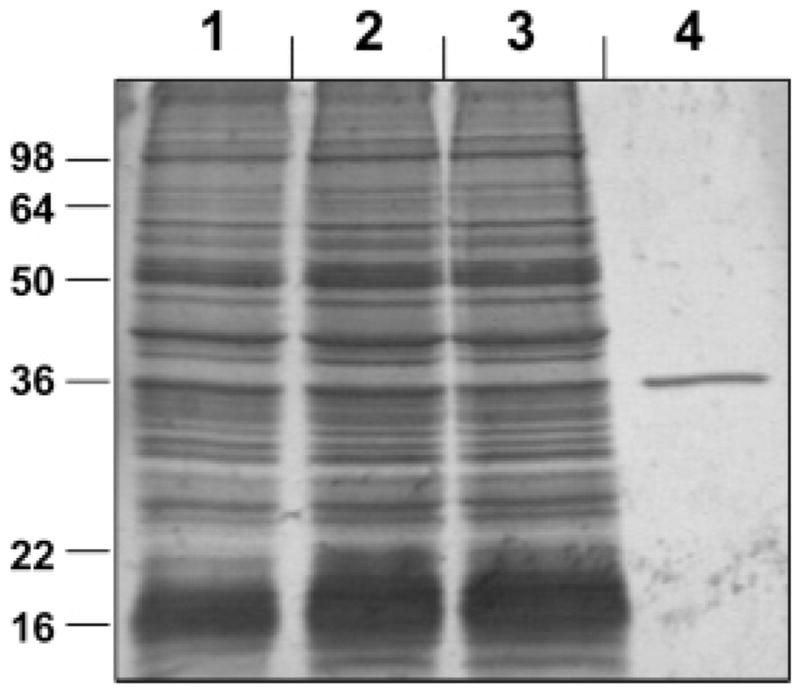
Purification of RDH12–His6. Samples of RDH12-containing fractions were analyzed by SDS–PAGE and stained by Coomassie R-250. Lane 1, Sf9 microsomes containing RDH12–His6 (16 μg); lane 2, DHPC extract of Sf9 microsomes (16 μg); lane 3, flow through the Ni2+–NTA resin (~16 μg); and lane 4, purified RDH12–His6 (0.42 μg). Positions of molecular weight markers are indicated on the left.
The apparent Km values of purified RDH12–His6 for retinoids and cofactors were similar to those of wild-type recombinant RDH12 expressed in Sf9 cells (Table 1), indicating that the addition of the His6-tag did not affect the properties of the enzyme. The Km values for NADP+ and NADPH were several 100-fold lower than those for NAD+ and NADH (Table 1).
Table 1.
Kinetic Constants of Microsomal and Purified RDH12 for Cofactorsa
| microsomal wtRDH12
|
purified RDH12–His6 |
|||
|---|---|---|---|---|
| cofactor | apparent KmμM | apparent Vmax (nmol/min)/mg | apparent KmμM | apparent Vmax (nmol/min)/mg |
| NADP+ | 1.2 ± 0.2 | 79.0 ± 2.5 | 3.2 ± 0.2 | 770 ± 30 |
| NAD+ | NDb | NDb | 7500 ± 2400 | 460 ± 80 |
| NADPH | 1.2 ± 0.3 | 98.0 ± 2.0 | 0.74 ± 0.06 | 1020 ± 20 |
| NADH | 2200 ± 130 | 180 ± 20 | 2400 ± 130 | 1600 ± 25 |
The apparent Km values for cofactors were determined at fixed saturating all-trans-retinal (5 μM) and all-trans-retinol (5 μM).
ND, not determined.
The Km values for substrates were determined using saturating concentrations of either NADP+ in the oxidative direction or NADPH in the reductive direction. The array of compounds included different stereoisomers of retinol and retinal, medium-chain (C9) aldehydes, and a variety of hydroxyl and ketosteroids. RDH12–His6 exhibited no significant 3α-, 3β,-, 17β-, or 11β-hydroxysteroid dehydrogenase or 3-, 17-, or 20-ketone reductase activity with any of the steroids tested. However, the enzyme was very active toward C9 aldehydes. The highest rate of conversion was observed with nonanal (Vmax of 1600 (nmol/min)/mg), followed by cis-6-nonenal (Vmax of 1300 (nmol/min)/mg) and trans-2-nonenal (Vmax of 810 (nmol/min)/mg). The Km values for nonanal and trans-2-nonenal were 3 and 20 μM, respectively (Table 2). The Km for cis-6-nonenal appeared to be lower than 1 μM, but the exact value could not be determined due to limited sensitivity of the spectrophotometric assay. 4-Hydroxynonenal was reduced with the Vmax of 180 (nmol/min)/mg and the Km value of less than 10 μM (Table 2).
Table 2.
Kinetic Constants of Purified RDH12 for Substratesa
| substrate | apparent KmμM | apparent kcat min−1 | kcat/Km min−1μM−1 |
|---|---|---|---|
| all-trans-retinal | 0.04 ± 0.01 | 36 ± 1.7 | 900 |
| 11-cis-retinal | 0.10 ± 0.05 | 45 ± 4 | 450 |
| 9-cis-retinal | 0.14 ± 0.03 | 14 ± 1 | 100 |
| all-trans-retinol | 0.40 ± 0.10 | 27 ± 2 | 68 |
| 11-cis-retinol | 0.16 ± 0.03 | 7.0 ± 0.7 | 44 |
| 9-cis-retinol | 0.16 ± 0.03 | 7.0 ± 0.3 | 44 |
| nonanal | 3.1 ± 0.7 | 56 ± 3 | 18 |
| cis-6-nonenal | <1b | 45 ± 1 | >45 |
| trans-2-nonenal | 20 ± 4 | 28 ± 2 | 1.4 |
| 4-hydroxynonenal | <10b | 6 ± 1 | – |
Kinetic constants for the reduction of retinals were determined in the presence of saturating NADPH (1 mM). Kinetic constants for the oxidation of retinols were determined at saturating NADP+ (1 mM). Kinetic constants were calculated using GraFit (Erithacus Software Ltd.) and expressed as the means ± SD.
The exact Km value could not be determined because of the limited sensitivity of the spectrophotometric detection.
Because the human ortholog (RDH11) of mouse SCALD has not been previously tested with C9 aldehydes as substrates, we included this enzyme in the present study for the purpose of comparison with the highly related RDH12. Surprisingly, human RDH11 exhibited little or no activity toward C9 aldehydes. Human RDH11–His6, purified as described previously (17), reduced cis-6-nonenal with the apparent Km value of 5 ± 1 μM and the Vmax value of only 19 ± 0.7 (nmol/min)/mg. The Km value of RDH11 for trans-2-nonenal could not be determined due to limitations of the detection method, but the reaction rate at 100 μM trans-2-nonenal was as low as 12.8 (nmol/min)/mg. Little or no RDH11 activity was observed with nonanal or 4-hydroxynonenal. These results suggested that, enzymatically, human RDH12 shared more similarity with the mouse RDH11 than the human RDH11.
