

Abstract
Post-transcriptional modification of ribosomal RNA occurs in all kingdoms of life. The S-adenosyl-L-me-thionine-dependent methyltransferase KsgA introduces the most highly conserved ribosomal RNA modification, the dimethylation of A1518 and A1519 of 16S rRNA. Loss of this dimethylation confers resistance to the antibiotic kasugamycin. Here, we report biochemical studies and high-resolution crystal structures of KsgA from Thermus thermophilus. Methylation of 30S ribosomal subunits by T. thermophilus KsgA is more efficient at low concentrations of magnesium ions suggesting that partially unfolded RNA is the preferred substrate. The overall structure is similar to other methyltransferases but contains an additional α-helix in a novel N-terminal extension. Comparison of the apo-enzyme with complex structures with 5’-methylthioadenosine or adenosine bound in the cofactor-binding site reveal novel features when compared to related enzymes. Several mobile loop regions are observed that restrict access to the cofactor-binding site. In addition, the orientation of residues in the substrate-binding site indicates that conformational changes are required for binding two adjacent residues of the substrate rRNA.
Keywords: ribosome modification, 16S rRNA, 30S ribosomal subunit, rRNA methyltransferase, kasugamycin
Introduction
KsgA is the S-adenosyl-L-methionine (AdoMet)-dependent methyltransferase responsible for producing the most highly conserved ribosome modification, the posttranscriptional N6, N6-dimethylation of two adjacent adenosines, A1518 and A1519 (Escherichia coli numbering), in the 3’ most helix of small subunit rRNAs. Loss of dimethylation of these residues in bacteria confers resistance to the antibiotic kasugamycin1; 2; 3 and creates an error prone phenotype in E. coli4. The yeast ortholog, Dim1p, is essential for viability5, although this is due to a second function of Dim1p in pre-rRNA processing that is distinct from its methyltransferase activity6.
The structural impact of N6, N6-dimethylation of A1518 and A1519 has been examined in some detail. Early biophysical studies indicated that dimethylation by KsgA destabilizes the conformation of the helix 45 GNRA tetraloop7; 8. The highly organized conformation of GNRA tetraloops is incompatible with N6, N6-dimethylation of these two adenosines9. NMR spectroscopy of a fully modified helix 45 oligonucleotide analog has confirmed that dimethylation by KsgA, in conjunction with N2-methylation of G1516, prevents formation of a canonical GNRA tetraloop fold10; 11. The crystal structure of the Thermus thermophilus 30S ribosomal subunit also shows a non-canonical fold for this loop in the context of the ribosome12. Further structural studies of the T. thermophilus 30S subunit13 and of the E. coli 70S ribosome14 in complex with kasugamycin revealed that the two bases modified by KsgA are not in direct contact with the antibiotic. In these structures, kasugamycin is observed bound in the mRNA channel and the location of the two modified bases A1518 and A1519 suggests that loss of dimethylation leads to an indirect destabilization of the antibiotic binding site. KsgA function and substrate recognition are highly conserved15; 16, indicating a more fundamental requirement for base methylation. A recent report proposes that these modifications serve as a checkpoint during ribosome biogenesis17.
The substrate for KsgA has also been the subject of extensive investigation. Early reconstitution studies showed that intact 30S subunits were refractory to methylation by KsgA, with the binding of a specific subset of ribosomal proteins being inhibitory to methylation18. These results suggested that an assembly intermediate is the true substrate for KsgA-catalyzed methylation. A subsequent study concluded that of these proteins, only S21 is directly inhibitory to methylation19. Interestingly, T. thermophilus lacks an S21 ortholog12. These observations have been reconciled recently with the demonstration that E. coli KsgA recognizes the intact 30S subunit in a translationally inactive conformation20. The crystal structure of the apo form of E. coli KsgA21 combined with tethered and solution hydroxyl radical probing, has led to a model for the KsgA binding mode on the 30S subunit17. Conformational differences in the two monomers in the asymmetric unit of the E. coli KsgA crystal structure led to the suggestion that a conformational change occurs upon binding AdoMet21. This hypothesis remains to be tested, as the E. coli KsgA crystal structure lacked a bound cofactor, a consequence of the enzyme’s inability to bind AdoMet from solution18.
Here, we report the identification of the KsgA ortholog from T. thermophilus HB8 and crystal structures of KsgA in the apo form and with 5’-methylthioadenosine or adenosine bound in the cofactor-binding site. We observed significant structural differences in the active site region between the apo-enzyme and the (5’-methylthio)adenosine-bound forms.
Results
Identification and characterization of T. thermophilus KsgA
The unpublished genome sequence of T. thermophilus HB8 (GenBank entry AP008226) annotates TTHA0083 as the likely open reading frame encoding KsgA. A BLASTp search of this genome sequence using the E. coli KsgA sequence (GenBank entry NP_414593) as the target corroborated this assignment (Fig.1, supplemental Fig. S1). To test this conclusion, we constructed a mutant in which TTHA0083 was replaced with the htk gene encoding a thermostable kanamycin adenyltransferase22. That the protein encoded by TTHA0083 is indeed responsible for dimethylation of A1518 and A1519 was established by primer extension of 16S rRNA using reverse transcriptase, whose elongation is strongly inhibited by N6, N6-dimethylation of adenosine23. As shown in Figure 2, primer extension using wild-type 16S rRNA as template produces a pair of strong stops at A1518 and A1519 (lane 1). In contrast, primer extension using 16S rRNA from the TTHA0083-deficient mutant results in complete read-through of these positions and downstream termination at m3U1498 (lane 2). Primer extension using 16S rRNA from subunits methylated in vitro by the TTHA0083-encoded enzyme, overexpressed in E. coli, shows restoration of reverse transcriptase stops at A1518 and A1519 (lanes 3 and 4). We conclude from these results that TTHA0083 encodes KsgA, and hereafter refer to this locus as ksgA.
Figure 1. Structure-based sequence alignment.
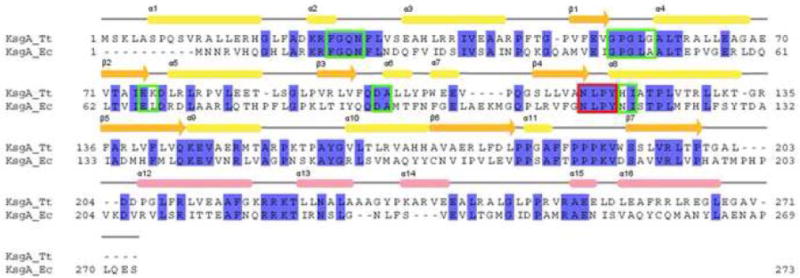
Alignment of KsgA from T. thermophilus with KsgA from E. coli (PDB code 1QYR). Secondary structure elements of T. thermophilus KsgA are indicated on top. The color scheme for secondary structure elements is as in Fig. 3b. Residues in the cofactor-binding site are indicated with green boxes, and the methyltransferase signature motif IV is indicated with a red box.
Figure 2. Identification of the T. thermophilus KsgA ortholog.
