

Abstract
The mucosal surfaces represent the main intersection between jawed vertebrates and the environment. The mucosal surface of the intestine alone forms the largest surface that is exposed to exogenous antigens as well as the largest collection of lymphoid tissue in the body. Therefore, a protective immune activity must coexist with efficient regulatory mechanisms in order to maintain a health status of these organisms. The discovery of a new lineage of helper T cells that produce interleukin (IL)-17 has provided valuable new insight into host defense and the pathogenesis of inflammatory diseases at the mucosal surfaces. Of particular interest for these surfaces, it has been reported that peripherally-induced regulatory T cells and Th17 effector cells arise in a mutually exclusive fashion, depending on whether they are activated in the presence of TGF-β or TGF-β plus inflammatory cytokines such as IL-6. This review will address the protective and pathogenic roles of Th17 cells in the mucosal surfaces and potential regulatory mechanisms that control their development.
Keywords: Intestine, Lung, Mucosal, TH17, T regulatory cells
The mucosa as the main environmental interface
The main intersection sites between the environment and our organism are the mucosal surfaces, represented by the gastrointestinal, respiratory, and genital tract. The majority of contacts established at the mucosal surfaces are with non-pathogenic microbial antigens, dietary antigens and air-borne antigens. The mucosal surfaces also contain a diverse and large immune system. For example, the gut-associated-lymphoid-tissue (GALT) harbors more lymphocytes than all remaining lymphoid tissues together 1. In contrast to the peripheral lymphoid tissue, the GALT consists of 70% constitutively activated T cells bearing an antigen-experienced phenotype (CD45RBlo, CD44hi, CD69hi, CD62Llo) 1. A large proportion of these T cells can be classified as memory T cells, based on their phenotype and functional capacity to display immediate cytotoxicity and the prompt ability to secrete cytokines such as IFN-γ, IL-4, and TNF-α 1. In the past few years, it has become clear that the GALT also naturally contains a population of T cells that constitutively produce pro-inflammatory cytokines such as IL-17A, IL-17F, TNF-α and IL-22.
Introduction to Th17 cells
Th17 cells were recently defined as a distinct lineage that does not share developmental pathways with either Th1 or Th2 cells 2. In fact, both IFN-γ and IL-4 are able to inhibit the differentiation of IL-17 producing T cells, while blocking antibodies to IFN-γ and IL-4 facilitate 3, 4. Moreover, overexpression of either T-bet (main Th1 transcription factor) or c-Maf, (transcription factor important for IL-4 expression), reduced IL-17 production 4, 5. Another cytokine involved in both Th1/Th2 and Treg development that negatively regulates Th17-differentiation is IL-2. IL-2 is essential for the TGF-β-mediated iTreg induction but its signaling via Stat5 constrains Th17 generation, while IL-2 deficiency was also shown to be associated with increased Th17 generation in vivo 6. Finally, the cytokine IL-27, another member of the IL-12 cytokine family, is composed by Epstein-Barr virus-induced gene 3 (EBI3) and p28 chains and also suppresses Th17 development 2. Although IL-27 signal activates several different pathways, including IL-6-related STAT3, the suppression of Th17 development is dependent on Stat1 7, 8.
In contrast to the Th1/Th2 subsets, which are inhibited by TGF-β, Th17 cells rather require TGF-β, along with the pro-inflammatory cytokines IL-6 or IL-21, for their differentiation from naïve precursors 2, 9–11. Data originated from Cua and Littman’s laboratory pointed to the retinoic acid-related orphan receptor (ROR)γt as the key transcription factor for generation of Th17 cells 2. Accordingly, RORγt-deficient naïve CD4 T cells have reduced ability to differentiate into IL-17-producing cells, whereas forced expression of RORγt in naïve CD4 T cells is sufficient to induce expression of IL-17, IL-17F and IL-22 12. Current dogma proposes that initial steps are driven by TGF-β and IL-6 and/or IL-21, while IL-23 plays a fundamental role in stabilizing the phenotypic features of the Th17 lineage 13. Similar TGF-β-dependent pathway also operates in human Th17 cell-differentiation from naïve CD4 T cells 14.
Th17 cells have been linked to several autoimmune disorders (reviewed elsewhere 2, 13) but are physiologically found in the lamina propria of the intestine. In the mucosal surfaces, the production of Th17-related cytokines such as IL-22 and IL-17 itself is crucial for host protection against several extracellular pathogens, but is also related to the development of pathological inflammatory disorders. In this review we will focus on the role of these cells in the mucosal surfaces of the gut and the lung and discuss the potential mechanisms that govern their efficient generation and regulation at these sites.