The Km values of RDH12 for all tested retinoids were much lower (0.04–0.4 μM) than those for C9 aldehydes, but the Vmax values were similar (Table 2), suggesting that at saturating concentrations of C9 aldehydes, the RDH12 activity would be as high as that with retinoids. In terms of catalytic efficiency (kcat/Km), all-trans-retinal appeared to be the best substrate (900 min−1 μM−1) followed by 11-cis-retinal (450 min−1 μM−1) and 9-cis-retinal (100 min−1 μM−1) (Table 2). In general, the catalytic efficiency of RDH12 was higher in the reductive direction than in the oxidative.
Effect of CRBPI on RDH12 Activity toward All-trans-Retinoids
Previously, it was shown that the activity of RDH12 toward all-trans-retinal and all-trans-retinol was severalfold lower in the presence of CRBPI than in its absence (1). However, without knowing the kinetic constants of RDH12 for these compounds, it was not entirely clear whether the enzyme utilized the available unbound retinoids in the reaction mixture or whether it was able to recognize and utilize the CRBPI-bound retinoids as substrates at a lower rate than free retinoids. To determine whether RDH12 could oxidize CRBPI-bound retinol, holo-CRBPI was prepared by saturating the binding protein with all-trans-retinol and purifying holo-CRBPI from free retinol by size exclusion chromatography. As shown in Figure 2, RDH12 produced small amounts of three isomers of retinal from holo-CRBPI. 13-cis-Retinaldehyde was a minor component and, most likely, was formed by spontaneous thermal isomerization of all-trans-retinal (31). However, 9-cis-retinaldehyde was clearly produced by the oxidation of 9-cis-retinol that was detected in holo-CRBPI (Figure 2B). At 1 μM total holo-CRBPI, all-trans-retinaldehyde (1.52 pmol) was the dominant peak compared to 9-cis-retinaldehyde (0.63 pmol) (Figure 2A). At 10 μM holo-CRBPI, 9-cis-retinaldehyde became the dominant peak (3.75 pmol versus 3.55 pmol for all-trans-retinal) (Figure 2C, inset). No product was formed in the absence of NADPH (Figure 2B,D). Since the kinetic parameters of RDH12 for the oxidation of 9-cis-retinol and all-trans-retinol were very close (Table 2), these results suggested that the amounts of all-trans-retinol and 9-cis-retinol available for the oxidation by RDH12 were similar despite the ~10-fold excess of the total all-trans-retinol over 9-cis-retinol in the preparation of holo-CRBPI. In other words, the majority of all-trans-retinol was sequestered from RDH12 by CRBPI, whereas 9-cis-retinol appeared to be readily available.
Figure 2.

Oxidation of holo-CRBPI by RDH12. Purified RDH12–His6 (0.7 μg) was incubated with the indicated concentrations of holo-CRBPI, and the products were extracted and analyzed by normal-phase HPLC as described under Experimental Procedures. HPLC traces were measured at 350 nm. Peaks were identified as follows: 1, 13-cis-retinal; 2, 9-cis-retinal; 3, all-trans-retinal; 4, 13-cis-retinol; 5, 9-cis-retinol; 6, all-trans-retinol. The result shown is representative of at least three independent experiments.
Previously, it was reported that 9-cis-retinol is a poor ligand for CRBPI (8–10). To determine just how well CRBPI binds 9-cis-retinol, we measured RDH12 activity toward 9-cis-retinol in the presence of varied amounts of apo-CRBPI versus bovine serum albumin (BSA) (Figure 3). A 6.25-fold molar excess of apo-CRBPI over 9-cis-retinol inhibited the oxidation of 9-cis-retinol 90% (Figure 3E). The same molar excess of BSA inhibited the oxidation of 9-cis-retinol 41% (Figure 3F). Thus, even though CRBPI exhibited a better binding of 9-cis-retinol than did BSA, still, a >6-fold molar excess of apo-CRBPI was required to fully sequester 9-cis-retinol from RDH12. These results confirmed that 9-cis-retinol binds to CRBPI with low affinity. At 10 μM holo-CRBPI, there was more free 9-cis-retinol available to RDH12 than at 1 μM holo-CRBPI; hence, 9-cis-retinaldehyde became the dominant product, whereas most of all-trans-retinol remained bound to CRBPI and protected from the oxidation.
Figure 3.

CRBPI inhibition of 9-cis-retinol oxidation by RDH12. Purified RDH12–His6 (0.02 μg) was incubated with 1.6 μM 9-cis-retinol and 1 mM NADP+ in the presence of 2 μM or 10 μM apo-CRBPI. Control reactions contained the same concentrations of BSA. Products were analyzed by normal-phase HPLC. Peaks were identified as follows: 1, 9-cis-retinal; 2, 9-cis-retinol; 3, all-trans-retinol.
Using the lowest estimated Kd value of CRBPI for retinol of 0.1 nM (10), we calculated that, at 1 μM holo-CRBPI, free all-trans-retinol concentration would be ~0.01 μM. On the basis of the Vmax of RDH12 for free retinol of 770 (nmol/min)/mg and the Km of 0.4 μM (Tables 1 and 2), the reaction rate at 0.01 μM retinol was calculated as ~19 (nmol/min)/mg. The actual rate of all-trans-retinaldehyde production from 1 μM holo-CRBPI was approximately 1.5 (nmol/min)/mg, below the theoretically possible rate based on the free retinol concentration. These calculations further indicated that RDH12 utilizes the free retinoids.
The appearance of 9-cis-retinol in the chromatographically purified holo-CRBPI was surprising because 9-cis-retinol was not present in the retinol stock used to saturate CRBPI. Previously, it was reported that bovine liver membranes can isomerize all-trans-retinoic acid into 9-cis-retinoic acid and 13-cis-retinoic acid and that these isomerization reactions appear to be mediated by thiol groups (32). Since the preparation of apo-CRBPI contained 50 mM DTT, which was used to elute the protein from the chitin column (Experimental Procedures), we investigated whether DTT had an effect on the isomerization of all-trans-retinol. All-trans-retinol was solubilized using BSA and incubated under various conditions to which holo-CRBPI was exposed during its preparation and purification (Figure 4). Under all conditions tested, samples with DTT (Figure 4E–H, peak 1) contained significantly more 13-cis-retinol than samples without DTT (Figure 4A–D, peak 1). 9-cis-Retinol was also formed, especially at room temperature (Figure 4G,H, peak 3) and in the light (Figure 4D,F,H, peak 3), but to a much lesser extent than 13-cis-retinol. These results showed that, in the presence of DTT, the BSA-solubilized all-trans-retinol isomerized primarily into 13-cis-retinol, not 9-cis-retinol.
Figure 4.

