

Abstract
Solid core polymeric particles are an attractive delivery vehicle as they can efficiently encapsulate drugs of different physical and chemical characteristics. However, the effective targeting of such particles for therapeutic purposes has been somewhat elusive. Here, we report novel polymeric particles comprised of poly(lactic acid) (PLA) with incorporated poly(ethylene glycol)-lipids (PEG-lipids). Particles are characterized for morphology, surface charge, and composition with field-emission scanning electron microscopy (FESEM), zeta potential measurements, and proton nuclear magnetic resonance (1H NMR) spectroscopy respectively. The surface densities of PEG lipids determined by 1H NMR and particle size distributions are consistent with scaling theory for adsorption of chains onto a surface. We observe significant binding of liganded PEG-lipid tethers when the molecular weight is greater than the unliganded PEG-lipids for significant binding events. Importantly, the binding is not completely lost when the unliganded PEG molecular weight is greater than the liganded PEG-lipid tether. We observe a similar trend for the lower affinity ligand (thioctic acid), but the degree of binding is significantly lower than the high affinity ligand (biotin). This novel technique used to fabricate these liganded particles combined with the lipid bilayer binding studies provides a platform for systematic optimization of particle binding.
Keywords: Drug delivery, Microsphere, Surface Modification, Polylactic Acid, Lipid
1. Introduction
A major aim of targeted drug delivery systems is to prevent the potentially harmful and serious side effects of various therapeutic agents on other organs. Polymeric micro- and nanoparticles, and liposomes are the major types of carriers that have been investigated extensively as particulate drug carriers. Microspheres are an attractive drug delivery vehicle as they can mimic leukocyte rolling and selectively adhere to inflamed endothelium(1). Additionally, microspheres can be used for pulmonary, subcutaneous and other nonsystemic forms of delivery(2) including delivery to M cells lining the respiratory and digestive tracts(3), vitreous fluid of the eye(4), and hair follicles and their sebaceous glands(5). Biodegradable polymer-based particles are advantageous as they allow for effective encapsulation of hydrophobic drugs, and have better control of drug release than liposomes(6). Multiple techniques are used to form polymeric particles, including organic phase separation, supercritical fluid, spray drying and double emulsion(7). The polymers most employed for the common double emulsion (W/O/W) technique are poly(lactic acid) (PLA)(8, 9) and poly(lactic-co-glycolic acid) (PLGA)(10–12), both of which are FDA-approved. Both polymer systems are biodegradable and biocompatible as they degrade into natural metabolites, thereby limiting toxic effects in the body(4, 13, 14); however, PLGA degrades faster than PLA. The degradation rate, which alters the drug release profiles of these polymer particles, is tunable by particle size, and polymer molecular weight(14).
An ultimate goal of drug delivery is to deliver a ligand-targeted drug payload specifically to the diseased tissue. The ligands can be either physically adsorbed to the surface of the preformed particles(15), or directly incorporated as co-polymer ligand conjugates into the particle during the formation process. For example, Gref et al.(8) incorporated biotin-PEG-poly(epsilon-caprolactone), a biotinylated copolymer, into the preparation of PLA and PEG-PLA particles to which they coupled avidin and biotinylated wheat germ agglutinin. These particles showed a specific binding interaction with Caco-2 cells in vitro. Recently, Fahmy et al. (16) developed a strategy to functionalize particles by incorporating avidin-fatty acid conjugates into PLGA particles to which biotinylated ligands could then be added. These physical incorporation methods are versatile alternatives to covalent chemical conjugation for functionalizing particles.
Ideally, ligand-targeted particles will reach their target site; however, rapid clearance of particles to the reticuloendothelial system (RES) prevents delivery vehicles from reaching their designated target site. In order to avoid this major obstacle, the particle surface must be modified to minimize nonspecific protein adsorption that leads to opsonization and RES clearance. To date, the most effective mitigation of RES-mediated particle clearance has been accomplished by surface grafting poly(ethylene glycol) (PEG) to build a sterically repulsive shield that protects the particle from recognition by the RES(17). Additionally, PEG spacers have been found to enhance the accessible range of PEG-tethered ligand binding(17, 18). Earlier studies modified particles by physically adsorbing a PEG-based copolymer (Poloxamer 338) onto the surface of preformed particles(15). More recently, a number of groups have incorporated a PEG copolymer in situ during particle formation. In this process a synthesized diblock copolymer such as PEG-PLA(19–21), PEG-PLGA(20, 22), or a triblock copolymer such as PLA-PEG-PLA(23) is directly incorporated during the particle preparation. Copolymers are advantageous for many applications, but release of acidic degradation products, processing difficulties and limited mechanical range do not make them an ideal choice (16, 24).
The design of targeted delivery systems is inherently complex as they require a surface architecture that simultaneously promotes specific and blocks nonspecific interactions. Some studies have noted that the presence of methoxy-PEG on the same surface as tethered ligands can cause steric hindrance of effective targeting(25) while others note that ligand density and ligand tether lengths can also alter binding effectiveness(17, 18, 26–28). Current approaches to optimizing and understanding these factors include modification of particle design, through varied ligand composition (29), surface charge, and particle size(30). Alternatively, others have modified the substrate and substrate components by using different cell types or lipid bilayers with varied receptor or ligand densities (31, 32). A combination of changes to particle design and altered substrate compositions would be a useful approach to elucidate the underlying complexity of effective targeting using PEG tethers.
Particle binding under flow, which models the conditions of vasculature, has been used in a few investigations. Eniola et al.(33) worked with PLGA microspheres coated with sialyl LewisX, a carbohydrate that allows particle binding to selectins to mimic leukocyte rolling. They investigated the effect of particle degradation on particle binding to selectin-coated slides under laminar flow and found that particles recognized the selectin surface under flow conditions. Previous research in our laboratory(31) has combined patterned substrates with a laminar flow chamber to screen receptor-ligand binding with multiple surface architectures.