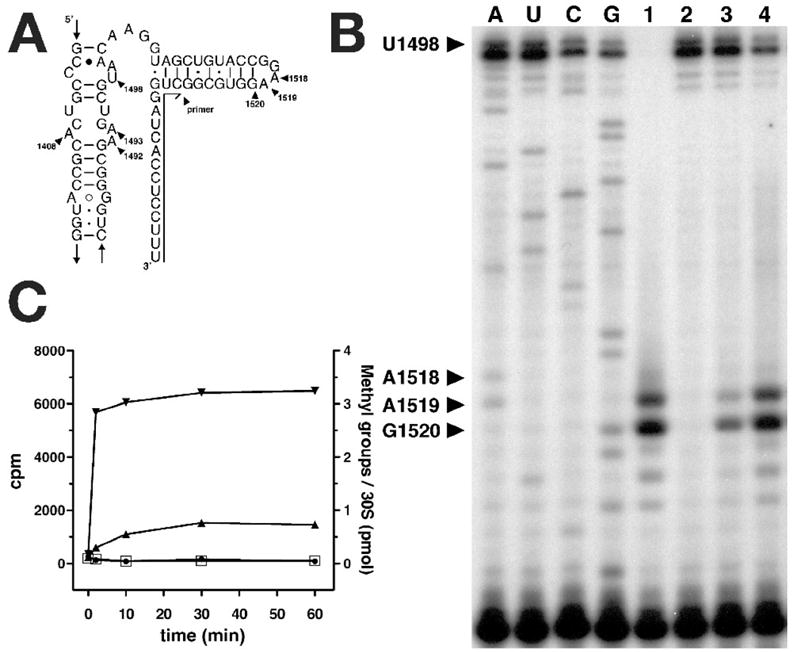
A, Secondary structure diagram of helix 45 in 16S rRNA. The annealing position of the oligonucleotide used for primer extension analysis is indicated. B, Primer extension analysis of Tth HB8 wt (wild type) and Tth HB8 ΔksgA∷htk 16S rRNA. A, U, C and G are sequencing lanes made using ΔksgA∷htk 16S rRNA as template for reverse transcription reaction. Lane 1, wt 16S rRNA; lane 2, ΔksgA∷htk 16S rRNA; lane 3, ΔksgA∷htk 16S rRNA after in vitro methylation of the 30S subunit at 10mM MgCl2; lane 4, ΔksgA∷htk 16S rRNA after in vitro methylation of the 30S subunit at 1mM MgCl2. C, 3H-AdoMet in vitro methylation assay of Tth HB8 wt and Tth HB8 ΔksgA∷htk 30S subunits. Methylation of wt 30S subunits at 10mM MgCl2( ) and 1mM MgCl2(
) and 1mM MgCl2( ); methylation of Tth HB8 ΔksgA∷htk 30S subunits at 10mM MgCl2(
); methylation of Tth HB8 ΔksgA∷htk 30S subunits at 10mM MgCl2( ) and 1mM MgCl2(
) and 1mM MgCl2( ).
).
Incorporation of 3H methyl groups into 30S subunits in vitro with cloned KsgA also shows that this enzyme methylates subunits from the ΔksgA∷htk strain but not those from wild-type T. thermophilus HB8 (Fig. 2c). This reaction is rapid at 70 °C, near the optimum growth temperature for T. thermophilus. Methylation of 30S subunits by T. thermophilus KsgA is much more effective at low Mg2+ ion concentration (1 mM) than at high Mg2+ concentration (10 mM) (Fig. 2b, lanes 3 & 4, Fig. 2c). This behavior is similar to that of the E. coli enzyme, for which the optimum Mg2+ concentration is around 3 mM20. The basis for the inhibitory effect of Mg2+ is suggested by the highest resolution (2.50 Å) crystal structure of the T. thermophilus 30S subunit24. In this and in all other 30S subunit structures, A1518 and A1519 are buried and interact with 16S rRNA helix 44. This interaction appears to be stabilized by a cluster of Mg2+ ions at the junction between helices 44 and 45 (Fig 3a). Loss of these Mg2+ ions could facilitate the disengagement of helix 45 from helix 44, making it more accessible to KsgA.
Figure 3. Overall structure of KsgA.
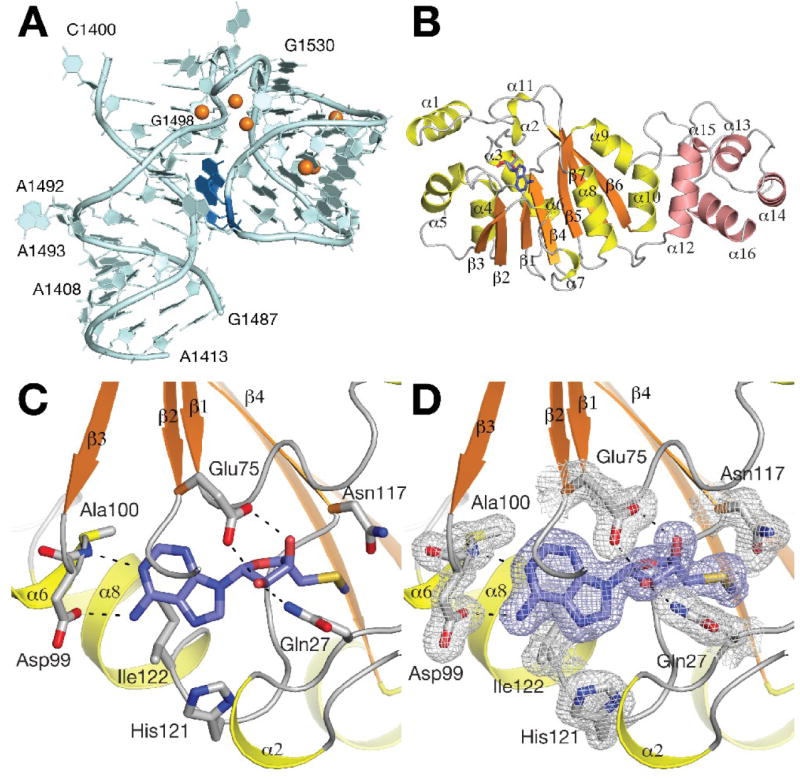
A, Schematic representation of helix 45 in 16S rRNA. The substrate bases A1518 and A1519 are colored in dark blue. Coordinated magnesium ions are shown as orange spheres. B, Schematic representation of the overall structure of KsgA. Secondary structure elements are colored in orange and yellow in the catalytic domain and in salmon in the substrate recognition domain. The bound 5’-methylthioadenosine molecule is shown as blue sticks. C, Cofactor-binding site in KsgA. 5’-methylthioadenosine is shown in purple sticks. Hydrogen bonds formed with the bound 5’-methylthioadenosine molecule are indicated. D, Final σA-weighted 2 FO - FC electron density map of the cofactor-binding site contoured at the 1 σ level.