Effect of DTT on the isomerization of free retinol. An aqueous 10 μM stock of all-trans-retinol was prepared from the ethanol stock by solubilization with equimolar BSA in the reaction buffer and subsequent sonication for 10 min. Equal amounts (500 μL) of solubilized all-trans-retinol were aliquoted into eight tubes. DTT was added to four out of eight tubes to the final concentration of 50 mM. Pairs of tubes (with and without DTT) were incubated for 24 h under different conditions as follows: (A) at 4 °C in the dark (covered with aluminum foil), (B) at 4 °C in the light (not covered), (C) at room temperature (RT) in the dark, and (D) at room temperature in the light. HPLC chromatograms were extracted at 325 nm. At this wavelength, 13-cis-retinol and 9-cis-retinol have an identical absorbance; therefore, the corresponding peaks can be compared directly. Peaks are numbered as follows: 1, 13-cis-retinol; 2, all-trans-retinol; 3, 9-cis-retinol.
To determine whether the presence of CRBPI played a role in accumulation of 9-cis-retinol, all-trans-retinol was incubated with apo-CRBPI or with BSA as a control. As shown in Figure 5A, the original ethanol stock solution of all-trans-retinol used in this experiment contained a small amount of 13-cis-retinol (peak 1), but little if any 9-cis-retinol. After 2 days of incubation in the light, the CRBPI-containing mixture had more 9-cis-retinol (Figure 5B, peak 3) than 13-cis-retinol. In contrast, the BSA-containing mixture contained more 13-cis-retinol than 9-cis-retinol (Figure 3C). After longer incubations, 9-cis-retinol constituted approximately half of all retinol in the CRBPI-containing mixture (Figure 5D). Thus, the accumulation of 9-cis-retinol in holo-CRBPI exposed to light appeared to be linked to the presence of CRBPI. CRBPI had no effect on isomerization of all-trans-retinaldehyde (data not shown).
Figure 5.

Isomerization of retinol in the presence of BSA or apoCRBPI. (A) HPLC spectrum of the ethanol stock of all-trans-retinol used in experiments B–D. (B) Twenty micromolar all-trans-retinol was solubilized using CRBPI and incubated at 4 °C in the light. After 2 days, retinoids were extracted with hexane and analyzed by HPLC. (C) Same as B, except CRBPI was substituted for BSA. (D) Same as B after 4 weeks of incubation. Retinoids in the control mixture were largely degraded after 4 weeks (data not shown). HPLC chromatograms were extracted at 325 nm. Peaks are numbered as follows: 1, 13-cis-retinol; 2, all-trans-retinol; 3, 9-cis-retinol. Inset: absorbance spectrum of peak 3 with a λmax of 322 nm, which matches the λmax of standard 9-cis-retinol in the mobile phase of HPLC.
The enhanced isomerization of all-trans-retinol to 9-cis-retinol in the presence of CRBPI was especially remarkable considering that, in ethanol solutions, it was 9-cis-retinol that isomerized to all-trans-retinol, not vice versa. For example, an all-trans-retinol stock solution did not contain any 9-cis-retinol (Figure 5), whereas the preparation of purified 9-cis-retinol rapidly accumulated up to ~10% of all-trans-retinol (Figure 3). Investigation of the molecular mechanism underlying the preferential formation of 9-cis-retinol in the presence of CRBPI was beyond the scope of this study. However, this finding suggested that the enzymes that recognize both all-trans- and 9-cis-retinols as substrates might appear to be active toward holo-CRBPI, while utilizing only the unbound 9-cis-retinol.
Next, we determined whether CRBPI inhibits the reduction of all-trans-retinal by RDH12. The Kd value of CRBPI for all-trans-retinaldehyde (~50 nM) is significantly higher than that for all-trans-retinol (<10 nM). Therefore, we expected that CRBPI will have a lesser effect on the reduction of retinal by RDH12 compared to the oxidation of retinol. We attempted to prepare the CRBPI-retinaldehyde complex following the protocol described above for holo-CRBPI preparation. However, HPLC analysis showed that purified CRBPI-bound retinaldehyde contained ~2.45-fold less retinal than would be expected from the 1:1 binding stoichiometry. It is possible that some retinal was lost during gel-filtration because of the lower binding affinity of CRBPI for all-trans-retinal compared to that of all-trans-retinol. To our knowledge, the extinction coefficient for CRBPI-bound retinal has not been reported, and other investigators used retinal premixed with CRBPI in the cuvette for activity measurements (33, 34).
To determine the effect of CRBPI on the reduction of all-trans-retinaldehyde, we incubated the microsomes containing wild-type RDH12 with all-trans-retinal and 0.5- to 20-fold molar excess of CRBPI (Figure 6). At a ~1:1 molar ratio of CRBPI to retinal, the initial rate of retinal reduction was as high as ~60% (48 (nmol/min)/mg) of the rate in the absence of CRBPI (Figure 6). At a 10-fold molar excess of CRBPI over retinal, RDH12 was still able to reduce retinaldehyde, albeit at a reduced rate (~19 (nmol/min)/mg). From the reported Kd value of CRBPI for retinaldehyde of 50 nM (9) and the concentration of retinaldehyde (2 μM) and CRBPI (20 μM), we calculated that the concentration of free retinaldehyde in the reaction was ~0.006 μM. This concentration of free retinaldehyde could support a reaction rate of ~13 (nmol/min)/mg. Thus, the activity of RDH12 in the presence of a 10-fold molar excess of CRBPI (~19 (nmol/min)/mg) could be largely explained by the presence of free retinaldehyde in the reaction mixture. These experiments suggested that RDH12 utilized unbound all-trans-retinoids in the reductive as well as the oxidative direction.
Figure 6.
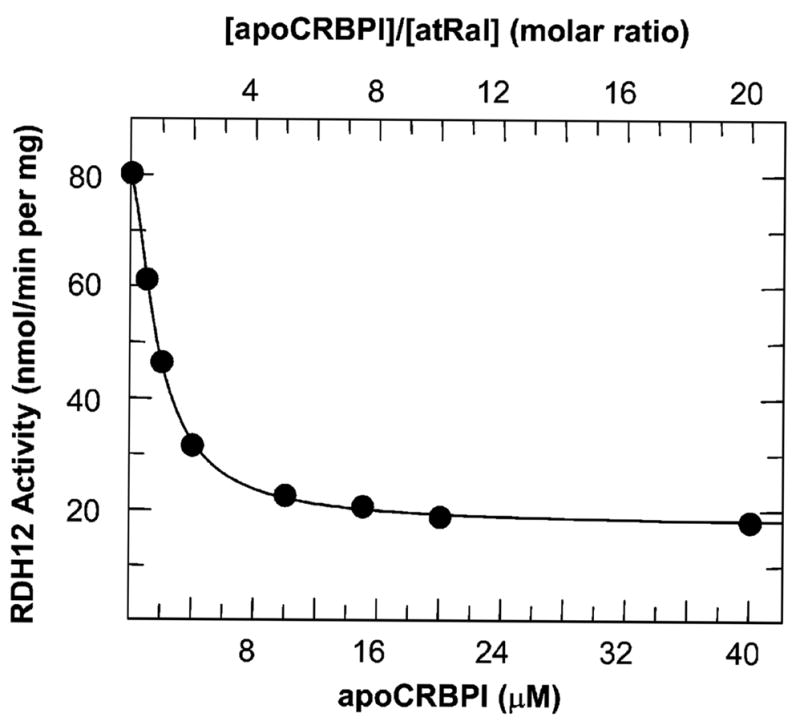
RDH12 activity toward all-trans-retinal in the presence of CRBPI. The formation of all-trans-retinol from 2 μM all-trans-retinal (atRal) was measured in the presence of increasing concentrations of CRBPI (0–40 μM). The 0.5-mL reactions were carried out with 0.3 μg of RDH12-containing Sf9 microsomes for 10 min at 37 °C. The result shown is representative of at least three independent experiments.