The objective of this study is to develop novel PEG-lipid based MPs with the long-term goal of achieving targeted binding under flow in vivo. In this study, lipid-PEG tethered model ligands are incorporated into the MPs with the lipid-methoxy PEG groups to balance the repulsive and binding forces. The well-studied model ligand, biotin (Ka ~ 1013 M−1) and the lower affinity ligand thioctic acid (Ka~7×107 M−1) are used toevaluate the applicability of this method to incorporate tethered ligands into MPs. Particle morphology is evaluated by FESEM, and the surface density of PEG-lipid is estimated through 1H NMR analysis. The specific binding of MP with different surface architectures to supported lipid bilayers is assessed under flow. These studies directly test the feasibility of PEG-lipid incorporation into PLA microspheres and serve to investigate the effects of particle surface architecture on binding efficacy.
2. Experimental
2.1 Materials
Poly (dl-lactide) (PLA) (Medisorb® 100DL High IV, Mw 109 kD), is purchased from Alkermes® (Cambridge, MA). Texas Red® 1,2-dihexadecanoyl-sn-glycero-3- phosphoethanolamine, triethylammonium salt (Texas Red-DHPE) is purchased from Molecular Probes (Eugene, OR). The lipids purchased from Avanti Polar Lipids (Alabaster, AL) include 1,2-Distearoyl-sn-Glycero-3-Phosphoethanolamine-N-z (mPEG2000-DSPE), 1,2-Distearoyl-sn-Glycero-3-Phosphoethanolamine-N-[Methoxy(Polyethylene glycol)-5000] (mPEG5000-DSPE), L-a-Phosphatidylcholine (eggPC) and 1,2-Dipalmitoyl-sn-Glycero-3-Phosphoethanolamine-N-(Cap Biotinyl) (biotin-DPPE). Biotin-PEG3350-DSPE is prepared as previously described (18, 34). Thioctic acid-PEG3350-DSPE is prepared as described below. Bovine serum albumin (BSA), streptavidin, reagent-grade chloroform, and methanol are purchased from Sigma. Phosphate buffered saline (PBS) is purchased from Gibco; and poly(dimethylsiloxane), Sylgard 184 (PDMS) from Dow Corning (Midland, MI). All chemicals are used without further purification.
2.2 Synthesis of thioctic acid-PEG-DSPE
Thioctic acid (31 mg, 0.15 mmol) is dissolved in dichloromethane (3 mL) and treated with hydroxybenzotriazole (20 mg, 0.15 mmol) in dimethylformamide (DMF) (200μL), followed by 1,3-dicyclohexylcarbodiimide (31 mg, 0.15 mmol). Due to the precipitation of 1,3-dicyclohexylurea, the reaction mixture turns cloudy as it is stirred at room temp (3 h). Then amino-PEG-DSPE (200 mg, 0.05 mmol (35)) is added to the activated thioctic acid solution, followed by tetraethylammonium (TEA) (84 μL, 0.6 mmol). After stirring at room temperature for 15 min, thin layer chromatography (TLC) reveals that the reaction is complete. The solvent is rotary evaporated and the ethyl acetate (5mL) added to the residue. The insolubles are filtered, the solvent removed, and the residue taken up with tert-butanol and then lyophilized. The crude product is dissolved in chloroform and chromatographed on a silica gel column using a methanol gradient (0 to 10%) in chloroform. Fractions containing the pure product are combined, evaporated, and lyophilized from tert-butanol. The white solid product is dried in vacuo over P2O5, yield (118 mg, 55%). 1H NMR (400 MHz, D6-DMSO): δ 0.88 (t, CH3, 6H.); 1.26 (s, CH2, 56H); 1.35–1.7 (m, CH2CH2C=O & CH2CH2CH2CHS, 10H from lipid and thioctic acid); 1.86 & 2.40 (two sextet, CH2CH2S-S, 2H); 2.06 (t, CH2COOH, 2H); 2.25 (m, CH2CO, 4H from lipid) 3.08–3.22 (m, CH2S-S, 2H); 3.50 (s, PEG, ≈ 300H); 3.6 (m, CHS-S, 1H); 4.03 (t, CH2O2CN, 2H); 4.08 & 4.28 (2x dd, OCH2CHCH2OP, 2H); 5.2 (m, PO4CH2CHCH2OCO 1H); 7.84 (t, CONH-PEG, 1H). Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) produces a bell–shaped distribution of ions characteristic to PEG derivatives spaced at equal ≈ 44 Da intervals and centered at 4324Da (calculated 4312 Da). This corresponds closely to the condensation product of thioctic acid (206 Da) and amino-PEG3350-DSPE starting lipopolymer (calculated 4124 Da, MALDI centered at 4092 Da).
2.3 Microparticle formation
A water-in-oil-in-water emulsion is used to form the four types of PEGylated MPs (Fig 1): mPEG2000-DSPE/Biotin-PEG3350-DSPE (mPEG2000-DSPE/B3350), mPEG5000-DSPE/Biotin-PEG3350-DSPE (mPEG5000-DSPE/B3350), mPEG2000-DSPE/Thioctic Acid -PEG3350-DSPE (mPEG-DSPE/TA3350), mPEG5000-DSPE/Thioctic Acid-PEG3350-DSPE (mPEG-DSPE/TA3350). Lipids for MP incorporation are combined in a vial, and chloroform is removed by a stream of argon gas, followed by at least 2 h under vacuum. Next, 20 mg of PLA and 2 mL ethyl acetate are added to the vial. The lipids and PLA are dissolved through bath sonication, and 4 mL deionized water is added to the vial and the solution is homogenized for 60 sec at 9,500 rpm with a homogenizer (High Shear Laboratory Mixer L4RT-A; Silverson). The resulting emulsion is then mixed with 50 mL of deionized water and stirred overnight to evaporate the ethyl acetate. The MPs are then rinsed 3x with deionized water at 8000 rpm and 4°C through centrifugation and supernatant replacement. For 1H NMR analysis, MPs are formed using 0.97 mg of mPEG2000-DSPE or 2.00 mg of mPEG5000-DSPE. The MPs used in binding studies are formed with the lipids indicated in Table 1 and 0.01 mg Texas Red-DHPE for quantification.
