

Abstract
Cellular APOBEC3G (A3G) protein is packaged into human immunodeficiency virus type 1 (HIV-1) virions in producer cells yet restricts viral replication in target cells. To characterize this restriction in target cells, the effect of A3G on generating various HIV-1 cDNA products was measured by quantitative real-time PCR. A3G decreased cDNA products from Vif-deficient HIV-1, with minor effects on early reverse transcripts and larger declines in late reverse transcripts. However, the greatest decline was typically observed in nuclear 2-LTR circles. Moreover, the magnitude of these declines varied with A3G dose. Adding integration inhibitor did not stop the A3G-mediated loss in 2-LTR circles. Moreover, obstructing HIV-1 nuclear entry using vesicular stomatitis virus matrix protein did not stop the A3G-mediated decline in late reverse transcripts. Collectively, these data suggest that A3G has important restriction activity in the cytoplasm and progressively diminishes viral cytoplasmic and nuclear cDNA forms with increasing magnitude during restriction.
Keywords: APOBEC3G, restriction, human immunodeficiency virus type 1, reverse transcription, 2-LTR circles
Introduction
Host cells possess innate defense mechanisms to obstruct invading pathogens. These defenses include the recently identified Apolipoprotein B mRNA-editing catalytic polypeptide-like 3 (APOBEC3) enzyme family (Bishop et al., 2004; Bogerd et al., 2006a; Bogerd et al., 2006b; Chen et al., 2006; Doehle, Schafer, and Cullen, 2005; Dutko et al., 2005; Langlois et al., 2005; Lochelt et al., 2005; OhAinle et al., 2006; Sheehy et al., 2002; Turelli et al., 2004; Wiegand et al., 2004; Yu et al., 2004a; Zheng et al., 2004). APOBEC proteins are found throughout the vertebrate lineage with more APOBEC genes observed in higher order vertebrates like mammals during evolution. In fact, the evolutionary advanced human primates contain APOBEC1, 2, 3A-3H and activation-induced deaminase (AID). These APOBEC proteins function as cytidine deaminases that remove the amino group from cytosine (C) bases in DNA and/or RNA creating uracil (U) [reviewed in (Cullen, 2006; Navaratnam and Sarwar, 2006)]. Various APOBEC3 proteins restrict the replication of pathogens involving a reverse transcription step, including retroviruses (Bishop et al., 2004; Doehle, Schafer, and Cullen, 2005; Harris et al., 2003; Langlois et al., 2005; Lochelt et al., 2005; Mangeat et al., 2003; OhAinle et al., 2006; Sheehy et al., 2002; Wiegand et al., 2004; Yu et al., 2004a; Zheng et al., 2004), retroelements like hepatitis B virus (Turelli et al., 2004) and both LTR and non-LTR type retrotransposons (Bogerd et al., 2006a; Bogerd et al., 2006b; Chen et al., 2006; Dutko et al., 2005; Esnault et al., 2005; Schumacher, Nissley, and Harris, 2005; Stenglein and Harris, 2006). However, the precise mechanism/s underlying APOBEC3 restriction remains unclear.
Many aspects of APOBEC3 interplay with reverse transcribing pathogens were first identified in studies of human APOBEC3G (A3G) restriction of Vif-deficient HIV-1. For instance, human A3G is packaged into assembling virions in producer cells but blocks retrovirus replication in target cells (Sheehy et al., 2002). This block in replication is coincident with dramatic cytosine deamination of reverse transcribed, single-stranded DNA that results in guanine (G) to adenine (A) mutations in the complementary DNA strand (Harris et al., 2003; Lecossier et al., 2003; Mangeat et al., 2003; Zhang et al., 2003a). To overcome this antiviral effect of A3G, HIV-1 encodes the viral infectivity factor (Vif) accessory protein. Vif targets A3G for proteasome degradation in the producer cell and prevents A3G packaging into assembling virions, thereby avoiding A3G effects in the target cell (Mariani et al., 2003; Marin et al., 2003; Sheehy, Gaddis, and Malim, 2003; Stopak et al., 2003; Yu et al., 2003). Similarities to this restriction system have since been described for other APOBEC3 restriction systems [recently reviewed in (Cullen, 2006)].
However, the precise mechanism/s allowing intra-virion APOBEC3 proteins to restrict retroviral replication in target cells remains controversial. During APOBEC3 restriction of retroviruses, intra-virion APOBEC3 proteins deaminate and thus mutate reverse transcribing viral cDNA (Bishop et al., 2004; Doehle, Schafer, and Cullen, 2005; Harris et al., 2003; Langlois et al., 2005; Lecossier et al., 2003; Lochelt et al., 2005; Mangeat et al., 2003; OhAinle et al., 2006; Wiegand et al., 2004; Yu et al., 2004a; Yu et al., 2004b; Zhang et al., 2003a; Zheng et al., 2004). Therefore, one restriction mechanism is this mutation disrupts viral gene expression through perturbing translation initiation codons (Yu et al., 2004b) and protein coding sequence to block viral replication. However, multiple studies indicate APOBEC3 proteins have extra restriction mechanism/s before gene expression as these proteins can also decrease various reverse transcribed viral cDNAs produced earlier in replication (Aires da Silva et al., 2004; Courcoul et al., 1995; Dornadula et al., 2000; Dutko et al., 2005; Esnault et al., 2006; Kaiser and Emerman, 2006; Langlois et al., 2005; Mangeat et al., 2003; Mariani et al., 2003; Nascimbeni et al., 1998; Sasada et al., 2005; Simon and Malim, 1996; Sova and Volsky, 1993; Stenglein and Harris, 2006; von Schwedler et al., 1993). This indicates APOBEC3 proteins can have multiple mechanisms to restrict reverse transcribing pathogens.
However, the mechanism/s causing APOBEC3 proteins to impair viral cDNAs generated earlier in replication remain unclear. One possibility is extensive deamination of the reverse transcribing viral cDNA by APOBEC3 causes this viral cDNA to decline using uracil DNA glycosylases (like UNG-2 or SMUG) and apurinic/apyrimidinic endonuclease (APE) base excision repair enzymes that are packaged into virions (Priet et al., 2005; Schrofelbauer et al., 2005; Yang et al., 2007a). These enzymes may cleave off the aberrant uracil bases and cleave the cDNA backbone during attempted repair but not completely repair the site, leaving the viral cDNA susceptible to decay (Harris et al., 2003; Yang et al., 2007a). However, separate work is at odds with this model (Kaiser and Emerman, 2006) and ultimately, this model remains controversial. In a second possibility, A3G-induced uracil in the reverse transcribing viral cDNA might perturb the reverse transcription process itself (Klarmann et al., 2003) to reduce viral cDNA levels. However, experimental evidence to confirm this possibility is lacking. Thirdly, recent work indicates APOBEC3 proteins also have antiviral activity independent of deamination as APOBEC3 deamination and antiviral activity can be separated. For instance, deamination-defective human A3G can still restrict infection by HIV-1 (Newman et al., 2005), human T-cell lymphotropic virus type 1 (Sasada et al., 2005) and hepatitis B virus replication (Turelli et al., 2004). Similarly, deamination-defective human APOBEC3F (A3F) and APOBEC3B (A3B) still inhibit HIV-1 infection (Holmes et al., 2007) and L1 retrotransposition (Stenglein and Harris, 2006), illustrating antiviral activity separate to deamination. Furthemore, A3G and A3F were recently reported to decrease tRNA initiation of HIV-1 reverse transcription ≥ 2-fold independent of deamination (Guo et al., 2006; Yang et al., 2007b). Moreover, separate work correlates A3G and A3F restriction of HIV-1 with their ability to deplete HIV-1 late RT products rather than deaminate (Bishop, Holmes, and Malim, 2006; Holmes et al., 2007). This suggests A3G and A3F have deamination-independent activity and that viral RT product depletion may be more important for HIV-1 restriction than deamination-dependent activities. Ultimately, APOBEC3 proteins appear to have multiple antiviral mechanisms, diminishing viral cDNAs early in replication via potential deamination dependent and/or independent means, as well as deaminating reverse transcribing genomes to inhibit downstream viral gene expression.
To gain more insight into this early antiviral activity of APOBEC3 protein in target cells, we studied human A3G restriction of Vif-deficient HIV-1. Specifically, we assessed the impact of intra-virion A3G on forming HIV-1 cDNA products during early viral replication in target cells and whether this antiviral activity occurred primarily in the cell cytoplasm or nucleus. The impact of A3G on various cDNA products varied with the A3G dose, but followed the same trend of minor declines in early reverse transcription (RT) products, larger declines in late RT but greatest declines typically measured in nuclear 2-LTR circles. Experiments adding an integration inhibitor did not prevent the A3G-mediated loss in 2-LTR circles. Importantly, late RT levels continued to decline when HIV-1 cDNA nuclear entry was impaired via the nuclear transport inhibitor, vesicular stomatitis virus (VSVM) matrix protein. Collectively, these data suggest intra-virion A3G has key restriction activity in the cytoplasm and ultimately depletes HIV-1 cytoplasmic and nuclear cDNA forms with increasing magnitude.
