

Abstract
Receptor for Activated C Kinase, RACK1, is an adaptor protein that regulates signaling via Src and PKC-dependent pathways, and has been implicated in cell migration. In this study we demonstrate novel functions for RACK1 in regulating adhesion dynamics during cell migration. We report that cells lacking RACK1 are less motile and show reduced dynamics of paxillin and talin at focal complexes. To investigate the role of the RACK1/Src interactions in adhesion dynamics, we used RACK1 in which the putative Src binding site has been mutated (RACK Y246F). RACK1-deficient cells showed enhanced c-Src activity that was rescued by expression of wild type RACK1, but not by RACK Y246F. Expression of wild type RACK1, but not RACK Y246F, was also able to rescue the adhesion and migration defects observed in the RACK1-deficient cells. Furthermore, our findings indicate that RACK1 functions to regulate paxillin phosphorylation and that its effects on paxillin dynamics requires the Src-mediated phosphorylation of tyrosine 31/118 on paxillin. Taken together, these findings support a novel role for RACK1 as a key regulator of cell migration and adhesion dynamics through the regulation of Src activity, and the modulation of paxillin phosphorylation at early adhesions.
Keywords: RACK1, Src, adhesion, cell migration
Introduction
Dynamic regulation of integrin-mediated adhesions is critical for cell migration. Integrins assemble into multi-protein adhesion complexes, called focal adhesions that contain a variety of cytoskeletal, adaptor and signaling proteins [1, 2]. These adhesion complexes are crucial for both the structural association between the extracellular matrix (ECM) and the actin cytoskeleton, and for the transduction of signals that regulate a variety of cellular functions, including cell survival, migration and differentiation [3, 4]. There has been substantial interest in dissecting the molecular mechanisms that regulate the composition and dynamics of focal adhesions. Direct correlations have been made between the organization and composition of adhesions and the ability of cells to migrate. For example, previous studies have shown that an absence of central focal adhesions and an increase in the size of peripheral adhesions is associated with impaired adhesion turnover and reduced cell migration. Accordingly, fibroblasts deficient in focal adhesion kinase [5, 6], Src kinases [5, 6] or the intracellular calcium-dependent protease calpain [7, 8] show an increase in the size of peripheral adhesions and reduced adhesion turnover and migration.
The scaffolding protein and regulator of c-Src activity, receptor for activated C kinase (RACK1) has also been implicated in the regulation of focal adhesion organization and cell migration [9, 10]. RACK1 is a 36 kDa cytosolic protein that contains seven WD repeats, and belongs to a superfamily of proteins that includes the β subunit of G-proteins. RACK1 was originally identified based on its ability to bind to and translocate the activated form of protein kinase C to specific regions within the cell [11]. RACK1 has also been reported to be a binding partner and inhibitor of Src tyrosine kinases [12], and the interaction between RACK1 and Src has been implicated in the regulation of cell growth and migration [10, 13]. RACK1 has many other binding partners involved in the organization of adhesions and cell migration, including the cytoplasmic tail of β integrins [9, 14], phospholipase C-γ and RasGAP [15], PTPμ [16], β-spectrin and dynamin [17] and Fyn [18]. These interactions support the role of RACK1 as a key scaffolding protein that mediates protein-protein interactions that regulate cell motility. However, much remains unknown about the functional role of RACK1 during cell migration.
RACK1 has been reported to bind and be a substrate of c-Src at least in part through tyrosine 246 [15] and negatively regulate Src activity [10, 12, 19]. Overexpression of RACK1 increases the number of central focal adhesions and reduces cell migration at least in part through its interaction with Src [10]. Endogenous pp60Src is one of the critical kinases involved in regulating phosphorylation events at cell adhesions by targeting specific focal adhesion proteins including the adaptor protein paxillin [20]. Tyrosine phosphorylation of focal adhesion proteins has been implicated in the formation and dynamics of focal adhesions by creating docking sites for proteins that contain phosphotyrosine (PY) binding domains [21]. Previous studies indicate that RACK1 localizes to a subset of peripheral adhesions that contain paxillin [10], suggesting that RACK1 is an attractive candidate to provide a mechanism to locally regulate paxillin phosphorylation and dynamics through its modulation of Src.
Using siRNA, we now show that RACK1 is required for efficient cell migration and the dynamic turnover of adhesions. Specifically, RACK1 is required for the efficient assembly and disassembly of paxillin into adhesions. RACK1-deficient cells displayed enhanced c-Src activity and phosphorylation of paxillin, suggesting that RACK1 is a critical negative regulator of endogenous c-Src activity. The effects of RACK1 on paxillin dynamics require tyrosine 31 and tyrosine 118 on paxillin, indicating that RACK1 modulates paxillin dynamics by regulating its tyrosine phosphorylation. To examine the dynamics of phosphoproteins at adhesion sites, we used GFP-tagged SH2 domain of Src as a reporter of tyrosine phosphorylation at adhesions [22]. Tyrosine phosphorylation of adhesion proteins was enhanced in RACK1-deficient cells, and our findings suggest that the turnover of phosphoproteins at adhesions may be reduced in cells that are deficient in RACK1. Taken together, the findings suggest that RACK1 is required for efficient assembly and disassembly of paxillin at adhesion sites, at least in part through its modulation of Src activity and the dynamic regulation of paxillin phosphorylation.
Materials and Methods
Antibodies and Reagents
Fibronectin was purified from human plasma by affinity chromatography as described previously [23]. Ham’s F12 was purchased from Invitrogen (Carlsbad, CA). Fatty-acid-free BSA was purchased from Sigma (St. Louis, MO). DMEM was purchased from Mediatech (Herndon, VA). Monoclonal anti-paxillin, anti-RACK1 and anti-FAK antibodies were purchased from BD Biosciences (Franklin Lakes, NJ). Anti-phosphospecific paxillin [pY118], anti-phosphospecific paxillin [pY31] and anti-phosphospecific FAK [pY576] antibodies were purchased from Biosource (Camarillo, CA). Anti-Src antibody (clone GD11) was obtained from Upstate Biotechnology (Lake Placid, NY). Anti-Src antibody was also obtained from Sigma (St. Louis, MO). Rhodamine-labeled phalloidin, rabbit anti-GFP antibody, Rhodamine Red-X goat-anti-mouse IgG and AlexaFluor 680 goat-anti-mouse IgG were purchased from Invitrogen (Carlsbad, CA). IRDye 800CW goat-anti-rabbit IgG was obtained from Rockland Immunochemicals (Gilbertsville, PA). Anti-mouse IgG (ChromaPure; whole molecule) was from Jackson ImmunoResearch Laboratories (West Grove, PA). Protein G-Sepharose beads were purchased from Amersham Biosciences (Piscataway, NJ). Enolase was purchased from Sigma-Aldrich (St. Louis, MO). Src inhibitor, PP2, was purchased from Axxora Life Sciences (San Diego, CA).