Figure 1.

Schematic diagram of the surface architectures of the microparticle formulations (a) mPEG2000-DSPE/Biotin-PEG3350-DSPE (b) mPEG2000-DSPE/Thioctic acid-PEG3350-DSPE (c) mPEG5000-DSPE/Biotin-PEG3350-DSPE (d) mPEG5000-DSPE/Thioctic acid-PEG3350-DSPE
Table 1.
Lipids added in MP formation. Note that when considering the PEG-lipids in any particular formulation, there is 5 mol% of liganded PEG-lipid. In addition, 0.01 mg Texas Red-DHPE is added to aid quantification.
| Amount of indicated lipid used in different particle formulations | ||||||||
|---|---|---|---|---|---|---|---|---|
| mPEG2000-DSPE | mPEG5000-DSPE | biotin-PEG3350-DSPE | thioctic acid-PEG3350-DSPE | |||||
| Microparticle type | mass (mg) | nmoles | mass (mg) | nmoles | mass (mg) | nmoles | mass (mg) | nmoles |
| mPEG2000/B3350 | 0.97 | 345 | _ | _ | 0.079 | 18.2 | _ | _ |
| mPEG5000/B3350 | _ | _ | 2.0 | 345 | 0.079 | 18.2 | _ | _ |
| mPEG2000/TA3350 | 0.97 | 345 | _ | _ | _ | _ | 0.078 | 18.2 |
| mPEG5000/TA3350 | _ | _ | 2.0 | 345 | _ | _ | 0.078 | 18.2 |
2.4 Particle Morphological Characterization
Particle shape and size is confirmed by a field emission scanning electron microscope (Zeiss Supra 40 FESEM). 5 μL of a microparticle suspension in water is dried on a Silicon wafer chip under vacuum. The shape morphology of the polymeric microspheres is imaged at 1000X, 3000X, and 5000X after the samples are dried.
2.5 Zeta potential Characterization
A microparticle suspension is diluted in 10−3M NaCl and added to the sample cell. The zeta potential is measured at 25°C with a Malvern Zetasizer NS. The results are reported as the average of five runs.
2.6 Characterization through 1H NMR
The NMR method of analysis is based on previous work(10, 36). MPs prepared with mPEG2000-DSPE, mPEG5000-DSPE, or unmodified PLA are freeze-dried and dissolved in 600 μL CDCl3. 1H NMR spectra are obtained with a Bruker AMX-300 NMR spectrometer equipped with a 7.05 T magnet with a 1H resonance frequency of 300.13 MHz. A 5-mm solution probe purchased from Bruker is used for all experiments. During a typical experiment, 64 to 1400 transients are acquired using a simple one-pulse with presaturation sequence. The typical 90° pulse is 6.25 μs; 8192 time domain data points are acquired with a dwell of 156 μs, and the recycle delay is 3s. The spectra are processed using the software supplied by Bruker. Peak integration and graph illustration are carried out with IGOR (Wavemetrics, Lake Oswego, Oregon). In order to quantify the amount of PEG-lipid in a given sample, known aliquots of mPEG-lipid are then doped into the sample and 1H NMR spectra are repeated. This yields a linear increase in the signal from the ethylene glycol protons (~3.6 ppm) with the added mPEG-lipid mass. The slope is then used to determine the initial mass of mPEG-lipid in the sample. For analysis of PEG density, the MP diameters are determined using phase contrast microscopy at 100x using ImageJ. In order to estimate the surface area per PEG-lipid the following formula is used:
where σPEG represents the area per PEG-lipid, NPEG is the number of PEG-lipids determined to be in the sample through NMR analysis, MPLA is the total mass of particles in the sample, mn is the average mass of a MP using the measured size distribution and assuming a density of 1.2 g/cm3, and sn is the average surface area of a MP calculated from the measured size distribution.
2.7 Assessment of binding in vitro
The method of substrate preparation has been previously reported in detail (31). Briefly, vesicle suspensions of eggPC with and without 5 mol % biotin-DPPE are formed via the sonication method. A PDMS stamp with individually addressable lanes is adhered to a glass slide. Vesicle suspensions mixed 1:1 with PBS supplemented with 140 mM NaCl are then injected into the microfluidic lanes, forming independent bilayers through vesicle fusion. Incubation with streptavidin forms a self-assembled streptavidin layer on the biotin-DPPE containing bilayers. Previous studies using fluorescein isothiocyanate (FITC)-streptavidin verify the homogeneous binding of streptavidin to the biotinylated bilayer and that the streptavidin withstands shear rates in excess of 1000 s−1.
A Glycotech™ flow chamber is then assembled onto the prepared substrate under deionized water in a crystallization dish. A steady flow of buffer (PBS with 1% BSA) and MP suspensions in buffer are then administered using an automated syringe pump (PHD 2000; Harvard Apparatus). The flow profile consists of 2 mL buffer at 0.6 mL/min (shear rate ~118 s−1), followed by 2 ml MP suspension at 0.03 mL/min (~6 s−1), and then 10 mL buffer at 6 ml/min (~1176 s−1). See Figure 2 for a schematic overview. The substrate is then imaged (40x) under an optical microscope (Zeiss Axiovert S100) equipped with fluorescence and a digital camera to obtain images for analysis. Fluorescence images are taken of the control eggPC bilayer and the streptavidin-coated lipid bilayers and analyzed with ImageJ software to determine the surface density of bound particles. The experiment is repeated three times with each MP formulation.
Figure 2.
