

Abstract
Background
Mutations in GJB2 are the most common molecular defects responsible for autosomal recessive nonsyndromic hearing impairment (NSHI). The mutation spectra of this gene vary among different ethnic groups.
Methods
In order to understand the spectrum and frequency of GJB2 mutations in the Chinese population, the coding region of the GJB2 gene from 2063 unrelated patients with NSHI was PCR amplified and sequenced.
Results
A total of 23 pathogenic mutations were identified. Among them, five (p.W3X, c.99delT, c.155_c.158delTCTG, c.512_c.513insAACG, and p.Y152X) are novel. Three hundred and seven patients carry two confirmed pathogenic mutations, including 178 homozygotes and 129 compound heterozygotes. One hundred twenty five patients carry only one mutant allele. Thus, GJB2 mutations account for 17.9% of the mutant alleles in 2063 NSHI patients. Overall, 92.6% (684/739) of the pathogenic mutations are frame-shift truncation or nonsense mutations. The four prevalent mutations; c.235delC, c.299_c.300delAT, c.176_c.191del16, and c.35delG, account for 88.0% of all mutantalleles identified. The frequency of GJB2 mutations (alleles) varies from 4% to 30.4% among different regions of China. It also varies among different sub-ethnic groups.
Conclusion
In some regions of China, testing of the three most common mutations can identify at least one GJB2 mutant allele in all patients. In other regions such as Tibet, the three most common mutations account for only 16% the GJB2 mutant alleles. Thus, in this region, sequencing of GJB2 would be recommended. In addition, the etiology of more than 80% of the mutant alleles for NSHI in China remains to be identified. Analysis of other NSHI related genes will be necessary.
Introduction
Hearing impairment is the most common neurosensory disorder in humans. The reported incidence varies from 1 in 300 to 1 in 1000 children [1-4]. Approximately half of cases have a genetic etiology, including syndromic and non-syndromic forms, with extraordinary genetic heterogeneity. Non-syndromic deafness accounts for 60–70% of inherited hearing impairment. It involves more than 100 different genes with autosomal dominant (DFNA), autosomal recessive (DFNB), X-linked (DFN), and maternal inheritance [5], with autosomal recessive being the most common. For many populations, the most common cause for non-syndromic autosomal recessive hearing loss is mutated Connexin 26, a gap junction protein encoded by the GJB2 gene (MIM 121011) [6-13]. There are a few specific mutations in GJB2 gene that are associated with the autosomal dominant syndromic forms of deafness, and typically present with skin abnormalities including keratitis-ichthyosis [14-16].
Connexins are transmembrane proteins. Six monomers of connexin proteins associate to form a transmembrane hexameric gap junction hemi-channel called a connexon. Connexons embedded in the surfaces of adjacent cells associate to form an intercellular channel [17,18]. In the inner ear, connexin 26 can be in association with other connexins to form heteromeric connexons. Gap junction channels can be homotypic or heterotypic. Connexin 26 gap junction channels recycle potassium ions as part of a mechanism of auditory signal transduction in inner ear [19].
Mutations in three connexin (Cx) genes, GJB2 (Cx26), GJB6 (Cx30), and GJB3 (Cx31), have been identified and are known to cause hearing impairment [18,19]. Sequence analysis of the GJB2 gene in subjects with autosomal recessive hearing impairment revealed that a high number of patients carried only one mutant allele. Some of these families showed clear evidence of linkage to the DFNB1 locus, which contains two genes, GJB2 and GJB6 [6,20]. Further analysis demonstrated that some GJB2 heterozygotes also carried a truncating deletion of the GJB6 gene, encoding connexin 30, in trans [21,22].
To date, more than 150 mutations, polymorphisms, and unclassified variants have been described in the GJB2 gene http://davinci.crg.es/deafness. The mutation spectrum and prevalence of mutations vary significantly among different ethnic groups. Three mutations, c.35delG, c.167delT, and c.235delC, are found to be the most frequent mutations in Caucasian, Ashkenazi Jewish, and Asian populations, respectively [6,7,9-13,20,23-26].
In China, it is estimated that 30,000 babies are born with congenital hearing impairment every year [27]. The mutation spectrum of the GJB2 gene in Chinese patients with nonsyndromic hearing impairment (NSHI) has not been analyzed. Our recent study by screening for just the most common mutation, c.235delC, in 3004 Chinese NSHI patients revealed that 488 (16.3%) patients carried at least one c.235delC mutant allele, with 233 (7.8%) homozygotes and 255 (8.5%) heterozygotes [28], though the frequencies of homozygote and heterozygote of c.235delC varied from 0% to 14.7% and from 1.7% to 16.1% respectively in the populations examined in this study. Among different Chinese sub-ethnic groups the c.235delC allele frequency was the lowest (0.8%) in the Tibetan and the highest (31.0%) in Maan. These results highlight the need to sequence the entire GJB2 gene in order to more accurately establish the actual mutation frequency and mutation spectrum of GJB2 gene within various Chinese sub-populations. Our preliminary results reveal that other GJB2 mutations account for an additional 7.1% of NSHI patients from Qinghai, where only 7.1% patients carried at least one c.235delC mutation. Nevertheless, sequencing analysis of the entire coding region of the GJB2 gene in patients from Guangxi where the frequency of the c.235delC mutation is 3.4% reveals only one other mutation in 87 deaf patients. These results have two important implications: that the GJB2 gene needs to be sequenced in its entirety; and that mutations in genes responsible for NSHI other than GJB2 should be searched in patients who do not harbor two mutant alleles in the GJB2 gene. In this study, we report the results of sequencing the GJB2 gene in 2063 patients with NSHI from 23 different regions of China (Figure 1).
Figure 1.
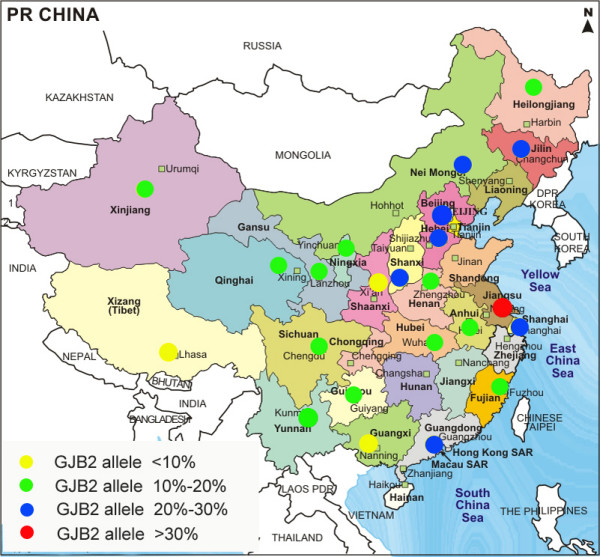
Geographic distribution and the proportion of patients carrying at least one GJB2 mutant allele in each region studied.
Materials and methods
Patients and DNA samples
A total of 2063 unrelated NSHI students from 23 different regions of China were included in this sequencing study. The selection of samples was random regardless of the c.235delC genotype. The patients consisted of 1179 males and 884 females ranging in age from 2 to 30 years with an average age of 13.7 ± 4.5. The majority of patients were Han Chinese (1640), followed by Tibetan (122), minorities in the Southwest region (119), Hui (79), minorities in Xinjiang (62), Mongolian (21), Maan (18) and Korean (2). Ethnic subgroup designations were based on permanent residency documentation.
This study was performed according to a protocol approved by the ethics committee of the Chinese PLA General Hospital. The subjects in this study were from deaf schools of each region and were recently described [28]. Only the unrelated patients with nonsyndromic hearing loss were included in this study. Parents were not included in this study. All patients showed moderate to profound bilateral sensorineural hearing impairment on audiograms and no pathient with mild hearing impairment was found in this cohort. In addition to the 2063 patients, 301 Han control individuals with normal hearing (either evaluated by pure tone audiometry or by self-assessment) from Beijing Capital (Northern) and Jiangsu Province (Eastern), two densely populated regions consisting of 98% Han Chinese, were also analyzed. DNA was extracted from peripheral blood leukocytes using a commercially available DNA extraction kit (Watson Biotechnologies Inc, Shanghai, China).