Effect of CRALBP on RDH12 Activity toward 11-cis-Retinoids
CRALBP was reported to have a mild inhibitory effect on the activity of RDH12 toward 11-cis-retinoids (1), suggesting that RDH12 might utilize CRALBP-bound retinoids as substrates. However, the concentrations of 11-cis-retinoids mixed with CRALBP in the assays were rather high (~20–30 μM) and, because of the high Kd values of CRALBP for 11-cis-retinoids (≥21 nM), the concentrations of unbound 11-cis-retinoids in the reactions might have been sufficient to support the observed rates.
To determine whether RDH12 could utilize CRALBP-bound 11-cis-retinoids, we assayed RDH12 activity toward 1 μM 11-cis-retinal or 11-cis-retinol in the presence of increasing concentrations of CRALBP. At a 1:1 ratio of CRALBP to substrate, the rate in the reductive direction was ~72% of the rate without CRALBP, and the rate in the oxidative direction was 82% of that without CRALBP. However, a 2-fold molar excess of CRALBP over retinoid strongly inhibited RDH12 activity in both directions (Figure 7).
Figure 7.

RDH12–His6 activity toward 11-cis-retinoids in the presence of CRALBP. Effect of CRALBP on the conversion of 11-cis-retinol (black bars) or 11-cis-retinal (white bars) by RDH12 was determined at 1 μM substrate and 0.5–4 μM CRALBP. The amount of enzyme used in the oxidative reactions was 0.04 μg. In the reductive direction, 0.01 μg of the purified RDH12–His6 was used. The reactions were performed in 1 mL volume with 1 mM NADP+ or NADPH as cofactors; samples were incubated at 37 °C for 10 min.
To exclude a possibility that the excess of apo-CRALBP acted as a competitive inhibitor of holo-CRALBP reduction, CRALBP was saturated by 11-cis-retinal, and holo-CRALBP was purified as described previously (11). From the A280/A425 absorbance spectral ratio, the CRALBP-binding stoichiometry was determined to be ~1:1 (11), indicating that holo-CRALBP did not contain any significant amount of apo-CRALBP. Nevertheless, the rate observed with 1 μM holo-CRALBP (~190 (nmol/min)/mg) was still much lower than the rate observed with 1 μM free 11-cis-retinal (~1200 (nmol/min)/mg). The higher rate of retinal reduction obtained in the reaction where CRALBP was mixed with 11-cis-retinal directly in the cuvette without further purification was, most likely, due to the greater concentration of unbound retinoid. Using the Kd value for CRALBP of 21 nM, we calculated that at 1 μM holo-CRALBP and ~1:1 binding stoichiometry (established from the ε280/ε425 ratio of 2.8 as described previously (11)), the concentration of unbound 11-cis-retinal in the reaction would be ~0.14 μM. At this concentration of 11-cis-retinal, the theoretical rate of RDH12 catalyzed reaction was estimated to be ~700 (nmol/min)/mg. Thus, the amount of unbound 11-cis-retinal in the reaction was sufficient to support the observed rate.
As a positive control for utilization of CRALBP-bound 11-cis-retinaldehyde, we used RDH5 (also known as 11-cis-retinol dehydrogenase) (35). A microsomal preparation of human RDH5 exhibited only a 5.3% reduction in the rate of 11-cis-retinol oxidation after the addition of 5 μM CRALBP to 5 μM 11-cis-retinol (360 (nmol/min)/mg) compared to the reaction in the absence of CRALBP (380 (nmol/min)/mg). The Km value of RDH5 for free 11-cis-retinol was reported to be ~7.5 μM (11). This Km value is much higher than the corresponding Km of RDH12 (0.16 μM). Binding to CRALBP would significantly lower the concentration of free 11-cis-retinol. Therefore, the lack of inhibition by CRALBP could be explained only if RDH5 utilized the CRALBP-bound 11-cis-retinol as substrate, as suggested previously (36, 11).
Expression Pattern of RDH12 mRNA in Human Tissues
To determine whether RDH12 is present in extraocular tissues and whether its activity in these tissues could be influenced by CRBPI, we determined the expression profile of RDH12 versus CRBPI using quantitative RT-PCR. Relative to the liver, RDH12 had the highest expression level in the retina followed by the kidney and pancreas (Figure 8). Much lower amounts of transcript were detected in the other tissues as indicated by high cycle threshold values (data not shown). No detectable RDH12 expression was observed in the heart and skeletal muscle. This expression pattern of RDH12 was quite distinct from that of other related SDRs (17, 37).
Figure 8.

Human tissue expression profile of RDH12 and CRBPI. SYBR green real-time quantitative RT-PCR was carried out using 200 pg of cDNA from the indicated tissues or 12.5 pg of retina cDNA and primers specific for human RDH12, CRBPI, and GAPDH. RDH12 and CRBPI expression was normalized to GAPDH and presented as a relative expression or fold difference (Δ) to the expression level in the liver for each gene, which was set to one. Data represent the average ± standard deviation of the triplicate real-time PCR within one experiment and are representative of three other experiments.
CRBPI was expressed in the majority of tissues at much higher levels than RDH12 based on the lower cycle threshold values (data not shown). The CRBPI transcript was most abundant in the ovary and pancreas and was not detectable in the peripheral blood leukocytes (Figure 8). The kidney tissue contained slightly more RDH12 than CRBPI relative to GAPDH as determined by the difference in cycle threshold values (ΔCTGAP–CRBPI or ΔCTGAP–RDH12). In contrast, ovary tissue contained considerably more CRBPI than RDH12.
Previously, it was shown that, at the level of protein, CRBPI was most abundant in the human ovary followed by testis, pituitary, adrenal gland, and liver (38, 39). Pancreas was not included in the previous studies (38, 39). In general, the distribution of CRBPI mRNA determined here was similar to the distribution of CRBPI protein reported earlier. Some discrepancy between the CRBPI mRNA and protein amounts could be due to tissue-specific differences in the regulation of its mRNA or protein levels (40). In general, CRBPI was found to be coexpressed with RDH12 in most human tissues.