Constructs and siRNA
DsRed-paxillin was a gift from A.F. Horwitz (University of Virginia) and have been previously described [6]. GFP-paxillin and GFP-paxillin Y31,118F were gifts from C.E. Turner (State University of New York Health Science Center) and have been previously described [24]. GFP-talin [8], GFP-RACK1 and GFP-RACK Y246F have been previously described [10]. pcDNA3.1 was purchased from Invitrogen (Carlsbad, CA). pcDNA3.1 Src-Y527F was created by digesting RcRSV-Src-Y527F (a gift from A.B. Reynolds, Vanderbilt) with ApaI and EcoRI and subcloning the resulting Src-Y527F cDNA into pcDNA3.1. RNA interference of RACK1 was performed using Dharmacon’s SMARTpool siRNA (Dharmacon, Lafayette, CO). As a control, siCONTROL NonTargeting siRNA Pool was used (Dharmacon) as described by manufacturer. For rescue experiments single siRNA RACK1 target sequence was used: 5′-CCAAGGATGTGCTGAGTGT-3′. GFP-RACK1 and GFP-RACK Y246F silent mutations were made using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) along with the following primers: forward, 5′-GGGCCATACCAAAGACGTCCTGAGCGTCGCCTTCTCC-3′; reverse, 5′-GGAGAAGGCGACGCTCAGGACGTCTTTGGTATGGCCCC-3′. All constructs were subjected to DNA sequencing to ensure accuracy. GFP-tagged dSH2 was a gift from T.M. Gomez and has been previously described [22, 25].
Cell Culture and Transfection
Human embryonic kidney (HEK)-293 cells were purchased from American Type Culture Collection (Manassas, VA) and cultured in complete growth media as described previously [8]. Transient co-transfection of the siRNA oligos and DNA constructs into HEK cells was carried out by a modified calcium phosphate precipitation protocol [26]. Briefly, HEK cells were seeded at 1.5 × 106 cells on 60mm tissue culture plates and transfected a day later with 3 μg of siRNA oligo and 1.5μg DNA constructs. Cells were used in experiments 48 h post-transfection unless otherwise noted. Analysis by flow cytometry showed transfection efficiencies between 80–90% 48 h post-transfection.
Immunoblot Analysis
HEK cells were plated in complete DMEM on tissue-treated dishes and incubated at 37°C under 5% CO2 for 48 h post-transfection. FN signaling studies were performed with cells plated on dishes coated with 10 μg ml−1 FN in DMEM containing 0.2% fatty-acid-free BSA and incubated at 37° C with 5% CO2 for 2 h. Cells were washed one time with DPBS, scraped into 1% NP-40 lysis buffer (1% NP-40, 10% glycerol, 75 mM NaCl, 50mM HEPES, supplemented with 1 μg/ml pepstatin A, 10 μg/ml leupeptin, 20 μg/ml aprotinin, 200 nM phenylmethylsulphonyl fluoride, 100 μM sodium vanadate, and Phospho-cocktail inhibitors) on ice and cleared of insoluble material by centrifugation. Protein concentrations were determined using a BCA protein assay kit (Pierce Biotechnology, Rockford, IL) according to manufacturer’s instructions. Equal amounts of total protein were denatured in SDS sample buffer, resolved on 4–20% gradient SDS-PAGE and transferred to nitrocellulose. For phospho-Src analysis, 48 h post-transfection, cells were plated on dishes coated with 10 μg ml−1 FN in DMEM containing 0.2% fatty-acid-free BSA and incubated at 37° C under 5% CO2 for 2 h. Cells were lysed in 50 mM Tris pH 7.5, 1% Triton-X, 5 mM EGTA, 150 mM NaCl, 10 mM sodium phosphate, 10 mM sodium fluoride, 5 mM iodoacetic acid, 1 mM benzamidine, 10 μg ml−1 aprotinin and 10 μg ml−1 leupeptin on ice, clarified by centrifugation and 10 mM DTT was added to the lysate after protein concentration determination [27]. Protein concentrations were determined using a BCA protein assay kit (Pierce Biotechnology, Rockford, IL) according to manufacturer’s instructions. Equal amounts of total protein were denatured in SDS sample buffer, resolved on 8% SDS-PAGE and transferred to nitrocellulose. Immunoblots were imaged and quantified with an Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE) as previously described [8].
Immunoprecipitation and Src Activity Assay
For the Src Activity Assay immunoprecipitations, 48 h post-transfection, HEK-293 cells were plated on 15-cm dishes coated with 10 μg ml−1 FN. Cells were incubated in DMEM containing 0.2% fatty-acid-free BSA for 2 h at 37°C in 5% CO2, washed one time with PBS and lysed with ice-cold RIPA buffer. Lysates were cleared of insoluble material by centrifugation and 500 μg of cleared lysate was used for the immunoprecipitation step. Next, 1 μg of anti-Src (Upstate) and 1 μg of anti-Src (Sigma) antibodies, or 2 μg of mouse IgG were added to the cleared lysates and incubated at 4°C with tilting for 2 hours. 20 μl of protein G-Sepharose (Amersham Biosciences, Piscataway, NJ) was then added to immunoprecipitations and incubated at 4°C with tilting for 1 hour. Samples were then washed three times with radioimmunoprecipitation assay buffer and split into equal volumes for immunoblot analysis or kinase assay. For immunoblot analysis, immunoprecipitated Src was extracted from beads with Laemmli SDS sample buffer and boiled for 10 min. Samples were run on an 8% SDS-polyacrylamide gel, and transferred to nitrocellulose. Immunoblots were imaged and quantified with an Odyssey Infrared Imaging System (LI-COR Biosciences, Omaha, NE). Kinase assays were performed as described previously [12] and were terminated with Laemmli SDS sample buffer and boiling for 10 minutes. Reactions were then run on an 8% SDS-polyacrylamide gel, dried and exposed to a phosphoimage screen for 15–20 hours.
Immunoprecipitation
For immunoprecipitation of dSH2, HEK cells were co-transfected with either pooled control-siRNA/GFP-dSH2 or pooled RACK1-siRNA/GFP-dSH2. 48 h post-transfection, cells were lysed with 1% NP-40 lysis buffer (same as above) and cleared of insoluble material by centrifugation. Protein concentrations were determined using a BCA protein assay kit according to manufacturer’s instructions. Lysates were incubated with either 1μl of anti-GFP antibody or 1 μg of rabbit IgG. Immune complexes were captured on 10μl of protein G-Sepharose (Amersham Biosciences, Piscataway, NJ), washed in lysis buffer, and analyzed by immunoblotting with anti-paxillin antibody.
Immunofluorescence
Cells were prepared for use in immunofluorescence cell staining experiments as described above. Glass coverslips were acid-washed, silanated, conjugated to 3 μg/ml fibronectin, and blocked with 2% BSA using a previously described method [28]. Transiently transfected HEK cells were plated onto coverslips in complete media at 5 × 104 per coverslip and allowed to adhere for 2 h. Cells were then fixed, permeabilized and stained for endogenous paxillin as described previously [29]. For the Src-Y527F overexpression studies, transiently transfected HEK cells were plated onto coverslips, conjugated with 3 μg/ml fibronectin, in complete media at 5 × 104 per coverslip and allowed to adhere for 2 h. Cells were then fixed with 3% formaldehyde, quenched in 0.15M Glycine and mounted onto slides. Coverslips were observed using an inverted fluorescent microscope (Nikon Eclipse TE300, Kanayawa, Japan).