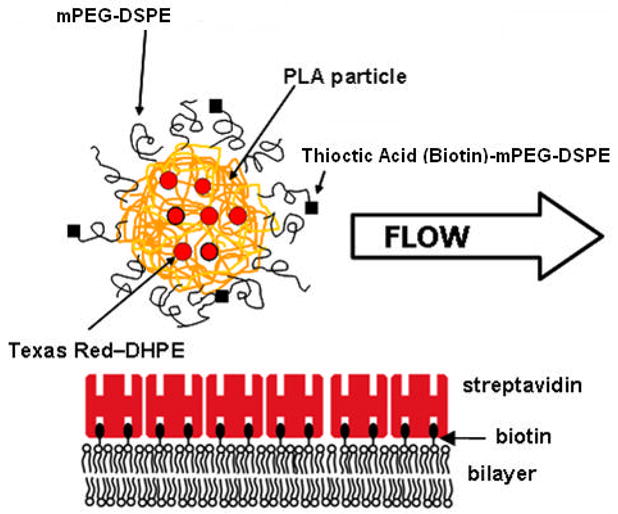
Schematic of binding study. The receptor surface consists of a solid-supported lipid bilayer (eggPC) that contains biotinylated lipid to enable self-assembly of a monolayer of streptavidin. mPEG-lipids and PEG-tethered ligands are incorporated into the PLA microparticle during the formation process.
3. Results and Discussion
3.1 Microparticle formation
The microparticle formation procedure developed in this study is unique in that an additional surfactant is not required for MP formation. We find that the addition of PEG-lipids lead to a stable emulsion and an easily-suspended final product. The absence of other surface active agents should ensure maximal PEG-lipid surface coverage. In contrast to conventionally-used detergents, good biocompatibility and low toxicity of PEG-based lipopolymers is well-documented in the literature(37). Although polymeric particles have been coated with PEG through a variety of processes, the adsorption of PEG-lipids has had limited use, and to our knowledge, this work represents the first investigation using ligand-conjugated PEG-lipids.
3.2 Characterization of MPs through SEM
The microparticles exhibit spherical morphology for each of the formulations studied (Fig 3). The size range of the populations of microparticles is slightly heterogeneous ranging from 0.5μm to 2.0 μm. The addition of PEG2000 or PEG5000 does not noticeably change the microparticle morphology.
Figure 3.

FESEM images of microparticles at 3000X (a)mPEG2000/B3350 (b) mPEG5000/B3350 (c)mPEG2000/TA3350 (d) mPEG5000/TA3350. Scale bar is 2μm.
3.3 Characterization of Particle Zeta Potential
The microparticles formed with PEG5000 have a less negative zeta potential value than the particles formed with PEG2000, i.e. ~ −30 mV vs. ~ −50 mV, respectively (Table 2). Zeta potential measures the charge at the plane of hydrodynamic shear. The surface charge of plain PLA nanoparticles and microparticles in ionic solution has been reported to be −50mV (38): this high negative surface charge can be explained by the presence of carboxyl end groups of the PLA (39–41). The value of −50 mV for our PLA particles formed with PEG2000 is not inconsistent with the reported value of −50 mV for plain PLA: we expect our zeta potential values to be more negative than plain PLA microparticles because in our case, we have incorporated DSPE-PEG (net negative charge) for steric stabilization. In contrast, the plain PLA particles discussed above (38) include sodium cholate as a surfactant stabilizer. Because the particles aggregate in the absence of DSPE-PEG, it is not possible to measure the zeta potential for PLA particles without DSPE-PEG.
Table 2.
Zeta potential values for the four particle formulations.
| Particle | Zeta Potential (mV) |
|---|---|
| PLA2000/B3350 | −48.96 |
| PLA2000/TA3350 | −46.46 |
| PLA5000/B3350 | −29.66 |
| PLA5000/TA3350 | −31.56 |
The changes we observe in the zeta potential measurements for different particle formulations suggest that the PEG portion of the lipid is situated on the surface of the particles. Previous work has shown that the use of PLA-PEG copolymers changes the zeta potential of plain PLA particles from −50mV to a less negative value of −30mV, indicating that PEG is shielding the particle surface charge (41). Similarly, it has been shown that increasing the length of the PEG chain of PLA-PEG copolymers further lowers negative zeta potential values, indicating the increased shift of the shearing plane away from the particle surface(38, 42). Our results indicate that the PEG 5000 is more effective in shielding the particle surface charge by moving the shearing plane outwards from the particle surface (43). These results are important as we find that PEG shielding influences the binding characteristics (described in section 3.5) by increasing the steric barrier for binding (20). We also observe that the zeta potential values are not significantly affected by the addition of either the biotin or thioctic acid ligand.
3.4 Characterization of MPs through 1H NMR analysis
1H NMR of pure samples consisting of PLA and mPEG-lipids show well-separated signals: from PLA (~1.6, ~5.1 ppm) and PEG-lipids (~3.6 ppm). The spectrum from mPEG2000-DSPE MPs reflects signals from both the PLA and mPEG-lipid components (Fig 4).
Figure 4.

(A) 1H NMR of mPEG5000-DSPE in CDCl 3. Note the peak at ~3.6 ppm which is from ethylene glycol -CH2- protons. This is the peak of interest that is integrated to determine the amount of PEG-lipids. (B) Spectra of PLA in CDCl3 confirm there is no interference with the PEG signal. (C) Spectra of mPEG2000-DSPE MPs dissolved in CDCl3 show the expected peaks from both PLA and PEG-lipid.
The method of doping in known amounts of PEG-lipid enables the semi-quantitative determination of the PEG-lipid content for MPs. By combining the determined PEG-lipid content by this method with the size distribution measured through microscopy, the PEG surface density can be estimated. It must be noted that this analysis assumes that the PEG-lipids are located exclusively on the surface of the MPs – an assumption that has been made by others (20). This is reasonable as it is unlikely that a significant portion of the long hydrophilic PEG chains would remain in the hydrophobic PLA core. This is further supported by our zeta potential measurements (see Section 3.3). Additionally, other studies have shown that analogous PEG-copolymers are not significantly internalized, and exist almost exclusively at the particle’s surface(44). A summary of the surface density calculations is presented in Table 3.
