

Abstract
Protease-activated receptor 1 (PAR1) is a G-protein coupled receptor that is expressed throughout the central nervous system. PAR1 activation by brain-derived as well as blood-derived proteases has been shown to have variable and complex effects in a variety of animal models of neuronal injury and inflammation. In this study, we have evaluated the effects of PAR1 on lesion volume in wild-type or PAR1−/− C57Bl/6 mice subjected to transient occlusion of the middle cerebral artery or injected with NMDA in the striatum. We found that removal of PAR1 reduced infarct volume following transient focal ischemia to 57% of control. Removal of PAR1 or application of a PAR1 antagonist also reduced the neuronal injury associated with intrastriatal injection of NMDA to 60% of control. To explore whether NMDA receptor potentiation by PAR1 activation contributes to the harmful effects of PAR1, we investigated the effect of NMDA receptor antagonists on the neuroprotective phenotype of PAR1−/− mice. We found that MK801 reduced penumbral but not core neuronal injury in mice subjected to transient middle cerebral artery occlusion or intrastriatal NMDA injection. Lesion volumes in both models were not significantly different between PAR1−/− mice treated with and without MK801. Use of the NMDA receptor antagonist and dissociative anesthetic ketamine also renders NMDA-induced lesion volumes identical in PAR1−/− mice and wild-type mice. These data suggest that the ability of PAR1 activation to potentiate NMDA receptor function may underlie its harmful actions during injury.
Keywords: N-methyl-D-aspartate, glutamate, thrombin, protease activated receptor, PAR, ischemia, stroke, neuronal death, neuroprotection
Introduction
Protease-activated receptors (PARs) are a family of four G-protein coupled receptors that are activated by proteolytic cleavage of the extracellular N-terminus by serine proteases such as thrombin. The new N-terminus revealed by this cleavage acts as a tethered ligand, activating a complex signaling cascade (Liu et al. 1991; Vu et al. 1991; Macfarlane et al. 2001; Trejo, 2003; Traynelis and Trejo, 2007). PAR1 couples to at least three different G-proteins, Gαi/o, Gα12/13, and Gαq/11, to initiate intracellular signaling (Smirnova et al. 2001; Macfarlane et al. 2001; Klarenbach et al. 2003; Junge et al. 2003, 2004;). Although first described for its role in the coagulation cascade, PAR1 is also expressed throughout the central nervous system. In situ hybridization studies in rats show that PAR1 is expressed in select neuronal populations, including motoneurons, dopaminergic neurons in the substantia nigra pars compacta (SNc), as well as a subset of cortical neurons (Weinstein et al. 1995; Niclou et al. 1998). Similarly, in human brain tissue, certain neuronal populations show PAR1 protein expression (Junge et al. 2004; Ishida et al. 2006). Glial expression of PAR1, particularly in astrocytes, is consistently strong in all regions (Weinstein et al. 1995; Wang et al. 2002; Junge et al. 2004; Hamill et al. 2005). Microglia also express functional PAR1 (Suo et al. 2002). PAR1 expression changes in response to injury in a manner dependent on cell-type and the nature of injury (Striggow et al., 2001; Riek-Burchardt et al., 2002; Rohatgi et al., 2004; Henrich-Noack et al., 2006).
The effects of PAR1 activation on neuronal health are complex (Gingrich and Traynelis, 2000; Vivien and Buisson, 2000; Matsuoka and Hamada, 2002; Xi et al., 2003; Ruf, 2003; Sheehan and Tsirka, 2005). Several in vitro models show thrombin activation of PAR1 to be neuroprotective. For example, thrombin administration protected cultured hippocampal neurons from glucose deprivation (Vaughan et al. 1995). Similarly, thrombin at concentrations lower than 50 nM (or other PAR1-specific agonists) protected organotypic hippocampal slices from oxygen-glucose deprivation (Striggow et al. 2000). Several in vivo studies also have found that administration of thrombin or PAR1 specific agonists several days prior to an insult, such as focal ischemia or 6-hydroxydopamine, is neuroprotective (Masada et al. 2000; Jiang et al. 2002; Xi et al. 2003; Cannon et al. 2006). Activated protein C protects brain endothelial cells from hypoxia and cultured hippocampal neurons from N-methyl-D-aspartate (NMDA)-induced toxicity in a PAR1-dependent manner (Shibata et al. 2001; Cheng et al. 2003; Mosnier and Griffin 2003; Ruf 2003; Guo et al. 2004). It has also been proposed that PAR1 activation by activated protein C administration is protective against neuronal damage in both a focal ischemia model and following NMDA microinjection into the parenchyma (Shibata et al. 2001; Cheng et al. 2003; Guo et al. 2004).
In contrast to these neuroprotective effects, several lines of evidence suggest that known PAR1 activators as well as PAR1 activation can be detrimental to neuronal health. A number of studies show that thrombin inhibitors can reduce damage in vitro and in vivo, with apparent contributions from both neuroprotective mechanisms and effects on cerebral blood flow (e.g. Friedmann et al., 2001; Jin et al., 2002; Karabiyikoglu et al., 2004; Cuomo et al., 2007). In addition, global inhibition of protease activity led to neurite extension in murine neuroblastoma cells (Monard et al. 1983; Snider and Richelson 1983; Snider et al. 1984), and thrombin was subsequently identified as the first chemorepellant to neurite outgrowth (Hawkins and Seeds 1986; Jalink and Moolenaar 1992; Suidan et al. 1992). Neurons that showed neurite retraction in response to thrombin subsequently died, whereas inhibition of thrombin by hirudin supported neurite outgrowth and neuronal survival (Jalink and Moolenaar 1992; Suidan et al. 1992; Turgeon et al. 1998). Although these studies did not implicate PAR1 in the actions of thrombin, subsequent studies that utilized a selective peptide mimic of PAR1’s new N-terminal following activation suggested that the changes in neuron shape and survival were dependent on PAR1 activation; neuronal death following PAR1 activation was at least in part apoptotic (Donovan et al. 1997; Turgeon et al. 1998). At high concentrations (>50 nM), thrombin is toxic to hippocampal cultures and organotypic slice cultures (Smith-Swintosky et al. 1995; Vaughan et al. 1995; Striggow et al. 2000; Xue et al., 2006; Fujimoto et al., 2008). Moreover, PAR1 activation appears to be proinflammatory (Suo et al. 2004). In vivo, thrombin pre-treatment exacerbates neuronal damage when administered close in time with an insult, such as transient ischemia (Henrich-Noack et al. 2006). Mice with PAR1 genetically removed (PAR1−/−) are protected from global ischemia/hypoxia as well as from 30 min intracerebral hemorrhage and transient focal ischemia (Junge et al. 2003; Olson et al. 2004; Xue et al. 2006). Similar levels of neuroprotection in both hypoxia/ischemia and transient focal ischemia can be achieved in wild-type animals by pretreatment with a selective PAR1 antagonist (Junge et al. 2003; Olson et al. 2004).
N-methyl-D-aspartate receptors mediate a slow Ca2+-permeable component of excitatory synaptic transmission, and are thought to be involved in learning and memory (Erreger et al., 2004). In addition, NMDA receptor overactivation is neurotoxic when studied in vitro (Koh and Choi 1987; Choi et al. 1988; Lei et al. 1992; Arundine and Tymianski 2003) as well as in vivo (Meldrum et al. 1987; Park et al. 1988; Swan and Meldrum 1990; Duncan et al. 1991; Uematsu et al. 1991; Miyabe et al. 1997; Dogan et al. 1999; Wang and Shuaib 2005). This toxicity is widely considered a leading cause of early neuronal death in situations in which extracellular glutamate is elevated, such as ischemia (Lipton 1993; Whetsell 1996; Dirnagl et al. 1999). PAR1 activation by a variety of agonists has been shown to potentiate neuronal responses to NMDA (Gingrich et al. 2000; Lee et al. 2007; Mannaioni et al. 2008) as well as increase spontaneous NMDA receptor mediated currents (Shigetomi et al. 2008). Given the prominent role of NMDA receptors in mediating excitotoxic injury during ischemia, potentiation of NMDA receptor responses by PAR1 activation, if it occurred during ischemia, should exacerbate neuronal damage.
One explanation for the apparent dual nature of PAR1 in neuronal injury is that PAR1 activation in vivo engages multiple signaling mechanisms in multiple cell types. We hypothesized that some of the harmful effects of PAR1 activation are due to its ability to potentiate NMDA receptor function in the ischemic penumbra (Gingrich et al. 2000; Henrich-Noack et al. 2006). If the harmful effects of PAR1 are primarily mediated through enhancement of NMDA receptor function, one would predict that the residual damage in the presence of NMDA receptor blockade will be insensitive to PAR1 antagonism or removal of the PAR1 gene. To test this idea we examined the lesion volume in PAR1−/− and wild-type mice both in the presence and absence of NMDA receptor blockers (MK801 or ketamine) in models of ischemia in which the NMDA receptor contribution to cell death is important. Our data are consistent with the idea that PAR1 activation exacerbates neuronal damage in the absence of NMDA receptor blockers. However, blockade of NMDA receptors removes the harmful effects exerted by PAR1. Previous reports that PAR1 activation potentiates NMDA receptor function (e.g. Gingrich et al., 2000; Mannaioni et al., 2008) and enhances glutamate release (Lee et al. 2007; Ramos-Mandujano et al. 2007) are consistent with an extensive literature showing NMDA receptor activation is harmful in animal models of ischemia (Koh and Choi 1987; Meldrum et al. 1987; Choi et al. 1988; Swan and Meldrum 1990; Duncan et al. 1991; Lei et al. 1992; Lees 1997; Miyabe et al. 1997; Dirnagl et al. 1999; Uematsu et al. 1991).
Methods
Ca2+ imaging in striatal cultures
All procedures involving animals were performed in accordance with international, national, and local standards on animal welfare, and were reviewed and approved by the Emory University Institutional Animal Care and Use Committee. Rat striatal neuronal cultures were obtained from E17–E18 pups (Charles River Laboratories, Inc, Wilmington, MA). Briefly, pregnant rats were sacrificed by CO2 asphyxiation followed by cervical dislocation. The brains were removed and placed in ice cold HEPES buffered saline. The striatum was dissected, and placed in fetal bovine serum and triturated through a fire polished pipette. Cultures were plated onto poly-D-lysine (10 μg/ml) coated glass coverslips in MEM media (Invitrogen, Carlsbad, CA) supplemented with B27 defined nutrients (Invitrogen). Cultures were maintained at 37°C in a humidified 5% CO2 incubator and the media replaced every 3 days. After 4–9 days, cells were placed (in mM) in 150 NaCl, 3 KCl, 10 HEPES, 2 CaCl2, 20 mannitol, 10 glucose, pH 7.3. Cells were subsequently incubated for 30–45 minutes in this solution supplemented with 0.1% pluronic acid, 0.5 % DMSO and 3 μM Fluo-3 acetoxymethyl ester (Molecular Probes, Eugene, OR), and transferred to the microscope stage for imaging. Images were recorded in response to 410 nm excitation and band-passed 500–550 nm emission at variable frequency (0.033 Hz for baseline and 0.5 Hz for drug application). After recording images for a three minute baseline period, cells were exposed to 30 μM of the PAR1 activating peptide TFLLR-NH2 (TFLLR, Emory Microchemical Facility) for three minutes in the presence of 1 mM Mg2+, 0.5 μM tetrodotoxin (TTX), and 50 μM D-2-amino-5-phosphonovalerate (D-APV), and the fluorescent responses were recorded. Following a three-minute wash, cultures were then exposed to 10 μM NMDA in order to identify neurons.