Sequence analysis
The coding exon (Exon2) and flanking intronic regions of GJB2 gene were PCR amplified with forward primer 5'TTGGTGTTTGCTCAGGAAGA 3' and reverse primer 5'GGCCTACAGGGGTTTCAAAT 3'. Among this study cohort, 851 patients from central China were also analyzed for mutations in Exon1 and flanking introns by PCR/sequencing. The PCR primers used are forward primer: 5'CTCATGGGGGCTCAAAGGAACTAGGAGATCGG3' and reverse primer 5'GGGGCTGGACCAACACACGTCCTTGGG3'. The PCR products were purified on Qia-quick spin columns (Qiagen, Valencia, CA) and sequenced using the BigDye Terminator Cycle Sequencing kit (version v3.1) and ABI 3130 automated DNA sequencers (Applied Biosystems, Foster City, CA, USA,) with Sequence Analysis Software (Sequencing Analysis version 3.7). DNA sequence variations were identified by comparison of subject DNA sequence to GJB2 reference sequences, Genebank Accession Number AY280971. Numbering of GJB2 begins with the nucleotide A of the ATG start codon in Exon2 as cDNA position number 1. The sequences were analyzed using Genetool Lite software and the GJB2 Genebank sequence. The presence of 309 kb deletion of GJB6 was analyzed by PCR method [21,22]. A positive control of this deletion provided by Balin Wu (Department of Laboratory Medicine, Children's Hospital and Harvard Medical School, USA.) was used for the detection of deletion in GJB6 gene.
Statistical analysis
The statistical analysis was performed using SAS 9.1.3 software (SAS, Cary, North Carolina, USA).
Results
Mutations in GJB2 gene
Sequencing of the coding region of the GJB2 gene revealed that at least 104 different genotypes were found in the 2063 patients (Table 1). Among them, 64 different genotypes harboring pathogenic mutations were found in 432 patients (Table 1). Three hundred and seven patients had two confirmed pathogenic mutations, including 178 homozygotes and 129 compound heterozygotes. One hundred twenty five patients carried one heterozygous pathogenic mutation without an identified second mutant allele. Thus, GJB2 mutant alleles account for 17.9% (739/4126) of the total alleles in 2063 NSHI patients. The most common genotype was homozygous c.235delC, followed by compound heterozygosity for c.235delC/c.299_300delAT, which accounted for 8.0% (164/2063) and 3.2% (66/2063) of NSHI patients respectively. The most common mutation c.235delC was in compound heterozygosity with 14 other different pathogenic mutations in 113 patients, and was present as a single heterozygous mutant allele in 68 patients. In addition, there were 23 different genotypes in patients carrying one allele of unclassified variants (Table 1). Twenty-three alterations were found, five (p.W3X, c.99delT, c.155_c.158delTCTG, p.Y152X, and c.512_c.513insAACG) of them were novel and pathogenic, and twelve (p,G21R, p,I30F, p.F31L, p.V37I, p.V63L, p.T123N, p.V153A, p.D159N, p.F191L, p.M195V, p.V198M, and p.I215N) are unclassified variants (Table 1 and Supplemental Table 1). The distribution of various genotypes in 23 regions (Figure 1) is detailed in Table 2 and Supplemental Table 2. The frequencies of the three most common GJB2 mutations in the 23 regions studied are listed in Table 2. The allele frequency of all mutations in the GJB2 gene in NSHI patients varied from 4.0% in Guangxi to 30.4% in Jiangsu (Table 2). Regions which appeared to have a higher frequency of the c.235delC mutation (Jiangsu, Inner Mongolia, Beijing, Hebei, Shanghai) also had a relatively high frequency of other GJB2 mutations (eg, the frequency of the c.235delC mutation in Jiangsu was as high as 20.6% and the frequencies of other mutations were also as high as 9.8%). Similarly, regions such as Shaanxi and Guangxi where the frequency of the c.235delC mutation is low (5.8 and 3.4% respectively), also had lower frequencies of other mutations (1.9 and 0.6% respectively). Patients from Tibet, Yunnan, Xinjiang, Heilongjiang, and Ningxia appear to have the most diverse mutation spectrum because uncommon mutations (except c.235delC, c.299_c.300delAT and c.176_c.191del16) comprise 84.2, 30.8, 26.1, 21.4, and 20.4%, respectively of overall GJB2 mutations in those regions.
Table 1.
GJB2 genotypes of 2063 Chinese NSHI patients
| Allele 1 | Allele 2 | |||||||
| nucleotide change | consequence or amino acid change | category | domain | nucleotide change | consequence or amino acid change | Category | domain | Number of patientsd |
| homozygous | ||||||||
| c.35delG | frame-shift | pathogenic | NT | c.35delG | frame-shift | Pathogenic | 2 | |
| c.176_c.191del16 | fram shift | pathogenic | EC1 | c.176_c.191del16 | frame-shift | Pathogenic | EC1 | 2 |
| c.235delC | frame-shift | pathogenic | TM2 | c.235delC | frame-shift | Pathogenic | TM2 | 164 |
| c.299_c.300delAT | frame-shift | pathogenic | CL | c.299_c.300delAT | frame-shift | pathogenic | CL | 8 |
| c.512_c.513insAACG | frame-shift | pathogenic | EC2 | c.512_c.513insAACG | frame-shift | pathogenic | EC2 | 1 |
| c.605_c.606ins46 | frame-shift | pathogenic | TM4 | c.605_c.606ins46 | frame-shift | pathogenic | TM4 | 1 |
| compound heterozygous | ||||||||
| c.9G>A, c.79G>A | p.W3X, p.V27I | pathogenic, polymophism | NT, TM1 | c.427C>T | p.R143W | pathogenic | TM3 | 1 |
| c.35delG | frame-shift | pathogenic | NT | c.299_c.300delAT | frame-shift | pathogenic | CL | 1 |
| c.35delG | frame-shift | pathogenic | NT | c.313_c.326del14 | frame-shift | pathogenic | CL | 1 |
| c.176_c.191del16 | frame-shift | pathogenic | EC1 | c.9G>A, c.79G>A | p.W3X, p.V27I | pathogenic, polymophism | NT+TM1 | 2 |
| c.176_c.191del16 | frame-shift | pathogenic | EC1 | c.299_c.300delAT | frame-shift | pathogenic | CL | 4 |
| c.176_c.191del16 | frame-shift | pathogenic | EC1 | c.388_c.397del10 | frame-shift | pathogenic | 1 | |
| c.235delC | frame-shift | pathogenic | TM2 | c.9G>A, c.79G>A | p.W3X, p.V27I | pathogenic, polymophism | NT+TM1 | 2 |
| c.235delC | frame-shift | pathogenic | TM2 | c.35delG | frame-shift | pathogenic | NT | 1 |
| c.235delC | frame-shift | pathogenic | TM2 | c.35insG | frame-shift | pathogenic | NT | 2 |
| c.235delC | frame-shift | pathogenic | TM2 | c.94C>T | p.R32C | pathogenic | TM1 | 1 |
| c.235delC | frame-shift | pathogenic | TM2 | c.99delT | frame-shift | pathogenic | TM1 | 1 |
| c.235delC | frame-shift | pathogenic | TM2 | c.139G>T | p.E47X | pathogenic | EC1 | 3 |
| c.235delC | frame-shift | pathogenic | TM2 | c.155_c.158delTCTG | frame-shift | pathogenic | EC1 | 2 |
| c.235delC | frame-shift | pathogenic | TM2 | c.176_191del16 | frame-shift | pathogenic | EC1 | 18 |
| c.235delC | frame-shift | pathogenic | TM2 | c.257C>G | p.T86R | pathogenic | TM2 | 6 |
| c.235delC | frame-shift | pathogenic | TM2 | c.299_c.300delAT | frame-shift | pathogenic | CL | 65 |
| c.235delC | frame-shift | pathogenic | TM2 | c.299_c.300delAT, c.79G>A | frame-shift, p.V27I | frame-shift, polymorphism | CL+TM1 | 1 |
| c.235delC | frame-shift | pathogenic | TM2 | c.313_c.326del14 | frame-shift | pathogenic | CL | 1 |
| c.235delC | frame-shift | pathogenic | TM2 | c.427C>T | p.R143W | pathogenic | TM3 | 3 |
| c.235delC | frame-shift | pathogenic | TM2 | c.512_c.513insAACG | frame-shift | pathogenic | EC2 | 6 |
| c.235delC | frame-shift | pathogenic | TM2 | c.605_c.606ins46 | frame-shift | pathogenic | TM4 | 1 |
| c.299_c.300delAT | frame-shift | pathogenic | CL | c.139G>A | p.E47K | pathogenic | EC1 | 1 |