Time-lapse microscopy
Fluorescence imaging of live cells was performed using a 60 X objective on an Olympus IX-70 inverted microscope housed in a closed system to maintain the temperature at 37°C and CO2 levels at 5%. Glass-bottom 35-mm dishes were acid-washed and coated with 3 μg/ml fibronectin. 48 h post-transfection, cells were plated in complete media at 7.5 × 104 and allowed to adhere for 2 h, after which time the media was replaced with Ham’s F12 supplemented with 0.2% fatty-acid-free BSA. Fluorescent images were then captured every 2 min for 2 h using MetaVue Imaging software (Universal Imaging Corporation, Downington, PA).
Dual fluorescence imaging of live cells was performed using a 60 X objective on a Nikon Eclipse TE300 microscope housed in a closed system to maintain the temperature at 37°C and CO2 levels at 5%, and imaged as above. Fluorescent images were captured every 2 min for 2 h using MetaMorph Imaging software (Universal Imaging Corporation, Downington, PA).
Quantification of Adhesion Dynamics
Time-lapse sequences from live fluorescence imaging were first subjected to high-pass filtration to remove diffuse background fluorescence [30]. The dynamics of fluorescently tagged constructs were quantified according to the described protocols [6, 8].
Fluorescence intensities of individual adhesion complexes from background-subtracted images were measured over time using MetaMorph Imaging software (Universal Imaging Corporation, Downington, PA) as previously described [8]. For each rate constant, measurements were made on at least 15 individual focal complexes throughout the cell, from three individual experiments. Duration measurements were made for these same complexes by counting the amount of time lapsed between the first and last frames in which an individual adhesion complex was observed. All measurements shown are the mean ± standard error of the mean (SEM).
Wound Healing Assay
For the wound healing assays, 24 h post-transfection, HEK cells were seeded onto 35mm tissue cultured dishes at 1.2 × 106 and allowed to adhere overnight to create a confluent monolayer. 48 h post-transfection, scratch wounds were made on the cell surface with a micropipette tip. The wounds were rinsed two times gently with DPBS and replaced with complete media. Migration was quantified 8 h after wounding, and images were either captured every 10 min, using a 10 X objective on an Olympus IX-70 inverted microscope housed in a closed system to maintain the temperature at 37°C and CO2 levels at 5%, or photographed at various time points after the wound. Wound healing was quantified by measuring the change in area of a wound from time 0 h to 8 h and graphed as the relative wound closure compared to controls cells. Measurements were done in three to four separate experiments. All measurements shown are the mean ± standard error of the mean (SEM). P values were calculated using Student’s t-test.
Results
RACK1-deficient cells have increased endogenous c-Src activity
Previous studies have indicated that RACK1 is able to bind and negatively regulate Src [12], and that this binding is at least in part through RACK1 tyrosine 246 [15]. To determine if RACK1 regulates endogenous c-Src activity, Src activity was analyzed in RACK1-deficient cells. We generated RACK1-deficient HEK cells using RNA interference (siRNA) with either pooled siRNA (Figure 1A) or single target siRNA (Figure 1B). RACK1 has been reported to have a long half-life (t1/2 = 40 h) [31], and we therefore determined the optimum time post-transfection for maximum knockdown of endogenous RACK1. We observed 80–90% knockdown efficiency 48 to 72 h post-transfection with pooled siRNA (Figure 1A), and single target siRNA, with only modest knockdown 24 h post-transfection. Therefore, all experiments using siRNA were analyzed 48–72 h after transfection. To further elucidate the role of RACK1-Src interactions in regulating Src activity, we generated silent mutations that produced mismatches in the region homologous to the single target siRNA sequence that allowed successful rescue with GFP-RACK1 and GFP-RACK Y246F in RACK1-deficient cells (Figure 1B). We observed a substantial increase in Src activity in the RACK1-deficient cells that was rescued by expression of wild type RACK1 but not the mutant, RACK Y246F (Figure 1C). Together, these findings indicate that RACK1 is a critical negative regulator of endogenous c-Src activity, and that this inhibitory effect requires tyrosine 246.
Figure 1.

RACK1 is a negative regulator of endogenous c-Src activity. (A) HEK cells were transiently transfected with pooled control siRNA (Csi) or pooled RACK1 siRNA (Rsi) for 24 h, 48 h and 72 h as indicated. Representative blot shows expression of RACK1. Actin was used as a loading control. (B) HEK cells were transiently transfected with single target control siRNA (Csi) or RACK1 siRNA (Rsi) co-expressed with GFP, GFP-RACK1 (RACK1) or GFP-RACK Y246F (Y246F) as described in Materials and Methods. Representative blot shows knockdown of endogenous RACK1 and expression of GFP constructs as indicated by immunoblotting with α-RACK1 or α-GFP. (C) HEK cells were transiently transfected with control siRNA (Csi) or RACK1 siRNA (Rsi) co-expressed with GFP, GFP-RACK1 (RACK1) or GFP-RACK Y246F (Y246F). Immunoprecipitation was performed on lysates using a Src-specific antibody. Kinase activity assays were performed with or without Src inhibitor PP2 as described in Materials and Methods and the autoradiograph is shown. Quantification of Src activity from three separate experiments is shown as the mean ± SEM.
RACK1-deficient cells have impaired cell migration
Previous studies have demonstrated that RACK1 regulates cell migration [9, 10], and that its interaction with Src may play a role in this regulation [32, 33]. To determine if RACK1 is required during cell migration, wound healing assays were performed in RACK1-deficient cells. RACK1-deficient cells showed reduced migration by wound healing (Figure 2A–B; supplemental video 1–2) and transwell assay (data not shown). Furthermore, the migration defect was rescued by expression of GFP-RACK1 but not by the expression of GFP-RACK Y246F, suggesting that tyrosine 246 is required for RACK1-mediated cell migration (Figure 2A–B; supplemental video 3–4). Interestingly, time-lapse movies (supplemental movies 1–4) showed that cells that are deficient in RACK1 displayed more transient protrusive activity and that this phenotype was rescued by expression of wild type RACK1, but not RACK Y246F. This is in agreement with our previous publication that indicated that RACK1 regulates transient protrusion through tyrosine 246 [10]. Furthermore, the movies suggest that the RACK1-Src interaction may modulate cell-cell contact during wound closure since the cells that expressed RACK Y246F displayed a substantial loss of cell-cell contact, which may contribute to the impaired rate of wound closure. In accordance with these findings, previous studies have shown that RACK1 associates with PTPμ at cell-cell contact sites and regulates cell-cell adhesion [16]. Taken together, our findings suggest that RACK1 is required for efficient cell migration at least in part through its regulation of Src activity.
Figure 2.