Results
A3G induces a large decline in HIV-1ΔVif 2-LTR circles
To explore how intra-virion human A3G blocks the early replication of Vif-deficient HIV-1 (HIV-1ΔVif), we aimed to clarify at what steps A3G impaired early HIV-1 replication and in which cellular compartment this occurred. Therefore, we measured the effect of A3G on HIV-1 early, late, and 2-LTR circle products of reverse transcription (RT) in target cells. Primers for early and late RT products detect viral cDNAs containing minus-strand strong-stop cDNA (Munk et al., 2002) or progressing beyond second strand transfer in reverse transcription (Butler, Hansen, and Bushman, 2001) respectively. These products arise in the cytoplasm. In contrast, the 2-LTR circle primers amplify the linkage region between the 5′ and 3′ LTR ends in 2-LTR circles (Butler, Hansen, and Bushman, 2001). As the non-homologous end joining enzymes forming these circles (Li et al., 2001) localize to the nucleus, these circles can mark nuclear entry of the viral cDNA (Brown, 1997). Vif negative and Vif positive HIV-1 were both examined for this work, which should be sensitive to or avoid A3G restriction respectively. For these studies, HIV-1 stocks were generated using the same 293T producer cells to avoid cell-type variation. However, A3G was introduced by cotransfecting HA-tagged human A3G plasmid (Mariani et al., 2003) with the proviral plasmid into 293Ts. Promoter-matched pCMplpa replaced the A3G plasmid when making virus without A3G. To maximize viral infection for accurate 2-LTR circle measurements, we continuously infected cells in experiments and also used HIV-1 lacking the native envelope (Env) surface protein but pseudotyped with VSVg protein for efficient entry. The infectivity of these VSVg pseudotyped virus stocks generated with increasing amounts of A3G was measured using MAGI reporter cells. Both A3G amounts tested impaired HIV-1ΔVif infection in contrast to Vif positive HIV-1, whose infection was not affected by A3G (Fig. 1A). Therefore, these viruses exhibited the expected infectivity in response to A3G and were used in subsequent experiments to explore the impact of A3G on viral cDNA.
Figure 1. Human A3G impairs HIV-1ΔVif 2-LTR circle formation.

(A) Human A3G impairs the infectivity of VSVg pseudotyped HIV-1 lacking Vif but not Vif positive HIV-1. VSVg pseudotyped HIV-1 or HIV-1ΔVif were produced with different human A3G amounts, as indicated on the graph by the molar ratio of A3G to provirus plasmid transfected. The infectivity of this virus on MAGI reporter cells, measured using a liquid β-gal assay (infection), per p24Gag in the virus stock (p24) is shown for a representative experiment. Error bars indicate standard deviation in triplicate values. HIV-1 without VSVg envelope acts a negative control. (B–D) A3G potently impairs 2-LTR circle production from HIV-1ΔVif. HOS cells were continuously infected 15 hours (h) with 7.94 ng of VSVg pseudotyped HIV-1 without or with A3G (2:1 molar ratio). The virus was removed, new media added and the intracellular DNA harvested at 15 h, 26 h or 48 h from the start of infection (light, medium and dark grey bars respectively). Viral early RT (B), late RT (C), 2-LTR circle (D) and cellular β-actin were quantified by real-time PCR and values shown per cell (β-actin). Error bars indicate standard deviation in triplicate values from a representative experiment. The negligible viral cDNA in cells infected with virus plus the Nevirapine (Nev) reverse transcription inhibitor, demonstrates viral reverse transcription products are measured in this assay.
The effect of A3G on forming various HIV-1 reverse transcription products was explored in HOS cells at different times after viral infection using quantitative real-time PCR (Fig. 1B–D). Virus generated at a 2:1 molar ratio of A3G to provirus was used for this analysis (Fig. 1A). For the Vif positive HIV-1 made with or without A3G, early and late RT products were at maximal levels near 4 copies per cell at the initial 15 hour (h) time point but decreased to approximately 2 copies per cell by 48 h (Fig. 1B, C). Furthermore, 2-LTR circles peaked around 26 h, but decreased again by 48 h (Fig. 1D). HIV-1ΔVif without A3G followed a similar trend (Fig. 1B, C). However, HIV-1ΔVif plus A3G differed in that peak levels of early and late RT only accumulated to 2 copies per cell at 15 h, which was roughly half the level seen for the other viruses (Fig. 1B, C). Furthermore, these early and late RT products from HIV-1ΔVif plus A3G declined to a larger extent by 48 h, decreasing 3.3 and 3.8 fold respectively from 15 h compared to the near 2 fold loss for the other viruses (Fig. 1B, C). However, the greatest change for HIV-1ΔVif plus A3G was observed in 2-LTR circles, which dramatically decreased at all times compared to the other viruses (Fig. 1D). Therefore, A3G potently reduced 2-LTR circles generated from HIV-1ΔVif compared to smaller losses in early and late RT. As 2-LTR circles can serve as a marker for nuclear localization of retroviral cDNA (Brown, 1997), this suggested A3G may interfere with the accumulation of nuclear HIV-1ΔVif cDNA.
A3G decline in HIV-1 cDNA is not an artifact of real-time PCR primers
In this study, a hot-start enzyme system (Bio-Rad) was chosen for real-time PCR to prevent non-specific products arising during assay setup, rather than an enzyme system relying on dUTP incorporation and UNG as we reasoned the latter system may obstruct and underestimate measurements of A3G-deaminated viral cDNAs. However, primers used for real-time PCR analysis could also underestimate measurements of A3G-deaminated viral cDNAs if the primers contained sequences sensitive to A3G hypermutation that reduced primer binding to hypermutated viral cDNA. Both 2-LTR circle primers used in the previous analysis contained sequence prone to A3G hypermutation [Fig. 2A, dark gray box, (Yu et al., 2004b)]. Further, the late RT reverse primer also contained a site favored for A3G hypermutation (Yu et al., 2004b). Therefore, to assess if our real-time PCR results were being influenced by A3G hypermutation reducing primer-binding efficiency, we repeated the real-time PCR assay using primers that removed or reduced primer sensitivity to A3G hypermutation (Fig. 2A, 2LTR primers or late RT reverse primer respectively). Irrespective of whether the 2-LTR or late RT primers contained A3G sensitive sites, similar viral 2-LTR (Fig. 2B) and late RT (Fig. 2C) cDNA products were measured. Therefore, the decline in cDNA products from HIV-1ΔVif plus A3G measured by real-time PCR could not be attributed to PCR primers, indicating instead that actual losses in viral cDNA products were being measured following A3G activity.
Figure 2. Primers with or without A3G hypermutation sites show similar decreases in HIV-1ΔVif cDNA products by A3G.

(A) Primer sequences prone (+) or not prone (−) to A3G hypermutation and used to amplify 2-LTR circle (upper) and late RT (lower) viral cDNA products are shown. F, forward (plus strand) primer; R, reverse (minus strand) primer. Dark and light grey boxes indicate sequence sensitive to A3G hypermutation at high and low frequency respectively (Yu et al., 2004b). (B–C) HOS cells were continuously infected for 14 h with 3.42 ng of HIV-1 or HIV-1ΔVif lacking or containing A3G (made at 1:1 molar ratio of A3G:provirus plasmid). Intracellular DNA was analysed for 2-LTR circles (B) or late RT (C) using primers containing sequence prone (dark grey) or not prone (light grey) to high frequency A3G hypermutation.
Declines in HIV-1 cDNA products vary with A3G dose
In these studies, we noticed the A3G induced losses in HIV-1ΔVif cDNA followed the same trend of minor decline in early RT, larger decline in late RT but greatest decline in 2-LTR circles (Fig. 1B–D). To extend these studies, we next evaluated whether different A3G levels altered the magnitude of HIV-1ΔVif cDNA decline. Consequently, VSVg pseudotyped HIV-1 was titrated with increasing amounts of A3G during virus production and effects on viral infection, early RT, late RT and 2-LTR circle output measured (Fig. 3). As expected, different levels of A3G had little impact on the infectivity of Vif positive HIV-1 in MAGI reporter cells given Vif was present in producer cells to prevent A3G incorporation into virions (Fig. 3A). In contrast, while low A3G levels did not alter HIV-1ΔVif infection, elevated A3G levels inhibited HIV-1ΔVif infection completely (Fig. 3A, HIV-Vif, 0.01 and 0.1 A3G versus 1, 2 and 5 respectively).