Intrastriatal microinjection of NMDA or TFLLR
Male C57Bl/6 wild-type or PAR1−/− mice (see below; 3–5 month) were anesthetized with chloral hydrate (400 mg/kg, Sigma, St. Louis, MO), isoflurane (5% in 100% oxygen for induction, 1–2% in 100% oxygen for maintenance) or ketamine (100 mg/kg, Allan Labs, Chicago, IL) and xylazine (10 mg/kg, Henry Schein Inc. Melville, NY). A subset of animals received 3 mg/kg of (−)MK801 (Tocris, Ellisville, MO). (−)MK801 is the less potent stereoisomer (Wong et al. 1986; Kant et al. 1991; Galbicka et al. 1994; Dravid et al. 2007) and is better tolerated by C57Bl/6 mice than (+)MK801. We have found that 1–3 mg/kg of either MK801 stereoisomer is equally effective at reducing infarct volume in C57Bl/6 mice following transient focal ischemia, with (−)MK801 showing a reduction in infarct volume to 55+10% of control (n=31) and (+)MK801 reducing infarct volume to 57+8% (n=26) of control. However administration of (+)MK801 causes higher mortality (41%) than (−)MK801 (10%). We subsequently utilized a well-tolerated dose (3 mg/kg) of the NMDA receptor antagonist (−)MK801 for experiments in PAR1−/− and littermate controls.
Following induction of anesthesia, animals were place on a homeothermic blanket and body temperature maintained between 36.5–37°C. The heads were immobilized in a small animal stereotaxic device (Model SAS 75, Cartesian Research Inc., Sandy, OR). A scalpel was used to expose the skull, a hole was drilled with a 0.0087 inch drill bit, and a 33 gauge needle was lowered into the striatum (rostral 1.000, lateral 2.150, ventral 3.450). The needle was left in place for two minutes prior to drug injection, then 1 μl of 20 mM NMDA (20 nmol; Sigma) in phosphate-buffered saline was injected at a rate of 0.1 μl per minute for 10 minutes or 0.5 μl of 20 mM TFLLR in phosphate-buffered saline with 0.1% bovine serum albumin was injected at a rate of 0.1 μl per minute for 5 minutes. The needle was left in place for five more minutes then slowly withdrawn and the wound closed with staples. For NMDA injections, the animals were allowed to recover for 24 hours, and then overdosed with sodium pentobarbital (390 mg/ml) and transcardially perfused with ice cold 4% paraformaldehyde in phosphate buffered saline (PBS) for 10 min. The brain was removed and placed in 4% paraformaldehyde for 24 hours and cryoprotected in 20% sucrose in PBS at 4°C for 24 hours. The brains were then frozen and 20 μm-thick coronal cryostat sections cut (Leica Model CM3050). Every tenth section was stained with cresyl violet, the lesion was identified by loss of staining, and the area was measured using Open Lab 3.08 (Improvision, Lexington, MA) and integrated to obtain the total lesion volume (Ayata et al. 1997; Guo et al. 2004). For TFLLR injections, mice were allowed to survive for five days and sacrificed in a similar manner. The sections from TFLLR-injected mice were blocked in 10% normal goat serum, and incubated overnight at 4°C in rabbit anti-GFAP antibody (Sigma, 1:5000). Sections were removed from primary antibody, washed in PBS, and incubated in secondary antibody for 1 hour at room temperature. Staining was visualized using a FITC-conjugated secondary antibody (goat-anti rabbit, Jackson ImmunoResearch, West Grove, PA, 1:200). All analysis was performed by a person blinded to genotype and condition (with or without MK801 or ketamine).
Intracerebroventricular injection of BMS200261
C57Bl/6 wild-type mice (3–5 month; Jackson Laboratory, Bar Harbor, ME) were anesthetized with isofluorane (5% in 100% oxygen for induction, 1–2% in 100% oxygen for maintenance). Animals were placed on a homeothermic blanket and body temperature was maintained between 36.5 and 37°C. The head was immobilized in a small animal stereotaxic device. A scalpel was used to expose the skull, a hole was drilled with a 0.04 inch carbide bit, and a 32-gauge needle was lowered into the brain (caudal, 2.000; lateral, 1.000; ventral, 3.000). One microliter of either the selective PAR1 antagonist BMS200261 (6 mM, Emory Microchemical Facility) or vehicle (10 mM HEPES-buffered saline) was delivered over 3 minutes. The needle was then slowly withdrawn, and the wound closed with staples.
Transient focal cerebral ischemia
Transient focal ischemia was performed and analyzed as previously described by persons blinded to the experimental conditions (Junge et al. 2003). Immediately prior to surgery, 3–5 month male wild-type or PAR1−/− C57Bl/6 mice (see below) were injected intraperitoneally (IP) with either 0.9% NaCl or 3 mg/kg of (−)MK801 in saline and subsequently anesthetized with isoflurane (5% in 100% oxygen for induction, 1–2% in 100% oxygen for maintenance). Mice were placed on a homeothermic blanket and body temperature was maintained between 36.5–37°C. Relative changes in regional cerebral blood flow were monitored with a laser Doppler flow meter (Perimed Periflux System 5000, Jarfalla, Sweden). The probe was placed directly on the skull in the area of the middle cerebral artery (1 mm posterior and 5 mm lateral from bregma). The tip of an 11 mm 5-0 dermalon nylon suture was smoothed by heat, and the suture introduced into the left internal carotid artery through the external carotid artery stump until blood flow dropped. After 30 min of middle cerebral artery occlusion, blood flow was restored by withdrawing the suture. The incisions were surgically closed, and the mice immediately placed in warmed cages post-surgery. All mice had at least an 80% drop in blood flow during middle cerebral artery occlusion, as measured by laser Doppler flow. Mice that showed either no reperfusion following suture removal or a subdural hematoma were not studied further. After 24 hours survival, the brain was removed and cut into 2 mm sections. The lesion was identified by incubating the tissue in 2% 2,3,5-triphenyltetrazolium chloride (TTC) in PBS at 37°C for 20 min. The infarct area of each section was manually measured using NIH IMAGE (Scion Corporation, Beta 4.0.2 release), multiplied by the section thickness, and summed across sections to give the infarct volume. A ratio of the contralateral to ipsilateral hemisphere section volume was multiplied by the corresponding infarct section volume to correct for edema.
PI Hydrolysis
Young mice (C57Bl/6, age P14–17) were deeply anesthetized with isoflurane and decapitated. The brain was rapidly removed and submerged in an ice-cold oxygenated artificial cerebrospinal fluid (ACSF) comprised of (in mM) 130 NaCl, 24 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 1 CaCl2, 3 MgCl2 and 10 glucose saturated with 95% O2/5% CO2, at pH 7.4. The hemisected brain was glued onto the stage of a vibrating microtome (Leica VT1000 S) and sections of 300 μm thickness were cut and stored in an incubation chamber at room temperature for about 1 hour before use. Slices containing the striatum were removed, the striatum dissected, and the tissue labeled for 30 minutes with 3H-inositol (1 μCi/well) at 35°C. Slices were subsequently incubated for 15 minutes in LiCl, and the accumulation of radioactive inositol phosphates was measured after a 20 minute incubation with thrombin (30 nM; 3U/ml, Gingrich et al. 2000) or the group I mGluR agonist dihydroxyphenylglycine (30 μM). Tissue samples were extracted with chloroform/methanol and the 3H-inositol monophosphate fraction was separated by anion exchange chromatography (Dowex 1 X 4 400) using increasing amounts of ammonium formate. 3H-inositol monophosphate content was assessed by liquid scintillation spectrometry. For each experiment, measurements were from three different reaction tubes (in triplicate), each of which had tissue samples from three different slices from an animal. Three separate experiments were performed using different animals.
Animals
Male PAR1+/− mice (Connolly et al. 1996) were provided by Dr. Shaun Coughlin (UCSF), from which a colony of PAR1−/− mice that were >99% C57Bl/6 (backcrossed >7 times) were derived. Wild-type male C57BL/6 mice were obtained either from Jackson Laboratory (Bar Harbor, ME; BMS200261 experiments) or from a C57Bl/6 colony that was derived from littermates of PAR1−/+ animals used to establish the PAR1−/− homozygous colony. All experiments involving PAR1 −/− animals were compared to mice from a littermate derived wild-type colony.
Statistics
All measurements are given as mean ± standard error of the mean (SEM) to 2 significant digits. Measured values were compared using t-test or ANOVA, as appropriate. Results were considered significant if p < 0.05.
Results
PAR1 is functionally expressed in the striatum of wild-type mice
Much of the damage induced by the injury models that we used in this study (middle cerebral artery occlusion, intrastriatal NMDA injection) is found in the striatum. Therefore, before examining the potential inter-dependence of PAR1 and NMDA receptors in these injury models, it was important to confirm that both receptors are expressed in striatal tissue. Although striatal neurons are well-known to express several subtypes of the NMDA receptors (Buller et al. 1994; Qin et al. 1996; Calabresi et al. 1998; Standaert et al. 1999; Kuppenbender et al. 2000; Cepeda et al. 2001; Dunah and Standaert, 2003), no data exists on PAR1 signalling in mouse striatum. We therefore initially examined whether PAR1 was also functionally expressed in the striatum of wild-type C57Bl/6 mice. PAR1 receptors are coupled to Gαq/11 and mediate robust increases in intracellular calcium. We evaluated the fluorescent response of mixed striatal cultures containing neurons and glia that had been loaded with the Ca2+ sensitive dye Fluo-3. We found that exposing cultures to a maximally effective concentrations of the selective PAR1 activating peptide TFLLR (30 μM) increased Fluo-3 fluorescence in both neurons and glia, which we interpret as an increase in intracellular Ca2+ (Fig. 1A, n=63 cells in three separate experiments). Several studies show that TFLLR induces Ca2+ signaling and ERK phosphorylation in astrocytes from wild type mice, however TFLLR has no effect on Ca2+ signaling and ERK phosphorylation in astrocytes prepared from PAR1−/− mice (Lee et al., 2007; Mannaioni et al., 2008), consistent with previous suggestions that TFLLR is a selective PAR1 antagonist (Hollenberg et al., 1997). Experiments were performed in the presence of TTX and the NMDA receptor antagonist D-APV to eliminate signals from synaptic transmission or NMDA receptor activation secondary to glutamate release (Lee et al., 2007). Approximately 15 percent of the cells responded to both NMDA (10 μM) and TFLLR, suggesting that PAR1 is functionally coupled to Ca2+ signaling in a subset of neurons. Astrocytes, which show strong PAR1 responses in cultures from other brain regions, likely comprise the remaining 85% of TFLLR-responsive cells in our preparation (Sorenson et al., 2003; Nicole et al., 2005; Lee et al., 2007).
Figure 1. PAR1 is functionally expressed in the striatum.