DISCUSSION
Enzymes that are active toward retinoids are found in the cytosolic and microsomal fractions of the cells (reviewed in ref 41). The cytosolic activity has been linked to the members of the alcohol dehydrogenase (ADH) (42–44) and aldoketo reductase (AKR) superfamilies (45). The microsomal activity has been linked to the members of the SDR superfamily (46–50). ADHs are NAD+-dependent enzymes that are believed to function in the oxidative direction in vivo, whereas the NADP+-dependent AKRs are believed to function in the reductive direction. The SDR superfamily includes both the NAD+- and NADP+-dependent enzymes. All of the currently known cytosolic ADH and AKR enzymes have been purified and characterized. In contrast, very few microsomal SDRs have been purified to homogeneity (11, 17, 28), which impedes the comparison of their catalytic properties with those of other retinoid-active SDRs, ADHs, and AKRs.
In this manuscript, we present the first biochemical characterization of purified human RDH12, an enzyme that belongs to the new subfamily of SDRs comprised of at least four proteins, RDH11/RalR1/PSDR1, RDH12, RDH13, and RDH14/PAN2 (1, 16, 37). RDH11 and RDH14 both recognize all-trans- and cis-retinoids as substrates, but RDH13 appears to be inactive toward any of the tested compounds (1). Similar to RDH11 and RDH14, RDH12 characterized in this study exhibits a much greater (~2000-fold) affinity for NADP+ and NADPH than for NAD+ and NADH. However, at saturating concentrations of cofactors (millimolar), the reaction rate of RDH12 in the reductive direction is ~2.6-fold higher with NADH than with NADPH. The same is true for RDH11 (17). Thus, if RDH12 and RDH11 activities were assayed in tissue samples at millimolar concentrations of cofactors, NADH would appear as the preferred cofactor for both enzymes.
RDH12 exhibits very low apparent Km values for retinoids and is most efficient in the reductive direction with all-trans-retinaldehyde as substrate followed by 11-cis-retinaldehyde. Thus far, RDH12 is the most efficient retinaldehyde reductase among the characterized SDRs with the kcat/Km value for the reduction of all-trans-retinaldehyde ~6-fold higher than that of RDH11 (ref 17 and this study).
RDH12 recognizes only the unbound forms of all-trans-and 11-cis-retinoids. CRBPI strongly inhibits the oxidation of all-trans-retinol by RDH12 but has little effect on the reduction of all-trans-retinaldehyde, in accord with the much higher binding affinity of CRBPI for all-trans-retinol than for all-trans-retinaldehyde. Similarly, CRALBP, which has a higher affinity for retinaldehyde, sequesters 11-cis-retinaldehyde from RDH12 more efficiently than 11-cis-retinol. The exclusive recognition of free 11-cis-retinoids as substrates distinguishes RDH12 from RDH5, which is believed to oxidize CRALBP-bound 11-cis-retinol (36).
Tissue distribution of the RDH12 mRNA suggests that this enzyme is most highly expressed in retina (this study), where it is localized specifically in photoreceptors (1). Photoreceptors lack either CRBPI (51) or CRALBP (52, 53). Although RDH12 is highly active toward 11-cis-retinoids and CRALBP cannot interfere with its activity in photoreceptors, it is unlikely that RDH12 is involved in the metabolism of 11-cis-retinoids, because according to the current model of the visual cycle, 11-cis-retinol is produced and oxidized in the retinal pigment epithelium (RPE) (reviewed in ref 54) and 11-cis-retinal that diffuses into photoreceptors from RPE is likely to be sequestered by opsin in the outer segments of photoreceptors before it can reach RDH12. The most obvious retinoid substrate for RDH12 in photoreceptors is all-trans-retinaldehyde, which forms in large quantities by the photoisomerization of 11-cis-retinal-dehyde.
In addition to RDH12, photoreceptors contain three other microsomal SDRs, retSDR1 (48), prRDH (49), and RDH14 (1), which can also reduce all-trans-retinaldehyde in vitro. The contribution of retSDR1 and RDH14 to the reduction of all-trans-retinaldehyde in vivo is not yet clear, but prRDH (RDH8) was recently shown to have a rather minor role (55). Ethanol-active ADHs do not appear to play a role in photoreceptors, because high doses of the ADH inhibitor 4-methylpyrazole are not toxic for these cells (56). The contribution of retinoid-active AKRs to retinoid metabolism in photoreceptors has not yet been investigated (57). Thus far, of all photoreceptor-associated retinoid oxidoreductases, only RDH12 has been genetically linked with retinal degeneration in humans (2, 3).
RDH12 mRNA is also expressed in several extraocular tissues such as kidney, pancreas, liver, and prostate. As shown in this and previous studies (38, 39), all of these tissues contain CRBPI, which can sequester all-trans-retinol but not all-trans-retinaldehyde from RDH12. Thus, RDH12 could catalyze the reduction of retinaldehyde in extraocular tissues. All-trans-retinaldehyde could come from the symmetrical cleavage of β-carotene catalyzed by the locally expressed β-carotene monooxygenase (reviewed in ref 58). The role of RDH12 in the reduction of retinaldehyde is consistent with its high affinity for NADPH as a cofactor, because in the liver cytosol and presumably other tissues with the exception of retina (59), the concentration of NADPH is about 100-fold higher than the concentration of NADP+ (60).
Like mouse RDH11/SCALD, human RDH12 efficiently reduces unsaturated medium-chain aldehydes cis-6-nonenal and trans-2-nonenal. This property distinguishes RDH12 from the human RDH11 (this study) and prRDH (18), the only other SDRs that were tested with C9 aldehydes. Unsaturated aldehydes are toxic to cells because they combine spontaneously with glutathione and with cysteine, histidine, and lysine residues in cellular proteins and display a variety of cytotoxic and genotoxic effects (19, 61). These aldehydes are produced by lipid peroxidation of unsaturated fatty acids and can reach high concentrations (up to the millimolar range (19)) in tissues undergoing oxidative stress. Photoreceptors are especially vulnerable to oxidative damage because they contain rhodopsin and the highest content of docosahexaenoic acid (22:6, n − 3) of any cell type (62, 63). Aldehydes can be detoxified through one of the following pathways: (1) enzymatic conjugation with glutathione catalyzed by glutathione S-transferase, (2) reduction to corresponding alcohols by aldoketoreductases or alcohol dehydrogenases, and (3) oxidation to acids by aldehyde dehydrogenases (64–66). The kinetic parameters of RDH12 for C9 aldehydes (Km < 1–20 μM and Vmax between 1000 and 2000 (nmol/min)/mg) are comparable to those of other enzymes implicated in detoxification of these aldehydes, such as human aldehyde dehydrogenase ALDH3A1 (Km values of 0.5–155 μM and Vmax values of ~500–5000 (nmol/min)/mg) (67). These observations suggest that RDH12 might contribute to detoxification of medium-chain aldehydes in the cells with active lipid peroxidation. It will be interesting to see whether RDH12 knockout mice have defects only in the visual cycle or whether they have elevated levels of peroxidic aldehydes as well.