| c.299_c.300delAT | frame-shift | pathogenic | CL | c.257C>G | p.T86R | pathogenic | TM2 | 1 |
| c.299_c.300delAT | frame-shift | pathogenic | CL | c.512_c.513insAACG | frame-shift | pathogenic | EC2 | 3 |
| c.456C>A | p.Y152X | pathogenic | TM3, CL | c.380G>A, c.79G>A, c.341A>G | p.R127H, p.V27I, E114G | pathogenic, polymophism | TM1+CL | 1 |
| heterozygous (one mutant allele only) | ||||||||
| c.11G>A | p.G4D | pathogenic | NT | c.109G>A | p.V37I | see note | TM1 | 1 |
| c.11G>A | p.G4D | pathogenic | NT | Nv | 2 | |||
| c.35delG | frame-shift | pathogenic | NT | c.79G>A, c.341A>G | p.V27I p,E114G | polymorphism | TM1+CL | 1 |
| c.35delG | frame-shift | pathogenic | NT | Nv | 4 | |||
| c.155_c.158delTCTG | frame-shift | pathogenic | EC1 | c.341A>G, c.644T>A | p.E114G, p.I215N | polymorphism, unclassified | CL+CT | 1 |
| c.176_c.191del16 | frame-shift | pathogenic | EC1 | Nv | 2 | |||
| c.235delC | frame-shift | pathogenic | TM2 | c.109G>A | p.V37I | see note | TM1 | 11 |
| c.235delC | frame-shift | pathogenic | TM2 | c.79G>A | p.V27I | polymorphism | TM1 | 6 |
| c.235delC | frame-shift | pathogenic | TM2 | c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | 5 |
| c.235delC | frame-shift | pathogenic | TM2 | c.341A>G | p.E114G | polymorphism | CL | 2 |
| c.235delC | frame-shift | pathogenic | TM2 | c.558G>A | p.T186T | polymorphism | EC2 | 1 |
| c.235delC | frame-shift | pathogenic | TM2 | Nv | 43 | |||
| c.253T>C | p.S85P | pathogenic | TM2 | Nv | 1 | |||
| c.299_c.300delAT | frame-shift | pathogenic | CL | c.109G>A | p.V37I | see note | TM1 | 1 |
| c.299_c.300delAT | frame-shift | pathogenic | CL | c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | 1 |
| c.299_c.300delAT | frame-shift | pathogenic | CL | Nv | 4 | |||
| c.380G>A, c.341A>G | p.R127H, p.E114G | pathogenic, polymophism | CL+CL | c.109G>A | p.V37I | see note | TM1 | 1 |
| c.380G>A | p.R127H | pathogenic | CL | c.109G>A | p.V37I | see note | TM1 | 1 |
| c.380G>A, c.109G>A | p.R127H, p.V37I | pathogenic, polymophism | TM1+CL | c.79G>A | p.V27I | polymorphism | TM1 | 1 |
| c.380G>A, c.79G>A | p.R127H, p.V27I | pathogenic, polymophism | TM1+CL | c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | 1 |
| c.380G>A | p.R127H | pathogenic | CL | c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | 9 |
| c.380G>A, c.147C>T | p.R127H, p.A49A | pathogenic, polymophism | EC1+CL | c.79G>A | p.V27I | polymorphism | TM1 | 1 |
| c.380G>A, c.608T>C | p.R127H, p.I203T | pathogenic, polymophism | CL+TM4 | c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | 1 |
| c.380G>A, c.608T>C | p.R127H, p.I203T | pathogenic, polymophism | CL+TM4 | c.79G>A | p.V27I | polymorphism | TM1 | 1 |
| c.380G>A | p.R127H | pathogenic | CL | c.79G>A | p.V27I | polymorphism | TM1 | 4 |
| c.380G>A | p.R127H | pathogenic | CL | c.457G>A | p.V153I | polymorphism | TM3 | 1 |
| c.380G>A | p.R127H | pathogenic | CL | Nv | 10 | |||
| c.416G>A | p.S139N | pathogenic | CL | c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | 1 |
| c.416G>A | p.S139N | pathogenic | CL | Nv | 1 | |||
| c.424_c.426del3 | p.del142F | pathogenic | TM3 | c.79G>A, c.341A>G, c.109G>A | p.V27I, p.E114G, p.V37I | polymorphisms, see note | TM1+CL | 3 |
| c.424_c.426del3 | p.del142F | pathogenic | TM3 | c.79G>A, c.109G>A | p.V27I, p.V37I | polymorphisms, see note | TM1 | 1 |
| c.512_c.513insAACG | frame-shift | pathogenic | EC2 | c.79G>A, c.368C>A | p.V27I, p.T123N | polymorphism, unclassified | TM1+CL | 1 |
| c.512_c.513insAACG | frame-shift | pathogenic | EC2 | Nv | 1 | |||
| unclassified variant | ||||||||
| c.61G>C, c.79G>A | p.G21R, p.V27I | unclassified, polymorphism | NT+TM1 | c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | 1 |
| c.88A>T | p.I30F | unclassified | TM1 | Nv | 1 | |||
| c.93T>G | p.F31L | unclassified | TM1 | c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | 1 |
| c.187G>T | p.V63L | unclassified | EC1 | Nv | 2 | |||
| c.368C>A, c.79G>A | p.T123N, p.V27I | unclassified, polymorphism | CL+TM1 | c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | 1 |
| c.368C>A, c.79G>A | p.T123N, p.V27I | unclassified, polymorphism | CL+TM1 | c.79G>A | p.V27I | polymorphism | TM1 | 3 |
| c.368C>A | p.T123N | unclassified | CL | c.79G>A | p.V27I | polymorphism | TM1 | 7 |
| c.368C>A, c.608T>C | p.T123N, p.I203T | unclassified, polymorphism | CL+TM4 | c.79G>A | p.V27I | polymorphism | TM1 | 1 |
| c.458T>C | p.V153A | unclassified | EC2 | c.608T>C | p.I203T | polymorphism | TM4 | 1 |
| c.571T>C, c.592G>A | p.F191L, p.V198M | unclassified | TM4+TM4 | c.79G>A | p.V27I | polymorphism | TM1 | 1 |
| c.583A>G | p.M195V | unclassified | TM4 | Nv | 1 | |||
| c.583A>G | p.M195V | unclassified | TM4 | c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | 1 |
| c.592G>A, c.79G>A, c.341A>G | p.V198M, p.V27I, p.E114G | unclassified, polymorphism | TM4+TM1+CL | c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | 1 |
| c.592G>A, c.79G>A | p.V198M, p.V27I | unclassified, polymorphism | TM4+TM1 | c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | 1 |
| c.592G>A | p.V198M | unclassified | TM4 | c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | 2 |
| c.475G>A | p.D159N | unclassified | EC2 | Nv | TM1+CL | 1 | ||
| c644T>A, c.79G>A, c.341A>G | p.I215N, p.V27I, p.E114G | unclassified, polymorphism | CT+TM1+CL | c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | 1 |
| c.644T>A | p.I215N | unclassified | CT | c.608T>C | p.I203T | polymorphism | TM4 | 1 |
| c.109G>A | p.V37I | see note | TM1 | c.109G>A | p.V37I | see note | TM1 | 23 |
| c.109G>A | p.V37I | see note | TM1 | c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | 29 |
| c.109G>A | p.V37I | see note | TM1 | c.79G>A | p.V27I | polymorphism | TM1 | 10 |
| c.109G>A | p.V37I | see note | TM1 | c.608T>C | p.I203T | polymorphism | TM4 | 3 |
| c.109G>A | p.V37I | see note | TM1 | Nv | 91 | |||
| polymorphism | ||||||||
| c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | 90 |
| c.79G>A | p.V27I | polymorphism | TM1 | c.79G>A | p.V27I | polymorphism | TM1 | 18 |
| c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | c.79G>A | p.V27I | polymorphism | TM1 | 42 |
| c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | c.341A>G | p.E114G | polymorphism | 2 | |
| c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | c.457G>A | p.V153I | polymorphism | TM3 | 1 |
| c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | c.608T>C | p.I203T | polymorphism | TM4 | 12 |
| c.79G>A, c.341A>G | p.V27I, p.E114G | polymorphism | TM1+CL | Nv | 387 | |||
| c.79G>A | p.V27I | polymorphism | TM1 | c.608T>C | p.I203T | polymorphism | TM4 | 5 |
| c.79G>A, c.608T>C | p.V27I | polymorphism | TM1+TM4 | c.608T>C | p.I203T | polymorphism | TM4 | 1 |
| c.79G>A | p.V27I | polymorphism | TM1 | Nv | 202 | |||
| c.147C>T | p.A49A | polymorphism | EC1 | Nv | 1 | |||
| c.181A>G | p.K61K | polymorphism | EC1 | Nv | 1 | |||
| c.341A>G | p.E114G | polymorphism | CL | Nv | 14 | |||
| c.438C>T | p.F146F | polymorphism | TM3 | Nv | 2 | |||
| c.608T>C | p.I203T | polymorphism | TM4 | c.608T>C | p.I203T | polymorphism | TM4 | 3 |
| c.608T>C | p.I203T | polymorphism | TM4 | Nv | 28 | |||
| nv | Nv | 638 | ||||||
| total | 2063 | |||||||
nv: no variant
Note: p.V37I is controversy variant, see the discussion.