Table 3.
Surface coverage of PEG-lipids determined through 1H NMR. The surface density σ is the inverse of the surface area per PEG-lipid.
| Particle type | Mass of sample (mg) | Calculated PEG-lipid mass (mg) | Mean MP diameter (μm) | Surface area (nm2) per PEG-lipid* |
|---|---|---|---|---|
| mPEG2000-DSPE | 3 | 0.025 | 1.22 | 1.84 ± 0.05 |
| mPEG5000-DSPE | 3.3 | 0.02 | 1.29 | 4.85 ± 0.15 |
Error is from the standard deviation of the slope in the linear regression used to determine PEG-lipid content.
The mPEG5000-DSPE MPs have a surface area per lipid of 4.85 nm2 and mPEG2000-DSPE MPs have a surface area per lipid of 1.84 nm2. This can be explained by the difference in PEG chain length, whereby a shorter PEG chain allows for tighter packing on the particle surface. Previous studies investigating the incorporation of mPEG-PLA block copolymers found that the surface area per PEG chain ranged from 0.9 nm2 to 6.6 nm2, depending on the formulation(45). This indicates that the surface coverage we achieve with PEG-lipids is similar to that achieved with the incorporation of copolymers.
Lipid-PEG molecules have also been studied extensively in monolayer (e.g air-water interface) and bilayer forms (e.g. supported lipid bilayers and liposomes). We can also compare our values to the packing density reported in the literature for PEG-lipids. As the PEG-lipid surface concentration increases, the structural organization of the chains transition from the non-overlapping ‘mushroom’ (~ 1.3 mol%) to the ‘weak-overlap’ (~4.5 mol%) to the strongly-overlapping ‘brush’ (~9 mol%) regime (46). The 4.5 mol% formulation has been shown to successfully block protein adsorption and enhance in vivo circulation times (35). Using the value for area per DSPE headgroup (0.43 nm2) (46), our area per molecule of PEG2000-DSPE and PEG5000-DSPE (Table 2) give surface coverages of 23.4 and 8.9 mol%, respectively, which are within the brush configurations.
Our results for the polymer surface coverage can also be understood by a simple Flory argument. We first recall the theoretical results from scaling theory for block copolymer adsorption onto a surface(47). In our case, the lipid anchor is attracted to the hydrophobic PLA, whereas the PEG chain is repelled from the surface of PLA. In a good solvent and taking into account excluded volume effects, the PEG chains will adopt a swollen coil conformation with Flory radius RF= N3/5a, where N is the degree of polymerization and a is the monomer size. If one assumes that each lipid provides an anchoring energy of δ kBT, then the simple adsorption balance in (47) gives a chain surface density σ = (δ/N)6/5. The surface chain densities of the two PEG molecular weights, 2000 and 5000, should therefore scale proportionally
The right hand side of the equation gives a value of 3.0, while the ratio of the measured surface densities gives us a value of 2.8 when including the standard error. We recognize that the determination of particle size is not exact; nevertheless, this limited data is in close agreement with the predicted value. A more in-depth study is required to verify experimental agreement with scaling theory to predict the surface coverage of particles, which in turn can aid in the optimization of surface functionalization. A key assumption is that the PEG-lipid is strongly associated with the surface. As described in the next section (Section 3.5), we examine the effect of sodium dodecyl sulfate (SDS) on MP binding and find that the PEG-lipids are strongly associated with the PLA microparticles.
3.5 Comparative binding studies
MPs are formed with 5 mol% of either biotin-PEG3350-DSPE or thioctic acid-PEG3350-DSPE, in either mPEG2000-DSPE or mPEG5000-DSPE. The results of MP binding indicate that both the type of unliganded mPEG-lipid and the ligand affinity have a significant effect on MP binding (Fig. 5). In addition, we find that the addition of SDS does not result in any significant change in binding with either 0.1 or 0.01% SDS (data not shown). This indicates that the surface-associated lipids are stable in up to 0.1 % SDS.
Figure 5.
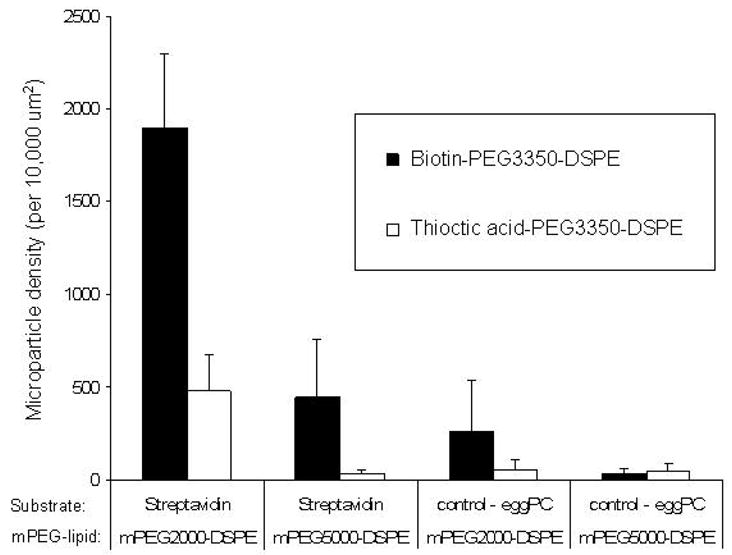
Comparison of binding for MPs containing biotin-PEG3350-DSPE or thioctic acid-PEG3350-DSPE. Binding studies indicate the effect of ligand affinity on MP binding. The binding of MPs containing biotin-PEG3350-DSPE (¦) is shown adjacent to the binding
The use of mPEG5000-DSPE as opposed to mPEG2000-DSPE reduces the binding of MPs containing biotin-PEG3350-DSPE four-fold, and restricts significant binding events when particles contain thioctic acid-PEG3350-DSPE. This indicates that the presence of mPEG chains that are longer than the liganded tether can prevent effective binding. We further observe that use of thioctic acid-PEG3350-DSPE rather than biotin-PEG3350-DSPE in equivalent mol %, significantly reduces MP binding: for MPs containing mPEG2000-DSPE binding is reduced about 75%, whereas for MPs containing mPEG5000-DSPE the binding is reduced to control levels.