Figure 3. Declines in HIV-1 cDNA products vary with A3G dose.
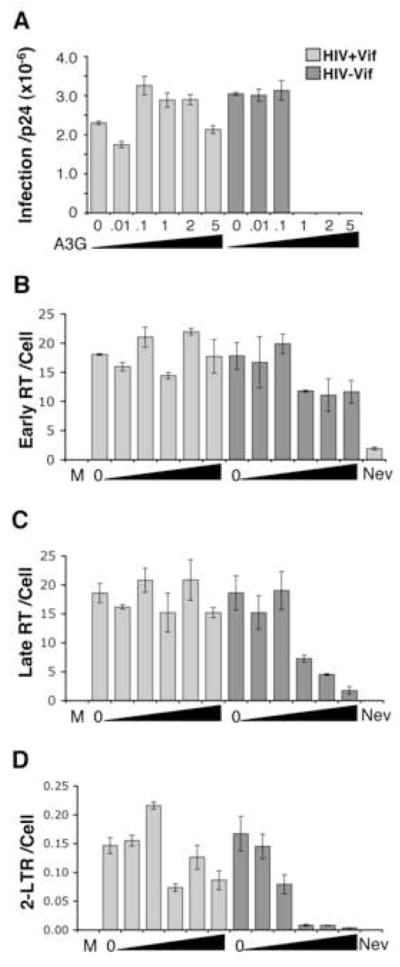
(A) VSVg pseudotyped HIV-1 with (light grey) or without (dark grey) Vif was produced with increasing levels of human A3G (indicated by the molar ratio of A3G per 1 provirus plasmid plus increasing size of the black triangle). The infectivity of these virus stocks on MAGI reporter cells per virion p24Gag concentration is shown for a representative experiment. (B–D) HOS cells were continuously infected with 29.77 ng of these viruses for 18 h and intracellular DNA measured for viral early RT (B), late RT (C) or 2-LTR circles (D) per cell (β-actin) using real-time PCR. Error bars indicate standard deviation of triplicate values. M, mock (no virus); 0, virus without A3G; Nev, Nevirapine. Increasing A3G during HIV-1ΔVif production correlates with larger declines in late RT and 2-LTR circles in target cells.
The impact of different A3G levels on viral cDNA output was subsequently measured 18 hours into infection of HOS cells. Primers lacking A3G hypermutation sites were used in real-time PCR analysis to ensure accurate cDNA measurement. For Vif positive HIV-1, early RT and late RT never deviated more than 1.3 fold with A3G relative to no A3G (Fig. 3B, C). Furthermore, these viruses all made 2-LTR circles, which declined near 1.5 fold in samples with highest A3G (Fig. 3D, HIV+Vif, 1, 2 and 5 A3G). For HIV-1ΔVif made with low A3G levels that did not change infection, early and late RT output also did not change (Fig. 3A–C, HIV-Vif, 0.01 and 0.1 A3G). These viruses also produced 2-LTR circles, although this level decreased 2 fold at 0.1 A3G (Fig. 3D). In contrast, for HIV-1ΔVif made with A3G levels that blocked infection, early RT output declined 1.5 fold (Fig. 3A and B, HIV-Vif, 1, 2 and 5 A3G). Furthermore, greater losses in late RT arose for these samples as A3G increased (Fig. 3C, HIV-Vif, 1, 2 and 5 A3G). However, dramatic losses in 2-LTR circles were measured for these samples coinciding with lost infection (Fig. 3A and D, HIV-Vif, 1, 2 and 5 A3G). Therefore, different A3G levels clearly influenced the output of HIV-1 cDNA products, with HIV-1ΔVif late RT diminishing in a dose-dependant manner. The 2-LTR circles were also most sensitive to inhibition by A3G, with the dramatic loss in 2-LTR circles correlating with the A3G threshold blocking infection. A similar effect of human A3G on HIV-1ΔVif late RT and 2-LTR circles was also observed in CD4 expressing HeLa cells infected with HIV-1ΔVif using native Env protein for viral entry (data not shown, Fig. 5). Hence, this phenomenon is observed in a second HIV-1 system closer to native viral infection.
Figure 5. A3G still depletes HIV-1 Late RT when VSV matrix protein impairs nuclear entry.

(A) HeLa cells 24 h after transfection with pEGFP-VSVM (green), and stained for nuclear pore complexes using anti-Nup414 nucleoporin antibody (red) and filamentous actin using texas-red phalloidin (blue). GFP-VSVM localizes to nuclear pore complexes. (B) HIV-1 with (grey) or without (white) Vif and containing wildtype envelope protein were produced with increasing levels of human A3G (molar ratio of A3G to 1 provirus plasmid shown). The infectivity of these virus stocks on MAGI reporter cells per p24Gag concentration is shown for a representative experiment. (C–F) A, A3G; Mock, no virus; Nev, Nevirapine. HeLa cells were transfected without (light grey), with pCD4 (medium grey) or with pEGFP-VSVM plus pCD4 (dark grey) and 24 h later, continuously infected with 36.74 ng of the above HIV-1ΔVif stocks a further 24 h. Intracellular DNA was then analysed for viral early RT (C), late RT (D) or 2-LTR circles (E) per cell, the latter acting as a surrogate for nuclear localization of viral cDNA. The % of late RT per early RT is also shown (F) to demonstrate when changes in early RT levels are accounted for, A3G still decreases HIV-1 late RT when viral cDNA import is impaired by VSVM. Error bars indicate standard deviation of triplicate values.
A3G impairs HIV-1ΔVif 2-LTR circle formation in cells treated with an integrase inhibitor
When viral cDNA enters the nucleus, it can either integrate into the host cell chromosome generating provirus or form aberrant circles, like 1-LTR or 2-LTR circles via homologous recombination or non-homologous end joining respectively (Brown, 1997; Li et al., 2001). Moreover, when HIV-1 integration is blocked, the level of 2-LTR circles increase presumably as there is more viral cDNA substrate available for 2-LTR circle formation (Butler, Hansen, and Bushman, 2001; Hazuda et al., 2000; Svarovskaia et al., 2004). To gain more insight into A3G-mediated effects on viral cDNA substrates in the nucleus, we next determined the impact of an integration inhibitor on the observed decline in different HIV-1 cDNA products by A3G. A monosubstitution azido-containing β-diketo acid derivative (MA-DKA, 118-D-24) was used to block integration. MA-DKA binds HIV-1 integrase enzyme and selectively inhibits the strand transfer step of integration (Svarovskaia et al., 2004; Zhang et al., 2003b).
HOS cells were pretreated with MA-DKA (or DMSO diluent in the no drug control) for 4 hours, infected with various VSVg pseudotyped HIV-1 plus drug where appropriate and viral cDNA products measured 24 hours into infection (Fig. 4). Virus produced at a 2:1 molar ratio of A3G to provirus was used for these experiments. In general, MA-DKA caused minor declines in late RT relative to the same samples without drug (Fig. 4A). However, A3G still decreased HIV-1ΔVif late RT near 2.8 fold irrespective of drug, indicating MA-DKA was not changing the A3G-mediated decline in late RT. 2-LTR circle analysis either per cell (Fig. 4B) or per late RT to account for the change in late RT levels (Fig. 4C) both revealed MA-DKA increased 2-LTR output from Vif positive viruses or HIV-1ΔVif without A3G. This is consistent with MA-DKA blocking integration and providing more substrate for 2-LTR circles. However, 2-LTR circle output remained poor for HIV-1ΔVif containing A3G during MA-DKA treatment (Fig. 4B, C). A possible explanation is A3G depletes levels of integrated provirus along with 2-LTR circles from HIV-1ΔVif, providing little viral cDNA substrate during the MA-DKA integration block to increase 2-LTR circles, causing the 2-LTR levels to remain poor. This is consistent with recent reports that A3G impairs provirus levels in addition to 2-LTR circles (Mariani et al., 2003; Luo et al., 2007; Mbisa et al., 2007). An alternative or additional explanation is that the HIV-1ΔVif cDNA substrate that makes it into the nucleus is not normal and cannot form more 2-LTR circles when integration is blocked with the MA-DKA, even if more viral cDNA substrate becomes available to make 2-LTR circles. Either possibility would have the same outcome of not increasing 2-LTR circles from A3G-restricted HIV-1ΔVif when integration is blocked via MA-DKA.
Figure 4. MA-DKA integration inhibitor reveals human A3G impairs HIV-1 provirus as well as 2-LTR circles.