Table 2.
Prevalence of GJB2 mutations in different areas of China
| Number of NSHI | c.235delC allele | c.299_c.300delAT allele | c.176_c.191del16 allele | Uncommon mutant allele | total number of mutant alleles(%) | ||||||||||||
| total | with two mutation | 1 allele with one mutaion | number with 1 mutant allele (%) | homo | het | total (%)a | homo | het | total (%)a | homo | het | total (%)a | homo | het | total (%)a | ||
| Jiangsu | 102 | 26 | 10 | 36 (35.3) | 12 | 18 | 42 (67.7) | 2 | 7 | 11 (17.7) | 1 | 7 | 9 (14.5) | 0 | 0 | 0 | 30.4 |
| Nei Mongol | 115 | 30 | 5 | 35 (30.4) | 14 | 18 | 46 (70.8) | 0 | 11 | 11 (16.9) | 0 | 3 | 3 (4.6) | 1 | 3 | 5 (7.7) | 28.3 |
| Beijing | 155 | 37 | 6 | 43 (27.7) | 24 | 13 | 61 (76.3) | 0 | 10 | 10 (12.5) | 0 | 0 | 0 | 0 | 9 | 9 (11.3) | 25.8 |
| Hebei | 64 | 14 | 3 | 17 (26.6) | 7 | 9 | 23 (74.2) | 0 | 3 | 3 (9.7) | 0 | 1 | 1 (3.2) | 0 | 4 | 4 (12.9) | 24.2 |
| Shanghai | 31 | 7 | 1 | 8 (25.8) | 3 | 5 | 11 (73.3) | 0 | 2 | 2 (13.3) | 0 | 1 | 1 (6.7) | 0 | 1 | 1 (6.7) | 24.2 |
| Heilongjiang | 36 | 5 | 4 | 9 (25.0) | 1 | 7 | 9 (64.3) | 0 | 2 | 2 (14.3) | 0 | 0 | 0 | 0 | 3 | 3 (21.4) | 19.4 |
| Guangdong | 77 | 15 | 4 | 19 (24.7) | 10 | 7 | 27 (79.4) | 0 | 4 | 4 (11.8) | 0 | 0 | 0 | 0 | 3 | 3 (8.8) | 22.1 |
| Sichuan | 109 | 17 | 8 | 25 (22.9) | 10 | 13 | 33 (78.6) | 0 | 3 | 3 (7.1) | 0 | 4 | 4 (9.5) | 0 | 2 | 2 (4.8) | 19.3 |
| Shanxi | 57 | 11 | 2 | 13 (22.8) | 4 | 9 | 17 (70.8) | 0 | 5 | 5 (20.8) | 0 | 1 | 1 (4.2) | 0 | 1 | 1 (4.2) | 21.1 |
| Gansu | 42 | 7 | 2 | 9 (21.4) | 3 | 5 | 11 (68.8) | 0 | 3 | 3 (18.8) | 0 | 0 | 0 | 0 | 2 | 2 (12.5) | 19 |
| Jilin | 57 | 12 | 0 | 12 (21.1) | 7 | 4 | 18 (75.0) | 0 | 5 | 5 (21.0) | 0 | 0 | 0 | 0 | 1 | 1 (4.0) | 21.1 |
| Fujian | 48 | 6 | 4 | 10 (20.8) | 5 | 4 | 14 (87.5) | 0 | 1 | 1 (6.3) | 0 | 0 | 0 | 0 | 1 | 1 (6.3) | 16.7 |
| Ningxia | 145 | 20 | 9 | 29 (20.0) | 8 | 14 | 30 (61.2) | 1 | 3 | 5 (10.2) | 0 | 4 | 4 (8.2) | 0 | 10 | 10 (20.4) | 16.9 |
| Xinjiang | 136 | 19 | 8 | 27 (19.9) | 9 | 5 | 23 (50.0) | 2 | 4 | 8 (17.4) | 0 | 3 | 3 (6.5) | 1 | 10 | 12 (26.1) | 16.9 |
| Hubei | 47 | 7 | 2 | 9 (19.1) | 6 | 2 | 14 (87.5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 2 (12.5) | 17 |
| Yunnan | 230 | 23 | 19 | 42 (18.3) | 11 | 14 | 36 (55.4) | 1 | 3 | 5 (7.7) | 1 | 2 | 4 (6.2) | 1 | 18 | 20 (30.8) | 14.1 |
| Guiyang | 138 | 23 | 2 | 25 (18.1) | 16 | 9 | 41 (85.4) | 0 | 6 | 6 (12.5) | 0 | 0 | 0 | 0 | 1 | 1 (2.1) | 17.4 |
| Henan | 126 | 16 | 5 | 21 (16.7) | 10 | 8 | 28 (75.7) | 0 | 5 | 5 (13.5) | 0 | 0 | 0 | 0 | 4 | 4 (10.8) | 14.7 |
| Tibet | 118 | 0 | 19 | 19 (16.1) | 0 | 2 | 2 (10.5) | 0 | 1 | 1 (5.3) | 0 | 0 | 0 | 0 | 16 | 16 (84.2) | 8.1 |
| Qinghai | 56 | 5 | 3 | 8 (14.3) | 1 | 3 | 5 (38.5) | 2 | 2 | 6 (46.2) | 0 | 0 | 0 | 0 | 2 | 2 (15.4) | 11.6 |
| Anhui | 35 | 3 | 2 | 5 (14.3) | 1 | 4 | 6 (75.0) | 0 | 1 | 1 (12.5) | 0 | 1 | 1 (12.5) | 0 | 0 | 0 | 11.4 |
| Shaanxi | 52 | 3 | 2 | 5 (9.6) | 1 | 4 | 6 (75.0) | 0 | 1 | 1 (12.5) | 0 | 0 | 0 | 0 | 1 | 1 (12.5) | 7.7 |
| Guangxi | 87 | 1 | 5 | 6 (6.9) | 1 | 4 | 6 (85.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 (14.3) | 4 |
| total | 2063 | 307 | 125 | 432 | 164 | 181 | 345 | 8 | 82 | 90 | 2 | 27 | 29 | 3 | 95 | 98 | 17.9 |
homo: homozygous; het: hetrozygous; apercentage of total mutant alleles identified.
Frame-shift and nonsense Pathogenic Mutations
The c.235delC is the most prevalent mutation in the Chinese NSHI population with a total of 509 alleles (164 homozygous, 113 compound heterozygous with other pathogenic mutant alleles, and 68 one heterozygous allele only), followed by 98 c.299_c.300delAT mutant alleles (8 homozygotes, 76 compound heterozygotes, and 6 one allele heterozygotes), 31 c.176_c.191del16 mutant alleles (2 homozygous, 25 compound heterozygous and 2 with only one allele), and 12 c.35delG mutant alleles(2 homozygous, 3 compound heterozygous and 2 with only one allele) (Supplemental Table 1). The four prevalent mutations account for 88.0% (650/739) of all mutant alleles identified. Five novel mutations were identified in 20 patients; including two nonsense; p.W3X, p.Y152X, and 4 frame-shift truncation mutations; c.99delT, c.155–c.158 delTCTG, and c.512–c.513 insAACG. Among these, c.512–c.513insAACG occurs in 12 patients, including one homozygous from Yunnan. The novel truncation mutations account for only about 3.1% (23/739, Supplemental Table 1) of the overall GJB2 mutant alleles. The most prevalent Caucasian mutation, c.35delG, was found in 2 homozygous, 3 compound heterozygous, and 5 single allele heterozygous patients. Among the patients with c.35delG, 70% of patients (7/10) are Uigur from Xinjiang area. The c.35insG mutation was found in 2 patients (both are Hui people) compound heterozygous with the c.235delC mutation. Other reported frame-shift mutations; 1 c.388–c.397del10 and 3 c.605–c.606ins46, as well as nonsense mutations; 3 p.E47X, account for a small fraction (1.0%) of GJB2 mutant alleles. Overall, 92.6% (684/739) of the pathogenic mutations are frame-shift truncation or nonsense mutations, and they are predicted to cause loss of function of connexin 26. Only 6.9% (51/739) of the mutant alleles are reported missense mutations.