On the other hand, by choosing the appropriate length PEG chain spacer, it is possible to enhance ligand receptor binding (18, 26, 48). Our results using a bidisperse surface layer are in agreement with studies by Chen et al. (49) reporting that a polymer layer consisting of short (nonfunctional) and long (functionalized with ligands) chains leads to optimal binding. The affinity of thioctic acid for streptavidin is (Ka~7×107 M−1) similar to biologically relevant receptor-ligand pairs such as antibodies to selectins (Ka~1×108−2×109)(50, 51). In this study, the particles are subjected to rinsing at a shear rate of ~1176 s−1, comparable to the maximal shear rates seen in human vasculature (52). This indicates the incorporated PEG-lipids are quite stable and offer a viable method for particle targeting.
Importantly, the ability to achieve binding with the lower affinity thioctic acid-PEG3350-DSPE/mPEG2000-DSPE formulation is significant.
The extremely high affinity between biotin and streptavidin underlies the versatility of MPs containing biotin-PEG-DSPE, as they can be easily modified with various ligands through biotin-avidin coupling. Furthermore, the patterned substrate used in the flow studies can be altered to accommodate various systems, including mammalian cells. Our novel PEG-lipid based particles and binding study method offer a platform for systematic investigation and optimization of binding characteristics.
4. Conclusions
We demonstrate that PEG-lipids can be incorporated successfully into PLA microparticles (0.3μm–2.2μm) through direct addition in formation of an oil-in-water emulsion. In fact, the PEG-lipids serve as a suitable surfactant, and no additional co-surfactants are required to form stable particles. Binding studies show that the in vitro flow chamber can reproducibly quantify binding efficacy, and significant variations are seen between the different formulations. Specifically, our results that longer mPEG chains with liganded PEG chains and a lower affinity ligand serve to decrease binding. These studies represent the basis for a systematic particle optimization using physiologically-relevant ligands.
Acknowledgments
We acknowledge financial support from ALZA Corporation and the National Institutes of Health (NIH HL72900S) to JYW, and the NIH P50 HL083801 (JAH, JYW). The authors thank Nasreen Mullah (ALZA) for her technical assistance in synthesis of lipopolymer conjugates. We also thank C. Marques (CNRS) and E. Amstad for helpful and insightful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sakhalkar HS, Dalal MK, Salem AK, Ansari R, Fu J, Kiani MF, et al. Leukocyte-inspired biodegradable particles that selectively and avidly adhere to inflamed endothelium in vitro and in vivo. Proc Natl Acad Sci U S A. 2003;100(26):15895–900. doi: 10.1073/pnas.2631433100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pfeifer BA, Burdick JA, Langer R. Formulation and surface modification of poly(ester-anhydride) micro- and nanospheres. Biomaterials. 2005;26(2):117–24. doi: 10.1016/j.biomaterials.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Ermak TH, Giannasca PJ. Microparticle targeting to M cells. Adv Drug Deliv Rev. 1998;34(2–3):261–283. doi: 10.1016/s0169-409x(98)00043-x. [DOI] [PubMed] [Google Scholar]
- 4.Moshfeghi AA, Peyman GA. Micro- and nanoparticulates. Adv Drug Deliv Rev. 2005;57(14):2047–52. doi: 10.1016/j.addr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Mordon S, Sumian C, Devoisselle JM. Site-specific methylene blue delivery to pilosebaceous structures using highly porous nylon microspheres: an experimental evaluation. Lasers Surg Med. 2003;33(2):119–25. doi: 10.1002/lsm.10204. [DOI] [PubMed] [Google Scholar]
- 6.Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303(5665):1818–22. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 7.Keegan ME, Falcone JL, Leung TC, Saltzman WM. Biodegradable Microspheres with Enhanced Capacity for Covalently Bound Surface Ligands. Macromolecules. 2004;37:9779–9784. [Google Scholar]
- 8.Gref R, Couvreur P, Barratt G, Mysiakine E. Surface-engineered nanoparticles for multiple ligand coupling. Biomaterials. 2003;24(24):4529–37. doi: 10.1016/s0142-9612(03)00348-x. [DOI] [PubMed] [Google Scholar]
- 9.Nobs L, Buchegger F, Gurny R, Allemann E. Poly(lactic acid) nanoparticles labeled with biologically active Neutravidin for active targeting. European Journal of Pharmaceutics and Biopharmaceutics: official journal of Arbeitsgemeinschaft fur Pharmazeutische Verfahrenstechnik e V. 2004;58(3):483–90. doi: 10.1016/j.ejpb.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 10.Csaba N, Gonzalez L, Sanchez A, Alonso MJ. Design and characterisation of new nanoparticulate polymer blends for drug delivery. J Biomat Sci Polym Ed. 2004;15(9):1137–51. doi: 10.1163/1568562041753098. [DOI] [PubMed] [Google Scholar]