RACK1 modulates cell migration through its regulation of Src activity. (A) HEK cells were co-transfected with control siRNA (Csi) or RACK1 siRNA (Rsi) with GFP, GFP-RACK1 (RACK1) or GFP-RACK Y246F (Y246F) as indicated. Wound assays were performed as described in Materials and Methods. Representative images from three separate experiments are shown at time 0 h and time 8 h after wounding. See supplemental movies 1–4. Bar, 50 μm. (B) Quantification of wound assay was performed by measuring the change in wound area from 0 h to 8 h post-wound relative to control cells as described in Materials and Methods. Data represent the average from three separate experiments, with error bars representing as the mean ± SEM. *p<0.01 in a Student’s t test in comparison to control.
RACK1 regulates paxillin morphology
Recent studies have demonstrated that RACK1 regulates focal adhesion organization [9, 10, 33], although the mechanisms of this regulation have not been clearly defined. Since RACK1 localizes to early adhesions, or focal complexes at the cell periphery, we examined the role of RACK1 in early focal complex formation. To determine if RACK1 affects early adhesions, we examined the morphology of paxillin-containing adhesions two hours after plating control and RACK1-deficient cells. RACK1-deficient cells displayed an enhancement of paxillin-containing adhesions at the cell periphery (Figure 3A). Since RACK1 has been implicated as a modulator of translation by its association with ribosomes [34], we performed immunoblotting to ensure that the increase in paxillin at adhesions was not secondary to a change in paxillin expression. There was no change in global expression of any of the focal adhesion proteins tested including paxillin, talin and focal adhesion kinase (data not shown). To determine if RACK1-Src interactions are important for RACK1 effects on paxillin morphology in early focal complexes, rescue experiments were performed with GFP-RACK1 and GFP-RACK Y246F (Figure 3B). Our findings suggest that wild type RACK1 but not RACK Y246F was sufficient to rescue the focal complex morphology observed in RACK1-deficient cells. Together, our findings suggest that RACK1 plays a role in regulating the morphology of paxillin-containing focal complexes through a Src-dependent pathway.
Figure 3.
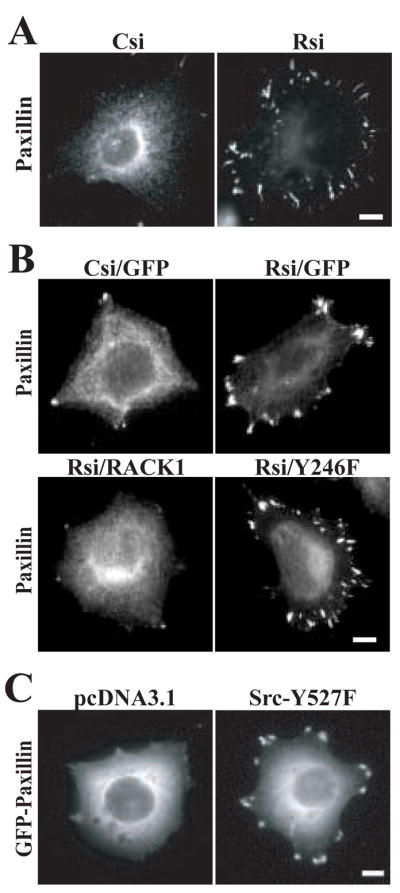
RACK1 regulates paxillin organization at focal complexes. (A) HEK cells were plated for 2 h on coverslips coated with 3 μg/ml fibronectin as described in the Materials and Methods. Representative images from more than four separate experiments show the distribution of paxillin in cells that express pooled control siRNA (Csi) or pooled RACK1 siRNA (Rsi). Bar, 10 μm. (B) HEK cells were co-transfected with single target control siRNA (Csi) or RACK1 siRNA (Rsi) with GFP, GFP-RACK1 (RACK1) or GFP-RACK Y246F (Y246F) as indicated. Representative images are shown from more than four separate experiments. Bar, 10 μm. (C) HEK cells were co-transfected with pcDNA3.1 or Src-Y527F with GFP-paxillin as indicated. Representative images are from three separate experiments. Bar, 10 μm.
Constitutively active Src regulates paxillin morphology
Previous studies have implicated Src activity in both the assembly and disassembly of adhesions [35, 36]. Our findings suggest that the enhanced Src activity in RACK1-deficient cells induces the formation of prominent paxillin-containing focal adhesions in HEK cells. To determine if enhanced Src activity alone is sufficient to induce formation of paxillin-containing focal adhesions, we co-expressed constitutively active Src-Y527F along with GFP-paxillin in HEK cells and assayed focal adhesion morphology. We found that active Src as compared to vector control induced the formation of paxillin-containing focal adhesions in HEK cells (Figure 3C), suggesting that active Src is sufficient to induce the formation of prominent paxillin-containing adhesions.
RACK1 regulates paxillin dynamics at peripheral adhesion complexes
To determine if RACK1 regulates paxillin dynamics at peripheral adhesions, live cell imaging was performed in control and RACK1-deficient cells that express GFP-paxillin. Live fluorescence imaging of GFP-paxillin in these cells revealed the persistence of paxillin at peripheral adhesion complexes for extended durations in the absence of RACK1 (Figure 4A,B; supplemental video 5 & 6). Using previously described methods for measuring the kinetics of adhesion components [6, 8], we measured both assembly and disassembly of paxillin in control and RACK1 siRNA-treated cells. Interestingly, we observed a reduction in both the average assembly and disassembly rates of paxillin at adhesions in RACK1-deficient cells (Figure 4B). Representative intensity graphs show that the dramatic increase in duration of paxillin at adhesions in RACK1-deficient cells is likely the result of a slow rate of adhesion assembly, a long transition period between assembly and disassembly, and reduced disassembly rate (Figure 4C). These results suggest that RACK1 modulates the rate at which paxillin-containing adhesions both assemble and disassemble, providing a novel role for RACK1 in regulating paxillin dynamics at early adhesions in adherent cells.
Figure 4.

Turnover of paxillin at focal complexes is reduced in the absence of RACK1. (A) GFP-paxillin was co-transfected with pooled control (Csi) or RACK1 (Rsi) siRNA. in HEK cells. Cells were plated on 3 μg/ml fibronectin and imaged by time-lapse fluorescence microscopy. Representative frames are shown from time-lapse sequences used to quantify duration and assembly/disassembly rates. See supplemental movies 5 and 6. For each sequence, T= 0 min is the first frame in which the adhesion marked by the arrow was observed. Bar, 3 μm. (B) Quantification of the persistence of GFP-paxillin in focal complexes demonstrates that paxillin remains at focal complexes for longer durations in the absence of RACK1. Duration measurements were made by counting the amount of time elapsed between the first and last frames in which an individual adhesion was observed in HEK cells that co-express GFP-paxillin and control siRNA (Csi) or RACK1 siRNA (Rsi). Rate constants for assembly and disassembly kinetics were calculated as described in the Materials and Methods. Quantification shows the mean ± SEM from three separate experiments (total of 45 adhesions quantified for each condition). (C) Representative fluorescence intensity graphs for HEK cells that express control (Csi) and RACK1 (Rsi) siRNA over time (min).