Role of Th17 cells in the gut
IL-17 producing cells are present at high numbers at steady state in the mucosal surfaces, being represented mostly by CD4+TCRαβ and CD8ααTCRγδ T cells in lamina propria of the small intestine 15. New studies have started to elucidate physiological factors that influence this spontaneous production of IL-17 in the intestinal lamina propria. In a recent study, Ivanov et al. reported that commensal bateria are required for IL-17 production in the small intestine, since germ-free mice from three different strains contained virtually no Th17 cells in the lamina propria. Surprisingly, neither Trif nor Myd88 were required for this “spontaneous” IL-17 production in the lamina propria, indicating that toll-like receptor signaling was not involved in this phenomenon 15. An explanation for these findings could be found in a recent report by Atarashi and coworkers, who have shown that adenosine 5′-triphosphate (ATP) derived from commensal bacteria can activate a subset of lamina propria antigen-presenting cells (F4/80+CD11b+CD70hiCD11clocells) that are able to produce IL-6, IL-23 and TGF-β, triggering the differentiation of Th17 cells 16.
Curiously, a different regulation seems to occur in large intestine. In contrast to what was described for the small intestine 15, Zaph and coworkers showed that large intestine from germ-free mice contains more IL-17 producing cells than mice reared in specific-pathogen-free (SPF) conditions 17. The authors found that the microbiota present in the large intestine is responsible for the production of IL-17-family cytokine IL-25 (IL-17E), which counter-regulates IL-17 production through inhibition of IL-23 production by lamina propria macrophages. Small and large intestines are indeed very different sites regarding the immune system. First, there are more IELs per epithelial cell and more LPLs in the small intestine and the lymphocyte migration to these sites is differentially regulated 1. Peyer’s patches and M cells are also mostly found in the small intestine, although the large intestine contains a similar structure, the cecal patch. The composition of lymphocytes is distinct between these two anatomical sites. For example, the so-called unconventional lymphocytes such as CD8αα TCRαβ and CD8ααTCRγδ IELs are more frequent in the small than in the large intestine 1. Additionally, the great majority of commensal bacteria are found in the large intestine and possibly the composition of bacteria species is distinct from the small intestine. Finally, “spontaneously” producing Th17 cells are present at higher frequency in the small intestine 15.
Therefore, different regulation of small and large intestine is plausible. Nonetheless, the findings by Atarashi and coworkers 16 differ from those by Zaph et al.17 since the first found a dramatic decrease while the latter found an increase in the frequency of IL-17 producing cells in the large intestine of germ-free mice. These discrepancies could be related to different genetic background, although consistent data with the conclusion reached in each of the three mentioned studies (either increased 17 or decreased 15, 16 IL-17 production in the lamina propria of mice maintained under germ-free conditions) were reported when comparing various conventional and germ-free mouse strains. Considering that germ-free mice should not harbor any microbiota, one reason for the divergent results might be related to alterations in the microbiota of conventional mice maintained in different facilities. For instance, Ivanov et al. described that mice originated from Taconic Farms contain significantly higher population of IL-17 producing cells in the lamina propria than from mice Jackson Laboratory 15.
The IL-23/IL-17 axis plays multiple roles in the intestinal immune system, being protective against certain extracellular pathogens while detrimental in different models of inflammatory bowel diseases 2. Genome-wide association studies from patients with inflammatory bowel diseases have pointed to IL-23R as one of the main genes associated with risk of developing Crohn’s disease and ulcerative colitis 18. IL-23 has been shown to mediate T-cell independent colitis and innate inflammatory responses induced by agonist CD40 antibodies or by Helicobacter hepaticus-infection of RAG-deficient mice 19, 20. Although Th17 cells were not required for the development of the disease in these models, predominantly mediated by IL-23 production, IL-17 expression by innate cells was found significantly inhibited upon IL-23p19 blockade 20. In T cell dependent models of colitis, induced either by Helicobacter hepaticus-infection associated with anti-IL10R treatment, or by naïve T cell transfer into RAG-deficient mice, IL-23 but not IL-12 was associated the intestinal pathology 20, 21. The involvement of IL-23 in the inflammatory process appeared to have strictly innate-immunity as well as a Th17-mediated components 20, 21. A recent study directly linked the Th17 lineage-differentiation with colitis development. Using the transfer model of colitis, Leppkes and coworkers showed that naïve CD4+T cells from wild type, but not from RORγ-deficient mice, induce colitis upon transfer into RAG-deficient mice. Additionally, by using blocking antibodies to IL-17A and naïve CD4+T cells from IL-17F-deficient mice, the authors showed that IL-17A and IL-17F redundantly trigger intestinal inflammation 22.