Structure determination and overall structure of KsgA
The structure of apo-KsgA (271 residues) was solved by single-wavelength anomalous dispersion from seleno-methionine labeled protein in space group P21212 to 1.51 Å resolution (data set KsgA1). The structures of the adenosine-bound form in space group P212121 (KsgA2, 1.53 Å resolution) and of the second apo-enzyme form in space group P43212 (KsgA3, 1.95 Å resolution) were subsequently solved by molecular replacement. Atomic coordinates of the KsgA2 structure were used for the initial refinement of the 5’-methylthioadenosine-bound complex structure in space group P212121 (KsgA4, 1.56 Å resolution). A second crystal form of the 5’-methylthioadenosine-bound complex was solved by molecular replacement in space group P212121 (KsgA5, 1.68 Å resolution). There are two molecules in the asymmetric unit in data set KsgA1, one molecule in data sets KsgA2 and KsgA4, and three molecules in data set KsgA3 and KsgA5. The crystallographic R / Rfree factors are 0.20/0.24, 0.21/0.26, 0.23/0.31, 0.20/0.23 and 0.20/0.24 for the five data sets, respectively. The majority of residues (94.1/95.5/92.3/95.9/95.7%) are in the most favored region of the Ramachandran plot and there are no residues in the disallowed regions. Electron density was well defined in the high-resolution data sets KsgA1, KsgA2, KsgA4 and KsgA5. The final model consists of residues 5-268 in chain A and residues 3-268 in chain B of KsgA1, residues 6-268 in KsgA2 and KsgA4, residues 5-268 in chains A and B, and residues 4-268 in chain C of KsgA5. Loop regions for residues 21 – 24, and 120 – 121 were disordered in chain B and not included in the KsgA1 model. The quality of data set KsgA3 was somewhat weaker. Electron density is well defined for chains A and B, but comparatively weak for chain C. The KsgA3 model consists of residues 6 – 271, 6 – 268, and 8-268 for the three monomers. Loop regions for residues A20 – 25, B20 – 26, and C19 – 27 were disordered and not included in the model. The data collection and refinement statistics are given in Table 1. The overall structure of KsgA consists of two domains. The N-terminal catalytic domain (residues 6 – 200) forms a canonical Rossmann-like methyltransferase fold, with a central seven-stranded β-sheet flanked by three helices on each side (Fig. 3b). Two additional N-terminal helices α1 and α2 and a loop region (including helix α11) that is inserted between strands β6 and β7 define the active site region. The smaller C-terminal domain spanning residues 206 – 268 consists of helices α12 to α16. The surface of the C-terminal domain contains many positively charged residues; a loop region between helices α12 and α13 (218KRRK221) creates a positively charged surface patch that may function in recognition and binding of the ribosomal rRNA substrate. A similar surface charge distribution was observed for KsgA from E. coli21 and for the structurally related ErmC’ methyltransferase25 (discussed below).
Table 1.
Data collection and refinement statistics
| KsgA1 Apo-enzyme, Se-Met | KsgA2 Adenosine | KsgA3 Apo-enzyme | KsgA4 5’-mt-adenosine | KsgA5 5’-mt-adenosine | |
|---|---|---|---|---|---|
| Data collection | |||||
| Space group | P21212 | P212121 | P43212 | P212121 | P212121 |
| Cell dimensions | |||||
| a, b, c (Å) | 137.0, 53.7, 69.4 | 53.2, 61.3, 82.7 | 85.1, 85.1, 215.9 | 53.4, 61.0, 82.5 | 56.1, 79.9, 186.4 |
| α, β, γ(°) | 90, 90, 90 | 90, 90, 90 | 90, 90. 90 | 90, 90, 90 | 90,90,90 |
| Resolution (Å) | 50-1.51 (1.56-1.51) | 25-1.53 (1.58-1.53) | 30-1.95 (2.02-1.95) | 30-1.55 (1.61-1.55) | 30-1.68 (1.74-1.68) |
| Rsym or Rmerge | 0.039 (0.25) | 0.077 (0.41) | 0.086 (0.42) | 0.063 (0.48) | 0.08 (0.38) |
| I / σI | 34.67 (4.27) | 18.27 (2.2) | 29.8 (3.36) | 26.67 (2.64) | 19.2 (2.03) |
| Completeness | 94.5 (71.8) | 98.3 (99.6) | 99.9 (100) | 97.0 (94.5) | 94.8 (95.4) |
| (%) Redundancy | 5.0 (3.5) | 4.3 (4.2) | 11.8 (7.9) | 6.9 (5.9) | 4.3 (3.2) |
| Refinement | |||||
| Resolution (Å) | 1.52 (1.56-1.52) | 1.53 (1.57-1.53) | 1.95 (2.0-1.95) | 1.56(1.60-1.56) | 1.68 (1.74-1.68) |
| No. reflections | 73668 (5001) | 38584 (2861) | 56012 (4001) | 36048 (2524) | 86748 (6303) |
| Rwork / Rfree | 0.20/0.24 (0.23/0.27) | 0.21/0.26 (0.29/0.33) | 0.23/0.31 (0.27/0.39) | 0.20/0.23 (0.23/0.24) | 0.20/0.24 |
| No. atoms | |||||
| Protein | 4188 | 2097 | 6063 | 2076 | 6241 |
| Ligands | 19 | 20 | 60 | ||
| Water | 825 | 408 | 594 | 374 | 733 |
| B-factors | |||||
| Protein | 15.13 | 23.59 | 37.48 | 18.01 | 29.44 |
| Ligands | 19.35 | 18.09 | 21.86 | ||
| Water | 27.27 | 35.15 | 44.06 | 29.58 | 37.12 |
| R.m.s. deviations | |||||
| Bond lengths | 0.010 | 0.011 | 0.024 | 0.010 | 0.014 |
| (Å) | |||||
| Bond angles (°) | 1.33 | 1.41 | 2.09 | 1.34 | 1.45 |
Coordination of adenosine and 5’-methylthioadenosine in the cofactor-binding site
Parallel to the crystallization experiments with apo-KsgA, we attempted to obtain ternary complex crystals with AdoMet in the cofactor-binding site and adenosine in the substrate binding site by co-crystallization of KsgA with 4 mM AdoMet and 4 mM adenosine (data set KsgA2). Unexpectedly, these crystals were found to have adenosine in the cofactor-binding site. The observation that adenosine outcompetes AdoMet for binding in the cofactor-binding site was unexpected considering the additional binding interactions formed with the cofactor methionine group. We therefore repeated the co-crystallization experiments with AdoMet but without adenosine. Data sets (KsgA4 and KsgA5) collected from these crystals showed that 5’-methylthioadenosine was bound in the cofactor-binding site instead of AdoMet. 5’-methylthioadenosine is the major hydrolysis product of AdoMet that is obtained by incubation at 30 °C at pH 4 to pH 7 and a minor hydrolysis product obtained at pH 8.226. KsgA complex crystals could only be obtained after incubation at 50 °C for 10 minutes prior to crystallization, and in both experiments the smaller compound was preferentially bound in the cofactor-binding site.