To determine if RACK1 affects the dynamics of other adhesion components at early focal complexes, we performed live dual imaging of GFP-talin and dsRed-paxillin co-expressed in control or RACK1-deficient cells. We found that both paxillin and talin-containing adhesions were larger and more stable in RACK1-deficient cells as compared to control cells (Figure 5; supplemental movies 7 & 8). These findings suggest that RACK1 is involved in regulating the dynamics of early adhesion complexes at the cell periphery. In agreement with this possibility, recent studies have reported that RACK1 and talin associate in a complex [37], suggesting that RACK1 may be involved in regulating talin at early focal complexes. Taken together, these findings suggest that RACK1 modulates the dynamics of early focal complexes formed at the cell periphery.
Figure 5.

RACK1 regulates the dynamics of both paxillin and talin at focal complexes. Cells were plated on 3 μg/ml fibronectin and imaged by time-lapse fluorescence microscopy as described in Materials and Methods. Representative frames are shown from time-lapse sequences collected every 2 min. See supplemental movies 7 and 8. Dual imaging of GFP-talin and dsRed-paxillin in HEK cells that expressed pooled control (Csi) and RACK1 (Rsi) siRNA. Bar, 10 μm.
RACK1 modulates paxillin phosphorylation through a Src-dependent pathway
To determine how RACK1 specifically modulates the dynamics of paxillin at adhesions, we characterized the effects of RACK1 on paxillin phosphorylation. Previous studies have demonstrated that phosphorylation of paxillin modulates cell migration [24]. One of the main kinases responsible for paxillin phosphorylation is Src [35], and our findings demonstrate a substantial increase in Src activity in RACK1-deficient cells. Therefore, we hypothesized that RACK1 may be modulating paxillin at focal complexes by regulating Src-mediated phosphorylation of paxillin. In accordance with this hypothesis, we observed enhanced phosphorylation of tyrosine 118 on paxillin in RACK1-deficient cells up to 72 h after treatment with RACK1 siRNA as compared to control siRNA (Figure 6A). This enhancement of paxillin phosphorylation required Src since the Src inhibitor PP2 blocked the increase in paxillin phosphorylation induced by knocking down RACK1 (Figure 6B). To determine whether expression of RACK1 rescues the phosphorylation phenotype, paxillin phosphorylation was characterized in cells rescued for RACK1 expression. Expression of wild type RACK1 but not RACK Y246F rescued paxillin phosphorylation to basal levels (Figure 6C,D). In accordance with the enhanced Src activity in RACK1-deficient cells, we also observed increased FAK phosphorylation on the Src-specific site (Y576) and on tyrosine 31 of paxillin (Figure 6E). Together, the data suggest that RACK1 regulates phosphorylation of focal adhesion proteins through a Src-dependent pathway.
Figure 6.

RACK1 regulates phosphorylation of focal adhesion proteins through a Src-dependent pathway. (A) HEK cells were transiently transfected with pooled control siRNA (Csi) or RACK1 siRNA (Rsi) for 24 h, 48 h and 72with h. Representative blots show paxillin phosphorylation at Y118. Total paxillin was used as a loading control. (B) HEK cells transiently transfected with pooled control (Csi) or RACK1 (Rsi) siRNA were treated with control vehicle (−) or the Src inhibitor, PP2. Cell lysates were collected and immunoblotted for phospho-paxillin (pY118), total paxillin and RACK1 as indicated. Representative blot from three experiments is shown. (C) HEK cells were transiently transfected with single target control siRNA (Csi) or RACK1 siRNA (Rsi) co-expressed with GFP, GFP- GFP-RACK1 (RACK1) or GFP-RACK Y246F (Y246F). Representative blot shows paxillin phosphorylation at Y118 from cells plated on fibronectin as described in Materials and Methods. Total paxillin was used as a loading control. (D) Quantification of paxillin phosphorylation relative to control lysates for conditions described in (C). Data represents the average from five separate experiments, with error bars represented as the mean ± SEM. *p<0.03 using a Student’s t test (in comparison with control). (E) HEK cells were transiently transfected with single target control siRNA (Csi) or RACK1 siRNA (Rsi) co-expressed with GFP, GFP-RACK1 (RACK1) or GFP-RACK Y246F (Y246F). Representative blot shows FAK phosphorylation at the Src-specific site Y576 and paxillin phosphorylation at Y31 from cells plated on fibronectin as described in Materials and Methods. Total FAK and paxillin were used as loading controls.
Phosphorylation at tyrosine 31, 118 is required for RACK1 effects on paxillin morphology and dynamics
Phosphorylation of paxillin tyrosine residues 31 and 118 has been shown to be essential for actin cytoskeleton-dependent cell spreading and motility in adherent cells [24, 38, 39]. Previous studies have also shown that phosphorylation on Y31, 118 are required for efficient disassembly of paxillin from adhesions [6]. We have shown that RACK1 modulates paxillin dynamics and in the absence of RACK1 there is an increase in paxillin phosphorylation. To determine whether phosphorylation at tyrosine 31 and 118 is required for RACK1 effects on paxillin morphology, GFP-paxillin Y31,118F was expressed in control and RACK1-deficient cells. In contrast to cells that express wild type GFP-paxillin, RACK1 siRNA had no effect on paxillin morphology in cells that express GFP-paxillin Y31,118F (Figure 7A). In addition, the duration of GFP-paxillinY31,118F was not affected by RACK1 siRNA (Figure 7B,C), further supporting a critical role for paxillin phosphorylation in mediating RACK1 effects on paxillin morphology and dynamics. Taken together, these findings suggest that RACK1 affects paxillin dynamics at least in part through the regulated phosphorylation of paxillin at tyrosines 31 and 118.
Figure 7.

Paxillin phosphorylation is required for RACK1 effects on paxillin morphology and dynamics. (A) HEK cells were co-transfected with control siRNA or RACK1 siRNA with either wild type GFP-paxillin or GFP-paxillin that can not be phosphorylated at tyrosine 31 or 118 (GFP-paxillin Y31,118F). Representative images show the localization of GFP-paxillin or GFP-paxillin Y31,118F. Bar, 10 μm. (B) Representative frames are shown from time-lapse sequences used to quantify duration of adhesions. For each sequence, T= 0 min is the first frame in which the adhesion marked by the arrow was observed. Bar, 3 μm. (C) Quantification of GFP-paxillin Y31,118F duration at focal complexes in control and RACK1-deficient cells. The graph represents the average from three separate experiments, with error bars represented as the mean ± SEM.