The above data underscores the importance of the IL-23/IL-17 axis in mediating detrimental intestinal inflammation, but in many cases inflammatory processes triggered by these cytokines is crucial for host protection. Mangan and coworkers have shown that although IL-23p19-deficient mice are able to mount a vigorous Th17 response after Citrobacter rodentium infection, this response is not efficient enough, since p19−/−, but not WT mice, succumb shortly after infection with uncontrolled bacterial growth and dissemination, suggesting that IL-23 is important for a full differentiation and for an efficient Th17 response against this intestinal pathogen 11. The production of IL-23 has been recently linked also with the induction of IL-22-producing mucosal NK-cells (NK-22) in mice and humans 23, 24. This population of CD56hiNK cells seems to constitute another important branch of the immune response against intestinal pathogens and also requires stimulus from the microbiota and RORγt expression for their development 23, 25, 26. Furthermore, although in some inflammatory contexts IL-22 production by Th17-cells can exert proinflammatory effects, IL-22 produced either by Th17 cells or by NK-22 cells was shown to protect mice from IBD, probably exerting protective effects on responding intestinal epithelial cell (IEC) in the colon 25.
A recent report extended the protective role of Th17 cells to oropharyngeal fungus (C. albicans) infection. Conti et al. showed that IL23p19 and IL-17RA-deficient mice, but not IL-22 or IL-12p35-deficient mice are susceptible to oral candidiasis 27. The role played by IL-17- producing T cells in controlling invading pathogens, including Candida infection, is of special interest in infections that cause immune deficiency. Both simian and human immunodeficiency virus (SIV and HIV, respectively) were shown to massively deplete intestinal memory CD4+T cells at all stages of infection 28. Raffatellu et al. extended these observations by demonstrating that SIV infection of rhesus macaques resulted in a severe depletion of Th17 cells in the ileal mucosa. As a consequence of deprived Th17 responses, the macaques displayed blunted responses against Salmonella typhimurium, resulting in bacterial dissemination 29.
Role of Th17 cells in the lungs
In addition to their role in the intestinal immune system, IL-17-producing CD4+ T cells also constitute an important arm of adaptive immune responses against airborne-pathogens 30. For instance, IL-17-producing cells play an important role in the establishment of effective immune responses to Mycobacterium tuberculosis mainly through the recruitment of protective IFN-γ-producing CD4+ T cells 31. Increasing evidence suggests that IL-17, acting either directly or indirectly, significantly stimulates neutrophil maturation, migration, and function in the lung tissue and airways. IL-17 induction of neutrophil activation and migration is important in defense against a variety of organisms that infect the lung 32, 33. It was reported that Klebsiella pneumoniae-pulsed dendritic cells led to IL-17 production in an IL-23-dependent manner 34. Accordingly, IL-23 p19-deficient and IL-17R-deficient mice where shown to be more susceptible to lung infection with Klebsiella pneumoniae 34, 35. The increased bacteremia and mortality observed in these mice was associated with reduced levels of CXCL1, CXCL2, G-CSF, and subsequent neutrophillic influx in the lung upon challenge with Klebsiella pneumoniae. Likewise, in mice infected with Mycoplasma pneumoniae, infiltration of the lungs by neutrophils is dependent upon IL-23-induced upregulation of IL-17 36. Another important respiratory pathogen Bordetella pertussis, the cause of whooping cough, employs multiple pathways to elicit IL-17 response. Protection of mice by pertussis vaccine requires production of IL-17, IL-23, and IL-1 37. In addition to regulation of chemokines and cytokines, IL-17 regulates antimicrobial peptide production of beta defensins in human bronchial epithelial cells 32, 37. Taken together, these data support the idea that Th17 cells are critical for normal immunity against bacterial infections in the airways.
In some circumstances, however, IL-23 and/or IL-17 may also be associated with an unfavorable outcome to infection. A recent study by Zelante et al. demonstrated that the Th17 pathway acted as a negative regulator of the Th1-mediated immune resistance to both Candida albicans and Aspergillus fumigatus, and played an inflammatory role previously attributed to uncontrolled Th1 responses 38. IL-23 acted as a molecular connection between uncontrolled fungal growth and inflammation, being produced by DC in response to high fungal burden and counter regulating IL-12p70 production. Both IL-23 and IL-17 impaired antifungal effector activities of neutrophils even in the presence of IFN-γ. In addition, both cytokines activated the inflammatory program of neutrophils by inducing the release of MMP9 and MPO, which likely contributes to the high inflammatory pathology and tissue destruction associated with Th17 cell activation. Together, these results imply that the relative contribution of IL-17 to host defense against respiratory pathogens might vary depending on the infectious setting.