Reported missense pathogenic mutations
There are 8 reported missense pathogenic mutations and 1 in-frame deletion of 1 single amino acid, c.424_c.426del3 (p.del142F), which occurs in 4 heterozygous patients (Supplemental Table 1). The 8 missense mutations are p.G4D (3 heterozygous patients), p.R32C (one patient in compound heterozygosity with c.235delC), p.R143W (4 compound heterozygotes), p.T86R (all compound heterozygous, 6 with c.235delC and 1 with c.299_c.300delAT), p.R127H (one compound with p.Y152X, 31 single heterozygotes), p.S139N (2 single heterozygotes), p.E47K (one compound with c.299_c.300delAT), p.S85P (single heterozygote). All occur in an evolutionarily highly conserved region (Figure 2) [26,29,30].
Figure 2.
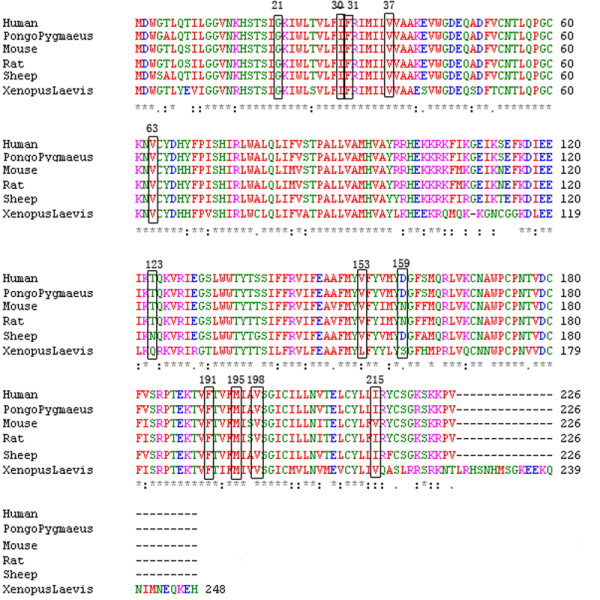
Amino acid alignment of Connexin26 in different species.
Unclassified Variants
Twelve unclassified missense variants were identified. The p.G21R is most likely to be pathogenic based on its highly evolutionarily conserved nature and the dramatic effect of the amino acid substitutions on structure and ionic strength. The p.I215N variant is located in the conserved region of C-terminal ion channel domain. Replacing the hydrophobic amino acid isoleucine with a hydrophilic amino acid asparagine in this conserved region is expected to cause detrimental effect. This variant is also in compound heterozygous with a novel pathogenic mutation, c.155_c.158delTCTG. Thus, it is likely to be pathogenic.
The missense variants, p.I30F, p.F31L, p.V63L, p.V153A, p.D159N, p.F191L, p.M195V, and p.V198M, do not involve drastic change in amino acid structure and polarity. They are all present as single heterozygous alleles without the presence of a second pathogenic mutant allele. Thus, their pathogenicity cannot be determined. Other changes of the same amino acids have been reported. For example, p.V63A has been reported as a novel variant, p.V153I and p.D159N were reported as a polymorphism [29]. The p.M195V and p.V198M, each occurs in two patients, without the second mutant allele. Each of the other variants occurs as heterozygous in one patient. None of these missense variants were detected in the control population.
Uncharacterized Novel Silent Variants
Several nucleotide substitutions do not result in amino acid change. These are p.A49A, p. K61K, p.F146F, and p.T186T (p.T186T is heterozygous with a single c.235delC). Although these nucleotide changes do not alter the encoded amino acids, we cannot exclude the possibility that they may activate an exonic splice enhancer and cause aberrant splicing. Alternatively, changes in triplet codon may affect the preference of codon usage or the stability of the mRNA, which in turn can affect the protein levels.
Genotypes and Carrier Frequency in the Normal Control Population
GJB2 is a small gene but harbors many mutations. Thus, the carrier frequency of GJB2 mutation in the Chinese population is not negligible. We sequenced the coding region of 301 normal control individuals of the Han ethnic group. Nine individuals were found to be heterozygous carriers of GJB2 pathogenic mutations; three had the c.235delC, three had the c.299_c.300delAT, and the c.512_c.513insAACG, c.35delG, and p.E47X mutation have been detected in single individuals (see Supplemental Table 3). Thus, the carrier frequency of GJB2 mutations in the control population is 3%.
Frequencies of missense variants in patient and control populations
The frequencies of common missense variants such as p.V37I, p.V27I, p.I203T, p.T123N, p.E114G in patients, control, and other Asian populations were compared (see Supplemental Table 4 and Table 5). The pathogenic role of p.V37I has been controversial [24-26,30-33]. It was found that the p.V37I allele frequency was significantly higher in the Han patient group (excluding all cases with two clearly pathogenic mutations) than in the control group (6.7% and 2.8% respectively,. p = 0.0003), supporting a pathogenic role of p.V37I. The allele frequencies of p.V27I, p.E114G, p.I203T, and p.T123N were higher in the control group than in the Han patient group (excluding all cases with two clearly pathogenic mutations), arguing against their pathogenic role (see Supplemental Table 4 and Table 5).
GJB2 mutation spectra among different sub-ethnic groups in China
As indicated in Table 2, the frequency of GJB2 mutations varies from 4% in Guangxi to 30.4% in Jiangsu. These results suggest that the variation in mutation frequencies may be due to ethnic diversity in various regions. The total population of China is 1.3 billion and sub-populations of Han, Tibetan, Hui, Man, Mon, minorities in Xinjiang, and minorities in South-western China are 1137.4 million, 5.4 million, 9.8 million, 10.7 million, 5.8 million, 10.8 million, and 57.1 million, respectively (http://www.cnmuseum.com/intro/renkou_intro.asp, http://www.xzqh.org/quhua/index.htm). We therefore analyzed the mutation frequencies in different sub-ethnic groups. As shown in Supplemental Table 6, Hui has the highest frequency of overall GJB2 mutations, followed by Han and minorities in Xinjiang with 20.3, 19.1, and 15.3% respectively. Tibetan and the minorities in the Southwest have lower mutation frequencies, 9.4 and 5.0% respectively, similar to the frequencies observed in corresponding regions. The majority of mutations found in this study were found in the Han patient group (1640 cases) only except c.35 insG that was in compound heterozygous with c.235delC found in two Hui patients. The common Caucasian mutation, c.35delG was mainly detected in the minorities of Xinjiang, and accounted for almost half of the GJB2 mutant alleles in minorities of Xinjiang (9 c.35delG/19 total mutant alleles). The finding of the c.35delG mutation in Xinjiang may be due in part to the close vicinity of Xinjiang to Russia and Eastern European countries, and possible admixture. The Maan sub-ethnic group also appears to have diverse GJB2 mutation spectrum because mutations other than c.235delC account for more than one third of the mutant alleles. The three most common mutations c.235delC, c.299_c.300delAT, and c.176_c.191del16 account for 100% of GJB2 mutations in 18 Mongolian individuals analyzed. However, the sample size is too small to be statistically significant.