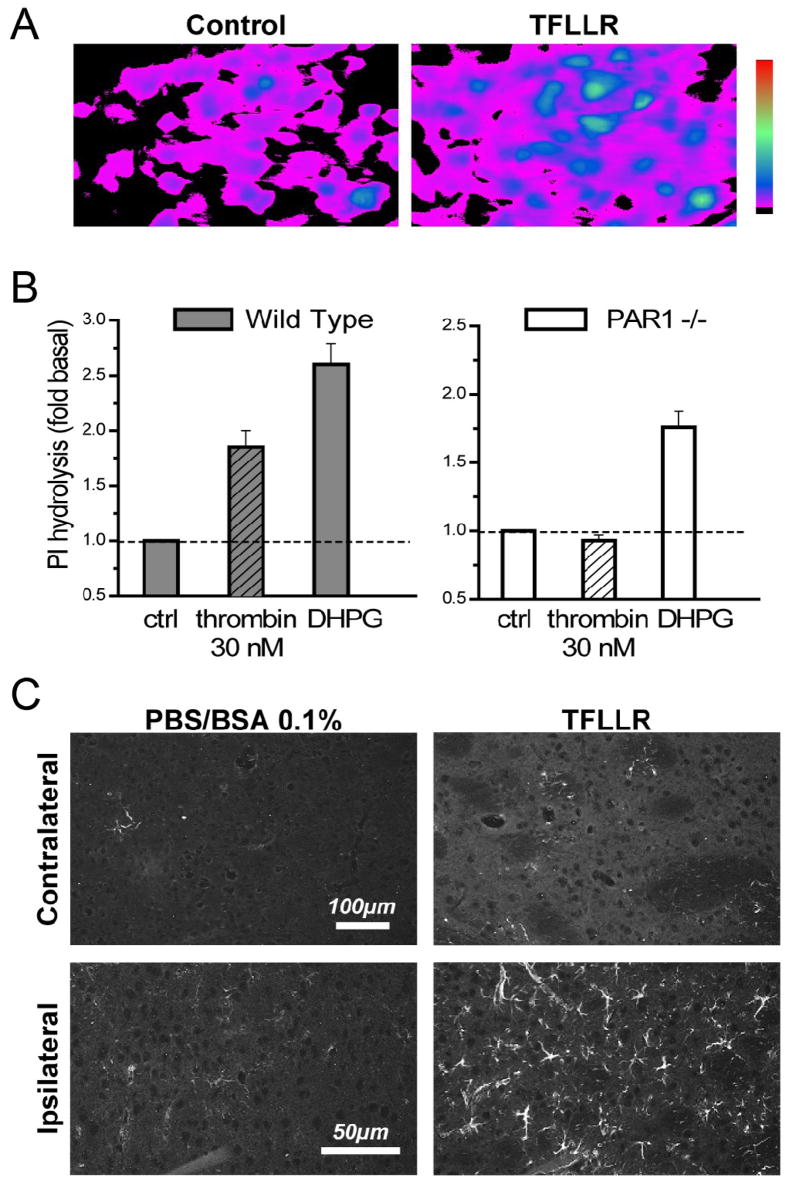
A) Striatal cultures containing both neurons and glia were loaded with the Ca2+ -sensitive fluorescent dye Fluo-3 and subsequently exposed to 30 μM TFLLR, which induced an increase in fluorescence, which we interpret to reflect an increase in intracellular calcium. Both neurons and glia responded to TFLLR. B) Striatal slices were metabolically labeled with 3H-myo-inositol, stimulated with 30 nM thrombin for 20 minutes, and accumulation of 3H-inositol phosphates was measured. The group I metabotropic glutamate receptor agonist dihydroxyphenylglycine (DHPG, 30 μM) was used as a positive control, and evoked robust response in both experiments, demonstrating that the slices were viable and signaling through the phosphoinositide pathway was functional. All measurements were performed in triplicate; data is the mean of 3 experiments; error bars are SEM. C) GFAP immunohistochemistry was performed on 20 μm mouse brain sections 5 days after an intrastriatal injection of TFLLR (right panels) or vehicle (PBS–BSA 0.1%; left panels) in a volume of 0.5 μl over 5 min using a Hamilton syringe (n=2 animals for PBS control, 4 animals for TFLLR). TFLLR injection caused increased GFAP reactivity in the ipsilateral striatum of all wild-type mice compared to the contralateral striatum or mice injected with vehicle.
Activation of PAR1 has previously been shown to stimulate inositol phosphate signaling in hippocampal cultures and slices (Macfarlane et al. 2001; Sorensen et al. 2003; Junge et al., 2003). Exposing intact striatal slices to 30 nM thrombin increased the accumulation of tritiated inositol phosphates in wild-type mice (n=3 experiments) but not in PAR1−/− mice (n=3 experiments), suggesting that functional PAR1 is expressed in intact striatum (Fig. 1B). The group I mGluR1 agonist DHPG was used as a positive control to show that cells in slices from PAR1−/− animals were capable of mediating signaling through the Gq/11 pathway. Previous work has shown that PAR1 activation will cause proliferation of cortical astrocytes in vitro (Wang et al. 2002; Sorensen et al. 2003) and increased expression of GFAP in vivo in wild-type but not PAR1−/− mice (e.g. Nicole et al. 2005). Therefore, we tested whether injection of the selective PAR1 activating peptide TFLLR caused astrogliosis in the striatum, as has been previously shown in the cortex (Nicole et al. 2005). Figure 1C shows that TFLLR injection causes strong astrogliosis in wild-type mice in the ipsilateral striatum (n=4). In contrast, no gliosis was observed on the contralateral side (n=4) or in the striata of mice injected with vehicle (n=2; Fig. 1C). Together these data suggest that PAR1 is functionally expressed in the striatum of C57Bl/6 mice.
Neuroprotective phenotype of PAR1−/− mice requires NMDA receptor activation in transient focal ischemia
Our working hypothesis is that the difference in the lesion volume previously observed between wild-type mice and PAR1−/− mice (Junge et al., 2003; Olson et al., 2004) was due in part to PAR1-mediated potentiation of NMDA-receptor signaling (Gingrich et al. 2000; Lee et al., 2007; Mannaioni et al. 2008), which exacerbates NMDA receptor-mediated cell death in the penumbra. PAR1 potentiation of NMDA function has been observed using several enzymatic PAR1 activators (e.g. thrombin, plasmin) and selective PAR1 peptide agonists (Gingrich et al. 2000; Lee et al. 2007; Mannaioni et al. 2008). Furthermore, NMDA receptor potentiation was reduced in PAR1−/− mice, and could be blocked using the PAR1 antagonist BMS200261 or direct inhibitors of the activating enzymes (Gingrich et al. 2000; Lee et al. 2007; Mannaioni et al. 2008). Our working hypothesis predicts that when NMDA receptor overactivation is blocked, there should be little difference in ischemia/hypoxia-induced lesion volume between PAR1−/− and wild-type mice.
In order to test this hypothesis, we evaluated the effects of an NMDA receptor antagonist in vivo in a model of transient focal ischemia in wild-type and PAR1−/− mice. Junge et al. (2003) observed that PAR1−/− mice were protected against ischemic damage following 30 min transient focal ischemia. In our hands, this duration of ischemia produces lesion volumes of less than 1/4 hemispheric volume, which approximate relative infarct volumes in human stroke. Prolonged periods of focal ischemia in C57Bl/6 mice (e.g. 1 hour) produce large lesion volumes of over 50% of hemispheric volume and engage additional molecular mechanisms that occlude the effects of PAR1 removal (Junge et al., 2003). The neuroprotective effect of PAR1 deletion following 30 min ischemia did not involve changes in blood flow, blood gases, blood-brain-barrier breakdown, or cerebral blood vessel anatomy between wild-type or PAR1−/− mice (Junge et al. 2003), which we interpreted as consistent with parenchymal effects of PAR1. If PAR1 activation during 30 min ischemia exacerbated NMDA receptor-mediated cell death, then we predict that administration of an NMDA antagonist would eliminate the harmful effects of PAR1 activation in wild-type mice, rendering lesion volumes in wild-type and PAR1−/− mice similar.
We evaluated the effect of the NMDA receptor antagonist MK801 (3 mg/kg, see Methods) on the neuroprotective phenotype observed in 30 min transient focal ischemia (Junge et al., 2003). MK801 is an uncompetitive open channel NMDA receptor blocker that has been shown to be neuroprotective in animal models of neuronal damage (Park et al. 1988; McDonald et al. 1989; Swan and Meldrum 1990; Warner et al. 1991; Roussel et al. 1992). In the absence of NMDA receptor blockade, PAR1−/− mice had significantly smaller lesions than their wild-type counterparts (Fig 2A, B; wild-type mice 57±9.4 mm3, PAR1−/− mice 32±8.4 mm3; p<0.05; ANOVA; n=11–12), consistent with previously published results (Junge et al., 2003). As expected, NMDA receptor blockade by MK801 administration produced significant neuroprotection in wild-type mice (Meldrum et al. 1987; Park et al. 1988; Swan and Meldrum 1990; Uematsu et al. 1991; Miyabe et al. 1997). However, lesion volumes in PAR1−/− knockout mice were not further reduced by injection with MK801 (Fig 2B). Wild-type mice treated with MK801 had a mean lesion volume of 27±5.8 mm3, which was virtually identical to the lesion volume in PAR1−/− mice treated with MK801 (27±7.8 mm3; p>0.05; ANOVA; n=10–12). These data support our working hypothesis that the harmful effects of PAR1 during transient focal ischemia require functional NMDA receptors. These findings are consistent with the idea that PAR1 potentiation of NMDA receptor function exacerbates neuronal damage. However, the alternative hypothesis that a ceiling effect for neuroprotection obscured potential additive effects of PAR1−/− mice and MK801 treatment cannot be ruled out.
Figure 2. PAR1−/− mouse neuroprotection is dependent on NMDA receptor activation in transient focal ischemia.