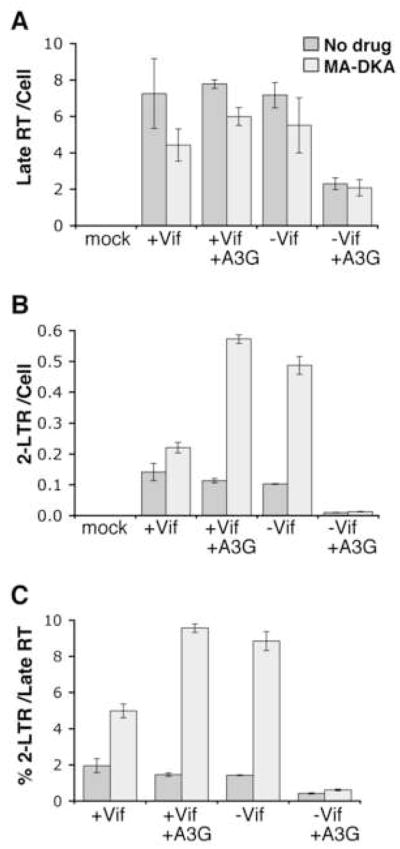
HOS cells were pretreated 4 h with MA-DKA (light grey) or DMSO (medium grey) as a no drug control and continuously infected 24 h with 16.27 ng of VSVg pseudotyped HIV-1 produced without or with A3G (2:1 molar ratio of A3G:provirus plasmid). Intracellular DNA was analysed for late RT (A) or 2-LTR circles (B) per cell (β-actin) by real-time PCR, with the percentage of 2-LTR circles per late RT also shown to account for changes in late RT between samples (C). Error bars indicate standard deviation in triplicate values from a representative experiment. As 2-LTR circle output from HIV-1ΔVif plus A3G remains poor with MA-DKA integration inhibitor indicates A3G impedes HIV-1ΔVif provirus as well as 2-LTR circle forms.
Impairing HIV-1 cDNA nuclear import does not prevent the A3G-mediated decline in late RT
To suppress 2-LTR circle and possibly provirus forms of nuclear HIV-1 cDNA, A3G may: a) act primarily in the cytoplasm with consequences that suppress these cDNAs forming in the nucleus and/or b) act on viral cDNA once in the nucleus to suppress these nuclear forms. To gain insight into these possibilities, we examined if A3G could still decrease late RT levels when nuclear import of viral cDNA was impaired. We reasoned that if A3G was primarily acting in the cytoplasm before or at nuclear entry, late RT should continue to decline even if we impaired viral cDNA import. Conversely, if A3G was primarily acting in the nucleus to reduce 2-LTR circles and provirus, preventing viral cDNA entry to the nucleus should prevent this block and allow late RT levels to increase. Therefore, we impaired HIV-1 cDNA import and measured if A3G still decreased late RT levels to gain insight into the cellular compartment in which A3G was likely obstructing nuclear 2-LTR circle and possibly provirus cDNAs.
Impairing HIV-1 cDNA import is challenging as no sole determinant for importing HIV-1 cDNAs into the nucleus has been identified [reviewed in (Bukrinsky, 2004; Nisole and Saib, 2004)]. However, vesicular stomatitis virus matrix protein (VSVM) interacts with the nucleoporin (Nup) 98 component of nuclear pore complexes (NPCs) and disrupts the import and export of most molecules through NPCs [reviewed in (Gustin, 2003)], including HIV-1 cDNA (Ebina et al., 2004). Consistent with being recruited to NPCs via Nup 98 to impair nuclear transport, HeLa cells transfected with plasmid expressing GFP-VSVM fusion protein and stained for the Nup 414 component of NPCs showed near identical overlap of GFP-VSVM with NPCs [Fig. 5A, (Petersen et al., 2000)].
To effectively impair HIV-1 cDNA import with VSVM, we used an approach that ensured only cells expressing VSVM were potential targets of infection. This was achieved by: a) cotransfecting HeLa cells with plasmids expressing GFP-VSVM and the primary receptor for HIV-1 entry, CD4; and b) infecting with HIV-1 containing native envelope protein to restrict HIV-1 entry to the transfected cells expressing CD4 and GFP-VSVM. Consequently, new HIV-1 stocks were prepared for this work containing native envelope protein for entry and titrated with increasing A3G. When the infectivity of these stocks was assessed (Fig. 5B), the infection of Vif positive HIV-1 reduced with increasing A3G. This is consistent with Vif not overcoming all the A3G during virion production, thereby allowing some A3G antiviral effect in target cells. In contrast, HIV-1ΔVif infection was far more sensitive to the A3G compared to Vif positive HIV-1 (Fig. 5B) reflecting the lack of functional Vif to overcome A3G during virion production.
When HeLa cells transiently expressing CD4 with or without GFP-VSVM were infected with these HIV-1ΔVif stocks, VSVM impaired viral cDNA products relative to cells not expressing VSVM in all cases (Fig. 5C–F, CD4 versus CD4+VSVM). However, for each virus, GFP-VSVM decreased viral early RT and late RT 3–5 fold but greater 15 fold declines in 2-LTR circles were observed (Fig. 5C–E, CD4 versus CD4+VSVM). This greater loss in 2-LTR circles by GFP-VSVM relative to early RT and late RT is expected if HIV-1 cDNA import is blocked, demonstrating that GFP-VSVM is impairing nuclear import in these samples. Importantly, in the presence of GFP-VSVM, A3G still decreased HIV-1ΔVif late RT in a dose dependent manner when corrected for changes in early RT (Fig. 5F), even though HIV-1 cDNA import was impaired (Fig. 5E). This indicates A3G still reduces HIV-1ΔVif late RT when viral cDNA is retained in the cytoplasm through a nuclear import block, suggesting A3G has important activity in the cytoplasm to impair early HIV-1 replication.
Discussion
By measuring the impact of intra-virion human A3G on HIV-1ΔVif cDNA products during replication in target cells, this study demonstrates A3G progressively depletes viral cytoplasmic and nuclear cDNA forms with increasing magnitude, causing minor losses in early RT, larger losses in late RT, and greatest losses in 2-LTR circles (Fig. 1–3). Moreover, the magnitude of these losses also varied with the A3G dose (Fig. 3). This data agrees with early studies before the identification of A3G that report non-permissive cells (now shown to contain A3G) impair Vif-deficient HIV-1 late RT but block 2-LTR circle levels (von Schwedler et al., 1993; Nascimbeni et al.,1998). Moreover, it is consistent with recent findings that intra-virion A3G impairs early RT, late RT and 2-LTR circles with increasing magnitude (Luo et al., 2007; Mbisa et al., 2007). Experiments using MA-DKA to inhibit integration did not stop the poor 2-LTR circle output from HIV-1ΔVif containing A3G (Fig. 4B, C). This may suggest that the HIV-1ΔVif cDNA substrate that makes it into the nucleus is not normal and cannot form more 2-LTR circles when integration is blocked with MA-DKA, even if more viral cDNA substrate becomes available to make 2-LTR circles. Recent work reports A3G can alter the ends of viral cDNA substrates (Mbisa et al., 2007). However, the extent that these abnormal ends contribute to losses in 2-LTR circles and provirus remains unclear, as discussed below. An alternative or additional interpretation of this MA-DKA data is that A3G depletes integrated provirus along with 2-LTR circles from HIV-1ΔVif, providing little viral cDNA substrate when integration is blocked to increase 2-LTR circles, causing 2-LTR circles to remain poor. This interpretation is consistent with previous reports that intra-virion A3G induces greater losses in HIV-1 integrated provirus than late RT (Aires da Silva et al., 2004; Mangeat et al., 2003; Mariani et al., 2003). Moreover, it agrees with recent work published during preparation of this manuscript, showing wildtype human A3G and A3F progressively impair early RT, late RT, 2-LTR circles and then provirus with increasing magnitude (Luo et al., 2007; Mbisa et al., 2007). Therefore, this data seems most consistent with A3G suppressing nuclear 2-LTR circle and provirus cDNAs with greater magnitude and thus in addition to RT product losses. Hence, A3G is clearly blocking viral replication at multiple steps.