Discussion
Previous reports have suggested that the prevalence of GJB2 mutations among different ethnic groups varies. In our patients, the most common Caucasian mutation, c.35delG was only found in 10 patients (seven of them were Uigur from Xinjiang). Instead, the c.235delC account for 68.9% of all GJB2 mutant alleles in our Chinese study population. These results support that the c.235delC mutation in connexin 26 gene is the most prevalent mutation in most Asian populations, including Han Chinese [11,24,30,34]. The results from this study indicate that analysis of four common mutations, c.235delC, c.299_c.300delAT, c.176_c.191del16, and 35delG can detect 88.0% (650/739) of GJB2 mutations. In 13 regions of China, by analyzing these four mutations, we were able to identified at least one mutant allele in all studied patients with one or two GJB2 mutations (see Table 2 and Supplemental Table 2). In contrast, mutations in the GJB2 gene account for a variable proportion of the molecular etiology of NSHI in different regions and sub-ethnic groups in China. Our results have tremendous impact on the design of molecular diagnostic and carrier testing of NSHI families in China. For example, in addition to the three most common mutations of c.235delC, c.299_c.300delAT, c.176_c.191del16, for minorities in Xinjiang, testing of Caucasian c.35delG mutation should be included. In patients with Maan ethnic background, sequencing of the GJB2 coding region should be offered, since the analysis of three common mutations detects only 71% of GJB2 mutant alleles. In minorities from Southwest provinces, although the three most common mutations account for >90% of all GJB2 mutations, defects in GJB2 gene account for only a small fraction (5%, Supplemental Table 2 and Table 6) of mutant alleles in NSHI patients. Thus, in these groups, analysis of other NSHI related genes should be pursued.
We recently reported that 7.8% of patients with autosomal recessive nonsyndromic hearing impairment in China were homozygous for the most common c.235delC mutation in GJB2 gene and 8.5% of them carried one mutant allele of the c.235delC mutation [28]. Sequencing of the coding region of the GJB2 gene reveals that 14.9% of the patients carry two pathogenic GJB2 mutation and 6.1% carry only one mutant allele. These results are comparable to other reported studies [7,11,13,24,29,30,33-35]. The proportions of patients with GJB2 mutations carrying only one mutant allele vary among different regions, different sub-ethnic groups, and different countries [7,11,13,24,29,30,33-35]. The observation that sequence analysis of GJB2 gene in subjects with autosomal recessive NSHI results in a high number of patients with only one GJB2 mutant allele has been puzzling [23]. Our unpublished data showed that no mutation were found in GJB2 Exon1 and its splicing sequence among 851 deaf individuals from Central China in this cohort which suggested extremely low detection rate of GJB2 Exon1 mutation among Chinese deaf population. For there is higher frequency of single heterozygous GJB2 mutation detected in the deaf population than in the normal population in this study, the further more extensive study of sequence change in GJB2 Exon1 or promoter area and 3'-UTR, fragment deletion neighboring GJB2 ORF region and digenic inheritance with other genes are already considered in this large Chinese deaf cohort for elucidating complex pathogenesis of GJB2 gene to hearing impairment. We already added a paragraph in discussion. Thus, a digenic hypothesis was proposed and mutations in two other connexin (Cx) genes, GJB6 for Cx30 and GJB3 for Cx31 were studied [21,22,36]. In families with clear evidence of linkage to the DFNB1 locus, which contains two genes, GJB2 and GJB6 [6,20], a common 309 kb deletion, involving the coding region GJB6 gene upstream of GJB2 gene has been identified and found to account for up to 10% of DFNB1 alleles in Caucasians [22]. We analyzed the deletion in GJB6 gene in 372 patients from Inner Mongolia and central China, and deletions in GJB6 gene were not detected. Similar studies of GJB6 mutations in Taiwanese prelingual NSHI patients carrying one GJB2 mutant allele also did not detect any deleterious mutations in GJB6, consistent with our results [30].
Although the spectrum of rare GJB2 mutations varies among sub-ethnic groups and in different regions of China, the same most common c.235delC mutation is shared. This observation is in agreement with the reports from the studies of other Asian NSHI patients [10,11,24,30,34]. However, instead of c.299_c.300delAT being the second most prevalent mutation, p.G45E accounts for 16% of the Japanese GJB2 mutations, while p.G4D accounts for 10.6% of Taiwanese GJB2 mutant alleles [10,30]. The p.G45E mutation was not detected in our patients. The p.G4D mutation accounts for only 0.3% of GJB2 mutant alleles in Chinese NSHI patients and was recently reported in a US study [29,30].
Among the 23 pathogenic mutations, 14 cause truncated connexin 26 proteins due to nonsense or frame-shift mutations, 8 are missense mutations, and one is a deletion of one amino acid. These mutations occur along the coding region. The truncation mutations account for 92.6% of the mutant alleles. Amino acids sequence homology alignment reveals that all missense mutations and unclassified variants occur at an evolutionarily conserved amino acid (Figure 2).
Three missense variants, p.V63L, p.V153A, and p.V198M, are located in extracelluar domain 1, 2, and transmembrane span 4, respectively, of connexin 26 protein. All these changes have not been reported in the Connexins and Deafness mutations database at http://davinci.crg.es/deafness. However, p.V63L has been found in 1 Taiwanese patient [30]. These three variants likely contribute to the pathogenesis of deafness, because (a) they were detected only in the patient group and not in 394 Japanese, 864 Taiwanese, 494 Korean and 301 Chinese (in this study) hearing normal subjects, and (b) they are evolutionarily conserved in xenopus, mouse, rat, sheep, orangutan, and human (Figure. 2). These variants were found in a heterozygous state in 4 unrelated patients who carried only one mutant allele.
The pathogenicity of p.V37I is controversial. In a recent multicenter study, the p.V37I mutation was found to be associated with mild to moderate hearing impairment (median 25–40 dB) [37]. Our study revealed that p.V37I with an allele frequency of 6.7% (185/2744) in the Han patient group (excluding all cases with two clearly pathogenic mutations) is significantly higher compared with that (2.8%;17/602) found in the control population (p = 0.0003, see Supplemental Table 4 and Table 5), supporting Wu's opinion to reassignment of p. V37I from an allele variant to a pathogenic mutation [38].
The p.T123N is an unclassified variant. It was counted as a mutation in Japanese group but a polymorphism in a Taiwanese study [10,30]. We found a higher p.T123N allele frequency in the control group than in the patient group, suggesting that it may be neutral variant. However, its clinical implication is not clear at this time.
The results of this study provide a great potential benefit for the clinical application of genetic testing for deafness. Based upon our preliminary data of molecular epidemiology of hearing impairment in China [28,39-41], Li has combined allele-specific PCR and universal array (ASPUA) methodologies for the detection of mutations causing hereditary hearing loss. It was employed for multiplex detection of 11 mutations in GJB2, GJB3, SLC26A4 and mitochondrial DNA causing hereditary hearing loss [42]. Although this simple screening chip only include probes and primers for the c.35delG, c.176_c.191del16, c.235delC, c.299_c.300delAT mutations of GJB2 gene, it can detect 88.0% (650/739) of GJB2 mutations among these 2063 deaf individuals, meanwhile, up to 88.9% (384/432) of 432 patients confirmed to carry at least one GJB2 mutation by sequencing in this study will be picked up by this fast screen method. The new methods for multiple mutation detection including ASPUA with capacity to test more gene loci have been under developed in our center, the data of this study will be crucial for the mutation selection in any new technology development for GJB2 gene testing in Chinese population.
In summary, this study revealed a unique GJB2 mutation spectrum in Chinese patients with nonsyndromic hearing impairment. The c.235delC mutation is the most frequent mutation in Chinese patients. Testing of four common mutations, c.235delC, c.299_c.300delAT, c.176_c.191del16, and c.35delG can detect 88.0% of the GJB2 mutant alleles. However, in some regions or sub-ethnic groups, the GJB2 mutations only account for a small fraction of the NSHI mutant alleles. In these regions, analysis of NSHI related genes is necessary. The molecular defects of more than 80% of the mutant alleles for NSHI in China remain to be identified.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PD, FY and BH carried out the molecular genetic studies, participated in the sequence alignment and drafted the manuscript. GW, QL, YY, XL, KY, JH, JH, YH, YW, QY, YY, HL, LL, WD, XZ, YY, JC, NH, XX, JZ, LT, RS, YL, SS, RZ, HW and YM carried out epidemiological survey.
DK and XZ participated in the sequence alignment. SZHY and DH participated in the design of the study and performed the statistical analysis. PD, DH, XL and BW conceived of the study, and participated in its design and coordination and helped to draft the manuscript. L-JW reviewed and interpreted the results, drafted and revised the manuscript.
Acknowledgments
Acknowledgements
The authors would like to thank Dr. Dennis Johnson and Dr. Raye L. Alford for their valuable suggestions. This work was supported by the Chinese National Nature Science Foundation Research Grant 30728030,30872862, and Chinese Capital Medical Development Scientific Funding 2005-1032 to Dongyi Han.