There is growing evidence from clinical studies that exaggerated recruitment and activation of neutrophils in the airways is linked to the clinical course of several inflammatory diseases in the airways and lungs, such as asthma, nonspecific bronchial hyperreactivity (BHR), chronic bronchitis, chronic obstructive pulmonary disease (COPD), cystic fibrosis, and acute respiratory distress syndrome 32, 39. The precise role of Th17 cells in lung diseases is still largely unclear, but there is growing clinical and experimental evidence to suggest that they may be important for neutrophilic influx in acute and chronic airway inflammation. In asthmatic patients, IL-17 expression was increased in the lungs, sputum, BALF, or sera, and severity of airway hypersensitivity in patients correlated with the level of IL-17 expression 40. Human bronchial fibroblasts cells, airway smooth muscle cells, and lung epithelial cells respond to stimulation with IL-17 in vitro by producing IL-6, IL-8, and GRO-α. IL-8 and GRO-α are known chemoattractants for neutrophils, and IL-6 is a neutrophil activating cytokine 41. Thus, the increased expression of IL-17 in the lung during allergic responses may explain the increased accumulation and activation of lung neutrophils. Consistent with this, Hellings et al. in a mouse model of allergic asthma showed that acute allergen provocation is followed by a rapid increase in IL-17 mRNA in mouse lungs, which correlated with an increase in neutrophilic influx in the airways 42. Neutralizing anti-IL-17 mAb treatment ablated bronchial neutrophilia in parallel with reduction of bone marrow and blood neutrophilia. Moreover, IL-23 and Th17 cells not only induce Th17-cell mediated neutrophil recruitment but can also upregulate Th2-mediated eosinophilic airway inflammation and hyperresponsiveness 43, 44. Recently, using a well-established murine model of hypersensitivity pneumonitis and pulmonary fibrosis, Simonian et al. repeatedly exposed C57BL/6 mice to Saccharopolyspora rectivirgula and demonstrated that CD4 T cells expressing IL-17 and IL-22 are recruited in to the lung 45. Importantly, after exposure of IL-17Ra−/− mice to S. rectivirgula, a significantly decreased T cell alveolitis was seen, with an associated decrease in collagen deposition. These data highlight a role for Th17 cells in the immune response directed against bacterial pathogens and for the subsequent development of lung fibrosis. Thus, in contrast to Th1 and Th2 cells, Th17 cells may be involved in the pathogenesis of various inflammatory and allergic responses in the lung in part by supporting neutrophil recruitment and survival and inducing proinflammatory cytokines by structural cells.
Similar to the intestine, IL-17 producing cells are present at high numbers at steady state in the airways, mostly represented by invariant NKT cells, TCRγδ T cells and non-T cells 15. Although many studies have characterized pathways that induce and regulate Th17-related cytokines in the intestine, very few studies have attempted to describe how these pathways operate in the airways. For example, are there microbial or endogenous stimuli required for the spontaneous production of IL-17 in the lungs? Since intestinal and lung-associated lymphoid tissue share characteristics typically found at mucosal sites, such as predominant induction of tolerance at steady state, it is plausible that parallel regulatory mechanisms to the intestine can be found in the airways (discussed below).
Regulation of Th17 cells in the mucosa: IL-25 and TSLP
The cytokine, IL-25, is expressed by lung epithelial cells and activated eosinophils and basophils. It mediates early T-cell differentiation towards a Th2-phenotype and development of airway hyperactivity and allergic diseases. This characteristic, associated with the ability to suppress Th17 differentiation suggested that IL-25 could be an important regulatory mechanism to suppress the development of different inflammatory diseases. Additionally, IL-25 promotes Th2-cell differentiation upon thymic stromal lymphopoietin (TSLP)-activated DC stimulation 46. This IL-25/TSLP pathway seems to operate in the intestinal mucosa as well. Mice with an IEC-specific deletion of the NF-κB-related kinase, IKKβ, show a reduced expression of TSLP in the intestine and fail to develop a protective Th2 response against the intestinal parasite Trichuris. Moreover, these mice develop exaggerated IFN-γ and IL-17 responses 47. These results underscore the importance of mechanisms that regulate Th17 responses under pathogen-induced or autoimmune and allergic diseases-induced inflammation in the mucosal surfaces.
Regulation of Th17 cells in the mucosa: Reciprocal development of Th17 and Treg cells
In addition to its effects on Th17-differentiation, TGF-β has multiple roles in both systemic and mucosal immune-regulation, among them is the ability to convert naïve CD4 cells into Foxp3-expressing regulatory T cells (iTreg) 48. This contrasting deviation underscores a central role of TGF-β in orchestrating the pro- and anti-inflammatory nature of adaptive immunity. This pathway has particular relevance at mucosal surfaces such as the intestine, where both intense microbial load and production of TGF-β are constant under physiological conditions. How is the balance of pro- and anti-inflammatory responses achieved in the highly stimulated surface of the intestinal mucosa?