- 11.Saxena V, Sadoqi M, Shao J. Indocyanine green-loaded biodegradable nanoparticles: preparation, physicochemical characterization and in vitro release. Int J Pharm. 2004;278(2):293–301. doi: 10.1016/j.ijpharm.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 12.Venier-Julienne MC, Benoit JP. Preparation, purification and morphology of polymeric nanoparticles as drug carriers. Pharmaceutica acta Helvetiae. 1996;71(2):121–8. doi: 10.1016/0031-6865(95)00059-3. [DOI] [PubMed] [Google Scholar]
- 13.El-Sherif DM, Wheatley MA. Development of a novel method for synthesis of a polymeric ultrasound contrast agent. J Biomed Mater Res A. 2003;66(2):347–55. doi: 10.1002/jbm.a.10586. [DOI] [PubMed] [Google Scholar]
- 14.Belbella A, Vauthier C, Fessi H, Devissaguet J-P, Puisieux F. In vitro degradation of nanospheres from poly(D,L-lactides) of different molecular weights and polydispersities. Int J Pharm. 1996;129(1–2):95–102. [Google Scholar]
- 15.Illum L, Davis SS. The organ uptake of intravenously administered colloidal particles can be altered using a non-ionic surfactant (Poloxamer 338) FEBS Lett. 1984;167(1):79–82. doi: 10.1016/0014-5793(84)80836-4. [DOI] [PubMed] [Google Scholar]
- 16.Fahmy TM, Samstein RM, Harness CC, Mark Saltzman W. Surface modification of biodegradable polyesters with fatty acid conjugates for improved drug targeting. Biomaterials. 2005;26(28):5727–36. doi: 10.1016/j.biomaterials.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 17.Gabizon A, Horowitz AT, Goren D, Tzemach D, Mandelbaum-Shavit F, Qazen MM, et al. Targeting folate receptor with folate linked to extremities of poly(ethylene glycol)-grafted liposomes: in vitro studies. Bioconjug Chem. 1999;10(2):289–98. doi: 10.1021/bc9801124. [DOI] [PubMed] [Google Scholar]
- 18.Wong JY, Kuhl TL, Israelachvili JN, Mullah N, Zalipsky S. Direct measurement of a tethered ligand-receptor interaction potential. Science. 1997;275(5301):820–2. doi: 10.1126/science.275.5301.820. [DOI] [PubMed] [Google Scholar]
- 19.Dong Y, Feng SS. Methoxy poly(ethylene glycol)-poly(lactide) (MPEG-PLA) nanoparticles for controlled delivery of anticancer drugs. Biomaterials. 2004;25(14):2843–9. doi: 10.1016/j.biomaterials.2003.09.055. [DOI] [PubMed] [Google Scholar]
- 20.Gref R, Luck M, Quellec P, Marchand M, Dellacherie E, Harnisch S, et al. ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids and Surfaces B, Biointerfaces. 2000;18(3–4):301–313. doi: 10.1016/s0927-7765(99)00156-3. [DOI] [PubMed] [Google Scholar]
- 21.Kim SY, Shin IG, Lee YM. Preparation and characterization of biodegradable nanospheres composed of methoxy poly(ethylene glycol) and DL-lactide block copolymer as novel drug carriers. Journal of Controlled Release: official journal of the Controlled Release Society. 1998;56(1–3):197–208. doi: 10.1016/s0168-3659(98)00083-2. [DOI] [PubMed] [Google Scholar]
- 22.Csaba N, Gonzalez L, Sanchez A, Alonso MJ. Design and characterisation of new nanoparticulate polymer blends for drug delivery. Journal of Biomaterials Science Polymer edition. 2004;15(9):1137–51. doi: 10.1163/1568562041753098. [DOI] [PubMed] [Google Scholar]
- 23.Ruan G, Feng SS. Preparation and characterization of poly(lactic acid)-poly(ethylene glycol)-poly(lactic acid) (PLA-PEG-PLA) microspheres for controlled release of paclitaxel. Biomaterials. 2003;24(27):5037–44. doi: 10.1016/s0142-9612(03)00419-8. [DOI] [PubMed] [Google Scholar]
- 24.Gunatillake P, Mayadunne R, Adhikari R. Recent developments in biodegradable synthetic polymers. Biotechnol Annu Rev. 2006;12:301–47. doi: 10.1016/S1387-2656(06)12009-8. [DOI] [PubMed] [Google Scholar]
- 25.Gabizon A, Shmeeda H, Horowitz AT, Zalipsky S. Tumor cell targeting of liposome-entrapped drugs with phospholipid-anchored folic acid-PEG conjugates. Advanced Drug Delivery Reviews. 2004;56(8):1177–92. doi: 10.1016/j.addr.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 26.Jeppesen C, Wong JY, Kuhl TL, Israelachvili JN, Mullah N, Zalipsky S, et al. Impact of polymer tether length on multiple ligand-receptor bond formation. Science. 2001;293(5529):465–8. doi: 10.1126/science.293.5529.465. [DOI] [PubMed] [Google Scholar]
- 27.DeFrees SA, Phillips LM, Guo LSS, Zalipsky S. Sialyl lewis X liposomes as a multivalent ligand and inhibitor of E-selectin mediated cellular adhesion. J Am Chem Soc. 1996;118:6101–6104. [Google Scholar]
- 28.Zalipsky S, Mullah N, Dibble A, Flaherty T. New chemoenzymatic approach to glyco-lipopolymers: Practical preparation of functionally active galactose-poly(ethylene glycol)-distearoylphosphatididic acid (Gal-PEG-DSPA) conjugate. Chem Commun. 1999;7:653–654. [Google Scholar]
- 29.Jolimaitre P, Poirier C, Richard A, Blanpain A, Delord B, Roux D, et al. Synthesis of versatile chemical tools toward a structure/properties relationships study onto targeting colloids. Eur J Med Chem. 2006 doi: 10.1016/j.ejmech.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 30.Deshpande D, Blezinger P, Pillai R, Duguid J, Freimark B, Rolland A. Target specific optimization of cationic lipid-based systems for pulmonary gene therapy. Pharm Res. 1998;15(9):1340–7. doi: 10.1023/a:1011933117509. [DOI] [PubMed] [Google Scholar]