The 5’-methylthioadenosine molecule in the cofactor-binding site is bound in a position similar to Ado-Met in other class I methyltransferases (Fig. 3c, d). The adenine N6 atom forms a hydrogen bond with Asp99 and the adenine N1 nitrogen atom interacts with the main chain of Ala100. The two ribose hydroxyl groups form hydrogen bonds with Glu75, while the 2’ hydroxyl group forms an additional hydrogen bond with the side chain of Gln27. Finally, Asn117 is located in a position to interact with the carboxylate of a bound AdoMet cofactor. All three residues (Glu75, Gln27 and Asn117) are strictly conserved in KsgA (Fig.1 and supplemental Fig. S1). Adenosine binding induces significant structural rearrangements in the N-terminal loop region including helix α2 and also in the region between residues Asn117 and His121 (see below). The structures with bound 5’-methylthioadenosine and adenosine were initially determined from the same crystal form (KsgA2 and KsgA4). The KsgA structure in the additional 5’-methylthioadenosine bound crystal form KsgA5 is highly similar to these two structures. The overall rmsd between KsgA2 and KsgA4 is 0.14 Å and 0.12 Å between KsgA2 and KsgA5 for all 263 Cα atoms. Side chain orientations in the cofactor-binding site of KsgA2 and KsgA4 remain unchanged.
Conformational differences between KsgA crystal forms
A comparison of the KsgA conformations observed in five crystal forms reveals significant global and local structural rearrangements. Interdomain movement was observed in the structure of E. coli KsgA21. To investigate a similar domain movement in T. thermophilus KsgA, we performed a least-squares super-position of the catalytic domains of all monomers. Comparison of the C-terminal domain orientation does indeed reveal a variable orientation of the C-terminal domain with residues in the loop region between α14 and α15 shifting by up to 4 Å (Fig. 4a). KsgA modifies two adjacent adenine bases and it is conceivable that interdomain movements between the catalytic domain and the C-terminal domain are required to place the substrate into two slightly different orientations with respect to the active site. This need for reorientation could be the basis for both the enhanced T. thermophilus KsgA activity at low Mg2+ concentration and the preference of E. coli KsgA for 30S subunits in an inactive conformation17.
Figure 4. Conformational flexibility of KsgA.
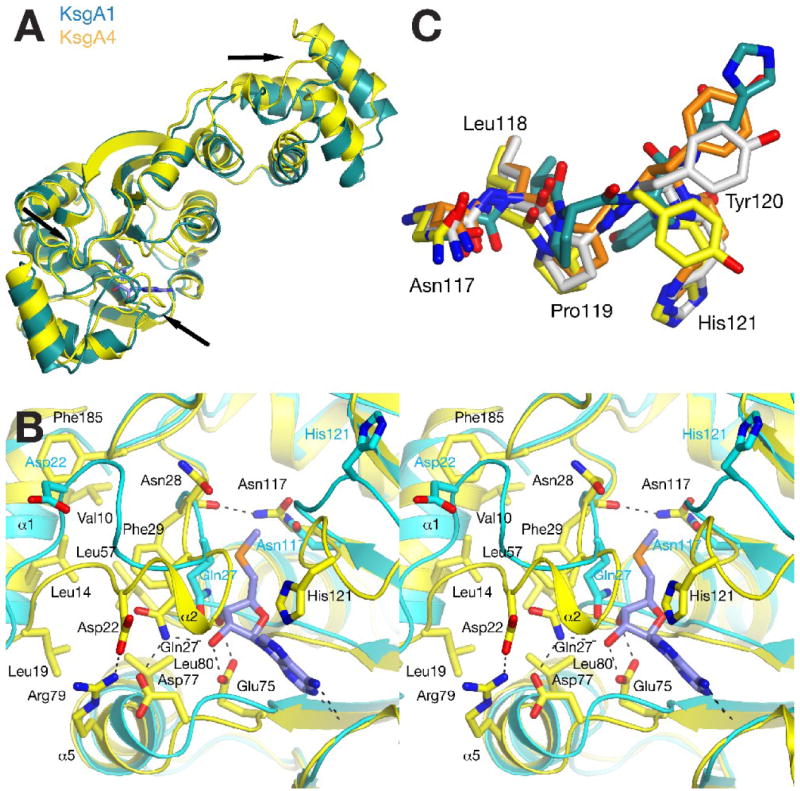
A, Comparison of conformational changes observed in data sets KsgA1 (chain A in teal) and KsgA4 (yellow). The position of major differences is indicated with black arrows. B, stereo diagram showing the different conformations observed for the cofactor-binding site of the apo-enzyme (KsgA1, cyan) and the 5’-methylthioadenosine-bound structure in KsgA4 (yellow). C, Comparison of four different conformations of the 117NLPYH121 loop in the active site region observed in data sets KsgA1 chain A (teal), KsgA2 (yellow), and KsgA3 chain A (orange) and chain C (grey).
In addition to these variations in interdomain orientation, the comparison of the active site shows significant local rearrangements. All KsgA models in the five data sets contain a well-defined first helix α1 that is held in place by hydrophobic interactions between residues Val10, Leu14, Leu19, Phe29, Leu57, and Phe185 (Fig. 4b). The loop region following helix α1 (residues 20 to 27), however, is disordered in four chains in the KsgA1 and KsgA3 apo-enzyme structures, indicating that this loop is likely mobile in solution and in the absence of cofactor. The length of the flexible region is well defined as both flanking residues Leu19 and Phe29 remain engaged in the hydrophobic interface.
Surprisingly, the loop assumes two significantly different ordered conformations in the other structures (Fig. 4b). In the apo-enzyme conformation of chain A in KsgA1, Gln27 partially obstructs the cofactor ribose position. In the 5’-methylthioadenosine-bound structure, this residue moves sideways to interact with Asp77 and a second hydrogen bond formed between Asp22 and Arg79 stabilizes the loop in a conformation that restricts the access to the cofactor-binding site. In contrast, Asp22 is located at a distance of 7.2 Å from Arg79 and interacts with Arg24 in the KsgA1 apo-enzyme structure. In addition, the position of helix α1 shifts significantly along the helix axis in the cofactor-bound structure to accommodate this loop movement.
A second rearrangement occurs in the loop region surrounding His121. Here, the functionally important Asn117 remains constant and the following loop region closes onto the 5’-methylthioadenosine molecule. In the complex structure, His121 is oriented towards the 5’-methylthioadenosine and Ile122 engages in a hydrophobic interaction with the adenine base. In combination, both loop rearrangements fully enclose the 5’methylthioadenosine molecule in the cofactor-binding site indicating that the observed loop flexibility is required for cofactor binding in the Thermus enzyme. In comparing the two conformations, it should however be noted that the loop conformation in the apo-structure is also stabilized by crystal contacts with Pro92 from another protein molecule in the KsgA1 crystal form. Therefore, the loop conformation in the absence of cofactor represents only one of many conformations available to this loop rather than a functionally significant conformation.
In addition to its interaction with cofactor, the conformational flexibility of the second loop region may be relevant for substrate binding. The loop contains the methyltransferase signature motif IV (117NLPY120). While Asn117 remains similar in all KsgA monomers, the Tyr120 side chain assumes several different orientations (Fig. 4c). Residues equivalent to Tyr120 were observed to coordinate the substrate base in other methyltransferases such as DNA adenine methyltransferase M.TaqI27 and the rRNA guanine methyltransferase RsmC28. The large degree of conformational flexibility for Tyr120 may therefore aid in positioning the substrate in two different orientations.