Footnotes
Supported by the National Institute on Alcohol Abuse and Alcoholism Grant AA12153 to N.Y.K. and the National Institute for Digestive Diseases and Kidney Grants DK59125 and DK65204 to P.S.N. T.K. was supported by a CIHR fellowship.
Abbreviations: SDR, short-chain dehydrogenase/reductase; CRBPI, cellular retinol-binding protein type I; RDH12, retinol dehydrogenase 12; RalR1, retinaldehyde reductase 1; SCALD, short-chain aldehyde dehydrogenase, mouse ortholog of human RDH11; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; SDS–PAGE, polyacrylamide gel electrophoresis in the presence of sodium dodecyl sulfate; DHPC, 1,2-diheptanoyl-sn-glycero-3-phosphocholine; DTT, dithiothreitol.
References
- 1.Haeseleer F, Jang GF, Imanishi Y, Driessen CAGG, Matsumura M, Nelson PS, Palczewski K. Dual-substrate specificity short chain retinol dehydrogenases from the vertebrate retina. J Biol Chem. 2002;277:45537–45546. doi: 10.1074/jbc.M208882200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janecke AR, Thompson DA, Utermann G, Becker C, Hubner CA, Schmid E, McHenry CL, Nair AR, Ruschendorf F, Heckenlively J, Wissinger B, Nurnberg P, Gal A. Mutations in RDH12 encoding a photoreceptor cell retinol dehydrogenase cause childhood-onset severe retinal dystrophy. Nat Genet. 2004;36:850–854. doi: 10.1038/ng1394. [DOI] [PubMed] [Google Scholar]
- 3.Perrault I, Hanein S, Gerber S, Barbet F, Ducroq D, Dollfus H, Hamel C, Dufier JL, Munnich A, Kaplan J, Rozet JM. Retinal dehydrogenase 12 (RDH12) mutations in Leber congenital amaurosis. Am J Hum Genet. 2004;75:639–646. doi: 10.1086/424889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noy N. Retinoid-binding proteins: mediators of retinoid action. Biochem J. 2000;348:481–495. [PMC free article] [PubMed] [Google Scholar]
- 5.Folli C, Calderone V, Ottonello S, Bolchi A, Zanotti G, Stoppini M, Berni R. Identification, retinoid binding, and X-ray analysis of a human retinol-binding protein. Proc Natl Acad Sci USA. 2001;98:3710–3715. doi: 10.1073/pnas.061455898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vogel S, Mendelsohn CL, Mertz JR, Piantedosi R, Waldburger C, Gottesman ME, Blaner WS. Characterization of a new member of the fatty acid-binding protein family that binds all-trans-retinol. J Biol Chem. 2001;276:1353–1360. doi: 10.1074/jbc.M005118200. [DOI] [PubMed] [Google Scholar]
- 7.Folli C, Calderone V, Ramazzina I, Zanotti G, Berni R. Ligand binding and structural analysis of a human putative cellular retinol-binding protein. J Biol Chem. 2002;277:41970–41977. doi: 10.1074/jbc.M207124200. [DOI] [PubMed] [Google Scholar]
- 8.MacDonald PN, Ong DE. Binding specificities of cellular retinol-binding protein and cellular retinol-binding protein, type II. J Biol Chem. 1987;262:10550–10556. [PubMed] [Google Scholar]
- 9.Levin MS, Locke B, Yang NC, Li E, Gordon JI. Comparison of the ligand binding properties of two homologous rat apocellular retinol-binding proteins expressed in Escherichia coli. J Biol Chem. 1988;263:17715–17723. [PubMed] [Google Scholar]
- 10.Li E, Qian SJ, Winter NS, d’Avignon A, Levin MS, Gordon JI. Fluorine nuclear magnetic resonance analysis of the ligand binding properties of two homologous rat cellular retinol-binding proteins expressed in Escherichia coli. J Biol Chem. 1991;266:3622–3629. [PubMed] [Google Scholar]
- 11.Golovleva I, Bhattacharya S, Wu Z, Shaw N, Yang Y, Andrabi K, West KA, Burstedt MS, Forsman K, Holmgren G, Sandgren O, Noy N, Qin J, Crabb JW. Disease-causing mutations in the cellular retinaldehyde binding protein tighten and abolish ligand interactions. J Biol Chem. 2003;278:12397–12402. doi: 10.1074/jbc.M207300200. [DOI] [PubMed] [Google Scholar]
- 12.Saari JC. Enzymes and proteins of the mammalian visual cycle. In: Osborn N, Chader G, editors. Progress in Retinal Research. Vol. 9. Pergamon Press; Oxford, U.K.: 1990. pp. 363–381. [Google Scholar]
- 13.Intres R, Goldflam S, Cook JR, Crabb JW. Molecular cloning and structural analysis of the human gene encoding cellular retinaldehyde-binding protein. J Biol Chem. 1994;269:25411–25418. [PubMed] [Google Scholar]
- 14.Napoli JL. Interactions of retinoid binding proteins and enzymes in retinoid metabolism. Biochim Biophys Acta. 1999;1440:139–162. doi: 10.1016/s1388-1981(99)00117-1. [DOI] [PubMed] [Google Scholar]
- 15.Lin B, White JT, Ferguson C, Wang S, Vessella R, Bumgarner R, True LD, Hood L, Nelson PS. Prostate short-chain dehydrogenase reductase 1 (PSDR1): a new member of the short-chain steroid dehydrogenase/reductase family highly expressed in normal and neoplastic prostate epithelium. Cancer Res. 2001;61:1611–1618. [PubMed] [Google Scholar]
- 16.Kedishvili NY, Chumakova OV, Chetyrkin SV, Belyaeva OV, Lapshina EA, Lin DW, Matsumura M, Nelson PS. Evidence that the human gene for prostate short-chain dehydrogenase/reductase (PSDR1) encodes a novel retinal reductase (RalR1) J Biol Chem. 2002;277:28909–28915. doi: 10.1074/jbc.M202588200. [DOI] [PubMed] [Google Scholar]
- 17.Belyaeva OV, Stetsenko AV, Nelson P, Kedishvili NY. Properties of short-chain dehydrogenase/reductase RalR1: characterization of purified enzyme, its orientation in the microsomal membrane, and distribution in human tissues and cell lines. Biochemistry. 2003;42:14838–14845. doi: 10.1021/bi035288u. [DOI] [PubMed] [Google Scholar]
- 18.Kasus-Jacobi A, Ou J, Bashmakov YK, Shelton JM, Richardson JA, Goldstein JL, Brown MS. Characterization of mouse short-chain aldehyde reductase (SCALD), an enzyme regulated by sterol regulatory element-binding proteins. J Biol Chem. 2003;278:32380–32389. doi: 10.1074/jbc.M304969200. [DOI] [PubMed] [Google Scholar]