To deplete 2-LTR circle and provirus forms of nuclear HIV-1 cDNA, A3G may act in the cytoplasm with consequences preventing 2-LTR circle and provirus formation in the nucleus, or act in the nucleus itself to suppress these viral cDNAs. Our experiments using VSVM to impair HIV-1 cDNA import did not stop A3G from decreasing viral late RT (Fig. 5). As the same decrease in viral late RT was seen when the viral cDNA was restricted to the cytoplasm, this suggests A3G likely acts primarily in the cytoplasm to decrease the HIV-1 cDNA products. Notably, while our study does not exclude potential, additional effects of A3G in the nucleus that could also suppress cDNA substrates forming 2-LTR circles or provirus, recent work supports our findings that A3G acts primarily in the cytoplasm to impede HIV-1ΔVif. Importantly, Mbisa and colleagues found that A3G impairs the in vitro integration activity of cytoplasmic HIV-1 preintegration complexes (PICs) that accounts for the provirus decline in their system. This infers that A3G acts on viral complexes in the cytoplasm to cause the subsequent provirus loss (Mbisa et al., 2007), consistent with our VSVM findings. Therefore collectively, these data are most consistent with intra-virion human A3G having major antiviral activity on HIV-1ΔVif in the cytoplasm of target cells, progressively depleting cytoplasmic RT products and then nuclear viral cDNA forms with increasing magnitude.
Overall, the ability of A3G to decrease the various HIV-1 cDNA products varied with the A3G dose (Fig. 3). However, the different cDNA products appeared to respond differently to the A3G dose. For instance, when A3G blocked viral infection, the early RT declined but routinely plateaued at a 1.5–2 fold loss despite increasing A3G (Fig. 3A and B; 1, 2 and 5 A3G). In contrast, late RT products declined in a dose-dependent manner with increasing A3G, diminishing 2.5, 4 and 10 fold with 1, 2 and 5 molar ratios of A3G respectively in the experiment shown (Fig. 3C; 1, 2 and 5 A3G). Furthermore, a dramatic, near complete loss of 2-LTR circles accompanied the block to infection, decreasing 21 to 42 fold as A3G increased (Fig. 3A and D; 1, 2 and 5 A3G). In fact, this dramatic loss in 2-LTR circles typically correlated with the A3G block to infection (Fig. 1A and D), suggesting 2-LTR circles may serve as a good indicator for the potency of A3G inhibition of HIV-1 infection.
These different behaviours of the various HIV-1 cDNA products to the A3G dose may represent different antiviral consequences of A3G. For instance, the approximate 1.5–2 fold loss in early RT that plateaued despite increasing A3G likely represents an initial antiviral activity of A3G that interferes with tRNA initiation of reverse transcription, as recently published for A3G and A3F (Guo et al., 2006; Yang et al., 2007b). In contrast, the dose-dependent loss in late RT with increasing A3G may arise from a second antiviral activity like impaired second strand transfer during reverse transcription (Klarmann et al., 2003; Mbisa et al., 2007), causing continual loss of late RT with more A3G despite the plateau in early RT. Although, it should be noted that the magnitude of the plus strand transfer defect does not account for the magnitude of late RT decline (Mbisa et al., 2007), indicating other antiviral activity is involved. Finally, the greater loss in 2-LTR circles may represent a culmination of the previous antiviral activities and a third antiviral activity.
The explanation for this third activity enhancing the loss of 2-LTR circles remains unclear. However, recently published work indicates A3G alters cytoplasmic HIV reverse transcription complexes (RTCs) and/or PICs. This is supported by reports that A3G: a) increases the sedimentation of HIV-1ΔVif RTCs coincident with increased viral cDNA mutation in these abnormal complexes (Carr et al., 2006); b) perturbs the in vitro integration activity of cytoplasmic PICs (Mbisa et al., 2007); c) interacts with viral integrase, nucleocapsid and the ribonucleoprotein complex inside virion cores (Luo et al., 2007; Soros, Yonemoto, and Greene, 2007); and d) A3G interaction with HIV-1 RNA in virions blocks A3G deamination that is restored by RNase H activity during reverse transcription (Soros, Yonemoto, and Greene, 2007). This links A3G to incoming HIV-1 RTCs/PICs and in a position to perturb the structure and/or behaviour of these viral complexes to then impair 2-LTR circle and provirus formation. One study correlates A3G and A3F interaction with integrase in virions to the efficient loss of HIV-1ΔVif provirus in their system, speculating this interaction may: a) interfere with the structural integrity of the PIC to inhibit PIC intracellular transport and integration or b) obstruct integrase function and integration (Luo et al., 2007). However, as 2-LTR circles rely on non-homologous end joining proteins to form (Li et al., 2001) and not integrase, if A3G just obstructs integrase function, this does not easily explain the 2-LTR circle loss. Hence, this result seemingly favors a model where changes to the HIV PIC integrity perturb it’s trafficking to impair nuclear entry and thus nuclear 2-LTR circles and provirus, plus cause extra integration defects to further impair provirus to a greater magnitude. A separate study posits A3G alters viral cDNA ends to cause the losses in provirus and 2-LTR circle levels (Mbisa et al., 2007). However, only 30% of the HIV-1ΔVif 2-LTR circles that formed in this study had the specific cDNA end alteration (Mbisa et al., 2007). Moreover, A3G antiviral activity did not significantly alter the proportion of 2-LTR circles with normal ends, insertions or deletions, which should change if A3G caused incomplete or aberrant cDNA ends (Mbisa et al., 2007). Therefore, it is unclear from this study the extent that altered viral cDNA ends actually contribute to losses in 2-LTR circles or provirus in the same experiment and additional studies are required.
Notably, A3G-mediated deamination may also contribute to losses in all of these viral cDNA products. This is supported by reports that the A3G C-terminal deaminase domain can enhance losses in viral early RT and late RT cDNAs (Guo et al., 2006; Yang et al., 2007b; Holmes et al., 2007), even if it is not required for A3G or A3F restriction (Holmes et al., 2007; Newman et al., 2005). Moreover, these C-terminal A3G mutants were recently shown to either reduce or block losses in 2-LTR circles and provirus in one study (Luo et al., 2007), or late RT, 2-LTR circle and provirus in another study (Mbisa et al., 2007). Therefore, while A3G and A3F can have antiviral activity independent of deamination (Holmes et al., 2007; Newman et al., 2005), these data indicate deamination clearly enhances losses in these viral cDNAs, particularly 2-LTR circles and provirus depending on the deamination mutant. How deamination contributes to these viral cDNA declines is unclear. Among the many possibilities, recent work indicates A3G deamination prevents viral cDNA integration as, in contrast to wildtype A3G, a C-terminal A3G mutant no longer restricted the in vitro integration of viral complexes and provirus formation (Mbisa et al., 2007). Hence, these data hint A3G may use deamination dependent and independent methods to impair viral cDNA, with deamination particularly impacting viral cDNA integration. However, more work is required to determine the precise role of deamination in depleting all the forms of viral cDNA.
Ultimately, based on these collective data, we speculate the additional losses in 2-LTR circles and provirus observed here and by others (Luo et al., 2007; Mbisa et al., 2007) arise from additional A3G activity. This activity potentially results from A3G-mediated changes to HIV-1 RTC/PIC integrity and behaviour, dependent or independent of deamination, which diminish RTC/PIC trafficking to/into the nucleus, viral cDNA end formation and/or integration ability to collectively deplete 2-LTR circles and provirus to a greater magnitude. Ultimately, our A3G titration experiments support the notion A3G can have multiple antiviral activities on early HIV replication and indicate the A3G dose may dictate the magnitude of the antiviral activities at play in the target cell.
Curiously, the magnitude of the A3G loss in 2-LTR circles relative to late RT products varied in our experiments. For instance, A3G caused a more complete loss in 2-LTR circles versus late RT in figure 1 and 3 compared to figure 2. As figure 3C and 3D show that the greater the A3G dose, the closer the declines in late RT and 2-LTR circles become, we speculate this difference in the extent of 2-LTR circle loss may arise from variation in A3G doses between different virus preparations. A difference in A3G dose also likely accounts for greater fold losses in overall late RT levels (Bishop, Holmes, and Malim, 2006; Holmes et al., 2007), or late RT versus early RT products (Guo et al., 2006; Yang et al., 2007b) seen in previous studies relative to our and other studies (Luo et al., 2007; Mbisa et al., 2007). Again, figure 3B and 3C indicate that despite the early RT decline plateauing around 1.5–2 fold with increasing A3G, late RT continues to decline with increasing A3G. Thus, greater losses in late RT versus early RT can be observed depending on the A3G dose. Ultimately, throughout all our experiments, A3G routinely caused losses in early RT, late RT and 2-LTR circles with increasing magnitude, indicating A3G has pleiotropic and accumulative antiviral effects on all of these HIV cDNA products.