Discussion
Structural comparison with related enzymes
A data base search with the SSM algorithm confirmed that the structures of E. coli KsgA (Pdb 1QYR21) and of the ErmC’ adenine N6-methyltransferase (Pdb 1QAO29) are most closely related to the T. thermophilus KsgA structure. The two KsgA homologs share 33 % sequence identity with each other and the two structures align with an rmsd of 1.4 Å for 216 Cα atoms. In contrast to the T. thermophilus structure, the N-terminal region of the E. coli enzyme is disordered and the first residue in the model is Gln17 (equivalent to T. thermophilus Gln27, Fig. 5a). Similar structural differences are observed when comparing KsgA with ErmC’. Both structures can be aligned with an rmsd of 1.7 Å for 197 Cα atoms (Fig. 5b). The first ordered residue in the ErmC’ N-terminus is Ser9 and the following Gln10 is equivalent to Gln27 in KsgA. Not surprisingly, residues in the cofactor-binding site other than the N-terminal region are well conserved between all three enzymes. His121, which indirectly coordinates to the adenosine molecule (in KsgA2), is the only variant residue in the T. thermophilus structure replacing asparagine side chains in E. coli KsgA and ErmC’. For ErmC’, Maravic et al. investigated a potential role of basic residues in the disordered N-terminal region for cofactor binding. They concluded that Lys4 and Lys7 are not essential for catalytic activity but instead may contribute to substrate binding30. Inspection of the N-terminal structure in KsgA shows that Lys23 and Arg24 could conceivably contribute to substrate recognition in a similar fashion. However, only Lys23 is moderately conserved in a sequence alignment of KsgA homologues suggesting that a functional role in substrate binding is less likely (supplemental Fig. S1).
Figure 5. Comparison with other methyltransferases.
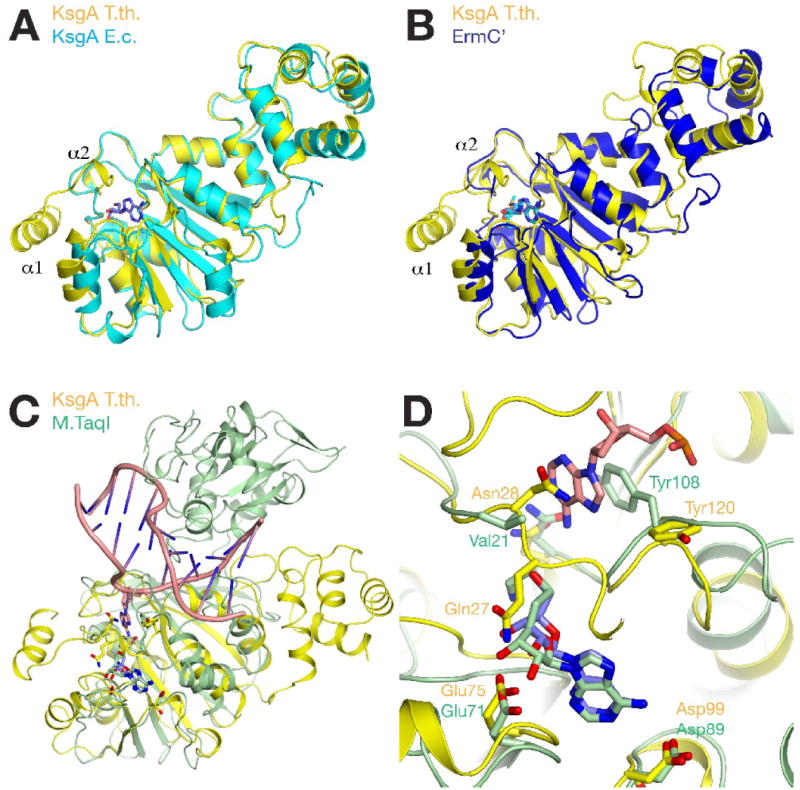
A, Differences between the overall structures of KsgA from T. thermophilus (yellow) and E. coli (cyan). Adenosine is shown with blue sticks. B, Structural differences between KsgA (yellow) and ErmC’ (blue). C, Comparison of the overall structure of KsgA with the DNA-bound complex structure of M.TaqI (PDB code 1G38). M.TaqI is colored in light green, DNA is colored in salmon. D, Comparison of the active site of KsgA with M.TaqI. KsgA is colored as before, the AdoMet analog bound in M.TaqI is shown in light green sticks, and the substrate adenosine is shown in salmon sticks.
Implications of structural rearrangements for substrate recognition
A structural comparison of KsgA with its E. coli ortholog and with ErmC’ reveals differences in the orientation of the C-terminal domains, in the conformation of the loops connecting both domains, and in several short loop regions between secondary structure elements (Fig. 5a, b). The comparison with the DNA adenine-N6 methyltransferase M.TaqI shows the strong conservation of the catalytic domain among class I methyltransferases. Similar to the closely related E. coli KsgA and ErmC’, the N-terminal region of M.TaqI is disordered and Val21 is the first ordered residue (Fig. 5c). Both enzymes contain unique additional domains, which in the case of M.TaqI, bind the substrate DNA and form extensive interactions with the non-substrate DNA strand. An analogous function was proposed for the C-terminal domain of KsgA based on two observations21. First, the interdomain movement observed in E. coli KsgA would be consistent with substrate binding in two orientations for modification of the two substrate bases. Second, the interface region between both domains is highly positively charged in the E. coli enzyme and this charge distribution is conserved in ErmC’25. In addition, a model of the interaction of KsgA with the 30S subunit places the C-terminal region in close proximity to rRNA, which would be consistent with a functional role in substrate binding17; 31. Similar to the E. coli structure, we observed an interdomain movement in the T. thermophilus KsgA structure and a positively charged surface patch in the domain interface region (residues Lys218, Arg219, Arg220, Lys221, and Arg249). However, the position of residues contributing to this charged surface region is not fully conserved between the E. coli and the T. thermophilus enzymes. Alternatively, the C-terminal domain may have an independent function. Inoue et al. reported that overexpression of KsgA in E. coli suppresses a cold-sensitive phenotype induced by a mutation in the Era GTP-binding protein. From mutation studies, the authors concluded that this KsgA function is mediated by the C-terminal domain of KsgA and independent of the methyltransferase activity15. Era is an essential ras-like protein in E. coli32 that binds to 16S rRNA and 30S subunits33. The protein was located in the region between the neck and the shoulder in direct contact with nucleotides 1530 – 1534 of the 16S rRNA in a cryo-electron microscopy study of an Era-30S complex34. Further experiments will be required to define the orientation of KsgA on the 30S subunit in order to understand this functional interaction between KsgA and Era.