The authors also would like to thank the Fuyang School for the Deaf and Dumb (Anhui province), Beijing No.3 School for the Deaf (Beijing), Pinggu Special Education School (Beijing), Beijing Children's Hospital(Beijing), Fuzhou Special Education School (Fujian province), Lanzhou Convalescent Center for Deaf Children (Gansu province), Gansu Convalescent Ccenter for Deaf Children (Gansu province), Foshan School for the Deaf and Dumb (Guangdong province), Liuzhou School for the Bblind Deaf and Dumb (Guangxi province), Guiyang School for the Blind, Deaf and Dumb (Guizhou Province), Zhuozhou and Gaobeidian School for the Ddeaf and Dumb (Hebei province), Mudanjiang Special Education School (Heilongjiang province), Anyang Special Education School (Henan province), Wuhan Yimeng Convalescent Center for Deaf Children (Hubei province), Chifeng Special Education School (Inner Mongolia), Nantong School for the Deaf and Dumb (Jiangsu province), Haian School for the Deaf and Dumb (Jiangsu province), Haimen School for the Deaf and Dumb (Jiangsu province), Rugao School for the Deaf and Dumb (Jiangsu province), Tongzhou School for the Deaf and Dumb (Jiangsu province), Jilin Special Education School (Jilin province), Yinchuan School for the Blind, Deaf and Dumb (Ningxia Province), Xining Special Education School (Qinghai province), Changan School for the Deaf and Dumb (Shaanxi province), Affiliated Pediatric Medical Center of Shanghai Jiao Tong University (Shanghai), Yuncheng School for the Deaf and Dumb (Shanxi province), Yuncheng Disabled Person's Federation (Shanxi province), Yuncheng Convalescent Center for Deaf Children (Shanxi province), Chengdu Special Education School (Sichuan province), Urumchi School for the Deaf and Dumb (Xinjiang province), Korla School for the Deaf and Dumb (Xinjiang province), Kunming Huaxia Secondary School (Yunnan province), Kunming Convalescent Center for Deaf Children (Yunnan province), Lincang Special Education School (Yunnan province), Kunming Convalescent Center for Deaf Children (Yunnan province) and Lhasa Special Education School (Tibet municipality area) for their fundamental support and contributions to this work.
Contributor Information
Pu Dai, Email: daipu301@vip.sina.com.
Fei Yu, Email: playufei@163.com.
Bing Han, Email: daisy9716@yahoo.com.cn.
Xuezhong Liu, Email: xliu@med.miami.edu.
Guojian Wang, Email: wjcmu@263.net.
Qi Li, Email: liqi52002101@163.com.
Yongyi Yuan, Email: yyymzh@163.com.
Xin Liu, Email: lxlxlpy@126.com.
Deliang Huang, Email: huangdl301@sina.com.cn.
Dongyang Kang, Email: kangdongyang33@yahoo.com.cn.
Xin Zhang, Email: zhangxin_615@163.com.
Huijun Yuan, Email: yuanhj301@yahoo.com.cn.
Kun Yao, Email: 66panhong@163.com.
Jinsheng Hao, Email: nervina@126.com.
Jia He, Email: hejia63@yahoo.com.cn.
Yong He, Email: heyong1971@163.com.
Youqin Wang, Email: wangyouqin1971@163.com.
Qing Ye, Email: yeqing-1971@163.com.
Youjun Yu, Email: y200076@163.com.
Hongyan Lin, Email: ly.9658@163.com.cn.
Lijia Liu, Email: liulijia1971@163.com.
Wei Deng, Email: dengwei-1971@163.com.
Xiuhui Zhu, Email: zhuxiuhui1971@163.com.
Yiwen You, Email: qiangwang71@sina.com.
Jinghong Cui, Email: cuijhong@163.com.
Nongsheng Hou, Email: hounongsheng@163.com.
Xuehai Xu, Email: xuxuehai1971@163.com.
Jin Zhang, Email: zhangjin--1971@163.com.
Liang Tang, Email: tangliang--1971@163.com.
Rendong Song, Email: songrendong1966@yahoo.com.cn.
Yongjun Lin, Email: linyongjun1971@163.com.
Shuanzhu Sun, Email: ssz701205@sohu.com.
Ruining Zhang, Email: sxzm@163.com.
Hao Wu, Email: wuhao622sh@163.com.
Yuebing Ma, Email: mayuebing1971@163.com.
Shanxiang Zhu, Email: zhusx_mail@163.com.
Bai-lin Wu, Email: bai-lin.wu@childrens.harvard.edu.
Dongyi Han, Email: hdy301@263.net.
Lee-Jun C Wong, Email: ljwong@bcm.edu.
References
- Cohen MM, Gorlin RJ. Epidemiology, etiology and genetic patterns. In: Gorlin RJ, Toriello HV, Cohen MM, editor. Hereditary hearing loss and its syndromes. Oxford: Academic Press; 1995. pp. 9–21. [Google Scholar]
- Downs MP. Universal newborn hearing screening – the Colorado story. Int J Pediatr Otorhinolaryngol. 1995;32:257–259. doi: 10.1016/0165-5876(95)01183-C. [DOI] [PubMed] [Google Scholar]
- Mehl AL, Thomson V. Newborn hearing screening: the great omission. Pediatrics. 1998;101:E4.23. doi: 10.1542/peds.101.1.e4. [DOI] [PubMed] [Google Scholar]
- Mehl AL, Thomson V. The Colorado newborn hearing screening project, 1992–1999: on the threshold of effective population-based universal newborn hearing screening. Pediatrics. 2002;109:E7. doi: 10.1542/peds.109.1.e7. [DOI] [PubMed] [Google Scholar]
- Bitner-Glindzicz M. Hereditary deafness and phenotyping in humans. Br Med Bull. 2002;63:73–94. doi: 10.1093/bmb/63.1.73. [DOI] [PubMed] [Google Scholar]
- Estivill X, Fortina P, Surrey S, Rabionet R, Melchionda S, D'Agruma L, et al. Connexin-26 mutations in sporadic and inherited sensorineural deafness. Lancet. 1998;351:394–398. doi: 10.1016/S0140-6736(97)11124-2. [DOI] [PubMed] [Google Scholar]
- Gabriel H, Kupsch P, Sudendey J, Winterhager E, Jahnke K, Lautermann J. Mutations in the connexin26/GJB2 gene are the most common event in non-syndromic hearing loss among the German population. Hum Mutat. 2001;17:521–522. doi: 10.1002/humu.1138. [DOI] [PubMed] [Google Scholar]
- Lench N, Houseman M, Newton V, Van Camp G, Mueller R. Connexin-26 mutations in sporadic non-syndromal sensorineural deafness. Lancet. 1998;351:415. doi: 10.1016/S0140-6736(98)24006-2. [DOI] [PubMed] [Google Scholar]
- Morell RJ, Kim HJ, Hood LJ, Goforth L, Friderici K, Fisher R, et al. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N Engl J Med. 1998;339:1500–1505. doi: 10.1056/NEJM199811193392103. [DOI] [PubMed] [Google Scholar]
- Ohtsuka A, Yuge I, Kimura S, Namba A, Abe S, Van Laer L, et al. GJB2 deafness gene shows a specific spectrum of mutations in Japan, including a frequent founder mutation. Hum Genet. 2003;112:329–333. doi: 10.1007/s00439-002-0889-x. [DOI] [PubMed] [Google Scholar]
- Park HJ, Hahn SH, Chun YM, Park K, Kim HN. Connexin26 mutations associated with nonsyndromic hearing loss. Laryngoscope. 2000;110:1535–1538. doi: 10.1097/00005537-200009000-00023. [DOI] [PubMed] [Google Scholar]
- Rabionet R, Zelante L, Lopez-Bigas N, D'Agruma L, Melchionda S, Restagno G, et al. Molecular basis of childhood deafness resulting from mutations in the GJB2 (connexin 26) gene. Hum Genet. 2000;106:40–44. doi: 10.1007/s004390051007. [DOI] [PubMed] [Google Scholar]
- Wilcox SA, Saunders K, Osborn AH, Arnold A, Wunderlich J, Kelly T, et al. High frequency hearing loss correlated with mutations in the GJB2 gene. Hum Genet. 2000;106:399–405. doi: 10.1007/s004390000273. [DOI] [PubMed] [Google Scholar]
- Denoyelle F, Lina-Granade G, Plauchu H, Bruzzone R, Chaib H, Levi-Acobas F, et al. Connexin 26 gene linked to a dominant deafness. Nature. 1998;393:319–320. doi: 10.1038/30639. [DOI] [PubMed] [Google Scholar]