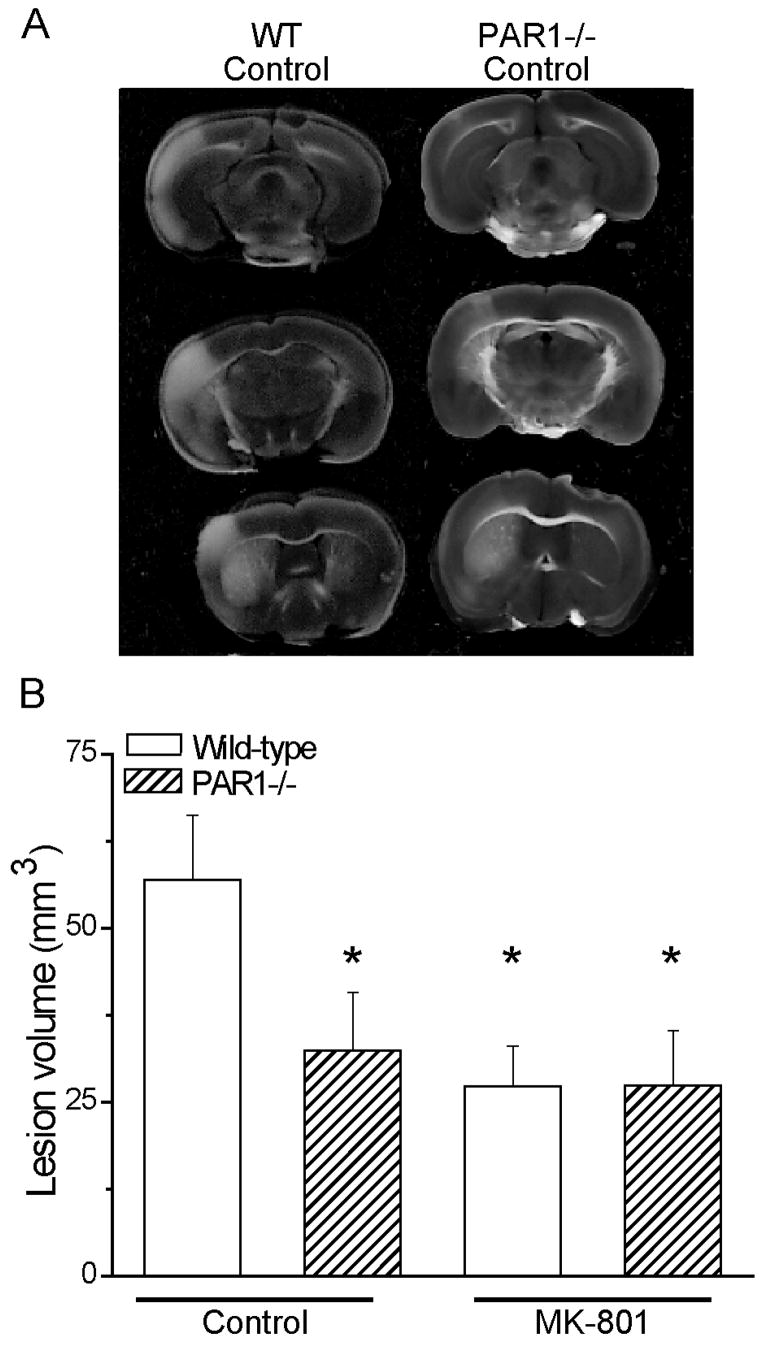
A) Representative TTC-stained brain sections from wild-type and PAR1−/− mice at 24 hours recovery following 30 min occlusion of the middle cerebral artery under isoflurane anesthesia. B) Quantification of lesion volumes is shown in both genotypes in the presence (first two bars) and absence (last two bars) of 3 mg/kg MK801. PAR1−/− mice are significantly protected from ischemia-induced damage in the absence of the NMDA receptor blocker. There is no difference between lesion volume of PAR1−/− mice and wild-type mice in the presence of MK801; * p<0.05 indicates measurements different than wild-type control (ANOVA, n=10–12 per condition). Error bars indicate SEM.
Neuroprotection in PAR1−/− mice requires NMDA receptor activation in a model of striatal toxicity
Because glutamate accumulation during brain ischemia can mediate neuronal damage through overactivation of NMDA receptors, a useful model for NMDA receptor-mediated damage has been intraparenchymal injection of the agonist NMDA, which selectively activates NMDA receptors. We applied the same experimental design described above to this model of intrastriatal NMDA injection in vivo to further examine whether NMDA receptor activity was involved in the actions of PAR1 (Portera-Cailliau et al. 1995; Ayata et al. 1997; Guo et al. 2004; Leavitt et al. 2006). PAR1−/− and wild-type mice either with or without 3 mg/kg MK801 pretreatment received intrastriatal injections of 20 nmol of NMDA or vehicle under isoflurane anesthesia. After 24 hours survival, mice were sacrificed, 20 μm sections were cut spanning the striatum, sections were stained with cresyl violet (Fig. 3A), and lesion volumes were measured. Injection of saline into the striatum did not produce detectable lesions in any condition (data not shown).
Figure 3. The neuroprotective phenotype of PAR1 −/− mice is dependent on NMDA receptor activation.
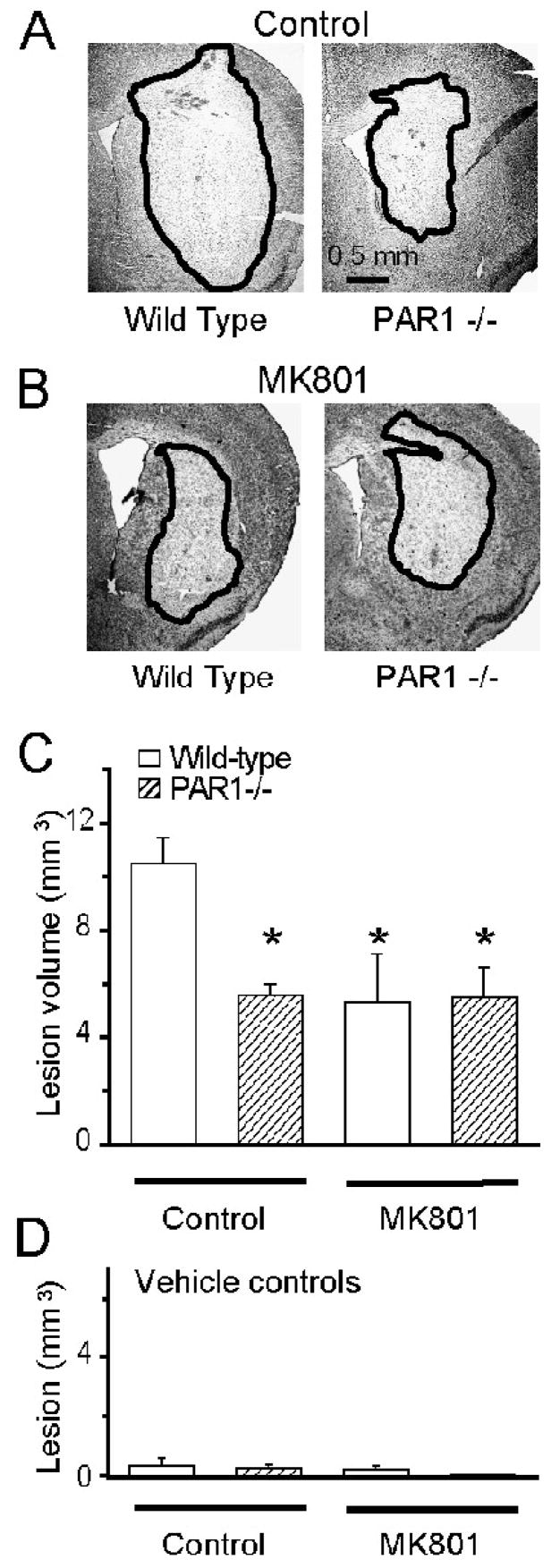
A,B) Cresyl violet staining is shown for striatal lesions 24 hours after intrastriatal NMDA injection (1 μl of 20 mM NMDA administered over 10 minutes) in wild-type and PAR1−/− mice under isoflurane anesthesia in the presence or absence of MK801. The lesion is indicated by pallor in staining and surrounded by a black line. C) Mean lesion volumes are shown from NMDA-injected animals. Error bars indicate SEM; * p<0.05 indicates measurements different than wild-type control (ANOVA, n=3–12 per condition). There was no significant difference between PAR1−/− animals treated with MK801, wild-type animals treated with MK801, or vehicle treated PAR1−/− animals. D) Intrastriatal injection of PBS did not produce measurable lesions (n=2–4 per condition).
Removal of PAR1 or administration of MK801 significantly reduced lesion volume in wild-type mice receiving intrastriatal NMDA injection (Fig 3AB). Wild-type mice without MK801 treatment had a lesion volume of 11±1.0 mm3 (n=12; Fig 3C), which was significantly higher than that of PAR1−/− mice (5.6+0.4 mm3; p<0.05; ANOVA; n=6) or that of wild-type mice receiving 3 mg/kg MK801 (5.3±1.8 mm3; p<0.05; ANOVA; n=3). Similar to results obtained with transient focal ischemia, PAR1−/− mice did not show any further protection following MK801 injection, and had a lesion of 5.5±1.1 mm3 (n=4), similar to the lesion volume for PAR1−/− mice without MK801 injection. There was no significant difference between lesion volumes in PAR1−/− mice, MK801-injected wild-type mice, and MK801-injected PAR1−/− mice (p>0.05; ANOVA; n=3–6; Fig 3C). These data are consistent with our findings in transient focal ischemia, and suggest that the harmful actions of PAR1 require functional NMDA receptors.
An important caveat to the use of knockout animals is the potential for compensatory mechanisms to alter gene regulation and animal response to various perturbations. One important control for this is to test whether a selective antagonist can produce the same effects as a receptor knockout. We have previously confirmed that pharmacological blockade of PAR1 is neuroprotective in transient focal ischemia (Junge et al., 2003). In order to insure that the results found in PAR1−/− mice receiving intrastriatal NMDA injection were not due to an epigenetic phenomena, we treated wild-type C57Bl/6 mice (Jackson Labs) with the selective PAR1 antagonist BMS200261, (Bernatowicz et al. 1996; Kawabata et al. 1999; Mannaioni et al. 2008) injected intracerebroventricularly (ICV) 30 minutes prior to the intrastriatal NMDA injection. The mice pretreated with the PAR1 antagonist had significantly smaller lesions than those pretreated with vehicle control (Fig 4). In these studies, mice receiving vehicle ICV injections showed a mean NMDA-induced lesion volume of 8.8±1.1 mm3 (n=6), compared to those receiving antagonist, which had a lesion volume of 4.8±0.3 mm3 (Fig. 4B; ANOVA; p<0.01; n=6). Injection of PBS into striatum alone produced no detectable lesion (data not shown). This result strengthens our interpretation that effects seen in PAR1−/− mice reflect the absence of the PAR1 receptor. The results further suggest that PAR1 activation, rather than just expression alone, is necessary to exacerbate NMDA-induced intrastriatal damage.
Figure 4. Wild-type mice treated with PAR1 antagonist are protected from NMDA-induced damage.