Catalytic mechanism of methylation by KsgA
The comparison of the active site of KsgA with the substrate bound complex structure of M.TaqI may provide a model for substrate placement in KsgA. The catalytic domains of both enzymes are quite similar with an overall rmsd of 1.4 Å for 108 Cα atoms. In the comparison between the two cofactor-binding sites, it can be seen that the ribose position is shifted in KsgA relative to the cofactor analog bound in the M.TaqI ternary complex structure because of the interaction with Gln27, which is not present in M.TaqI. In the substrate-binding region, it can be seen that the side chain of the flexible Tyr120 in KsgA is oriented away from the substrate-binding site whereas the equivalent Tyr108 in M.TaqI is engaged in a base-stacking interaction with the substrate adenine. We did not observe a comparable inward orientation among the different Tyr120 orientations in KsgA, but it seems quite likely that this residue will assume a similar orientation in the substrate-bound form. In E. coli KsgA, the side chain of the equivalent Tyr116 was not modeled presumably because it was disordered in this structure21. The orientation of the equivalent Tyr104 in ErmC’ corresponds to the orientation of Tyr120 as observed in the KsgA3 crystal form. Tyr104 was found to be indispensable for ErmC’ function35 and computational docking calculations with ErmC’ indicated that a similar rearrangement of Tyr104 is likely to occur in this enzyme36. Together, these observations suggest that the conformational flexibility that we observed for the loop region near Tyr120 may have a function in coordinating the substrate base analogous to Tyr108 in M.TaqI.
Similar to other methyltransferases, there is no basic residue present in the active site of KsgA that could serve to deprotonate the substrate amino group during catalysis. In the M.TaqI complex structure, the adenine amino group forms hydrogen bonds to the side chain of Asn105 and to the main chain carbonyl group of Pro106 of the methyltransferase signature motif IV. Goedecke et al. proposed that deprotonation might occur after methylgroup transfer via either Asn105 or the main chain carbonyl group in this enzyme27. The sequence and position of motif IV residues in the active site are highly conserved and similar interactions have also been observed for other enzymes. For example, the coordination of the substrate amino group with a main chain carbonyl has been observed in complex structures of the N5-glutamine methyltransferase PrmC37 and of the protein lysine methyltransferase PrmA38. The main chain carbonyls of Leu118 in KsgA, Pro198 in HemK, and Leu192 in PrmA are all in positions equivalent to that of Pro106 in M.TaqI, and might interact with the substrate amino group in a similar manner. Interestingly, PrmA contains a unique truncated motif IV (191NLY193) that results in an outward orientation of Tyr193 similar to the Tyr120 orientation in KsgA. The Tyr193 side chain is observed in different orientations in PrmA – substrate complex structures, reminiscent of the mobility of Tyr120 reported in this study. In addition, the Leu192 carbonyl group rotates by about 120 degrees in two PrmA structures in complex with a dimethylated and a trimethylated substrate amino group suggesting that rotation of a carbonyl group that contacts the substrate amino group may occur during catalysis38. The catalytic mechanism for multiple methylation (processive or consecutive) of both KsgA and PrmA is currently unknown, and a sequential mechanism, which would not require amino group rotation in the active site, was found for ErmC’39. Further structural and functional studies will be required to investigate the catalytic mechanism of dimethylation by KsgA.
Materials and Methods
Growth and genetic manipulation of T. thermophilus
T. thermophilus HB8 (ATCC27634) was cultivated aerobically at 72 °C using Thermus Enhanced Medium (TEM), ATCC Medium 1598. Two segments of T. thermophilus HB8 chromosomal DNA surrounding TTHA0083 were amplified by PCR using Pfu DNA polymerase (Stratagene) and cloned into pUC18. The upstream segment was amplified using the oligonucleotide primers Tth ksgA-1 (5’- GCTCCGCAACCTTCTGAAGGGATGAGG-3’) and Tth ksgA-2 (5’- AGGCTGCAGGCGAGCGAGCTTACTCATGGAGG-3’) and cloned as a BamHI/PstI fragment while the downstream segment was amplified using the primers Tth ksgA-3 (5’- CGCCTGCAGCCTCGAGGCCTTCCGCCGGCTGAGG-3’) and Tth ksgA-4 (5’- CGAAGCTTCCAGCTCCTGGACGTCCCGCTC-3’) and cloned as a HindIII / PstI fragment. The htk gene, amplified using primers HTK-18 (5’- GAACTGCAGTACCCGTTGACGGCGGATATGG-3’) and HTK-19 (5’- GCTTGCATGCCTGCAGCGTAACCAAC-ATG-3’), was inserted into the PstI site between the two cloned chromosomal DNA fragments. Transformation with plasmid or chromosomal DNA was performed by the method of Koyama40. Recombinants were selected on TEM plates containing 25 μg/ml kanamycin sulfate (Sigma). Transformants were purified by restreaking, and chromosomal DNA was used to re-transform T. thermophilus HB8. The structure of the null allele was confirmed by PCR and DNA sequencing.
Preparation of 30S ribosomal subunits, primer extension analysis and in vitro methylation assays
30S subunits were prepared by dissociation of 70S ribosomes (prepared as described previously41) in 1 mM MgCl2 buffer followed by fractionation on 10-35 % sucrose gradients in an SW28 rotor at 18,000 rpm for 18 hrs. Primer extension was carried out using the 32P-end labeled oligonucleotide primer Tth 16S-Z2 (5’- AAAGAGGTGATCCAG -3’) complementary to positions 1542 to 1527 of T. thermophilus 16S rRNA. Extension was performed using AMV reverse transcriptase (Promega). Extension products were resolved on 13 % acrylamide 8 M urea gels. 150 pmol of 30S subunits were methylated with 6 pmol of KsgA using 2 μl of [3H]-S-adenosyl-L-methionine (2 mM; 100 cpm/pmol) at 70 °C in 150 μl of a buffer containing 20 mM HEPES-KOH pH 7.6, 200 mM NH4Cl and MgCl2 (1 mM - 10 mM). At each time point (0, 2, 10, 30 and 60 minutes) 20 μl aliquots were removed and the reactions were terminated by addition of 10% trichloroacetic acid. Samples were filtered through fiberglass filters (Whatman) and washed three times with 1ml of 5% trichloroacetic acid. The filters were dried and counted in a Beckman scintillation counter.
Protein expression and purification
The full-length ksgA gene (Genbank NC_006461) from T. thermophilus HB8 was cloned into the expression vector pET26b (Novagen) and over-expressed in E. coli strain BL21Star (Invitrogen). Bacterial cells were grown to mid-log phase in LB medium at 37 °C in the presence of 35 μg/ml kanamycin. Protein expression was induced at 20 °C with 400 μM IPTG. Cells were pelleted after 18 hours by centrifugation at 4000 rpm for 20 minutes at 4 °C. Bacterial cells were lysed by ultrasonification on ice in a buffer containing 20 mM Tris-HCl (pH 6.8), 5 mM β-mercaptoethanol, 0.1% Triton-X 100 and 5% glycerol. Cell debris and membranes were pelleted by centrifugation at 15000 rpm for 30 minutes at 4 °C. The lysate was heat treated at 65 °C for 30 minutes and precipitated E. coli proteins were removed by centrifugation at 15000 rpm at 4 °C for 30 minutes. The soluble native T. thermophilus KsgA was further purified by cation exchange chromatography (SP) (GE healthcare) at pH 6.8, using a linear gradient of 10 mM to 1 M NaCl concentration. KsgA fractions were concentrated and applied to a size-exclusion S200 column equilibrated with buffer containing 20 mM Tris-HCl (pH 6.8) and 200 mM NaCl. The purified KsgA was buffer exchanged in 20 mM Tris-HCl (pH 8.0) and concentrated to 14 mg/ml for crystallization trials. For the production of selenomethionyl proteins, the expression construct was transformed into B834 (DE3) cells (Novagen). Cells were grown in defined LeMaster medium42, and the protein was purified using the same protocol as for the native protein. To form the KsgA-adenosine complex, purified KsgA was mixed with 4 mM adenosine and 4 mM AdoMet chloride (Sigma), incubated at 50 °C for 10 minutes, and slowly cooled to room temperature. For the formation of KsgA-5’-methylthioadenosine complex, purified KsgA was incubated with 4 mM AdoMet chloride at 50 °C for 10 minutes and slowly cooled to room temperature. Incubation at 50 °C was the only experimental approach to obtain complex crystals. Co-crystallization with AdoMet, S-adenosyl-homocysteine, or sinefungin at 4 °C without prior incubation at 50 °C or soaking of KsgA crystals from different crystallization conditions was not successful.