- 19.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radical Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 20.Winkler BS, Boulton ME, Gottsch JD, Sternberg P. Oxidative damage and age-related macular degeneration. Mol Vision. 1999;5:32–42. [PMC free article] [PubMed] [Google Scholar]
- 21.Bok D. New insights and new approaches toward the study of age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99:14619–14621. doi: 10.1073/pnas.242607899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–134. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 23.Nowak M, Swietochowska E, Wielkoszynski T, Marek B, Karpe J, Gorski J, Glogowska-Szelag J, Kos-Kudla B, Ostrowska Z. Changes in blood antioxidants and several lipid peroxidation products in women with age-related macular degeneration. Eur J Ophthalmol. 2003;13:281–286. doi: 10.1177/112067210301300307. [DOI] [PubMed] [Google Scholar]
- 24.Gough WH, VanOoteghem S, Sint T, Kedishvili NY. cDNA cloning and characterization of a new human microsomal NAD+-dependent dehydrogenase that oxidizes all-trans-retinol and 3α-hydroxysteroids. J Biol Chem. 1998;273:19778–19785. doi: 10.1074/jbc.273.31.19778. [DOI] [PubMed] [Google Scholar]
- 25.Chetyrkin SV, Belyaeva OV, Gough WH, Kedishvili NY. Characterization of a novel type of human microsomal 3α-hydroxysteroid dehydrogenase: unique tissue distribution and catalytic properties. J Biol Chem. 2001;276:22278–22286. doi: 10.1074/jbc.M102076200. [DOI] [PubMed] [Google Scholar]
- 26.Chetyrkin SV, Hu J, Gough WH, Dumaual N, Kedishvili NY. Further characterization of human microsomal 3α-hydroxysteroid dehydrogenase. Arch Biochem Biophys. 2001;386:1–10. doi: 10.1006/abbi.2000.2203. [DOI] [PubMed] [Google Scholar]
- 27.Lowry OH, Rosebrough NH, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 28.Lapshina EA, Belyaeva OV, Chumakova OV, Kedishvili NY. Differential recognition of the free versus bound retinol by human microsomal retinol/sterol dehydrogenases: characterization of the holo-CRBP dehydrogenase activity of RoDH-4. Biochemistry. 2003;42:776–784. doi: 10.1021/bi026836r. [DOI] [PubMed] [Google Scholar]
- 29.Leatherbarrow RJ. GraFit user’s guide. Erithacus Software Limited; London, UK: 1989–1992. [Google Scholar]
- 30.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 31.McBee JK, Van Hooser JP, Jang GF, Palczewski K. Isomerization of 11-cis-retinoids to all-trans-retinoids in vitro and in vivo. J Biol Chem. 2001;276:48483–48493. doi: 10.1074/jbc.M105840200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Urbach J, Rando RR. Isomerization of all-trans-retinoic acid to 9-cis-retinoic acid. Biochem J. 1994;299:459–465. doi: 10.1042/bj2990459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Penzes P, Napoli JL. Cloning of a cDNA encoding an aldehyde dehydrogenase and its expression in Escherichia coli. Recognition of retinal as substrate. J Biol Chem. 1996;271:16288–16293. doi: 10.1074/jbc.271.27.16288. [DOI] [PubMed] [Google Scholar]
- 34.Posch KC, Burns RD, Napoli JL. Biosynthesis of all-trans-retinoic acid from retinal. Recognition of retinal bound to cellular retinol binding protein (type I) as substrate by a purified cytosolic dehydrogenase. J Biol Chem. 1992;267:19676–19682. [PubMed] [Google Scholar]
- 35.Wang J, Chai X, Eriksson U, Napoli JL. Activity of human 11-cis-retinol dehydrogenase (Rdh5) with steroids and retinoids and expression of its mRNA in extraocular human tissue. Biochem J. 1999;338:23–27. [PMC free article] [PubMed] [Google Scholar]
- 36.Saari JC. Retinoids in photosensitive system. In: Sporn MB, Roberts AB, Goodman DS, editors. The Retinoids: Biology, Chemistry and Medicine. Raven Press; New York: 1994. pp. 351–385. [Google Scholar]
- 37.Belyaeva OV, Kedishvili NY. Human pancreas protein 2 (PAN2) has a retinal reductase activity and is ubiquitously expressed in human tissues. FEBS Lett. 2002;531:489–493. doi: 10.1016/s0014-5793(02)03588-3. [DOI] [PubMed] [Google Scholar]
- 38.Fex G, Johannesson G. Radioimmunological determination of cellular retinol-binding protein in human tissue extracts. Cancer Res. 1984;44:3029–3032. [PubMed] [Google Scholar]
- 39.Ong DE, Page DL. Quantitation of cellular retinol-binding protein in human organs. Am J Clin Nutr. 1986;44:425–430. doi: 10.1093/ajcn/44.3.425. [DOI] [PubMed] [Google Scholar]
- 40.Eskild W, Troen G, Blaner WS, Nilsson A, Hansson V. Evidence for independent control at the mRNA and protein levels of cellular retinol binding protein 1 in rat Sertoli cells. J Reprod Fertil. 2000;119:101–109. doi: 10.1530/jrf.0.1190101. [DOI] [PubMed] [Google Scholar]
- 41.Kedishvili NY. Multifunctional nature of human retinol dehydrogenases. Curr Org Chem. 2002;6:1247–1257. [Google Scholar]
- 42.Julia P, Farres J, Pares X. Purification and partial characterization of a rat retina alcohol dehydrogenase active with ethanol and retinol. Biochem J. 1983;213:547–550. doi: 10.1042/bj2130547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang ZN, Davis GJ, Hurley TD, Stone CL, Li TK, Bosron WF. Catalytic efficiency of human alcohol dehydrogenases for retinol oxidation and retinal reduction. Alcohol : Clin Exp Res. 1994;18:587–591. doi: 10.1111/j.1530-0277.1994.tb00914.x. [DOI] [PubMed] [Google Scholar]
- 44.Molotkov A, Deltour L, Foglio MH, Cuenca AE, Duester G. Distinct retinoid metabolic functions for alcohol dehydrogenase genes Adh1 and Adh4 in protection against vitamin A toxicity or deficiency revealed in double null mutant mice. J Biol Chem. 2002;277:13804–13811. doi: 10.1074/jbc.M112039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crosas B, Hyndman DJ, Gallego O, Martras S, Pares X, Flynn TG, Farres J. Human aldose reductase and human small intestine aldose reductase are efficient retinal reductases: consequences for retinoid metabolism. Biochem J. 2003;373:973–979. doi: 10.1042/BJ20021818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simon A, Hellman U, Wernstedt C, Eriksson U. The retinal pigment epithelial-specific 11-cis retinol dehydrogenase belongs to the family of short chain alcohol dehydrogenases. J Biol Chem. 1995;270:1107–1112. [PubMed] [Google Scholar]