RACK1 modulates the dynamics of tyrosine phosphorylation at adhesions
Previous studies have indicated that tyrosine phosphorylation of paxillin is required for its efficient turnover at adhesions [6]. However, our findings suggest that the increase in paxillin phosphorylation observed in RACK1-deficient cells may be associated with reduced adhesion turnover, suggesting that constitutive phosphorylation of paxillin may lead to less dynamic adhesions. Phosphorylation of many focal adhesion proteins provides docking sites for additional molecules that contain phosphotyrosine (PY)-binding domains such as Src-homology 2 (SH2) domains [40, 41]. To determine whether RACK1 affects phosphorylation at focal complexes, we utilized a GFP-tagged phosphotyrosine reporter construct consisting of tandem repeats of the PY-binding SH2 domains of pp60c-Src (GFP-dSH2) [22, 25] an established reporter of tyrosine phosphorylation at adhesions. Expression of GFP-dSH2 labeled focal complexes in HEK cells and showed enhancement at peripheral adhesions in cells that express RACK1 siRNA as compared to control siRNA (Figure 8A). Furthermore, the duration of GFP-dSH2 was increased in RACK1-deficient cells (Figure 8B), suggesting that RACK1 is involved in the dynamics of phosphoproteins at adhesion sites. To determine if there is an increased association of dSH2 with paxillin in RACK1-deficient cells as compared to control cells, co-immunoprecipitation experiments were performed. We observed an increase in paxillin pulldown from RACK1-deficient cells, suggesting that there is an increase in dSH2 association with phosphorylated paxillin in the absence of RACK1 (Figure 8C). Taken together, our findings suggest that RACK1 regulates the dynamics of phosphoproteins at adhesions through Src-mediated phosphorylation of paxillin. It also suggests that constitutive phosphorylation of paxillin may be associated with its stabilization at focal complexes.
Figure 8.

Turnover of phosphoproteins at focal complexes is reduced in the absence of RACK1. (A) GFP-dSH2 was co-transfected with pooled control (Csi) or RACK1 (Rsi) siRNA in HEK cells. Cells were plated on 3 μg/ml fibronectin and imaged by time-lapse fluorescence microscopy. Representative frames are shown from time-lapse sequences used to quantify duration of adhesions. For each sequence, T= 0 min is the first frame in which the adhesion marked by the arrow was observed. Bar, 3 μm. (B) Quantification of the persistence of GFP-dSH2 in focal complexes demonstrates that phosphoproteins remain at focal complexes for longer durations in the absence of RACK1. Duration measurements were made by counting the amount of time elapsed between the first and last frames in which an individual adhesion was observed in HEK cells that express control siRNA (Csi) or RACK1 siRNA (Rsi). The graph represents the average from at least three separate experiments, with error bars represented as the mean ± SEM. (C) HEK cells were transiently transfected with dSH2-GFP and either control (Csi) or RACK1 (Rsi) siRNA for 48 h. Immunoprecipitation for GFP or nonspecific IgG control was performed as described in Materials and Methods and immunoblotted for paxillin. Total cell lysates from control and RACK1-deficient cells were immunoblotted for phosphopaxillin (pY118), total paxillin and RACK1 as indicated. Representative blots from three separate experiments are shown.
Discussion
In this study we demonstrate novel functions for RACK1 in regulating adhesion dynamics and cell migration. We report that cells deficient in RACK1 are less motile and show reduced turnover of paxillin and talin at adhesions. Our findings indicate that RACK1 functions as a negative regulator of endogenous c-Src activity through tyrosine 246. Accordingly, in the absence of RACK1 there is constitutive phosphorylation of paxillin, and reduced dynamics of phosphoproteins at adhesions. We also show that RACK1 effects on paxillin dynamics required tyrosine 31/118 on paxillin. Taken together, these findings support a novel role for RACK1 as a key regulator of cell migration through its association with Src, and modulation of paxillin phosphorylation and dynamics at adhesions.
It is well established that RACK1 is a negative regulator of endogenous Src kinase activity, and may also have inhibitory effects on transforming v-Src [19]. Direct binding between Src and RACK1 has previously been shown to be mediated at least in part through tyrosine 246 of RACK1 [15]. The dramatic increase in Src activity in RACK1-deficient cells supports RACK1 as a critical negative regulator of endogenous c-Src activity. Furthermore, our findings demonstrate that the putative Src binding site, tyrosine 246 on RACK1, is required for this inhibitory effect.
Recent studies indicate that RACK1 reduces cell cycle progression and growth of colon carcinoma cells by negatively regulating endogenous Src kinase activity, suggesting that RACK1 may be an attractive therapeutic target to treat cancer [13]. On the other hand, our findings suggest that RACK1 can positively regulate cell migration through its association with Src, since cells that are deficient in RACK1 display reduced migration that can be rescued by expression of wild type RACK1 but not RACK Y246F. Interestingly, RACK1 is overexpressed in conditions that may be associated with enhanced motility in vivo, including human cancers and under conditions of tissue regeneration [42]. However, it is likely that RACK1 may also function to negatively regulate cell migration since previous studies indicate that overexpression of RACK1 can be associated with reduced migration [9, 10]. In agreement with this possibility, we also found that overexpression of RACK1 reduced HEK migration (data not shown), supporting a biphasic relationship between RACK1 expression and cell migration speed.
Our findings indicate that tyrosine 246 is a key determinant of RACK1 effects during cell migration, suggesting that the inhibitory effect of RACK1 on Src is critical for efficient cell migration. Accordingly, we observed enhanced phosphorylation of paxillin in RACK1-deficient cells that was dependent on Src kinase activity. In addition, the effects of RACK1 on paxillin dynamics required the ability of tyrosine 31 and 118 to be phosphorylated, indicating that RACK1 exerts its effects on paxillin dynamics through the regulation of paxillin tyrosine phosphorylation. Previous studies have shown that paxillin phosphorylation is required for the disassembly of paxillin from adhesions, in that cells that express GFP-paxillin Y31, 118F showed reduced adhesion disassembly [6]. Our findings however suggest that enhanced phosphorylation at these sites through the constitutive activation of Src activity may also be associated with reduced assembly and disassembly of paxillin-containing adhesions, thereby leading to stabilized adhesion complexes. In support of this possibility, we observed enhanced duration of phosphoproteins at adhesions in the absence of RACK1, supporting a critical role for dynamic assembly and disassembly of phosphoproteins at adhesions for efficient cell migration.
It is important to consider that although RACK1-Src interaction is likely an important component of RACK1 function during migration, that there are numerous other binding partners that likely participate in RACK1 effects during migration. RACK1 interacts with many other proteins that are involved in cell migration including the β integrin cytoplasmic domain [9, 14], PKCs [32, 43], Fyn [18], phospholipase Cγ and the transporter NHE5 [44]. Furthermore, RACK1 has been implicated in the regulation of eukaryotic translation by its direct association with ribosomes, where it can recruit activated PKC to ribosomes [37, 45]. The association of RACK1 with both focal adhesion proteins and ribosomes raises the intriguing possibility that RACK1 may also function to recruit ribosomes to early adhesions to allow local translation. Although this function may contribute to RACK1 effects on adhesion dynamics, we observed no global changes in expression of focal adhesion proteins including talin, FAK and paxillin in RACK1-deficient cells. Interestingly, RACK1 also interacts with tyrosine phosphatases, such as SHP-2 [46] and PTPμ [16], providing an alternative mechanism that contributes to RACK1 effects on adhesion dynamics. A key challenge for future investigation will be to dissect the temporal and spatial interactions between RACK1 and specific effector proteins during cell migration in vivo. This will provide critical information to aid in the identification of the key effectors of RACK1 during cell migration.