The finding that human A3G impairs HIV-1ΔVif 2-LTR circles and provirus to greater magnitudes than early and late RT products both here and other studies (Aires da Silva et al., 2004; Luo et al., 2007; Mangeat et al., 2003; Mariani et al., 2003; Mbisa et al., 2007; von Schwedler et al., 1993; Nascimbeni et al.,1998) collectively indicate A3G reduces nuclear forms of HIV-1 cDNA in addition to cytoplasmic RT products. This is also observed in recent studies of A3F impact on HIV-1 cDNA forms (Luo et al., 2007). Previous work with MLV vectors demonstrate human A3G, A3F and A3C also impair MLV cDNA levels (unintegrated and/or integrated) in infected cells (Langlois et al., 2005). Furthermore, human A3G impedes HTLV-1 integrated DNA levels in target cells (Sasada et al., 2005). Human A3G also impairs integrated cDNA levels for the Ty1 LTR retrotransposon (Dutko et al., 2005), as do human A3B and A3F that impede integrated cDNA accumulation for the L1 non-LTR retrotransposon (Stenglein and Harris, 2006). Finally, human A3G or A3F, African Green Monkey A3G and murine A3 all diminish integrated cDNA levels for the MusD LTR retrotransposon (Esnault et al., 2006). Therefore, these studies collectively indicate losses in integrated viral cDNA maybe a common aspect to the antiviral mechanism of APOBEC3 proteins on reverse transcribing pathogens. Intriguingly, resting CD4+ T cells rich in low molecular mass (LMM) A3G also reduce HIV-1 late RT levels with little impact on early RT (Chiu et al., 2005). Therefore, antiviral LMM A3G in target cells might also impede HIV-1 at a comparable stage early in replication similar to the intra-virion A3G assessed here.
In conclusion, our work demonstrates that A3G has important activity in the cytoplasm and progressively impairs viral early RT, late RT and 2-LTR circles with increasing magnitude. Thus, A3G restricts HIV-1ΔVif replication via multiple activities, impairing multiple steps early in viral replication in addition to downstream effects of deamination on viral gene expression to presumably ensure that invading reverse transcribing pathogens are completely restricted in target cells. Our laboratory previously developed systems that allow the localization and trafficking of individual HIV-1 genomes to be visualized in situ (McDonald et al., 2002). Because earlier studies revealed A3G acts to perturb the early events of HIV-1 replication after entry (Aires da Silva et al., 2004; Mangeat et al., 2003; Mariani et al., 2003), we thought these systems might be useful to study the inhibitory action of this protein. At the time, a shortcoming of these systems was our inability to follow the genome into the nucleus using the GFP-Vpr tag (McDonald et al., 2002). Therefore, the impetus for the studies presented here was to determine if intra-virion A3G could inhibit HIV-1 in the cytoplasm. Both our data (Fig. 5) and a recent paper (Mbisa et al., 2007) infer intra-virion A3G inhibits HIV-1 replication primarily in the cytoplasm, likely at multiple stages, causing progressively greater defects in viral cytoplasmic RT products and then nuclear cDNA forms. Therefore, as A3G appears to be perturbing early HIV-1ΔVif replication primarily in the cytoplasm, we will now use microscopy to gain insight into how intra-virion A3G impedes early HIV-1ΔVif replication in the cytoplasm of target cells.
Materials and Methods
Plasmids
The HIV-1 pIIIB+/Δenv+/Δvif proviral plasmids that contained or lacked the env and/or vif gene (Simon et al., 1998; Simon et al., 1995), were provided by Dr. Michael Malim (King’s College London, United Kingdom). Dr. Nathaniel Landau (New York University, New York, USA) provided the phuA3G-HA plasmid expressing human A3G fused to an influenza A virus haemagglutinin tag [HA, (Mariani et al., 2003)]. Dr. James Dahlberg and Dr. Elsebet Lund (University of Wisconsin-Madison, Madison, USA) provided pEGFP-VSVM expressing enhanced green fluorescent protein (EGFP) fused to vesicular stomatitis virus matrix protein [VSVM, (Petersen et al., 2000)]. The pEGFPVpr (McDonald et al., 2002), pVSVg and pCD4 plasmids express EGFP fused to HIV-1 Vpr, VSV glycoprotein or human CD4 protein respectively. pCMplpa contains a CMV promoter and SV40 polyadenylation signal but lacks a gene for expression in mammalian cells.
Cells and pharmaceuticals
MAGI reporter cells (Kimpton and Emerman, 1992), human osteosarcoma cells (HOS), human epithelial cervical adenocarcinoma cells (HeLa) and human embryonic kidney cells expressing SV40 large T antigen (293T) were propagated at 37°C, 7% CO2 in Dulbecco’s modified Eagle’s medium (HyClone) supplemented with 10% fetal bovine serum (Gibco), penicillin (100 U/ml; Gibco), streptomycin (100 μg/ml; Gibco), L-glutamine (292 μg/ml; Gibco) and ciprofloxacin (10 μg/ml; Cellgro). The 118-D-24 MA-DKA integration inhibitor and Nevirapine reverse transcription inhibitor were obtained from the NIH AIDS Research and Reference Reagent Program, dissolved in dimethyl sulfoxide (DMSO) and stored at −20°C as a 25 mM or 10 mM stock respectively.
Virus stocks
Virus stocks were generated by cotransfecting 293T cells with plasmid mixes via calcium phosphate. To produce VSVg pseudotyped viruses (Fig. 1–4), plasmid mixes contained 8 μg pIIIBΔenv+/Δvif provirus plasmid, 8 μg pVSVg, 4 ug pEGFPVpr and either 0, 0.0375, 0.375, 3.75, 7.5 or 18.75 μg of phuA3G-HA (for a 0:1, 0.01:1, 0.1:1, 1:1, 2:1 or 5:1 molar ratio of A3G:provirus plasmid respectively). To produce native envelope viruses (Fig. 5), plasmid mixes contained 16 μg pIIIB+/Δvif provirus plasmid and either 0, 0.075, 0.75 or 3.75 μg of phuA3G-HA (for a 0:1, 0.01:1, 0.1:1 or 0.5:1 molar ratio of A3G:provirus plasmid respectively). The promoter-matched plasmid, pCMplpa, was used to normalize DNA amount between transfection samples. Producer cell transfection efficiencies of 60–80% were typically achieved. Virus-containing media was collected 36–48 h post transfection, filtered (0.45 μm), stored at −80°C and characterized for infectivity plus yield before subsequent use. To measure infectivity, MAGI reporter cells were typically infected for 16 h and 30 h after infection, β-galactosidase activity quantified in a liquid assay as previously described (Campbell, Nunez, and Hope, 2004; McDonald et al., 2003). Virus yield was measured using a HIV-1 p24Gag ELISA kit (Perkin Elmer).
Quantitative real-time PCR
For real-time PCR experiments, 12 well plates were seeded with ≈1.5×105 HOS cells per well. The next day, equal p24Gag amounts of VSVg pseudotyped HIV-1 in the same volume were treated with 20 U/ml DNaseI (Roche) in 10 mM MgCl2 for 60 min at 20–25°C before adding to cell monolayers. Cells were continuously infected in these experiments to maximize 2-LTR circle output for accurate quantification. This involved adding 500 μl of virus containing equal p24Gag amount to the cells, incubating for 2 h at 37°C, adding an equal volume of media to increase the infection volume and returning the cells to 37°C to continue infection. At various timepoints after adding virus, cells were collected, treated with RNase A and genomic DNA extracted (Qiagen DNeasy Tissue Kit). Genomic DNA was digested with 1 U/μl Dpn I (New England Biolabs) for 4 h at 37°C and 50 ng samples typically analysed for various DNA products using real-time PCR. Cellular β-actin and viral early RT, late RT and 2-LTR circle DNAs were measured using published primers (Butler, Hansen, and Bushman, 2001; Campbell, Nunez, and Hope, 2004; Munk et al., 2002) or primers designed to lack sequence prone to A3G deamination: 2LTRF- (5′-CACTGCTTAAGCCTCAATAAAGC-3′), 2LTRR- (5′-ACAGATCAAGGATATCTTGTCTTCG-3′) and LateRT- (5′-TCTTGCCGTGCGCGCTTCAG-3′). Samples were analysed in triplicate using iQ SYBR Green Supermix and the iCycler iQ Real-Time PCR Detection System (Bio-Rad). Ten fold dilutions of genomic DNA, proviral plasmid or plasmid cloned with 2-LTR junction PCR product, in 30ng/ml tRNA diluent, were used to quantitate β-actin, early and late RT, or 2-LTR circles respectively.
For studies with MA-DKA integration inhibitor, similar experiments were performed except HOS cell monolayers were preincubated with 25 μM MA-DKA or 0.001% DMSO control 4 h before infection and these compounds also added to the virus inoculum and additional media.
Similar experiments were also performed for the VSVM studies except 12 well plates were seeded with ≈1.8×105 HeLa cells per well, monolayers transfected the next day with pCD4 and pEGFP-VSVM using Effectene (Qiagen) and cells infected 24 h later with equal p24Gag amounts of DNaseI treated HIV-1 containing wildtype envelope protein.