Crystallization
All crystals were obtained using the microbatch technique under oil at 4 °C. To obtain the KsgA1 crystal form, 1 μl of protein solution was mixed with a reservoir solution containing 200 mM di-ammonium hydrogen phosphate and 20% w/v polyethylene glycole 3350. Crystals grew over the course of 3 - 6 weeks with maximum dimensions of 600 × 600 × 600 μm. To obtain the KsgA2 crystal form, 1 μl of protein solution was mixed with a reservoir solution containing 50 mM magnesium chloride hexahydrate, 100 mM HEPES (pH 7.5) and 30% v/v polyethylene glycol monomethyl ether 550. Initial crystals grew over the course of 3 - 6 weeks with maximum dimensions of 500 × 500 × 500 μm. To obtain the KsgA3 crystal form, 1 μl of protein solution was mixed with a reservoir solution containing 160 mM magnesium chloride hexahydrate, 80 mM TRIS (pH 8.5), 24% w/v polyethylene glycol 4000 and 20% v/v anhydrous glycerol. Crystals grew over the course of 3-6 weeks with maximum dimensions of 300 × 300 × 300 μm. To obtain the KsgA4 crystal form, 1 μl protein solution was mixed with a reservoir solution containing 17% w/v polyethylene glycol 4000, 85 mM HEPES-NaOH (pH 8.5), 8.5% v/v isopropanol and 15% v/v anhydrous glycerol. Crystals grew over the course of 3 - 4 days with maximum dimensions of 300 × 300 × 200 μm. To obtain the KsgA5 crystal form, 1 μl of protein solution was mixed with reservoir solution containing 20% v/v polyethylene glycol 300, 100 mM TRIS-HCl (pH 8.5), 5% w/v polyethylene glycol 8000, 10% v/v anhydrous glycerol. Crystals grew over the course of 3-6 weeks with maximum dimensions of 400 × 400 × 300 μm. Crystals of KsgA1 were cryo-protected by rapid soaking in mother liquor supplemented with 30% v/v glycerol before freezing. Crystals of KsgA2 were cryo-protected by rapid soaking in a solution containing mother liquor supplemented with the addition of 30% w/v ethylene glycol before freezing. KsgA3, KsgA4 and KsgA5 crystals were flash-frozen by plunging into liquid nitrogen directly from their mother liquor.
Data collection
X-ray diffraction data for KsgA1, KsgA2, KsgA3, KsgA4 and KsgA5 crystals were collected on a MAR CCD detector at the X4C beamline of the National Synchrotron Light Source in Brookhaven. For the initial structure determination, a selenomethionyl single wavelength anomalous dispersion data set (KsgA1) to 1.51 Å resolution was collected at a wavelength of 0.979 Å at -180 °C. The crystals belong to the space group P21212, with cell dimensions of a = 137.0 Å, b = 53.7 Å, and c = 69.4 Å. The diffraction images were processed and scaled with the HKL2000 package43. Diffraction data to 1.53 Å for KsgA2 were collected in space group P212121 with cell dimensions a = 53.2 Å, b = 61.3 Å and c = 82.7 Å. Diffraction data for KsgA3 in space group P43212 were collected to 1.95 Å resolution with cell dimensions a = 85.1 Å, b = 85.1 Å and c = 215.9 Å. Diffraction data to 1.56 Å for KsgA4 were collected in space group P212121 with cell dimensions a = 53.4 Å, b = 61.0 Å and c = 82.5 Å. Diffraction data to 1.68 Å for KsgA5 were collected in space group P212121 with cell dimensions a = 79.9 Å, b = 186.4 Å and c = 56.1 Å. A single crystal was used for each data set. The data processing statistics are summarized in Table 1.
Structure determination and refinement
The locations of two selenium atoms (out of four expected selenium atoms) were determined with the program Solve44 based on the anomalous differences in the single wavelength anomalous KsgA1 data set. Reflection phases to 1.51 Å were calculated with Solve. An initial model was built with Resolve45 and ARP/wARP46 was used for subsequent model building. The KsgA2, KsgA3 and KsgA5 structures were solved by molecular replacement with the programs Phaser47 and Como48, respectively. The atomic coordinates from the KsgA2 model were then used as initial models for refinement against KsgA4. All models were checked and completed with Coot49. Crystallographic refinement was performed with the program Refmac50 from the CCP4 package51. The stereochemical quality of the models was assessed with Procheck52. The Ramachandran statistics (most favored / additionally allowed / generously allowed / disallowed) are 94.1/5.9/0.0/0.0% for KsgA1, 95.5/4.5/0.0/0.0% for KsgA2, 92.3/7.5/0.2/0.0% for KsgA3, 95.9/4.1/0.0/0.0% for KsgA4 and 95.7/4.3/0.0/0.0% for KsgA5. The refinement statistics are summarized in Table 1. Figures were generated using Pymol (http://pymol.org) and JalView53. Sequence alignments were generated with ClustalW254, MultiProt55 and Staccato56.
Accession codes
Atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 3FUT, 3FUU, 3FUV, 3FUW, 3FUX for data sets KgsA1 - 5.