In summary, our studies demonstrate a critical role for RACK1 during cell migration at least in part through its modulation of Src activity. Although it is possible that tyrosine 246 mediates interactions with other effector proteins, including other Src family members such as Fyn, our findings suggest that Src-mediated phosphorylation of paxillin is an important mediator of RACK1 effects on paxillin dynamics during migration. It is intriguing to speculate that the adaptor function of RACK1 targets specific effector proteins, including Src, to local adhesions, thereby contributing to the temporal and spatial dynamics of adhesions during migration. Future investigations will focus on characterizing the contribution of specific RACK1 interactions to the spatial and temporal regulation of adhesion turnover during migration.
Supplementary Material
Acknowledgments
We would like to thank Mary Lokuta for useful discussions and technical assistance. We would also like to acknowledge funding from American Cancer Society and NCI (R01CA85862-06) to AH and funding from Molecular and Cellular Pharmacology and PhRMA Foundation to AD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jockusch BM, Bubeck P, Giehl K, Kroemker M, Moschner J, Rothkegel M, Rudiger M, Schluter K, Stanke G, Winkler J. The molecular architecture of focal adhesions. Annu Rev Cell Dev Biol. 1995;11:379–416. doi: 10.1146/annurev.cb.11.110195.002115. [DOI] [PubMed] [Google Scholar]
- 2.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 3.Burridge K, Fath K, Kelly T, Nuckolls G, Turner C. Focal adhesions: transmembrane junctions between the extracellular matrix and the cytoskeleton. Annu Rev Cell Biol. 1988;4:487–525. doi: 10.1146/annurev.cb.04.110188.002415. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz MA, Schaller MD, Ginsberg MH. Integrins: emerging paradigms of signal transduction. Annu Rev Cell Dev Biol. 1995;11:549–99. doi: 10.1146/annurev.cb.11.110195.003001. [DOI] [PubMed] [Google Scholar]
- 5.Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–44. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- 6.Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 2004;6:154–61. doi: 10.1038/ncb1094. [DOI] [PubMed] [Google Scholar]
- 7.Dourdin N, Bhatt AK, Dutt P, Greer PA, Arthur JS, Elce JS, Huttenlocher A. Reduced cell migration and disruption of the actin cytoskeleton in calpain-deficient embryonic fibroblasts. J Biol Chem. 2001;276:48382–8. doi: 10.1074/jbc.M108893200. [DOI] [PubMed] [Google Scholar]
- 8.Franco SJ, Rodgers MA, Perrin BJ, Han J, Bennin DA, Critchley DR, Huttenlocher A. Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat Cell Biol. 2004;6:977–83. doi: 10.1038/ncb1175. [DOI] [PubMed] [Google Scholar]
- 9.Buensuceso CS, Woodside D, Huff JL, Plopper GE, O’Toole TE. The WD protein Rack1 mediates protein kinase C and integrin-dependent cell migration. J Cell Sci. 2001;114:1691–8. doi: 10.1242/jcs.114.9.1691. [DOI] [PubMed] [Google Scholar]
- 10.Cox EA, Bennin D, Doan AT, O’Toole T, Huttenlocher A. RACK1 regulates integrin-mediated adhesion, protrusion, and chemotactic cell migration via its Src-binding site. Mol Biol Cell. 2003;14:658–69. doi: 10.1091/mbc.E02-03-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ron D, Chen CH, Caldwell J, Jamieson L, Orr E, Mochly-Rosen D. Cloning of an intracellular receptor for protein kinase C: a homolog of the beta subunit of G proteins. Proc Natl Acad Sci U S A. 1994;91:839–43. doi: 10.1073/pnas.91.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang BY, Conroy KB, Machleder EM, Cartwright CA. RACK1, a receptor for activated C kinase and a homolog of the beta subunit of G proteins, inhibits activity of src tyrosine kinases and growth of NIH 3T3 cells. Mol Cell Biol. 1998;18:3245–56. doi: 10.1128/mcb.18.6.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mamidipudi V, Dhillon NK, Parman T, Miller LD, Lee KC, Cartwright CA. RACK1 inhibits colonic cell growth by regulating Src activity at cell cycle checkpoints. Oncogene. 2006 doi: 10.1038/sj.onc.1210091. [DOI] [PubMed] [Google Scholar]
- 14.Liliental J, Chang DD. Rack1, a receptor for activated protein kinase C, interacts with integrin beta subunit. J Biol Chem. 1998;273:2379–83. doi: 10.1074/jbc.273.4.2379. [DOI] [PubMed] [Google Scholar]
- 15.Chang BY, Chiang M, Cartwright CA. The interaction of Src and RACK1 is enhanced by activation of protein kinase C and tyrosine phosphorylation of RACK1. J Biol Chem. 2001;276:20346–56. doi: 10.1074/jbc.M101375200. [DOI] [PubMed] [Google Scholar]
- 16.Mourton T, Hellberg CB, Burden-Gulley SM, Hinman J, Rhee A, Brady-Kalnay SM. The PTPmu protein-tyrosine phosphatase binds and recruits the scaffolding protein RACK1 to cell-cell contacts. J Biol Chem. 2001;276:14896–901. doi: 10.1074/jbc.M010823200. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez MM, Ron D, Touhara K, Chen CH, Mochly-Rosen D. RACK1, a protein kinase C anchoring protein, coordinates the binding of activated protein kinase C and select pleckstrin homology domains in vitro. Biochemistry. 1999;38:13787–94. doi: 10.1021/bi991055k. [DOI] [PubMed] [Google Scholar]
- 18.Yaka R, Thornton C, Vagts AJ, Phamluong K, Bonci A, Ron D. NMDA receptor function is regulated by the inhibitory scaffolding protein, RACK1. Proc Natl Acad Sci U S A. 2002;99:5710–5. doi: 10.1073/pnas.062046299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mamidipudi V, Chang BY, Harte RA, Lee KC, Cartwright CA. RACK1 inhibits the serum- and anchorage-independent growth of v-Src transformed cells. FEBS Lett. 2004;567:321–6. doi: 10.1016/j.febslet.2004.03.125. [DOI] [PubMed] [Google Scholar]
- 20.Schaller MD, Hildebrand JD, Parsons JT. Complex formation with focal adhesion kinase: A mechanism to regulate activity and subcellular localization of Src kinases. Mol Biol Cell. 1999;10:3489–505. doi: 10.1091/mbc.10.10.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Webb DJ, Parsons JT, Horwitz AF. Adhesion assembly, disassembly and turnover in migrating cells -- over and over and over again. Nat Cell Biol. 2002;4:E97–100. doi: 10.1038/ncb0402-e97. [DOI] [PubMed] [Google Scholar]