- 47.Chai X, Boerman MH, Zhai Y, Napoli JL. Cloning of a cDNA for liver microsomal retinol dehydrogenase. A tissue-specific, short-chain alcohol dehydrogenase. J Biol Chem. 1995;270:3900–3904. doi: 10.1074/jbc.270.8.3900. [DOI] [PubMed] [Google Scholar]
- 48.Haeseleer F, Huang J, Lebioda L, Saari JC, Palczewski K. Molecular characterization of a novel short-chain dehydrogenase/reductase that reduces all-trans-retinal. J Biol Chem. 1998;273:21790–21799. doi: 10.1074/jbc.273.34.21790. [DOI] [PubMed] [Google Scholar]
- 49.Rattner A, Smallwood PM, Nathans J. Identification and characterization of all-trans-retinol dehydrogenase from photoreceptor outer segments, the visual cycle enzyme that reduces all-trans-retinal to all-trans-retinol. J Biol Chem. 2000;275:11034–11043. doi: 10.1074/jbc.275.15.11034. [DOI] [PubMed] [Google Scholar]
- 50.Wu BX, Chen Y, Chen Y, Fan J, Rohrer B, Crouch RK, Ma JX. Cloning and characterization of a novel all-trans retinol short-chain dehydrogenase/reductase from the RPE. Invest Ophthalmol Visual Sci. 2002;43:3365–3372. [PubMed] [Google Scholar]
- 51.Bok D, Ong DE, Chytil F. Immunocytochemical localization of cellular retinol binding protein in the rat retina. Invest Ophthalmol Visual Sci. 1984;25:877–883. [PubMed] [Google Scholar]
- 52.Bunt-Milam AH, Saari JC. Immunocytochemical localization of two retinoid-binding proteins in vertebrate retina. J Cell Biol. 1983;97:703–712. doi: 10.1083/jcb.97.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saari JC, Bunt-Milam AH, Bredberg DL, Garwin GG. Properties and immunocytochemical localization of three retinoid-binding proteins from bovine retina. Vision Res. 1984;24:1595–603. doi: 10.1016/0042-6989(84)90317-1. [DOI] [PubMed] [Google Scholar]
- 54.Gonzalez-Fernandez F. Evolution of the visual cycle: the role of retinoid-binding proteins. J Endocrinol. 2002;175:75–88. doi: 10.1677/joe.0.1750075. [DOI] [PubMed] [Google Scholar]
- 55.Maeda A, Maeda T, Imanishi Y, Kuksa V, Alekseev A, Bronson DJ, Zhang H, Zhu L, Sun W, Saperstein DA, Rieke F, Baehr W, Palczewski K. Role of photoreceptor-specific retinol dehyrogenase (PRRDH) in the retinoid cycle in vivo. J Biol Chem. 2005 doi: 10.1074/jbc.M501757200. published online Mar 8, http://www.jbc.org/cgi/reprint/M501757200v1. [DOI] [PMC free article] [PubMed]
- 56.Blomstrand R, Ingemansson SO, Jensen M, Hedstrom CG. Normal electroretinogram and no toxicity signs after chronic and acute administration of the alcohol dehydrogenase inhibitor 4-methylpyrazole to the cynomolgus monkey (Macaca fascicularis)—a possible new treatment of methanol poisoning. Drug Alcohol Depend. 1984;13:9–20. doi: 10.1016/0376-8716(84)90028-0. [DOI] [PubMed] [Google Scholar]
- 57.Akagi Y, Yajima Y, Kador PF, Kuwabara T, Kinoshita JH. Localization of aldose reductase in the human eye. Diabetes. 1984;33:562–566. doi: 10.2337/diab.33.6.562. [DOI] [PubMed] [Google Scholar]
- 58.Wyss A. Carotene oxygenases: a new family of double bond cleavage enzymes. J Nutr. 2004;134:246S–250S. doi: 10.1093/jn/134.1.246S. [DOI] [PubMed] [Google Scholar]
- 59.Matschinsky FM. Quantitative histochemistry of nicotinamide adenine nucleotides in retina of monkey and rabbit. J Neurochem. 1968;15:643–657. doi: 10.1111/j.1471-4159.1968.tb08963.x. [DOI] [PubMed] [Google Scholar]
- 60.Veech RL, Eggleston LV, Krebs HA. The redox state of free nicotinamide-adenine dinucleotide phosphate in the cytoplasm of rat liver. Biochem J. 1969;115:609–619. doi: 10.1042/bj1150609a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Esterbauer H. Cytotoxicity and genotoxicity of lipid-oxidation products. Am J Clin Nutr. 1993;57:779S–786S. doi: 10.1093/ajcn/57.5.779S. [DOI] [PubMed] [Google Scholar]
- 62.Bazan NG. Supply of n-3 polyunsaturated fatty acids and their significance in the central nervous system. In: Wurtman RJ, Wurtman JJ, editors. Nutrition and the Brain. Vol. 8. Raven; New York: 1990. pp. 1–24. [Google Scholar]
- 63.Anderson RE, Maude MB, McClellan M, Matthes MT, Yasumura D, LaVail MM. Low docosahexaenoic acid levels in rod outer segments of rats with P23H and S334ter rhodopsin mutations. Mol Vision. 2002;8:351–358. [PubMed] [Google Scholar]
- 64.Luckey SW, Petersen DR. Metabolism of 4-hydroxynonenal by rat Kupffer cells. Arch Biochem Biophys. 2001;389:77–83. doi: 10.1006/abbi.2001.2307. [DOI] [PubMed] [Google Scholar]
- 65.Srivastava S, Chandra A, Wang LF, Seifert WE, Jr, DaGue BB, Ansari NH, Srivastava SK, Bhatnagar A. Metabolism of the lipid peroxidation product, 4-hydroxy-trans-2-nonenal, in isolated perfused rat heart. J Biol Chem. 1998;273:10893–10900. doi: 10.1074/jbc.273.18.10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Srivastava S, Dixit BL, Cai J, Sharma S, Hurst HE, Bhatnagar A, Srivastava SK. Metabolism of lipid peroxidation product, 4-hydroxynonenal (HNE) in rat erythrocytes: role of aldose reductase. Free Radical Biol Med. 2000;29:642–651. doi: 10.1016/s0891-5849(00)00351-8. [DOI] [PubMed] [Google Scholar]
- 67.Pappa A, Estey T, Manzer R, Brown D, Vasiliou V. Human aldehyde dehydrogenase 3A1 (ALDH3A1): biochemical characterization and immunohistochemical localization in the cornea. Biochem J. 2003;376:615–623. doi: 10.1042/BJ20030810. [DOI] [PMC free article] [PubMed] [Google Scholar]