It was recently found that Foxp3 can directly inhibit Th17 differentiation by direct interaction with RORγt 49–51. Actually, RORγt and Foxp3 may coexist in the same cell 50, 52. Eberl’s group reported that RORγt-expressing T cells contain a sizeable population of cells with regulatory properties, including IL-10 producing Foxp3+Treg cells in vivo 52. The balance between IL-17 producing RORγt+ T cells and IL-10 producing Foxp3+ cells depends on factors such as inflammatory cytokines and the expression of Foxp3 itself. IL-6-deficient mice show decreased IL-17 producing RORγt cells in various tissues, including the airways and mesenteric lymph nodes. Remarkably, the relative frequency of IL-17-producing RORγt+ or IL-10-producing Foxp3+ RORγt+ T cells remained constant during chronic intestinal inflammation as well as after lung infection with influenza A-virus 52. The authors proposed the existence of a robust mechanism maintaining the equilibrium between Th17 and Tregs within RORγt cells during infection 52. Keeping the balance of IL-17 versus IL-10 production would promote inflammation while limiting collateral damage, a necessary compromise between maintaining effective immunity and tissue integrity.
The constant exposure to luminal antigens creates a so-called “physiological chronic inflammation” in the gut, which could easily favor the TGF-β driven effector differentiation at the expense of iTreg. The high amounts of IL-17 producing T cells at steady state correlates with the fact that inflammatory cytokines are produced physiologically in the intestine 12, 15, 52. At the same time, the intestine is highly regulated, containing several different mechanisms to control pathological inflammation. In normal circumstances, previous exposure to antigens via oral or nasal routes efficiently inhibits the development of immune responses to subsequent challenges with the same antigen 53, 54. The intestinal and airway mucosa are highly effective at inducing iTregs 48, 54. Lafaille’s group has shown that Tregs are induced after oral or nasal exposure to the cognate antigen and these iTregs are sufficient and required for mucosal tolerance in this system 48, 54. Belkaid and Powrie’s groups confirmed and extended these findings by demonstrating that iTregs were preferentially induced in MLN and lamina propria by a subpopulation of DCs, rather than in spleen or peripheral lymph nodes, reinforcing that the intestine is a privileged site for Treg induction 55, 56. Importantly, these intestinal DC subpopulations specifically produce a “co-factor” for Treg development, the vitamin-A metabolite retinoic acid 55–58.
Regulation of Th17 cells in the mucosa: Retinoic acid
Mucosal APCs, especially CD11b+CD11c+ or CD103−CD11c+ DCs, are able to help the development of the Th17 cells in the lamina propria of the intestine 55, 58. On the other hand, important regulatory functions can also be ascribed to DCs present in the mucosal sites. For example, antigen-presentation by mucosal DCs in the MLN plays crucial roles in the development of oral tolerance 59. Also, mucosal plasmacytoid CD8αα+ DCs are inefficient at inducing CD4 T helper proliferation but instead can induce their differentiation to IL-10-producing CD4 cells 60. Recently, the CD103+ DC-population was shown to be the main population involved in the production of retinoic acid (RA). Importantly, the production of RA by mucosal APCs, including lamina propria macrophages, was associated with their high efficiency in converting naïve CD4+ T cells into FoxP3+ T cells in a TGF-β-dependent fashion 55–58. In contrast to the effect on Treg cells, RA potently constrains Th17 conversion 57. Different groups have shown that RA signaling through RAR receptors in the T cells is able to block the inhibitory effects of inflammatory cytokines, such as IL-6, on the TGF-β mediated Foxp3 induction and consequently suppressing the development of IL-17-producing CD4 and CD8 T cells 57, 61. Additionally, RA directly inhibits TGF-β and IL-6-induced RORγt on naïve CD4+ T cells 57, 62, possibly through the inhibition of IL-6Rα and IL-23R expression 63.
The dose of RA associated with toll-like receptor (TLR) agonists was also suggested to play a role on the suppression of Th17 differentiation. Although previous studies have described that RA has suppressive effects on Th17 development in all doses examined, including spontaneous production of RA by mucosal DCs in T/DC co-cultures 57, 62, Uematsu and coworkers have recently suggested that production of RA by CD11chiCD11bhi LP DCs, when stimulated by TLR5-ligand flagellin, promoted a modest differentiation of antigen-specific Th17 and Th1 cells, suggesting that innate stimuli may induce contrasting effects of RA on either DCs or T cells.