- 31.Burridge KA, Figa MA, Wong JY. Patterning adjacent supported lipid bilayers of desired composition to investigate receptor-ligand binding under shear flow. Langmuir. 2004;20(23):10252–9. doi: 10.1021/la0489099. [DOI] [PubMed] [Google Scholar]
- 32.Farokhzad OC, Khademhosseini A, Jon S, Hermmann A, Cheng J, Chin C, et al. Microfluidic system for studying the interaction of nanoparticles and microparticles with cells. Anal Chem. 2005;77(17):5453–9. doi: 10.1021/ac050312q. [DOI] [PubMed] [Google Scholar]
- 33.Eniola AO, Hammer DA. Characterization of biodegradable drug delivery vehicles with the adhesive properties of leukocytes II: effect of degradation on targeting activity. Biomaterials. 2005;26(6):661–70. doi: 10.1016/j.biomaterials.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 34.Zalipsky S, Mullah N, Qazen M. Preparation of poly(ethylene glycol)-grafted liposomes with ligands at the extremities of polymer chains. Methods Enzymol. 2004;387:50–69. doi: 10.1016/S0076-6879(04)87004-6. [DOI] [PubMed] [Google Scholar]
- 35.Zalipsky S, Brandeis E, Newman MS, Woodle MC. Long circulating, cationic liposomes containing amino-PEG-phosphatidylethanolamine. FEBS Lett. 1994;353(1):71–4. doi: 10.1016/0014-5793(94)01013-7. [DOI] [PubMed] [Google Scholar]
- 36.Hrkach JS, Peracchia MT, Domb A, Lotan N, Langer R. Nanotechnology for biomaterials engineering: structural characterization of amphiphilic polymeric nanoparticles by 1H NMR spectroscopy. Biomaterials. 1997;18(1):27–30. doi: 10.1016/s0142-9612(96)00077-4. [DOI] [PubMed] [Google Scholar]
- 37.Working PK, Newman MS, Johnson J, Cornacoff JB. Safety of PEG and PEG derivatives. In: Harris JM, Zalipsky S, editors. Poly(ethylene Glycol): Chemistry and Biological Applications. Washington, DC.: ACS Symposium Series; 1997. pp. 45–57. [Google Scholar]
- 38.Gref R, Miralles G, Dellacherie E. Polyoxyethylene-coated nanospheres: effect of coating on zeta potential and phagocytosis. Polymer International. 1999;48(4):251–256. [Google Scholar]
- 39.E; GRMGD. Polyoxyethylene-coated nanospheres: effect of coating on zeta potential and phagocytosis. Polymer International. 1999;48(4):251–256. [Google Scholar]
- 40.Gref RAD, Quellec P, Blunk T, Müller RH, Verbavatz JM, Langer R. The controlled intravenous delivery of drugs using PEG-coated sterically stabilized nanospheres. Advanced Drug Delivery Reviews. 1995;16(2–3):215–233. doi: 10.1016/0169-409X(95)00026-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tobio M, Gref R, Sanchez A, Langer R, Alonso MJ. Stealth PLA-PEG nanoparticles as protein carriers for nasal administration. Pharm Res. 1998;15(2):270–5. doi: 10.1023/a:1011922819926. [DOI] [PubMed] [Google Scholar]
- 42.Hawley AE, Illum L, Davis SS. Preparation of biodegradable, surface engineered PLGA nanospheres with enhanced lymphatic drainage and lymph node uptake. Pharm Res. 1997;14(5):657–61. doi: 10.1023/a:1012117531448. [DOI] [PubMed] [Google Scholar]
- 43.Sahoo SK, Panyam J, Prabha S, Labhasetwar V. Residual polyvinyl alcohol associated with poly (D,L-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J Control Release. 2002;82(1):105–14. doi: 10.1016/s0168-3659(02)00127-x. [DOI] [PubMed] [Google Scholar]
- 44.Vila A, Gill H, McCallion O, Alonso MJ. Transport of PLA-PEG particles across the nasal mucosa: effect of particle size and PEG coating density. J Control Release. 2004;98(2):231–44. doi: 10.1016/j.jconrel.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 45.Bazile D, Prud’homme C, Bassoullet MT, Marlard M, Spenlehauer G, Veillard M, Stealth Me. PEG-PLA nanoparticles avoid uptake by the mononuclear phagocytes system. J Pharm Sci. 1995;84(4):493–8. doi: 10.1002/jps.2600840420. [DOI] [PubMed] [Google Scholar]
- 46.Kuhl TL, Leckband DE, Lasic DD, Israelachvili JN. Modulation of interaction forces between bilayers exposing short-chained ethylene oxide headgroups. Biophys J. 1994;66(5):1479–88. doi: 10.1016/S0006-3495(94)80938-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marques CM, Joanny JF. Block Copolymer Adsorption in a Nonselective Solvent. Macromolecules. 1989;22(3):1454–1458. [Google Scholar]
- 48.Longo G, Szleifer I. Ligand-receptor interactions in tethered polymer layers. Langmuir. 2005;21(24):11342–51. doi: 10.1021/la051685p. [DOI] [PubMed] [Google Scholar]
- 49.Chen CC, Dormidontova EE. Architectural and structural optimization of the protective polymer layer for enhanced targeting. Langmuir. 2005;21(12):5605–15. doi: 10.1021/la047109v. [DOI] [PubMed] [Google Scholar]
- 50.Stibenz D, Grafe M, Debus N, Hasbach M, Bahr I, Graf K, et al. Binding of human serum amyloid P componentto L-selectin. Eur J Immunol. 2006;36(2):446–56. doi: 10.1002/eji.200425360. [DOI] [PubMed] [Google Scholar]
- 51.Swers JS, Widom A, Phan U, Springer TA, Wittrup KD. A high affinity human antibody antagonist of P-selectin mediated rolling. Biochem Biophys Res Commun. 2006;350(3):508–13. doi: 10.1016/j.bbrc.2006.08.197. [DOI] [PubMed] [Google Scholar]
- 52.Whitmore RL. Rheology of the Circulation. Oxford: Pergamon Press; 1968. [Google Scholar]