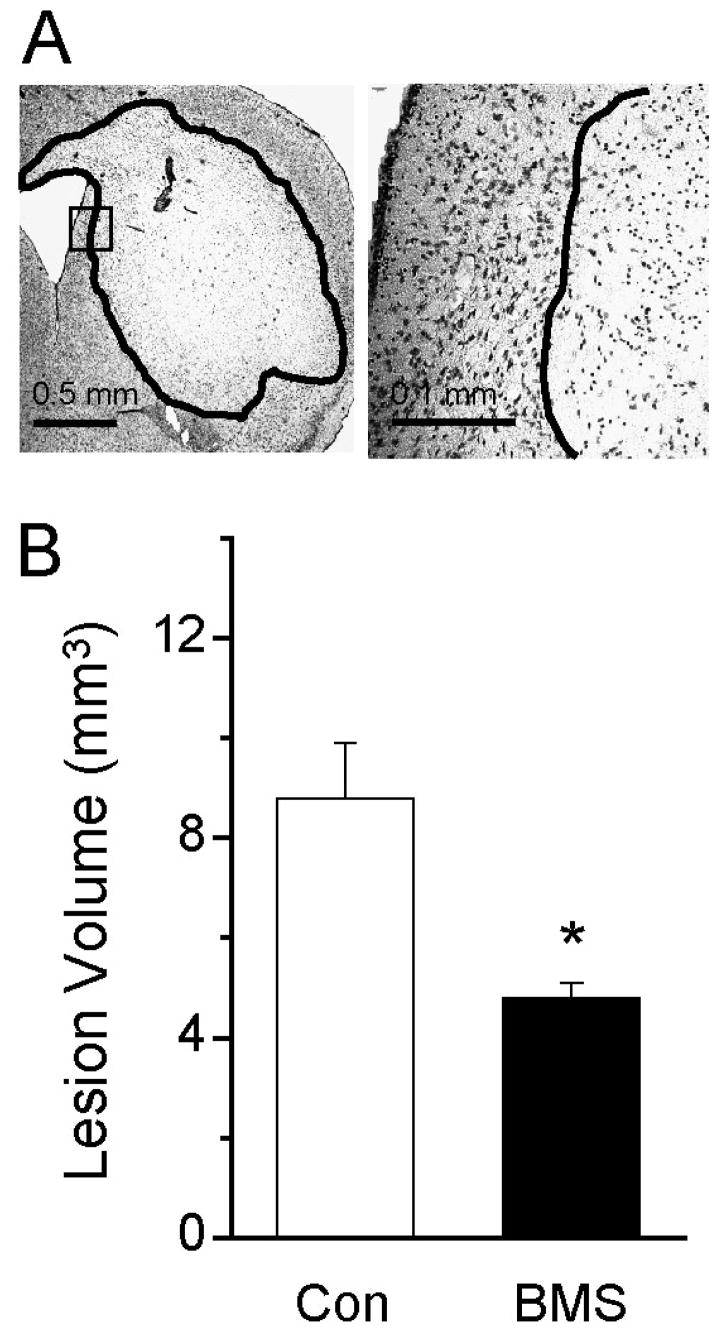
A) Cresyl violet staining is shown for wild-type mouse striatal sections 24 hours after NMDA injection. B) Mean lesion volume induced by intrastriatal NMDA injection into wild-type mice pretreated with either vehicle or PAR1 antagonist delivered ICV (1 μl of PBS or 6 mM antagonist in PBS given over 3 minutes); * p<0.01 (ANOVA n=6 animals per condition). Lesion volumes from mice injected with phosphate-buffered saline (PBS) were undetectable (not shown). Error bars indicate SEM.
The NMDA antagonist ketamine eliminates neuroprotection in PAR1−/− mice in a striatal model of toxicity
The dissociative anesthetic ketamine is a well known use-dependent uncompetitive NMDA receptor antagonist at all NMDA receptor subtypes (Harrison and Simmonds 1985; Thomson et al. 1985; Beal et al. 1988; Brady and Swann 1986; Dravid et al. 2007). Moreover, the anesthetic effect of ketamine is related to its ability to block NMDA receptor function, suggesting that NMDA receptor block will persist as long as animals show evidence of anesthesia. To assess whether ketamine can also reduce lesion volume in wild-type animals, we made intrastriatal injections of NMDA or vehicle in wild-type and PAR1−/− mice anesthetized with ketamine/xylazine. We found that the lesion volume in wild-type mice receiving ketamine (5.9±1.2 mm3; n=9) was significantly reduced compared to wild-type mice anesthetized with isoflurane (11±1.0 mm3; p<0.05; ANOVA; n=12). The lesion volume we found in ketamine-anesthetized wild-type mice (5.9 mm3; Fig 5) was identical to previously reported values (6 mm3; Guo et al. 2004), and similar to the volume observed in mice treated with the well-known NMDA receptor antagonist MK801 (see Fig 3). Consistent with data described above for MK801, ketamine had no effect on lesion volume in PAR1−/− mice (7.3±0.6 mm3; n=5; Fig 5), which in our hands was indistinguishable to previously reported findings (7 mm3; Guo et al. 2004) and not significantly different from wild-type mice receiving ketamine (Fig 5; p>0.05; ANOVA). These data are consistent with our working hypothesis that the harmful effects of PAR1 activation require functional NMDA receptors.
Figure 5. The NMDA antagonist ketamine eliminates the neuroprotective phenotype of PAR1 −/− mice.

A) Typical lesion that develops 24 hours after intrastriatal NMDA injection (see Methods) in mice anesthetized with ketamine/xylazine is shown. The lesion, indicated by pallor in staining, is surrounded by a black line showing how the volume was measured. B) Mean lesion volumes 24 hours following intrastriatal NMDA or PBS injection are shown; error bars indicate SEM. ns indicates that there is no significant difference between wild-type and PAR1−/− mice treated with ketamine/xylazine (p>0.05; ANOVA n=3–12).
Isoflurane, like most anesthetics, also shows a modest level of NMDA receptor blockade itself and some level of neuroprotection (Criswell et al. 2004; Kawaguchi et al. 2005; Ogata et al. 2006). The IC50 value for isoflurane inhibition of neuronal NMDA receptor function was 1 mM, or about 3 times the minimum alveolar concentration (MAC) of 0.32 mM (Yang and Zorumski, 1991; Yamakura et al., 2005; Ogata et al. 2006). Ketamine inhibits recombinant NMDA receptor function with an IC50 in the 1–3 μM range (Dravid et al. 2007), which is roughly equal to 1 MAC. These IC50 and MAC values suggest that anesthetic concentrations of isoflurane will have significantly less affect on NMDA signaling than those of ketamine. Consistent with this idea, we found no significant difference on lesion volume between mice treated with isoflurane and those treated with chloral hydrate, one of the few anesthetics not thought to be neuroprotective. Wild-type mice treated with isoflurane had an NMDA-induced lesion volume of 11+1.4 mm3 (n=8), which was identical to the NMDA-induced lesion volume of wild-type mice treated with chloral hydrate 11+0.7 mm3 (p=0.85; n=3; unpaired t-test). We used isoflurane for these studies because chloral hydrate may not fully prevent pain transmission (Ozden and Isenmann 2004; Silverman and Muir 1993).
Discussion
Our working hypothesis at the outset of these studies was that PAR1 activation, through its complex and multi-faceted signaling properties (Coughlin 1999; Macfarlane et al. 2001; Trejo 2003; Traynelis and Trejo, 2007), has both positive and negative effects on neuronal survival in various models of ischemia and excitotoxicity (Shibata et al., 2001; Ruf 2003; Cheng et al., 2003; Guo et al. 2004; Junge et al. 2003; Suo et al. 2004). Here we have shown that the pro-neurodegenerative effects of PAR1 activation are dependent on the activation of the NMDA receptor in two separate in vivo models of neuronal injury. The results of these studies clarify the relationship between PAR1 and the NMDA receptors during ischemia, and suggest that the ability of PAR1 to potentiate NMDA receptor function may be one important factor exacerbating neuronal death during ischemia.
PAR1 signaling and NMDA receptor function
Gingrich et al. (2000) were the first to predict that extravasation of blood-derived serine proteases during ischemia could trigger PAR1-mediated NMDA receptor potentiation, which could exacerbate neuronal damage (see also Gingrich and Traynelis 2000; Mannaioni et al., 2008). The data described here are consistent with this prediction, and suggest that NMDA receptor potentiation by PAR1 has detrimental effects during ischemia in vivo. In particular, the lack of further neuroprotection by the NMDA receptor antagonist MK801 in PAR1−/− mice subjected to transient focal ischemia or intrastriatal NMDA injection suggests that PAR1 activation facilitates penumbral damage in a manner that involves NMDA receptor activation. Although the mechanism by which PAR1 activation potentiates NMDA receptor function requires additional study, the voltage- and Mg2+-dependence described in the initial report (Gingrich et al., 2000) suggest that PAR1 may trigger a depolarization-induced relief of Mg2+ blockade. This idea is supported by a recent study showing that PAR1 stimulates release of glutamate which can subsequently potentiate postsynaptic NMDA receptor function through depolarization-mediated relief of voltage-dependent Mg2+ blockade (Lee et al., 2007; Ramos-Mandujano et al. 2007). This would facilitate the NMDA receptor response of penumbral neurons to rising levels of extracellular glutamate that occur during ischemia.
One alternative hypothesis to this interpretation is the idea that PAR1 activation sensitizes neurons to NMDA receptor-mediated toxicity, without directly potentiating the NMDA receptor responses. This could occur by a number of mechanisms, including altered divalent cation homeostasis, activation of pro-apoptotic pathways, stimulation of glutamate release during ischemia, or inhibition of glutamate uptake. Some of these possibilities, although not directly involving potentiation or NMDA receptor function, would still have the effect of enhancing NMDA receptor signaling. Although the in vivo models we employed do not allow us to rule out these alternative hypotheses, we should note that PAR1 potentiation of synaptically-activated NMDA receptors has recently been described (Lee et al., 2007). These electrophysiological studies suggest that PAR1 activation is likely to potentiate NMDA receptor function in vivo. Thus, the most parsimonious interpretation at present is that PAR1 activation, perhaps secondary to blood-brain-barrier breakdown and entry into brain parenchyma of PAR1 activators, potentiates synaptic and non-synaptic NMDA receptor function in penumbral tissue, which exacerbates excitotoxicity. The effect of this would be the enhancement of neuronal death in the earliest phases following ischemia; the 24 hours survival time in this study does not allow evaluation of the mechanisms of cell death that are active at later times.
It is important to point out that PAR1−/− mice and PAR1-antagonist-treated mice are not fully protected from either NMDA toxicity or from transient focal ischemia. Rather, a portion of the lesion is independent of PAR1 activation. For direct intrastriatal injections of NMDA, we hypothesize that this reflects a threshold effect of NMDA receptor overactivation on individual neuronal health, as shown in Figure 6. Following intrastriatal NMDA injection, a concentration gradient of NMDA is established centered at the injection site. This concentration gradient subsequently sets up a gradient of NMDA receptor activation. In regions close to the injection site, the degree of NMDA receptor activation is supramaximal with respect to neurotoxicity, and leads to rapid and perhaps immediate cytotoxic accumulation of divalent ions, cell swelling, and death. In regions distributed from this core, the concentration of NMDA falls as does the intensity of NMDA receptor activation. We predict that neurons outside this core area undergo modest NMDA receptor activation that is partially blunted by voltage-dependent channel block of NMDA receptors by extracellular Mg2+. Furthermore, their health and survival likely hang in the balance, as the level of NMDA receptor activation hovers near the threshold required to induce cell death. If PAR1 is present and becomes activated during injury, for example by brain-derived or blood-derived proteases (Gingrich and Traynelis 2000; Junge et al. 2003), the neurons in this peripheral region would show enhanced responses to extracellular glutamate. In this distal portion of the developing lesion, enhancement of NMDA receptor function would tip the balance towards neuronal death, expanding the lesion volume. By contrast, in the core of the lesion, the degree of NMDA receptor activation is supramaximal in terms of its cytotoxic processes. Thus, any effect of PAR1 activation on NMDA receptor function would have little effect on the survival of neurons in the core, since they are destined to die regardless of PAR1 activation.
Figure 6. Working model of exacerbation of excitotoxin damage by PAR1 potentiation of NMDA receptor function.