- Richard G, White TW, Smith LE, Bailey RA, Compton JG, Paul DL, Bale SJ. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum Genet. 1998;103:393–399. doi: 10.1007/s004390050839. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Huang D, Yu F, Zhu X, Kang D, Yuan H, Han D, Dai P. A De Novo GJB2 (Connexin 26) Mutation, R75W, in a Chinese Pedigree With Hearing Loss and Palmoplantar Keratoderma. Am J Med Genet. 2008. [DOI] [PubMed]
- Goodenough DA, Goliger JA, Paul DL. Connexins, connexons, and intercellular communication. Annu Rev Biochem. 1996;65:475–502. doi: 10.1146/annurev.bi.65.070196.002355. [DOI] [PubMed] [Google Scholar]
- Kumar NM, Gilula NB. The gap junction communication channel. Cell. 1996;84:381–388. doi: 10.1016/S0092-8674(00)81282-9. [DOI] [PubMed] [Google Scholar]
- Bruzzone R, White TW, Paul DL. Connections with connexins: the molecular basis of direct intercellular signaling. Eur J Biochem. 1996;238:1–27. doi: 10.1111/j.1432-1033.1996.0001q.x. [DOI] [PubMed] [Google Scholar]
- Scott DA, Kraft ML, Carmi R, Ramesh A, Elbedour K, Yairi Y, et al. Identification of mutations in the connexin 26 gene that cause autosomal recessive nonsyndromic hearing loss. Hum Mutat. 1998;11:387–394. doi: 10.1002/(SICI)1098-1004(1998)11:5<387::AID-HUMU6>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Tellería D, et al. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med. 2002;346:243–249. doi: 10.1056/NEJMoa012052. [DOI] [PubMed] [Google Scholar]
- Del Castillo I, Moreno-Pelayo MA, Del Castillo FJ, Brownstein Z, Marlin S, Adina Q, et al. Prevalence and evolutionary origins of the del(GJB6-D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: a multicenter study. Am J Hum Genet. 2003;73:1452–1458. doi: 10.1086/380205. Epub 2003 Oct 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Mueller RF, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387:80–83. doi: 10.1038/387080a0. [DOI] [PubMed] [Google Scholar]
- Abe S, Usami S, Shinkawa H, Kelley PM, Kimberling WJ. Prevalent connexin 26 gene (GJB2) mutations in Japanese. J Med Genet. 2000;37:41–43. doi: 10.1136/jmg.37.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballana E, Morales E, Rabionet R, Montserrat B, Ventayol M, Bravo O, et al. Mitochondrial 12S rRNA gene mutations affect RNA secondary structure and lead to variable penetrance in hearing impairment. Biochem Biophys Res Commun. 2006;341:950–957. doi: 10.1016/j.bbrc.2006.01.049. [DOI] [PubMed] [Google Scholar]
- Roux AF, Pallares-Ruiz N, Vielle A, Faugere V, Templin C, Leprevost D, et al. Molecular epidemiology of DFNB1 deafness in France. BMC Med Genet. 2004;5:5. doi: 10.1186/1471-2350-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai P, Liu X, Yu F, Zhu QW, Yuan YY, Yang SZ, et al. Molecular etiology of patients with nonsyndromic hearing loss from deaf-muta schools in 18 provinces of China. Chin J Otol. 2006;4:1–5. [Google Scholar]
- Dai P, Yu F, Han B, Yuan YY, Li Q, Wang GJ, et al. The prevalence of the 235delC GJB2 Mutation in a Chinese Deaf Population. Genet Med. 2007;9:283–289. doi: 10.1097/gim.0b013e31804d2371. [DOI] [PubMed] [Google Scholar]
- Tang HY, Fang P, Ward PA, Schmitt E, Darilek S, Manolidis S, et al. DNA sequence analysis of GJB2, encoding connexin 26: observations from a population of hearing impaired cases and variable carrier rates, complex genotypes, and ethnic stratification of alleles among controls. Am J Med Genet A. 2006;140:2401–2415. doi: 10.1002/ajmg.a.31525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwa HL, Ko TM, Hsu CJ, Huang CH, Chiang YL, Oong JL, et al. Mutation spectrum of the connexin 26 (GJB2) gene in Taiwanese patients with prelingual deafness. Genet Med. 2003;5:161–165. doi: 10.1097/01.GIM.0000066796.11916.94. [DOI] [PubMed] [Google Scholar]
- Bason L, Dudley T, Lewis K, Shah U, Potsic W, Ferraris A, et al. Homozygosity for the V37I Connexin 26 mutation in three unrelated children with sensorineural hearing loss. Clin Genet. 2002;61:459–464. doi: 10.1034/j.1399-0004.2002.610611.x. [DOI] [PubMed] [Google Scholar]
- Cryns K, Orzan E, Murgia A, Huygen PL, Moreno F, del Castillo I, et al. A genotype-phenotype correlation for GJB2 (connexin 26) deafness. J Med Genet. 2004;41:147–154. doi: 10.1136/jmg.2003.013896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi GZ, Gong LX, Xu XH, Nie WY, Lin Q, Qi YS. GJB2 gene mutations in newborns with non-syndromic hearing impairment in Northern China. Hear Res. 2004;197:19–23. doi: 10.1016/j.heares.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Wang YC, Kung CY, Su MC, Su CC, Hsu HM, Tsai CC, et al. Mutations of Cx26 gene (GJB2) for prelingual deafness in Taiwan. Eur J Hum Genet. 2002;10:495–498. doi: 10.1038/sj.ejhg.5200838. [DOI] [PubMed] [Google Scholar]
- Pollak A, Mueller-Malesinska M, Skórka A, Kostrzewa G, O dak M, Korniszewski L, et al. GJB2 and hearing impairment: promoter defects do not explain the excess of monoallelic mutations. J Med Genet. 2008;45:607–608. doi: 10.1136/jmg.2008.059873. [DOI] [PubMed] [Google Scholar]
- Liu XZ, Yuan Y, Yan D, Ding EH, Ouyang XM, Fei Y, Tang W, Yuan H, Chang Q, Du LL, Zhang X, Wang G, Ahmad S, Kang DY, Lin X, Dai P. Digenic inheritance of non-syndromic deafness caused by mutations at the gap junction proteins Cx26 and Cx31. Hum Genet. 2009;25:53–63. doi: 10.1007/s00439-008-0602-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoeckx RL, Huygen PL, Feldmann D, Marlin S, Denoyelle F, Waligora J, et al. GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet. 2005;77:945–957. doi: 10.1086/497996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu BL, Lindeman N, Lip V, Adams A, Amato RS, Cox G, et al. Effectiveness of sequencing connexin 26(GJB2) in familial or sporadic childhood deafness referred for molecular diagnostic testing. Genet Med. 2002;4:279–288. doi: 10.1097/00125817-200207000-00006. [DOI] [PubMed] [Google Scholar]
- Dai P, Yuan Y, Huang D, Zhu X, Yu F, Kang D, et al. Molecular Etiology of Hearing Impairment in Inner Mongolia: mutations in SLC26A4 gene and relevant phenotype analysis. J Transl Med. 2008;6:74. doi: 10.1186/1479-5876-6-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai P, Li Q, Huang D, Yuan Y, Kang D, Miller DT, et al. SLC26A4 c.919-2A>G varies among Chinese ethnic groups as a cause of hearing loss. Genet Med. 2008;10:586–592. doi: 10.1097/gim.0b013e31817d2ef1. [DOI] [PubMed] [Google Scholar]
- Dai P, Liu X, Han D, Qian Y, Huang D, Yuan H, et al. Extremely low penetrance of deafness associated with the mitochondrial 12S rRNA mutation in 16 Chinese families: implication for early detection and prevention of deafness. Biochem Biophys Res Commun. 2006;340:194–199. doi: 10.1016/j.bbrc.2005.11.156. [DOI] [PubMed] [Google Scholar]
- Li CX, Pan Q, Guo YG, Li Y, Gao HF, Zhang D, et al. Construction of a multiplex allele-specific PCR-based universal array (ASPUA) and its application to hearing loss screening. Hum Mutat. 2008;29:306–314. doi: 10.1002/humu.20622. [DOI] [PMC free article] [PubMed] [Google Scholar]