Clinically, vitamin-A derivatives have been used to treat certain types of cancer and skin-related diseases such as psoriasis 64. Interestingly, the production of IL-6 and IL-23 and differentiation of IL-17 and IL-22-producing Th17 cells has been implicated with the pathogenesis of autoimmune psoriasis 65. Several clinical trials currently investigate potential application of the inhibition of Th17 development, for example, using antibodies against IL-23, in the treatment of autoimmune diseases 66. In fact, retinoic acid and other vitamin-A derivatives were previously shown to suppress experimental models of autoimmune diseases recently linked to a Th17 phenotype, such as experimental autoimmune encephalomyelitis 67. Is possible that part of the mechanism of action of retinoids in the treatment of psoriasis and EAE is related to suppression of Th17 development. Therefore, it is of utmost interest to dissect the mechanism of action of retinoic acid-mediated suppression of these inflammatory processes as well as to evaluate possible therapeutic applications for retinoic acid in Th17-related diseases.
Concluding remarks
The discovery of Th17 cells represents a major step in our understanding of protective and pathological responses at the mucosal surfaces. However, there are still many unanswered questions, such as how endogenous and/or microbiota-derived stimuli trigger RORγt and consequently Th17 development in the mucosa. Additionally, it is not know which type/strains of commensal bacteria trigger natural production of Th17 in the intestine. A great deal of work needs to be done with regards to the precise regulatory mechanisms that govern the efficient generation, persistence, and reactivation of Th17 cells at different mucosal sites. The relationship between Tregs and Th17 cells as well as the possible pro-inflammatory role of Tregs that lose Foxp3 expression or co-express Foxp3 and RORγt are also of great interest. A largely unexplored topic is also the role of IL-17 production by cells other than CD4+ T cells. IL-17 can also be produced by CD8+ T cells, γδT cells, and in some cases even natural killer T (NKT) cells, macrophages, neutrophils, and eosinophils. Lastly, a critical direction for future studies of Th17 cells will be to determine what roles these cells play in human disease, and ultimately how useful or safe they will be as a therapeutic target.
Figure 1. Generation and regulation of Th17 responses in the intestine.
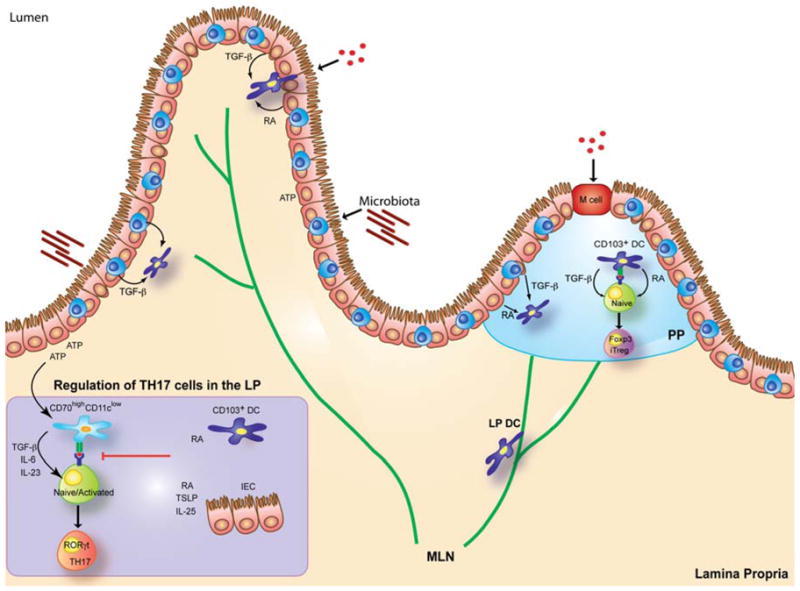
Under the influence of microbial-derived stimuli and ATP, lamina propria (LP) CD70high and/or CD103− DCs induce Th17 differentiation through the production of TGF-β, IL-6 and IL-23. Production of TGF-β by LP cells also promote the anti-inflammatory Foxp3+ induced regulatory T cells (iTreg). The balance between Th17 and Treg cell development is favored towards Treg cells upon the influence of retinoic acid (RA) produced by LP and Peyer’s patch CD103+ DCs, LP macrophages and intestinal epithelial cells (IEC). Besides RA, thymic stromal lymphopoietin (TSLP) expressed by IEC and the cytokine IL-25 were reported to suppress Th17 development in the intestine.
Acknowledgments
We thank Hilde Cheroutre and Alexandre Basso for their valuable suggestions for this manuscript.