A) Injection of NMDA creates a concentration gradient focused at the injection site. This concentration gradient creates two regions of toxicity—a region of assured cell death, where NMDA receptor activation is sufficient to be nearly uniformly toxic, and a region in which NMDA receptor activation may or may not cause cell death. In the presence of PAR1 activation, the cell death in the core of the lesion is unchanged. However, PAR1 increases NMDA signaling in the more peripheral regions thereby increasing toxicity. B) In the presence of submaximal levels of NMDA receptor blockers, neurotoxicity in the core of the lesion is unchanged. However, in surrounding areas, blockade of the NMDA receptor protects neurons from toxicity. By blocking NMDA receptor signaling, the channel blockers favor cell survival and prevent PAR1 activation from exacerbating the toxic effects of injected NMDA.
PAR1 signaling and neuroprotection with NMDA receptor antagonists
How can application of an NMDA receptor antagonist block the hypothesized role of PAR1 without blocking the full effect of intrastriatal NMDA? If an NMDA receptor antagonist was administered at a high enough concentration, the lesion volume including the core region of damage immediately adjacent to the injection site should be virtually eliminated. However, if a submaximally effective level of the uncompetitive antagonist is present, one would expect differential effects within the lesion core and distal regions. This is because the level of NMDA receptor activation is supramaximal near the injection site with respect to its ability to cause neurotoxicity. Removal of a portion of the active NMDA receptors by submaximal concentration of an uncompetitive channel blocker like ketamine would not prevent neuronal death in this core region; there is already more than enough NMDA receptor activation to cause neuronal death. However, in the penumbral regions, a submaximal dose of ketamine (or any NMDA receptor antagonist) would partially block NMDA receptor signaling in all neurons. We predict that NMDA receptor blockade in these penumbral regions could protect the neurons whose survival hangs in the balance from reaching a threshold level of NMDA receptor-induced intracellular divalent load, which is thought to drive neurotoxicity (Fig. 6). Thus, partial NMDA receptor block would effectively reduce the degree of penumbral cell death, which is a threshold event. Our data suggest that partial blockade of NMDA receptor activation in the penumbral region is sufficient to reduce lesion volume to a level observed in mice lacking PAR1. The similarity in the lesion volume between ketamine/MK801-treated mice and PAR1−/− mice further suggest that PAR1 potentiation of NMDA receptor function is a critical factor in tipping the balance towards cell death in penumbral regions.
Conclusions
Multiple reports have previously shown that PAR1 activation can potentiate NMDA receptor function in acutely prepared hippocampal slices (Gingrich et al., 2000; Lee et al., 2007; Mannaioni et al., 2008). Given the well known contribution of NMDA receptor overactivation to cell death in ischemia, we have focused in this study on the interaction of PAR1 and NMDA in two in vivo brain injury models. The data presented in this report are consistent with the idea that PAR1 activation during transient focal ischemia can exacerbate neuronal damage through enhancement of NMDA receptor signaling.
Acknowledgments
The authors thank Robert McKeon for assistance with image analysis, Sudar Alagarsamy and John Hepler for assistance with analysis of phosphoinositide hydrolysis, and Anna Orr for critical comments on the manuscript. This work was supported by the NIH-NINDS NS039419 (SFT), NS053062 (CEH), NARSAD (SFT, GM), and PRIN 2007 (GM). The authors declare no competing financial interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arundine M, Tymianski M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium. 2003;34:325–337. doi: 10.1016/s0143-4160(03)00141-6. [DOI] [PubMed] [Google Scholar]
- Ayata C, Ayata G, Hara H, Matthews RT, Beal MF, Ferrante RJ, Endres M, Kim A, Christie RH, Waeber C, Huang PL, Hyman BT, Moskowitz MA. Mechanisms of reduced striatal NMDA excitotoxicity in type I nitric oxide synthase knock-out mice. J Neurosci. 1997;17:6908–6917. doi: 10.1523/JNEUROSCI.17-18-06908.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, Kowall NW, Swartz KJ, Ferrante RJ, Martin JB. Systemic approaches to modifying quinolinic acid striatal lesions in rats. J Neurosci. 1988;8:3901–3908. doi: 10.1523/JNEUROSCI.08-10-03901.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernatowicz MS, Klimas CE, Hartl KS, Peluso M, Allegretto NJ, Seiler SM. Development of potent thrombin receptor antagonist peptides. J Med Chem. 1996;39:4879–4887. doi: 10.1021/jm960455s. [DOI] [PubMed] [Google Scholar]
- Brady RJ, Swann JW. Ketamine selectively suppresses synchronized afterdischarges in immature hippocampus. Neurosci Lett. 1986;69:143–149. doi: 10.1016/0304-3940(86)90593-8. [DOI] [PubMed] [Google Scholar]
- Buller AL, Larson HC, Schneider BE, Beaton JA, Morrisett RA, Monaghan DT. The molecular basis of NMDA receptor subtypes: native receptor diversity is predicted by subunit composition. J Neurosci. 1994;14:5471–5484. doi: 10.1523/JNEUROSCI.14-09-05471.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Pisani A, Sancesario G, Gubellini P, Marfia GA, Bernardi G. Striatal spiny neurons and cholinergic interneurons express differential ionotropic glutamatergic responses and vulnerability: implications for ischemia and Huntington’s disease. Ann Neurol. 1998;43:586–597. doi: 10.1002/ana.410430506. [DOI] [PubMed] [Google Scholar]
- Cannon JR, Keep RF, Schallert T, Hua Y, Richardson RJ, Xi G. Protease-activated receptor-1 mediates protection elicited by thrombin preconditioning in a rat 6-hydroxydopamine model of Parkinson’s disease. Brain Res. 2006;1116:177–186. doi: 10.1016/j.brainres.2006.07.094. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Ariano MA, Calvert CR, Flores-Hernandez J, Chandler SH, Leavitt BR, Hayden MR, Levine MS. NMDA receptor function in mouse models of Huntington disease. J Neurosci Res. 2001;66:525–539. doi: 10.1002/jnr.1244. [DOI] [PubMed] [Google Scholar]
- Cheng T, Liu D, Griffin JH, Fernandez JA, Castellino F, Rosen ED, Fukudome K, Zlokovic BV. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. 2003;9:338–342. doi: 10.1038/nm826. [DOI] [PubMed] [Google Scholar]
- Choi DW, Koh JY, Peters S. Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci. 1988;8:185–196. doi: 10.1523/JNEUROSCI.08-01-00185.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly AJ, Ishihara H, Kahn ML, Farese RV, Jr, Coughlin SR. Role of the thrombin receptor in development and evidence for a second receptor. Nature. 1996;381:516–519. doi: 10.1038/381516a0. [DOI] [PubMed] [Google Scholar]
- Coughlin SR. How the protease thrombin talks to cells. Proc Natl Acad Sci USA. 1999;96:11023–11027. doi: 10.1073/pnas.96.20.11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criswell HE, Ming Z, Pleasant N, Griffith BL, Mueller RA, Breese GR. Macrokinetic analysis of blockade of NMDA-gated currents by substituted alcohols, alkanes and ethers. Brain Res. 2004;1015:107–113. doi: 10.1016/j.brainres.2004.04.050. [DOI] [PubMed] [Google Scholar]
- Cuomo O, Pignataro G, Gala R, Scorziello A, Gravino E, Piazza O, Tufano R, Di Renzo G, Annunziato L. Antithrombin reduces ischemic volume, ameliorates neurologic deficits, and prolongs animal survival in both transient and permanent focal ischemia. Stroke. 2007;38:3272–3279. doi: 10.1161/STROKEAHA.107.488486. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Dogan A, Eras MA, Rao VL, Dempsey RJ. Protective effects of memantine against ischemia-reperfusion injury in spontaneously hypertensive rats. Acta Neurochir (Wien) 1999;141:1107–1113. doi: 10.1007/s007010050491. [DOI] [PubMed] [Google Scholar]
- Donovan FM, Pike CJ, Cotman CW, Cunningham DD. Thrombin induces apoptosis in cultured neurons and astrocytes via a pathway requiring tyrosine kinase and RhoA activities. J Neurosci. 1997;17:5316–5326. doi: 10.1523/JNEUROSCI.17-14-05316.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dravid S, Yuan H, Erreger K, Lyuboslavsky P, Le P, Almonte A, Barber J, Nicholson K, French A, Balster R, Murray TF, Traynelis SF. Proton sensitivity of NMDA receptor channel blockers. J Physiol. 2007;581:107–28. doi: 10.1113/jphysiol.2006.124958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Standaert DG. Subcellular segregation of distinct heteromeric NMDA glutamate receptors in the striatum. J Neurochem. 2003;85:935–43. doi: 10.1046/j.1471-4159.2003.01744.x. [DOI] [PubMed] [Google Scholar]
- Duncan CP, Seidler FJ, Slotkin TA. Effects of MK-801 on DNA synthesis in neonatal rat brain regions under normoxic and hypoxic conditions. Brain Res Dev Brain Res. 1991;58:67–71. doi: 10.1016/0165-3806(91)90238-e. [DOI] [PubMed] [Google Scholar]
- Erreger K, Chen P, Wyllie DJA, Traynelis SF. Glutamate receptor gating. Critical Reviews in Neurobiology. 2004;16:187–224. doi: 10.1615/critrevneurobiol.v16.i3.10. [DOI] [PubMed] [Google Scholar]
- Friedmann I, Yoles E, Schwartz M. Thrombin attenuation is neuroprotective in the injured rat optic nerve. JNeurochem. 2001;76:641–649. doi: 10.1046/j.1471-4159.2001.00001.x. [DOI] [PubMed] [Google Scholar]
- Fujimoto S, Katsuki H, Ohnishi M, Takagi M, Kume T, Akaike A. Plasminogen potentiates thrombin cytotoxicity and contributes to pathology of intracerebral hemorrhage in rats. JCereb Blood Flow Metab. 2008;28:506–515. doi: 10.1038/sj.jcbfm.9600547. [DOI] [PubMed] [Google Scholar]
- Galbicka G, Kautz MA, Jagers T. Behavioral effects of enantiomers of dizocilpine under two “counting” procedures in rats. Pharmacol Biochem Behav. 1994;49:943–948. doi: 10.1016/0091-3057(94)90247-x. [DOI] [PubMed] [Google Scholar]