Representative results selected from 4 independent experiments are shown for each figure except figure 3, which is representative of 2 independent experiments.
Immunofluorescence
To visualize intracellular EGFP-VSVM, fibronectin coated coverslips were seeded with ≈0.75×105 HeLa cells and transfected the next day with pEGFP-VSVM using Effectene (Qiagen). Cells were fixed 24 h later with 3.7% formaldehyde (Polysciences) in 0.1M PIPES pH 6.8, washed in PBS and treated 5 min with 0.1% triton X-100 and 10% normal donkey serum in PBS. The permeabilised cells were stained for filamentous actin using Texas red-labeled phalloidin (Molecular Probes) and nuclear pores using 414 monoclonal antibody (BabCO) directly conjugated to Alexa Fluor 647 via a monoclonal antibody labeling kit (Molecular Probes). Mounted coverslips were imaged in z-series using a charge-coupled device digital camera on a DeltaVision system. Out-of-focus light was digitally removed using Softworks deconvolution software and volume projections generated via Softworks software (Applied Precision Inc).
Acknowledgments
We thank Dr. Michael Malim (King’s College London, United Kingdom), Dr. Nathaniel Landau (New York University, New York, USA), and Dr. James Dahlberg plus Dr. Elsebet Lund (University of Wisconsin-Madison, Madison, USA) for generously providing reagents. The integrase inhibitor (118-D-24) and Nevirapine reverse transcription inhibitor were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. This work was supported by a National Institutes of Health grant RO1 AI47770 to T.J.H. T.J.H. is an Elizabeth Glaser Scientist.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aires da Silva F, Santa-Marta M, Freitas-Vieira A, Mascarenhas P, Barahona I, Moniz-Pereira J, Gabuzda D, Goncalves J. Camelized rabbit-derived VH single-domain intrabodies against Vif strongly neutralize HIV-1 infectivity. J Mol Biol. 2004;340(3):525–42. doi: 10.1016/j.jmb.2004.04.062. [DOI] [PubMed] [Google Scholar]
- Bishop KN, Holmes RK, Malim MH. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J Virol. 2006;80(17):8450–8. doi: 10.1128/JVI.00839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol. 2004;14(15):1392–6. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- Bogerd HP, Wiegand HL, Doehle BP, Lueders KK, Cullen BR. APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 2006a;34(1):89–95. doi: 10.1093/nar/gkj416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O’Shea KS, Moran JV, Cullen BR. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc Natl Acad Sci U S A. 2006b;103(23):8780–5. doi: 10.1073/pnas.0603313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PO. Integration. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor Laboratory Press; New York, USA: 1997. pp. 161–203. [PubMed] [Google Scholar]
- Bukrinsky M. A hard way to the nucleus. Mol Med. 2004;10(1–6):1–5. [PMC free article] [PubMed] [Google Scholar]
- Butler SL, Hansen MS, Bushman FD. A quantitative assay for HIV DNA integration in vivo. Nat Med. 2001;7(5):631–4. doi: 10.1038/87979. [DOI] [PubMed] [Google Scholar]
- Campbell EM, Nunez R, Hope TJ. Disruption of the actin cytoskeleton can complement the ability of Nef to enhance human immunodeficiency virus type 1 infectivity. J Virol. 2004;78(11):5745–55. doi: 10.1128/JVI.78.11.5745-5755.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr JM, Davis AJ, Coolen C, Cheney K, Burrell CJ, Li P. Vif-deficient HIV reverse transcription complexes (RTCs) are subject to structural changes and mutation of RTC-associated reverse transcription products. Virology. 2006;351(1):80–91. doi: 10.1016/j.virol.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, Landau NR, Weitzman MD. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr Biol. 2006;16(5):480–5. doi: 10.1016/j.cub.2006.01.031. [DOI] [PubMed] [Google Scholar]
- Chiu YL, Soros VB, Kreisberg JF, Stopak K, Yonemoto W, Greene WC. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature. 2005;435(7038):108–14. doi: 10.1038/nature03493. [DOI] [PubMed] [Google Scholar]
- Courcoul M, Patience C, Rey F, Blanc D, Harmache A, Sire J, Vigne R, Spire B. Peripheral blood mononuclear cells produce normal amounts of defective Vif- human immunodeficiency virus type 1 particles which are restricted for the preretrotranscription steps. J Virol. 1995;69(4):2068–74. doi: 10.1128/jvi.69.4.2068-2074.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR. Role and mechanism of action of the APOBEC3 family of antiretroviral resistance factors. J Virol. 2006;80(3):1067–76. doi: 10.1128/JVI.80.3.1067-1076.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehle BP, Schafer A, Cullen BR. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology. 2005;339(2):281–8. doi: 10.1016/j.virol.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Dornadula G, Yang S, Pomerantz RJ, Zhang H. Partial rescue of the Vif-negative phenotype of mutant human immunodeficiency virus type 1 strains from nonpermissive cells by intravirion reverse transcription. J Virol. 2000;74(6):2594–602. doi: 10.1128/jvi.74.6.2594-2602.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutko JA, Schafer A, Kenny AE, Cullen BR, Curcio MJ. Inhibition of a yeast LTR retrotransposon by human APOBEC3 cytidine deaminases. Curr Biol. 2005;15(7):661–6. doi: 10.1016/j.cub.2005.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebina H, Aoki J, Hatta S, Yoshida T, Koyanagi Y. Role of Nup98 in nuclear entry of human immunodeficiency virus type 1 cDNA. Microbes Infect. 2004;6(8):715–24. doi: 10.1016/j.micinf.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Esnault C, Heidmann O, Delebecque F, Dewannieux M, Ribet D, Hance AJ, Heidmann T, Schwartz O. APOBEC3G cytidine deaminase inhibits retrotransposition of endogenous retroviruses. Nature. 2005;433(7024):430–3. doi: 10.1038/nature03238. [DOI] [PubMed] [Google Scholar]
- Esnault C, Millet J, Schwartz O, Heidmann T. Dual inhibitory effects of APOBEC family proteins on retrotransposition of mammalian endogenous retroviruses. Nucleic Acids Res. 2006;34(5):1522–31. doi: 10.1093/nar/gkl054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Cen S, Niu M, Saadatmand J, Kleiman L. Inhibition of formula-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J Virol. 2006;80(23):11710–22. doi: 10.1128/JVI.01038-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin KE. Inhibition of nucleo-cytoplasmic trafficking by RNA viruses: targeting the nuclear pore complex. Virus Res. 2003;95(1–2):35–44. doi: 10.1016/S0168-1702(03)00165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113(6):803–9. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science. 2000;287(5453):646–50. doi: 10.1126/science.287.5453.646. [DOI] [PubMed] [Google Scholar]
- Holmes RK, Koning FA, Bishop KN, Malim MH. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J Biol Chem. 2007;282(4):2587–95. doi: 10.1074/jbc.M607298200. [DOI] [PubMed] [Google Scholar]
- Kaiser SM, Emerman M. Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase Apobec3G. J Virol. 2006;80(2):875–82. doi: 10.1128/JVI.80.2.875-882.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimpton J, Emerman M. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated beta-galactosidase gene. J Virol. 1992;66(4):2232–9. doi: 10.1128/jvi.66.4.2232-2239.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarmann GJ, Chen X, North TW, Preston BD. Incorporation of uracil into minus strand DNA affects the specificity of plus strand synthesis initiation during lentiviral reverse transcription. J Biol Chem. 2003;278(10):7902–9. doi: 10.1074/jbc.M207223200. [DOI] [PubMed] [Google Scholar]