This work was supported by a Career Development Award from The Crohn’s and Colitis Foundation of America (CCFA) to D.M, and NIH grant AI77079 to S.S.A. This is publication #1101 from the La Jolla Institute for Allergy and Immunology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cheroutre H. Starting at the beginning: new perspectives on the biology of mucosal T cells. Annu Rev Immunol. 2004;22:217–46. doi: 10.1146/annurev.immunol.22.012703.104522. [DOI] [PubMed] [Google Scholar]
- 2.Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol. 2007;25:221–42. doi: 10.1146/annurev.immunol.22.012703.104758. [DOI] [PubMed] [Google Scholar]
- 3.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 4.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol. 2007;19:409–17. doi: 10.1016/j.smim.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–81. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 7.Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, et al. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7:929–36. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- 8.Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, Johnson LM, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–45. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 9.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 10.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 11.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–4. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 12.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 13.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–48. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 14.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641–9. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4:337–49. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008 doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- 17.Zaph C, Du Y, Saenz SA, Nair MG, Perrigoue JG, Taylor BC, et al. Commensal-dependent expression of IL-25 regulates the IL-23-IL-17 axis in the intestine. J Exp Med. 2008;205:2191–8. doi: 10.1084/jem.20080720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25:309–18. doi: 10.1016/j.immuni.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 20.Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. 2006;203:2473–83. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med. 2006;203:2485–94. doi: 10.1084/jem.20061082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leppkes M, Becker C, Ivanov II, Hirth S, Wirtz S, Neufert C, et al. RORgamma-Expressing Th17 Cells Induce Murine Chronic Intestinal Inflammation via Redundant Effects of IL-17A and IL-17F. Gastroenterology. 2008 doi: 10.1053/j.gastro.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 23.Sanos SL, Bui VL, Mortha A, Oberle K, Heners C, Johner C, et al. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46(+) cells. Nat Immunol. 2008 doi: 10.1038/ni.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2008 doi: 10.1038/nature07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947–57. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 2008;29:958–70. doi: 10.1016/j.immuni.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 27.Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson RP. How HIV guts the immune system. N Engl J Med. 2008;358:2287–9. doi: 10.1056/NEJMcibr0802134. [DOI] [PubMed] [Google Scholar]
- 29.Raffatellu M, Santos RL, Verhoeven DE, George MD, Wilson RP, Winter SE, et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat Med. 2008;14:421–8. doi: 10.1038/nm1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Infante-Duarte C, Horton HF, Byrne MC, Kamradt T. Microbial lipopeptides induce the production of IL-17 in Th cells. J Immunol. 2000;165:6107–15. doi: 10.4049/jimmunol.165.11.6107. [DOI] [PubMed] [Google Scholar]
- 31.Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–77. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 32.Linden A, Laan M, Anderson GP. Neutrophils, interleukin-17A and lung disease. Eur Respir J. 2005;25:159–72. doi: 10.1183/09031936.04.00032904. [DOI] [PubMed] [Google Scholar]
- 33.Linden A, Adachi M. Neutrophilic airway inflammation and IL-17. Allergy. 2002;57:769–75. doi: 10.1034/j.1398-9995.2002.02164.x. [DOI] [PubMed] [Google Scholar]
- 34.Happel KI, Dubin PJ, Zheng M, Ghilardi N, Lockhart C, Quinton LJ, et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med. 2005;202:761–9. doi: 10.1084/jem.20050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–27. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu Q, Martin RJ, Rino JG, Breed R, Torres RM, Chu HW. IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect. 2007;9:78–86. doi: 10.1016/j.micinf.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zelante T, De Luca A, Bonifazi P, Montagnoli C, Bozza S, Moretti S, et al. IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur J Immunol. 2007;37:2695–706. doi: 10.1002/eji.200737409. [DOI] [PubMed] [Google Scholar]
- 39.Dubin PJ, McAllister F, Kolls JK. Is cystic fibrosis a TH17 disease? Inflamm Res. 2007;56:221–7. doi: 10.1007/s00011-007-6187-2. [DOI] [PubMed] [Google Scholar]
- 40.Oboki K, Ohno T, Saito H, Nakae S. Th17 and allergy. Allergol Int. 2008;57:121–34. doi: 10.2332/allergolint.R-07-160. [DOI] [PubMed] [Google Scholar]