- Gingrich MB, Junge CE, Lyuboslavsky P, Traynelis SF. Potentiation of NMDA receptor function by the serine protease thrombin. J Neurosci. 2000;20:4582–4595. doi: 10.1523/JNEUROSCI.20-12-04582.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingrich MB, Traynelis SF. Serine proteases and brain damage - is there a link? Trends Neurosci. 2000;23:399–407. doi: 10.1016/s0166-2236(00)01617-9. [DOI] [PubMed] [Google Scholar]
- Guo H, Liu D, Gelbard H, Cheng T, Insalaco R, Fernandez JA, Griffin JH, Zlokovic BV. Activated protein C prevents neuronal apoptosis via protease activated receptors 1 and 3. Neuron. 2004;41:563–572. doi: 10.1016/s0896-6273(04)00019-4. [DOI] [PubMed] [Google Scholar]
- Hamill CE, Goldshmidt A, Nicole O, Brat D, Traynelis SF. Glial reactivity following damage: implications for scar formation and neuronal recovery. Clinical Neurosurgery. 2005;52:29–44. [PubMed] [Google Scholar]
- Harrison NL, Simmonds MA. Quantitative studies on some antagonists of N-methyl D-aspartate in slices of rat cerebral cortex. Br J Pharmacol. 1985;84:381–391. doi: 10.1111/j.1476-5381.1985.tb12922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RL, Seeds NW. Effect of proteases and their inhibitors on neurite outgrowth from neonatal mouse sensory ganglia in culture. Brain Res. 1986;398:63–70. doi: 10.1016/0006-8993(86)91250-3. [DOI] [PubMed] [Google Scholar]
- Henrich-Noack P, Riek-Burchardt M, Baldauf K, Reiser G, Reymann KG. Focal ischemia induces expression of protease-activated receptor1 (PAR1) and PAR3 on microglia and enhances PAR4 labeling in the penumbra. Brain Res. 2006;1070:232–241. doi: 10.1016/j.brainres.2005.10.100. [DOI] [PubMed] [Google Scholar]
- Hollenberg MD, Saifeddine M, al-Ani B, Kawabata A. Proteinase-activated receptors: structural requirements for activity, receptor cross-reactivity, and receptor selectivity of receptor-activating peptides. Can J Physiol Pharmacol. 1997;75:832–841. [PubMed] [Google Scholar]
- Ishida Y, Nagai A, Kobayashi S, Kim SU. Upregulation of protease-activated receptor-1 in astrocytes in Parkinson disease: astrocyte-mediated neuroprotection through increased levels of glutathione peroxidase. J Neuropathol Exp Neurol. 2006;65:66–77. doi: 10.1097/01.jnen.0000195941.48033.eb. [DOI] [PubMed] [Google Scholar]
- Jalink K, Moolenaar WH. Thrombin receptor activation causes rapid neural cell rounding and neurite retraction independent of classic second messengers. J Cell Biol. 1992;118:411–419. doi: 10.1083/jcb.118.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Wum J, Hua Y, Keep RF, Xiang J, Hoff JT, Xi G. Thrombin-receptor activation and thrombin-induced brain tolerance. J Cereb Blood Flow Metab. 2002;22:404–410. doi: 10.1097/00004647-200204000-00004. [DOI] [PubMed] [Google Scholar]
- Jin YJ, Mima T, Raicu V, Park KC, Shimizu K. Combined argatroban and edaravone caused additive neuroprotection against 15 min of forebrain ischemia in gerbils. Neurosci Res. 2002;43:75–79. doi: 10.1016/s0168-0102(02)00019-6. [DOI] [PubMed] [Google Scholar]
- Junge CE, Lee CJ, Hubbard KB, Zhang Z, Olson JJ, Hepler JR, Brat DJ, Traynelis SF. Protease-activated receptor-1 in human brain: localization and functional expression in astrocytes. Exp Neurol. 2004;188:94–103. doi: 10.1016/j.expneurol.2004.02.018. [DOI] [PubMed] [Google Scholar]
- Junge CE, Sugawara T, Mannaioni G, Alagarsamy S, Conn PJ, Brat DJ, Chan PH, Traynelis SF. The contribution of protease-activated receptor 1 to neuronal damage caused by transient focal cerebral ischemia. Proc Natl Acad Sci USA. 2003;100:13019–13024. doi: 10.1073/pnas.2235594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kant GJ, Wright WL, Robinson TN, 3rd, D’Angelo CP. Effects of MK-801 on learning and memory as assessed using a novel water maze. Pharmacol Biochem Behav. 1991;39:479–485. doi: 10.1016/0091-3057(91)90212-k. [DOI] [PubMed] [Google Scholar]
- Karabiyikoglu M, Hua Y, Keep RF, Ennis SR, Xi G. Intracerebral hirudin injection attenuates ischemic damage and neurologic deficits without altering local cerebral blood flow. JCereb Blood Flow Metab. 2004;24:159–166. doi: 10.1097/01.WCB.0000100062.36077.84. [DOI] [PubMed] [Google Scholar]
- Kawabata A, Saifeddine M, Al-Ani B, Leblond L, Hollenberg MD. Evaluation of proteinase-activated receptor-1 (PAR1) agonists and antagonists using a cultured cell receptor desensitization assay: activation of PAR2 by PAR1-targeted ligands. J Pharmacol Exp Ther. 1999;288:358–370. [PubMed] [Google Scholar]
- Kawaguchi M, Furuya H, Patel PM. Neuroprotective effects of anesthetic agents. J Anesth. 2005;19:150–156. doi: 10.1007/s00540-005-0305-5. [DOI] [PubMed] [Google Scholar]
- Klarenbach SW, Chipiuk A, Nelson RC, Hollenberg MD, Murray AG. Differential actions of PAR2 and PAR1 in stimulating human endothelial cell exocytosis and permeability: the role of Rho-GTPases. Circ Res. 2003;92:272–278. doi: 10.1161/01.res.0000057386.15390.a3. [DOI] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- Kuppenbender KD, Standaert DG, Feuerstein TJ, Penney JB, Jr, Young AB, Landwehrmeyer GB. Expression of NMDA receptor subunit mRNAs in neurochemically identified projection and interneurons in the human striatum. J Comp Neurol. 2000;419:407–421. doi: 10.1002/(sici)1096-9861(20000417)419:4<407::aid-cne1>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Leavitt BR, Raamsdonk JM, Shehadeh J, Fernandes H, Murphy Z, Graham RK, Wellington CL, Raymond LA, Hayden MR. Wild-type huntingtin protects neurons from excitotoxicity. J Neurochem. 2006;96:1121–1129. doi: 10.1111/j.1471-4159.2005.03605.x. [DOI] [PubMed] [Google Scholar]
- Lee CJ, Mannaioni G, Yuan H, Woo DH, Gingrich MB, Traynelis SF. Astrocytic control of synaptic NMDA receptors. J Physiol. 2007;581:1057–81. doi: 10.1113/jphysiol.2007.130377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees KR. Cerestat and other NMDA antagonists in ischemic stroke. Neurology. 1997;49:S66–69. doi: 10.1212/wnl.49.5_suppl_4.s66. [DOI] [PubMed] [Google Scholar]
- Lei SZ, Zhang D, Abele AE, Lipton SA. Blockade of NMDA receptor-mediated mobilization of intracellular Ca2+ prevents neurotoxicity. Brain Res. 1992;598:196–202. doi: 10.1016/0006-8993(92)90183-a. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Molecular mechanisms of trauma-induced neuronal degeneration. Curr Opin Neurol Neurosurg. 1993;6:588–596. [PubMed] [Google Scholar]
- Liu LW, Vu TK, Esmon CT, Coughlin SR. The region of the thrombin receptor resembling hirudin binds to thrombin and alters enzyme specificity. J Biol Chem. 1991;266:16977–16980. [PubMed] [Google Scholar]
- Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–282. [PubMed] [Google Scholar]
- Mannaioni G, Goldshmidt A, Hamill CE, Yuan H, Mullasseril P, Hubbard KB, Junge CE, Lee CJ, Yepes M, Hepler JR, Traynelis SF. Plasmin potentiates synaptic NMDA receptor function in rat hippocampal neurons through activation of PAR1. JBiol Chem. 2008;283:20600–11. doi: 10.1074/jbc.M803015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masada T, Xi G, Hua Y, Keep RF. The effects of thrombin preconditioning on focal cerebral ischemia in rats. Brain Res. 2000;867:173–179. doi: 10.1016/s0006-8993(00)02302-7. [DOI] [PubMed] [Google Scholar]
- Matsuoka H, Hamada R. Role of thrombin in CNS damage associated with intracerebral haemorrhage: opportunity for pharmacological intervention? CNS Drugs. 2002;16:509–516. doi: 10.2165/00023210-200216080-00001. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Roeser NF, Silverstein FS, Johnston MV. Quantitative assessment of neuroprotection against NMDA-induced brain injury. Exp Neurol. 1989;106:289–296. doi: 10.1016/0014-4886(89)90162-3. [DOI] [PubMed] [Google Scholar]
- Meldrum BS, Evans MC, Swan JH, Simon RP. Protection against hypoxic/ischaemic brain damage with excitatory amino acid antagonists. Med Biol. 1987;65:153–157. [PubMed] [Google Scholar]
- Miyabe M, Kirsch JR, Nishikawa T, Koehler RC, Traystman RJ. Comparative analysis of brain protection by N-methyl-D-aspartate receptor antagonists after transient focal ischemia in cats. Crit Care Med. 1997;25:1037–1043. doi: 10.1097/00003246-199706000-00022. [DOI] [PubMed] [Google Scholar]
- Monard D, Niday E, Limat A, Solomon F. Inhibition of protease activity can lead to neurite extension in neuroblastoma cells. Prog Brain Res. 1983;58:359–364. doi: 10.1016/S0079-6123(08)60037-0. [DOI] [PubMed] [Google Scholar]
- Mosnier LO, Griffin JH. Inhibition of staurosporine-induced apoptosis of endothelial cells by activated protein C requires protease-activated receptor-1 and endothelial cell protein C receptor. Biochem J. 2003;373:65–70. doi: 10.1042/BJ20030341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niclou SP, Suidan HS, Pavlik A, Vejsada R, Monard D. Changes in the expression of protease-activated receptor 1 and protease nexin-1 mRNA during rat nervous system development and after nerve lesion. Eur J Neurosci. 1998;10:1590–1607. doi: 10.1046/j.1460-9568.1998.00183.x. [DOI] [PubMed] [Google Scholar]
- Nicole O, Goldshmidt A, Hamill CE, Sorensen SD, Sastre A, Lyuboslavsky P, Hepler JR, McKeon RJ, Traynelis SF. Activation of protease-activated receptor-1 triggers astrogliosis after brain injury. J Neurosci. 2005;25:4319–4329. doi: 10.1523/JNEUROSCI.5200-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata J, Shiraishi M, Namba T, Smothers CT, Woodward JJ, Harris RA. Effects of anesthetics on mutant N-methyl-D-aspartate receptors expressed in Xenopus oocytes. J Pharmacol Exp Ther. 2006;318:434–443. doi: 10.1124/jpet.106.101691. [DOI] [PubMed] [Google Scholar]
- Olson EE, Lyuboslavsky P, Traynelis SF, McKeon RJ. PAR-1 deficiency protects against neuronal damage and neurologic deficits after unilateral cerebral hypoxia/ischemia. J Cereb Blood Flow Metab. 2004;24:964–971. doi: 10.1097/01.WCB.0000128266.87474.BF. [DOI] [PubMed] [Google Scholar]
- Ozden S, Isenmann S. Neuroprotective properties of different anesthetics on axotomized rat retinal ganglion cells in vivo. J Neurotrauma. 2004;21:73–82. doi: 10.1089/089771504772695968. [DOI] [PubMed] [Google Scholar]