- Langlois MA, Beale RC, Conticello SG, Neuberger MS. Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic Acids Res. 2005;33(6):1913–23. doi: 10.1093/nar/gki343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003;300(5622):1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- Li L, Olvera JM, Yoder KE, Mitchell RS, Butler SL, Lieber M, Martin SL, Bushman FD. Role of the non-homologous DNA end joining pathway in the early steps of retroviral infection. Embo J. 2001;20(12):3272–81. doi: 10.1093/emboj/20.12.3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochelt M, Romen F, Bastone P, Muckenfuss H, Kirchner N, Kim YB, Truyen U, Rosler U, Battenberg M, Saib A, Flory E, Cichutek K, Munk C. The antiretroviral activity of APOBEC3 is inhibited by the foamy virus accessory Bet protein. Proc Natl Acad Sci U S A. 2005;102(22):7982–7. doi: 10.1073/pnas.0501445102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo K, Wang T, Liu B, Tian C, Xiao Z, Kappes J, Yu XF. Cytidine Deaminases APOBEC3G and APOBEC3F Interact with Human Immunodeficiency Virus Type 1 Integrase and Inhibit Proviral DNA Formation. J Virol. 2007;81(13):7238–48. doi: 10.1128/JVI.02584-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424(6944):99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003;114(1):21–31. doi: 10.1016/s0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003;9(11):1398–403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- Mbisa JL, Barr R, Thomas JA, Vandegraaff N, Dorweiler IJ, Svarovskaia ES, Brown WL, Mansky LM, Gorelick RJ, Harris RS, Engelman A, Pathak VK. Human Immunodeficiency Virus Type 1 cDNAs Produced in the Presence of APOBEC3G Exhibit Defects in Plus-Strand DNA Transfer and Integration. J Virol. 2007;81(13):7099–110. doi: 10.1128/JVI.00272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Vodicka MA, Lucero G, Svitkina TM, Borisy GG, Emerman M, Hope TJ. Visualization of the intracellular behavior of HIV in living cells. J Cell Biol. 2002;159(3):441–52. doi: 10.1083/jcb.200203150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Wu L, Bohks SM, KewalRamani VN, Unutmaz D, Hope TJ. Recruitment of HIV and its receptors to dendritic cell-T cell junctions. Science. 2003;300(5623):1295–7. doi: 10.1126/science.1084238. [DOI] [PubMed] [Google Scholar]
- Munk C, Brandt SM, Lucero G, Landau NR. A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc Natl Acad Sci U S A. 2002;99(21):13843–8. doi: 10.1073/pnas.212400099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimbeni M, Bouyac M, Rey F, Spire B, Clavel F. The replicative impairment of Vif- mutants of human immunodeficiency virus type 1 correlates with an overall defect in viral DNA synthesis. J Gen Virol. 1998;79 (Pt 8):1945–50. doi: 10.1099/0022-1317-79-8-1945. [DOI] [PubMed] [Google Scholar]
- Navaratnam N, Sarwar R. An overview of cytidine deaminases. Int J Hematol. 2006;83(3):195–200. doi: 10.1532/IJH97.06032. [DOI] [PubMed] [Google Scholar]
- Newman EN, Holmes RK, Craig HM, Klein KC, Lingappa JR, Malim MH, Sheehy AM. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol. 2005;15(2):166–70. doi: 10.1016/j.cub.2004.12.068. [DOI] [PubMed] [Google Scholar]
- Nisole S, Saib A. Early steps of retrovirus replicative cycle. Retrovirology. 2004;1(1):9. doi: 10.1186/1742-4690-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OhAinle M, Kerns JA, Malik HS, Emerman M. Adaptive evolution and antiviral activity of the conserved mammalian cytidine deaminase APOBEC3H. J Virol. 2006;80(8):3853–62. doi: 10.1128/JVI.80.8.3853-3862.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen JM, Her LS, Varvel V, Lund E, Dahlberg JE. The matrix protein of vesicular stomatitis virus inhibits nucleocytoplasmic transport when it is in the nucleus and associated with nuclear pore complexes. Mol Cell Biol. 2000;20(22):8590–601. doi: 10.1128/mcb.20.22.8590-8601.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priet S, Gros N, Navarro JM, Boretto J, Canard B, Querat G, Sire J. HIV-1-Associated Uracil DNA Glycosylase Activity Controls dUTP Misincorporation in Viral DNA and Is Essential to the HIV-1 Life Cycle. Mol Cell. 2005;17(4):479–90. doi: 10.1016/j.molcel.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Sasada A, Takaori-Kondo A, Shirakawa K, Kobayashi M, Abudu A, Hishizawa M, Imada K, Tanaka Y, Uchiyama T. APOBEC3G targets human T-cell leukemia virus type 1. Retrovirology. 2005;2(1):32. doi: 10.1186/1742-4690-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrofelbauer B, Yu Q, Zeitlin SG, Landau NR. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J Virol. 2005;79(17):10978–87. doi: 10.1128/JVI.79.17.10978-10987.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher AJ, Nissley DV, Harris RS. APOBEC3G hypermutates genomic DNA and inhibits Ty1 retrotransposition in yeast. Proc Natl Acad Sci U S A. 2005;102(28):9854–9. doi: 10.1073/pnas.0501694102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418(6898):646–50. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 2003;9(11):1404–7. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- Simon JH, Gaddis NC, Fouchier RA, Malim MH. Evidence for a newly discovered cellular anti-HIV-1 phenotype. Nat Med. 1998;4(12):1397–400. doi: 10.1038/3987. [DOI] [PubMed] [Google Scholar]
- Simon JH, Malim MH. The human immunodeficiency virus type 1 Vif protein modulates the postpenetration stability of viral nucleoprotein complexes. J Virol. 1996;70(8):5297–305. doi: 10.1128/jvi.70.8.5297-5305.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JH, Southerling TE, Peterson JC, Meyer BE, Malim MH. Complementation of vif-defective human immunodeficiency virus type 1 by primate, but not nonprimate, lentivirus vif genes. J Virol. 1995;69(7):4166–72. doi: 10.1128/jvi.69.7.4166-4172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soros VB, Yonemoto W, Greene WC. Newly synthesized APOBEC3G is incorporated into HIV virions, inhibited by HIV RNA, and subsequently activated by RNase H. PLoS Pathog. 2007;3(2):e15. doi: 10.1371/journal.ppat.0030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sova P, Volsky DJ. Efficiency of viral DNA synthesis during infection of permissive and nonpermissive cells with vif-negative human immunodeficiency virus type 1. J Virol. 1993;67(10):6322–6. doi: 10.1128/jvi.67.10.6322-6326.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenglein MD, Harris RS. APOBEC3B and APOBEC3F inhibit L1 retrotransposition by a DNA deamination-independent mechanism. J Biol Chem. 2006;281(25):16837–41. doi: 10.1074/jbc.M602367200. [DOI] [PubMed] [Google Scholar]
- Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell. 2003;12(3):591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- Svarovskaia ES, Barr R, Zhang X, Pais GC, Marchand C, Pommier Y, Burke TR, Jr, Pathak VK. Azido-containing diketo acid derivatives inhibit human immunodeficiency virus type 1 integrase in vivo and influence the frequency of deletions at two-long-terminal-repeat-circle junctions. J Virol. 2004;78(7):3210–22. doi: 10.1128/JVI.78.7.3210-3222.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli P, Mangeat B, Jost S, Vianin S, Trono D. Inhibition of hepatitis B virus replication by APOBEC3G. Science. 2004;303(5665):1829. doi: 10.1126/science.1092066. [DOI] [PubMed] [Google Scholar]
- von Schwedler U, Song J, Aiken C, Trono D. Vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. J Virol. 1993;67(8):4945–55. doi: 10.1128/jvi.67.8.4945-4955.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. Embo J. 2004;23(12):2451–8. doi: 10.1038/sj.emboj.7600246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Chen K, Zhang C, Huang S, Zhang H. Virion-associated uracil DNA glycosylase-2 and apurinic/apyrimidinic endonuclease are involved in the degradation of APOBEC3G-edited nascent HIV-1 DNA. J Biol Chem. 2007a;282(16):11667–75. doi: 10.1074/jbc.M606864200. [DOI] [PubMed] [Google Scholar]
- Yang Y, Guo F, Cen S, Kleiman L. Inhibition of initiation of reverse transcription in HIV-1 by human APOBEC3F. Virology. 2007b;365(1):92–100. doi: 10.1016/j.virol.2007.03.022. [DOI] [PubMed] [Google Scholar]
- Yu Q, Chen D, Konig R, Mariani R, Unutmaz D, Landau NR. APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J Biol Chem. 2004a;279(51):53379–86. doi: 10.1074/jbc.M408802200. [DOI] [PubMed] [Google Scholar]
- Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, Richman D, Coffin JM, Landau NR. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat Struct Mol Biol. 2004b;11(5):435–42. doi: 10.1038/nsmb758. [DOI] [PubMed] [Google Scholar]
- Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302(5647):1056–60. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003a;424(6944):94–8. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Pais GC, Svarovskaia ES, Marchand C, Johnson AA, Karki RG, Nicklaus MC, Pathak VK, Pommier Y, Burke TR. Azido-containing aryl beta-diketo acid HIV-1 integrase inhibitors. Bioorg Med Chem Lett. 2003b;13(6):1215–9. doi: 10.1016/s0960-894x(03)00059-3. [DOI] [PubMed] [Google Scholar]
- Zheng YH, Irwin D, Kurosu T, Tokunaga K, Sata T, Peterlin BM. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J Virol. 2004;78(11):6073–6. doi: 10.1128/JVI.78.11.6073-6076.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]