- 41.Aujla SJ, Dubin PJ, Kolls JK. Interleukin-17 in pulmonary host defense. Exp Lung Res. 2007;33:507–18. doi: 10.1080/01902140701756604. [DOI] [PubMed] [Google Scholar]
- 42.Hellings PW, Kasran A, Liu Z, Vandekerckhove P, Wuyts A, Overbergh L, et al. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am J Respir Cell Mol Biol. 2003;28:42–50. doi: 10.1165/rcmb.4832. [DOI] [PubMed] [Google Scholar]
- 43.McKinley L, Alcorn JF, Peterson A, Dupont RB, Kapadia S, Logar A, et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol. 2008;181:4089–97. doi: 10.4049/jimmunol.181.6.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wakashin H, Hirose K, Maezawa Y, Kagami S, Suto A, Watanabe N, et al. IL-23 and Th17 cells enhance Th2-cell-mediated eosinophilic airway inflammation in mice. Am J Respir Crit Care Med. 2008;178:1023–32. doi: 10.1164/rccm.200801-086OC. [DOI] [PubMed] [Google Scholar]
- 45.Simonian PL, Roark CL, Wehrmann F, Lanham AK, Diaz del Valle F, Born WK, et al. Th17-polarized immune response in a murine model of hypersensitivity pneumonitis and lung fibrosis. J Immunol. 2009;182:657–65. [PMC free article] [PubMed] [Google Scholar]
- 46.Wang YH, Angkasekwinai P, Lu N, Voo KS, Arima K, Hanabuchi S, et al. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J Exp Med. 2007;204:1837–47. doi: 10.1084/jem.20070406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zaph C, Troy AE, Taylor BC, Berman-Booty LD, Guild KJ, Du Y, et al. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature. 2007;446:552–6. doi: 10.1038/nature05590. [DOI] [PubMed] [Google Scholar]
- 48.Mucida D, Kutchukhidze N, Erazo A, Russo M, Lafaille JJ, Curotto de Lafaille MA. Oral tolerance in the absence of naturally occurring Tregs. J Clin Invest. 2005;115:1923–33. doi: 10.1172/JCI24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ichiyama K, Yoshida H, Wakabayashi Y, Chinen T, Saeki K, Nakaya M, et al. Foxp3 inhibits RORgamma t-mediated IL-17A mRNA transcription through direct interaction with RORgamma t. J Biol Chem. 2008 doi: 10.1074/jbc.M801286200. [DOI] [PubMed] [Google Scholar]
- 50.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–40. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lochner M, Peduto L, Cherrier M, Sawa S, Langa F, Varona R, et al. In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORgamma t+ T cells. J Exp Med. 2008;205:1381–93. doi: 10.1084/jem.20080034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vaz NM, Maia LC, Hanson DG, Lynch JM. Inhibition of homocytotropic antibody responses in adult inbred mice by previous feeding of the specific antigen. J Allergy Clin Immunol. 1977;60:110–5. doi: 10.1016/0091-6749(77)90035-5. [DOI] [PubMed] [Google Scholar]
- 54.Curotto de Lafaille MA, Kutchukhidze N, Shen S, Ding Y, Yee H, Lafaille JJ. Adaptive Foxp3+ regulatory T cell-dependent and -independent control of allergic inflammation. Immunity. 2008;29:114–26. doi: 10.1016/j.immuni.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 55.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–64. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. 2007;204:1775–85. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–60. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 58.Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8:1086–94. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 59.Worbs T, Bode U, Yan S, Hoffmann MW, Hintzen G, Bernhardt G, et al. Oral tolerance originates in the intestinal immune system and relies on antigen carriage by dendritic cells. J Exp Med. 2006;203:519–27. doi: 10.1084/jem.20052016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bilsborough J, George TC, Norment A, Viney JL. Mucosal CD8alpha+ DC, with a plasmacytoid phenotype, induce differentiation and support function of T cells with regulatory properties. Immunology. 2003;108:481–92. doi: 10.1046/j.1365-2567.2003.01606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schambach F, Schupp M, Lazar MA, Reiner SL. Activation of retinoic acid receptor-alpha favours regulatory T cell induction at the expense of IL-17-secreting T helper cell differentiation. Eur J Immunol. 2007;37:2396–9. doi: 10.1002/eji.200737621. [DOI] [PubMed] [Google Scholar]
- 62.Elias KM, Laurence A, Davidson TS, Stephens G, Kanno Y, Shevach EM, et al. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111:1013–20. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, et al. Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol. 2008;181:2277–84. doi: 10.4049/jimmunol.181.4.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Albanesi C, Scarponi C, Pallotta S, Daniele R, Bosisio D, Madonna S, et al. Chemerin expression marks early psoriatic skin lesions and correlates with plasmacytoid dendritic cell recruitment. J Exp Med. 2009;206:249–58. doi: 10.1084/jem.20080129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–81. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boniface K, Blom B, Liu YJ, de Waal Malefyt R. From interleukin-23 to T-helper 17 cells: human T-helper cell differentiation revisited. Immunol Rev. 2008;226:132–46. doi: 10.1111/j.1600-065X.2008.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Massacesi L, Castigli E, Vergelli M, Olivotto J, Abbamondi AL, Sarlo F, et al. Immunosuppressive activity of 13-cis-retinoic acid and prevention of experimental autoimmune encephalomyelitis in rats. J Clin Invest. 1991;88:1331–7. doi: 10.1172/JCI115438. [DOI] [PMC free article] [PubMed] [Google Scholar]