- Park CK, Nehls DG, Graham DI, Teasdale GM, McCulloch J. The glutamate antagonist MK-801 reduces focal ischemic brain damage in the rat. Ann Neurol. 1988;24:543–551. doi: 10.1002/ana.410240411. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau C, Hedreen JC, Price DL, Koliatsos VE. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J Neurosci. 1995;15:3775–3787. doi: 10.1523/JNEUROSCI.15-05-03775.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin ZH, Wang Y, Chase TN. Stimulation of N-methyl-D-aspartate receptors induces apoptosis in rat brain. Brain Res. 1996;725:166–176. doi: 10.1016/0006-8993(96)00200-4. [DOI] [PubMed] [Google Scholar]
- Ramos-Mandujano G, Vazquez-Juarez E, Hernandez-Benitez R, Pasantes-Morales H. Thrombin potently enhances swelling-sensitive glutamate efflux from cultured astrocytes. Glia. 2007;55:917–925. doi: 10.1002/glia.20513. [DOI] [PubMed] [Google Scholar]
- Riek-Burchardt M, Striggow F, Henrich-Noack P, Reiser G, Reymann KG. Increase of prothrombin-mRNA after global ischemia in rats, with constant expression of protease nexin-1 and protease-activated receptors. Neurosci Lett. 2002;329:181–184. doi: 10.1016/s0304-3940(02)00645-6. [DOI] [PubMed] [Google Scholar]
- Rohatgi T, Henrich-Noack P, Sedehizade F, Goertler M, Wallesch CW, Reymann KG, Reiser G. Transient focal ischemia in rat brain differentially regulates mRNA expression of protease-activated receptors 1 to 4. JNeurosci Res. 2004;75:273–279. doi: 10.1002/jnr.10847. [DOI] [PubMed] [Google Scholar]
- Roussel S, Pinard E, Seylaz J. Effect of MK-801 on focal brain infarction in normotensive and hypertensive rats. Hypertension. 1992;19:40–46. doi: 10.1161/01.hyp.19.1.40. [DOI] [PubMed] [Google Scholar]
- Ruf W. PAR1 signaling: more good than harm? Nat Med. 2003;9:258–260. doi: 10.1038/nm0303-258. [DOI] [PubMed] [Google Scholar]
- Sheehan JJ, Tsirka SE. Fibrin-modifying serine proteases thrombin, tPA, and plasmin in ischemic stroke: a review. Glia. 2005;50:340–350. doi: 10.1002/glia.20150. [DOI] [PubMed] [Google Scholar]
- Shibata M, Kumar SR, Amar A, Fernandez JA, Hofman F, Griffin JH, Zlokovic BV. Anti-inflammatory, antithrombotic, and neuroprotective effects of activated protein C in a murine model of focal ischemic stroke. Circulation. 2001;103:1799–1805. doi: 10.1161/01.cir.103.13.1799. [DOI] [PubMed] [Google Scholar]
- Shigetomi E, Bowser DN, Sofroniew MV, Khakh BS. Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. JNeurosci. 2008;28:6659–6663. doi: 10.1523/JNEUROSCI.1717-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman J, Muir WW., 3rd A review of laboratory animal anesthesia with chloral hydrate and chloralose. Lab Anim Sci. 1993;43:210–216. [PubMed] [Google Scholar]
- Smirnova IV, Citron BA, Arnold PM, Festoff BW. Neuroprotective signal transduction in model motor neurons exposed to thrombin: G-protein modulation effects on neurite outgrowth, Ca2+ mobilization, and apoptosis. J Neurobiol. 2001;48:87–100. [PubMed] [Google Scholar]
- Smith-Swintosky VL, Zimmer S, Fenton JW, 2nd, Mattson MP. Protease nexin-1 and thrombin modulate neuronal Ca2+ homeostasis and sensitivity to glucose deprivation-induced injury. J Neurosci. 1995;15:5840–5850. doi: 10.1523/JNEUROSCI.15-08-05840.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider RM, McKinney M, Fenton JW, 2nd, Richelson E. Activation of cyclic nucleotide formation in murine neuroblastoma N1E-115 cells by modified human thrombins. J Biol Chem. 1984;259:9078–9081. [PubMed] [Google Scholar]
- Snider RM, Richelson E. Thrombin stimulation of guanosine 3′,5′-monophosphate formation in murine neuroblastoma cells (clone N1E-115) Science. 1983;221:566–568. doi: 10.1126/science.6306770. [DOI] [PubMed] [Google Scholar]
- Sorensen SD, Nicole O, Peavy RD, Montoya LM, Lee CJ, Murphy TJ, Traynelis SF, Hepler JR. Common signaling pathways link activation of murine PAR-1, LPA, and S1P receptors to proliferation of astrocytes. Mol Pharmacol. 2003;64:1199–1209. doi: 10.1124/mol.64.5.1199. [DOI] [PubMed] [Google Scholar]
- Standaert DG, Friberg IK, Landwehrmeyer GB, Young AB, Penney JB., Jr Expression of NMDA glutamate receptor subunit mRNAs in neurochemically identified projection and interneurons in the striatum of the rat. Brain Res Mol Brain Res. 1999;64:11–23. doi: 10.1016/s0169-328x(98)00293-9. [DOI] [PubMed] [Google Scholar]
- Striggow F, Riek-Burchardt M, Kiesel A, Schmidt W, Henrich-Noack P, Breder J, Krug M, Reymann KG, Reiser G. Four different types of protease-activated receptors are widely expressed in the brain and are up-regulated in hippocampus by severe ischemia. Eur J Neurosci. 2001;14:595–608. doi: 10.1046/j.0953-816x.2001.01676.x. [DOI] [PubMed] [Google Scholar]
- Striggow F, Riek M, Breder J, Henrich-Noack P, Reymann KG, Reiser G. The protease thrombin is an endogenous mediator of hippocampal neuroprotection against ischemia at low concentrations but causes degeneration at high concentrations. Proc Natl Acad Sci USA. 2000;97:2264–2269. doi: 10.1073/pnas.040552897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suidan HS, Stone SR, Hemmings BA, Monard D. Thrombin causes neurite retraction in neuronal cells through activation of cell surface receptors. Neuron. 1992;8:363–375. doi: 10.1016/0896-6273(92)90302-t. [DOI] [PubMed] [Google Scholar]
- Suo Z, Citron BA, Festoff BW. Thrombin: a potential proinflammatory mediator in neurotrauma and neurodegenerative disorders. Curr Drug Targets Inflamm Allergy. 2004;3:105–114. doi: 10.2174/1568010043483953. [DOI] [PubMed] [Google Scholar]
- Suo Z, Wu M, Ameenuddin S, Anderson HE, Zoloty JE, Citron BA, Andrade-Gordon P, Festoff BW. Participation of protease-activated receptor-1 in thrombin-induced microglial activation. J Neurochem. 2002;80:655–666. doi: 10.1046/j.0022-3042.2001.00745.x. [DOI] [PubMed] [Google Scholar]
- Swan JH, Meldrum BS. Protection by NMDA antagonists against selective cell loss following transient ischaemia. J Cereb Blood Flow Metab. 1990;10:343–351. doi: 10.1038/jcbfm.1990.63. [DOI] [PubMed] [Google Scholar]
- Thomson AM, West DC, Lodge D. An N-methylaspartate receptor-mediated synapse in rat cerebral cortex: a site of action of ketamine? Nature. 1985;313:479–481. doi: 10.1038/313479a0. [DOI] [PubMed] [Google Scholar]
- Trejo J. Protease-activated receptors: new concepts in regulation of G protein-coupled receptor signaling and trafficking. J Pharmacol Exp Ther. 2003;307:437–442. doi: 10.1124/jpet.103.052100. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Trejo J. Protease receptor signaling: new roles and regualtory mechanisms. Current Opinion Haematology. 2007;14:230–5. doi: 10.1097/MOH.0b013e3280dce568. [DOI] [PubMed] [Google Scholar]
- Turgeon VL, Lloyd ED, Wang S, Festoff BW, Houenou LJ. Thrombin perturbs neurite outgrowth and induces apoptotic cell death in enriched chick spinal motoneuron cultures through caspase activation. J Neurosci. 1998;18:6882–6891. doi: 10.1523/JNEUROSCI.18-17-06882.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uematsu D, Araki N, Greenberg JH, Sladky J, Reivich M. Combined therapy with MK-801 and nimodipine for protection of ischemic brain damage. Neurology. 1991;41:88–94. doi: 10.1212/wnl.41.1.88. [DOI] [PubMed] [Google Scholar]
- Vaughan PJ, Pike CJ, Cotman CW, Cunningham DD. Thrombin receptor activation protects neurons and astrocytes from cell death produced by environmental insults. J Neurosci. 1995;15:5389–5401. doi: 10.1523/JNEUROSCI.15-07-05389.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivien D, Buisson A. Serine protease inhibitors: novel therapeutic targets for stroke. JCereb Blood Flow Metab. 2000;20:755–764. doi: 10.1097/00004647-200005000-00001. [DOI] [PubMed] [Google Scholar]
- Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- Wang CX, Shuaib A. NMDA/NR2B selective antagonists in the treatment of ischemic brain injury. Curr Drug Targets CNS Neurol Disord. 2005;4:143–151. doi: 10.2174/1568007053544183. [DOI] [PubMed] [Google Scholar]
- Wang H, Ubl JJ, Reiser G. Four subtypes of protease-activated receptors, co-expressed in rat astrocytes, evoke different physiological signaling. Glia. 2002;37:53–63. doi: 10.1002/glia.10012. [DOI] [PubMed] [Google Scholar]
- Warner MA, Neill KH, Nadler JV, Crain BJ. Regionally selective effects of NMDA receptor antagonists against ischemic brain damage in the gerbil. J Cereb Blood Flow Metab. 1991;11:600–610. doi: 10.1038/jcbfm.1991.110. [DOI] [PubMed] [Google Scholar]
- Weinstein JR, Gold SJ, Cunningham DD, Gall CM. Cellular localization of thrombin receptor mRNA in rat brain: expression by mesencephalic dopaminergic neurons and codistribution with prothrombin mRNA. J Neurosci. 1995;15:2906–2919. doi: 10.1523/JNEUROSCI.15-04-02906.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whetsell WO., Jr Current concepts of excitotoxicity. J Neuropathol Exp Neurol. 1996;55:1–13. doi: 10.1097/00005072-199601000-00001. [DOI] [PubMed] [Google Scholar]
- Wong EH, Kemp JA, Priestley T, Knight AR, Woodruff GN, Iversen LL. The anticonvulsant MK-801 is a potent N-methyl-D-aspartate antagonist. Proc Natl Acad Sci USA. 1986;83:7104–7108. doi: 10.1073/pnas.83.18.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi G, Reiser G, Keep RF. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective? J Neurochem. 2003;84:3–9. doi: 10.1046/j.1471-4159.2003.01268.x. [DOI] [PubMed] [Google Scholar]
- Xue M, Hollenberg MD, Yong VW. Combination of thrombin and matrix metalloproteinase-9 exacerbates neurotoxicity in cell culture and intracerebral hemorrhage in mice. J Neurosci. 2006;26:10281–10291. doi: 10.1523/JNEUROSCI.2806-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakura T, Askalany AR, Petrenko AB, Kohno T, Baba H, Sakimura K. The NR3B subunit does not alter the anesthetic sensitivities of recombinant N-methyl-D-aspartate receptors. Anesth Analg. 2005;100:1687–92. doi: 10.1213/01.ANE.0000152324.30272.49. [DOI] [PubMed] [Google Scholar]
- Yang J, Zorumski CF. Effects of isoflurane on N-methyl-D-aspartate gated ion channels in cultured rat hippocampal neurons. Ann NY Acad Sci. 1991;625:287–289. doi: 10.1111/j.1749-6632.1991.tb33851.x. [DOI] [PubMed] [Google Scholar]