- 22.Kirchner J, Kam Z, Tzur G, Bershadsky AD, Geiger B. Live-cell monitoring of tyrosine phosphorylation in focal adhesions following microtubule disruption. J Cell Sci. 2003;116:975–86. doi: 10.1242/jcs.00284. [DOI] [PubMed] [Google Scholar]
- 23.Ruoslahti E, Hayman EG, Pierschbacher M, Engvall E. Fibronectin: purification, immunochemical properties, and biological activities. Methods Enzymol. 1982;82(Pt A):803–31. doi: 10.1016/0076-6879(82)82103-4. [DOI] [PubMed] [Google Scholar]
- 24.Petit V, Boyer B, Lentz D, Turner CE, Thiery JP, Valles AM. Phosphorylation of tyrosine residues 31 and 118 on paxillin regulates cell migration through an association with CRK in NBT-II cells. J Cell Biol. 2000;148:957–70. doi: 10.1083/jcb.148.5.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robles E, Woo S, Gomez TM. Src-dependent tyrosine phosphorylation at the tips of growth cone filopodia promotes extension. J Neurosci. 2005;25:7669–81. doi: 10.1523/JNEUROSCI.2680-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jordan M, Schallhorn A, Wurm FM. Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res. 1996;24:596–601. doi: 10.1093/nar/24.4.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang F, Lee SY, Liang J, Lawrence DS, Zhang ZY. The role of protein-tyrosine phosphatase 1B in integrin signaling. J Biol Chem. 2005;280:24857–63. doi: 10.1074/jbc.M502780200. [DOI] [PubMed] [Google Scholar]
- 28.Crowley E, Horwitz AF. Tyrosine phosphorylation and cytoskeletal tension regulate the release of fibroblast adhesions. J Cell Biol. 1995;131:525–37. doi: 10.1083/jcb.131.2.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huttenlocher A, Ginsberg MH, Horwitz AF. Modulation of cell migration by integrin-mediated cytoskeletal linkages and ligand-binding affinity. J Cell Biol. 1996;134:1551–62. doi: 10.1083/jcb.134.6.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zamir E, Katz BZ, Aota S, Yamada KM, Geiger B, Kam Z. Molecular diversity of cell-matrix adhesions. J Cell Sci. 1999;112(Pt 11):1655–69. doi: 10.1242/jcs.112.11.1655. [DOI] [PubMed] [Google Scholar]
- 31.Hermanto U, Zong CS, Li W, Wang LH. RACK1, an insulin-like growth factor I (IGF-I) receptor-interacting protein, modulates IGF-I-dependent integrin signaling and promotes cell spreading and contact with extracellular matrix. Mol Cell Biol. 2002;22:2345–65. doi: 10.1128/MCB.22.7.2345-2365.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Besson A, Wilson TL, Yong VW. The anchoring protein RACK1 links protein kinase Cepsilon to integrin beta chains. Requirements for adhesion and motility. J Biol Chem. 2002;277:22073–84. doi: 10.1074/jbc.M111644200. [DOI] [PubMed] [Google Scholar]
- 33.Kiely PA, O’Gorman D, Luong K, Ron D, O’Connor R. Insulin-like growth factor I controls a mutually exclusive association of RACK1 with protein phosphatase 2A and beta1 integrin to promote cell migration. Mol Cell Biol. 2006;26:4041–51. doi: 10.1128/MCB.01868-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Link AJ, Eng J, Schieltz DM, Carmack E, Mize GJ, Morris DR, Garvik BM, Yates JR., 3rd Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol. 1999;17:676–82. doi: 10.1038/10890. [DOI] [PubMed] [Google Scholar]
- 35.Volberg T, Romer L, Zamir E, Geiger B. pp60(c-src) and related tyrosine kinases: a role in the assembly and reorganization of matrix adhesions. J Cell Sci. 2001;114:2279–89. doi: 10.1242/jcs.114.12.2279. [DOI] [PubMed] [Google Scholar]
- 36.Westhoff MA, Serrels B, Fincham VJ, Frame MC, Carragher NO. SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Mol Cell Biol. 2004;24:8113–33. doi: 10.1128/MCB.24.18.8113-8133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Hoog CL, Foster LJ, Mann M. RNA and RNA binding proteins participate in early stages of cell spreading through spreading initiation centers. Cell. 2004;117:649–62. doi: 10.1016/s0092-8674(04)00456-8. [DOI] [PubMed] [Google Scholar]
- 38.Yano H, Uchida H, Iwasaki T, Mukai M, Akedo H, Nakamura K, Hashimoto S, Sabe H. Paxillin alpha and Crk-associated substrate exert opposing effects on cell migration and contact inhibition of growth through tyrosine phosphorylation. Proc Natl Acad Sci U S A. 2000;97:9076–81. doi: 10.1073/pnas.97.16.9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakamura K, Yano H, Uchida H, Hashimoto S, Schaefer E, Sabe H. Tyrosine phosphorylation of paxillin alpha is involved in temporospatial regulation of paxillin-containing focal adhesion formation and F-actin organization in motile cells. J Biol Chem. 2000;275:27155–64. doi: 10.1074/jbc.M000679200. [DOI] [PubMed] [Google Scholar]
- 40.Pawson T, Gish GD, Nash P. SH2 domains, interaction modules and cellular wiring. Trends Cell Biol. 2001;11:504–11. doi: 10.1016/s0962-8924(01)02154-7. [DOI] [PubMed] [Google Scholar]
- 41.Zamir E, Geiger B. Molecular complexity and dynamics of cell-matrix adhesions. J Cell Sci. 2001;114:3583–90. doi: 10.1242/jcs.114.20.3583. [DOI] [PubMed] [Google Scholar]
- 42.Berns H, Humar R, Hengerer B, Kiefer FN, Battegay EJ. RACK1 is up-regulated in angiogenesis and human carcinomas. Faseb J. 2000;14:2549–58. doi: 10.1096/fj.99-1038com. [DOI] [PubMed] [Google Scholar]
- 43.Ron D, Luo J, Mochly-Rosen D. C2 region-derived peptides inhibit translocation and function of beta protein kinase C in vivo. J Biol Chem. 1995;270:24180–7. doi: 10.1074/jbc.270.41.24180. [DOI] [PubMed] [Google Scholar]
- 44.Onishi I, Lin PJ, Diering GH, Williams WP, Numata M. RACK1 associates with NHE5 in focal adhesions and positively regulates the transporter activity. Cell Signal. 2006 doi: 10.1016/j.cellsig.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 45.Nilsson J, Sengupta J, Frank J, Nissen P. Regulation of eukaryotic translation by the RACK1 protein: a platform for signalling molecules on the ribosome. EMBO Rep. 2004;5:1137–41. doi: 10.1038/sj.embor.7400291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kiely PA, Sant A, O’Connor R. RACK1 is an insulin-like growth factor 1 (IGF-1) receptor-interacting protein that can regulate IGF-1-mediated Akt activation and protection from cell death. J Biol Chem. 2002;277:22581–9. doi: 10.1074/jbc.M201758200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.