Supplementary Material
Acknowledgments
We thank John Schwanof and Randy Abramowitz for access to the X4C beamline at the National Synchrotron Light Source, and Hua Li for help with data collection at the synchrotron. This work was supported by grant GM19756 from the US National Institutes of Health to A.E.D. and by Brown University to G. J.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Van Buul CP, Damm JB, Van Knippenberg PH. Kasugamycin resistant mutants of Bacillus stearothermophilus lacking the enzyme for the methylation of two adjacent adenosines in 16S ribosomal RNA. Mol Gen Genet. 1983;189:475–8. doi: 10.1007/BF00325912. [DOI] [PubMed] [Google Scholar]
- 2.Helser TL, Davies JE, Dahlberg JE. Mechanism of kasugamycin resistance in Escherichia coli. Nature New Biol. 1972;235:6–9. doi: 10.1038/newbio235006a0. [DOI] [PubMed] [Google Scholar]
- 3.Helser TL, Davies JE, Dahlberg JE. Change in methylation of 16S ribosomal RNA associated with mutation to kasugamycin resistance in Escherichia coli. Nature New Biol. 1971;233:12–14. doi: 10.1038/newbio233012a0. [DOI] [PubMed] [Google Scholar]
- 4.van Buul CP, Visser W, van Knippenberg PH. Increased translational fidelity caused by the antibiotic kasugamycin and ribosomal ambiguity in mutants harbouring the ksgA gene. FEBS Lett. 1984;177:119–24. doi: 10.1016/0014-5793(84)80994-1. [DOI] [PubMed] [Google Scholar]
- 5.Lafontaine D, Delcour J, Glasser AL, Desgres J, Vandenhaute J. The DIM1 gene responsible for the conserved m6(2)Am6(2)A dimethylation in the 3’-terminal loop of 18 S rRNA is essential in yeast. J Mol Biol. 1994;241:492–7. doi: 10.1006/jmbi.1994.1525. [DOI] [PubMed] [Google Scholar]
- 6.Lafontaine D, Vandenhaute J, Tollervey D. The 18S rRNA dimethylase Dim1p is required for pre-ribosomal RNA processing in yeast. Genes Dev. 1995;9:2470–81. doi: 10.1101/gad.9.20.2470. [DOI] [PubMed] [Google Scholar]
- 7.Heus HA, Van Kimmenade JM, van Knippenberg PH, Hinz HJ. Calorimetric measurements of the destabilisation of a ribosomal RNA hairpin by dimethylation of two adjacent adenosines. Nucleic Acids Res. 1983;11:203–10. doi: 10.1093/nar/11.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heus HA, van Kimmenade JM, van Knippenberg PH, Haasnoot CA, de Bruin SH, Hilbers CW. High-resolution proton magnetic resonance studies of the 3’-terminal coli-cin fragment of 16 S ribosomal RNA from Escherichia coli. Assignment of iminoproton resonances by nuclear Overhauser effect experiments and the influence of adenine dimethylation on the hairpin conformation. J Mol Biol. 1983;170:939–56. doi: 10.1016/s0022-2836(83)80197-1. [DOI] [PubMed] [Google Scholar]
- 9.Pley HW, Flaherty KM, McKay DB. Model for an RNA tertiary interaction from the structure of an intermolecular complex between a GAAA tetraloop and an RNA helix. Nature. 1994;372:111–3. doi: 10.1038/372111a0. [DOI] [PubMed] [Google Scholar]
- 10.Rife JP, Moore PB. The structure of a methylated tetraloop in 16S ribosomal RNA. Structure. 1998;6:747–56. doi: 10.1016/s0969-2126(98)00076-8. [DOI] [PubMed] [Google Scholar]
- 11.Rife JP, Cheng CS, Moore PB, Strobel SA. N-2-methylguanosine is iso-energetic with guanosine in RNA duplexes and GNRA tetraloops. Nucleic Acids Res. 1998;26:3640–3644. doi: 10.1093/nar/26.16.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wimberly BT, Brodersen DE, Clemons WM, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. Structure of the 30S ribosomal subunit. Nature. 2000;407:327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 13.Schluenzen F, Takemoto C, Wilson DN, Kaminishi T, Harms JM, Hanawa-Suetsugu K, Szaflarski W, Kawazoe M, Shirouzu M, Nierhaus KH, Yokoyama S, Fucini P. The antibiotic kasugamycin mimics mRNA nucleotides to destabilize tRNA binding and inhibit canonical translation initiation. Nat Struct Mol Biol. 2006;13:871–8. doi: 10.1038/nsmb1145. [DOI] [PubMed] [Google Scholar]
- 14.Schuwirth BS, Day JM, Hau CW, Janssen GR, Dahlberg AE, Cate JH, Vila-Sanjurjo A. Structural analysis of kasugamycin inhibition of translation. Nat Struct Mol Biol. 2006;13:879–86. doi: 10.1038/nsmb1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inoue K, Basu S, Inouye M. Dissection of 16S rRNA methyltransferase (KsgA) function in Escherichia coli. J Bacteriol. 2007;189:8510–8518. doi: 10.1128/JB.01259-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Farrell HC, Pulicherla N, Desai PM, Rife JP. Recognition of a complex substrate by the KsgA/Dim1 family of enzymes has been conserved throughout evolution. RNA. 2006;12:725–33. doi: 10.1261/rna.2310406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Z, O’Farrell HC, Rife JP, Culver GM. A conserved rRNA methyltransferase regulates ribosome biogenesis. Nat Struct Mol Biol. 2008;15:534–6. doi: 10.1038/nsmb.1408. [DOI] [PubMed] [Google Scholar]
- 18.Thammana P, Held W. Methylation of 16S RNA during ribosome assembly in vitro. Nature. 1974;251:682–686. doi: 10.1038/251682a0. [DOI] [PubMed] [Google Scholar]
- 19.Poldermans B, Van Buul CP, Van Knippenberg PH. Studies on the function of two adjacent N6,N6-dimethyladenosines near the 3’ end of 16 S ribosomal RNA of Escherichia coli. II. The effect of the absence of the methyl groups on initiation of protein biosynthesis. J Biol Chem. 1979;254:9090–3. [PubMed] [Google Scholar]
- 20.Desai PM, Rife JP. The adenosine dimethyltransferase KsgA recognizes a specific conformational state of the 30S ribosomal subunit. Arch Biochem Biophys. 2006;449:57–63. doi: 10.1016/j.abb.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 21.O’Farrell HC, Scarsdale JN, Rife JP. Crystal structure of KsgA, a universally conserved rRNA adenine dimethyltransferase in Escherichia coli. J Mol Biol. 2004;339:337–353. doi: 10.1016/j.jmb.2004.02.068. [DOI] [PubMed] [Google Scholar]
- 22.Hashimoto Y, Yano T, Kuramitsu S, Kagamiyama H. Disruption of Thermus thermophilus genes by homologous recombination using a thermostable kanamycin-resistant marker. FEBS Lett. 2001;506:231–234. doi: 10.1016/s0014-5793(01)02926-x. [DOI] [PubMed] [Google Scholar]
- 23.Hagenbuchle O, Santer M, Steitz JA, Mans RJ. Conservation of the primary structure at the 3’ end of 18S rRNA from eucaryotic cells. Cell. 1978;13:551–63. doi: 10.1016/0092-8674(78)90328-8. [DOI] [PubMed] [Google Scholar]
- 24.Kurata S, Weixlbaumer A, Ohtsuki T, Shimazaki T, Wada T, Kirino Y, Takai K, Watanabe K, Ramakrishnan V, Suzuki T. Modified uridines with C5-methylene substituents at the first position of the tRNA anticodon stabilize U.G wobble pairing during decoding. J Biol Chem. 2008;283:18801–11. doi: 10.1074/jbc.M800233200. [DOI] [PubMed] [Google Scholar]
- 25.Bussiere DE, Muchmore SW, Dealwis CG, Schluckebier G, Nienaber VL, Edalji RP, Walter KA, Ladror US, Holzman TF, Abad-Zapatero C. Crystal structure of ErmC ’, an rRNA methyltransferase which mediates antibiotic resistance in bacteria. Biochemistry. 1998;37:7103–7112. doi: 10.1021/bi973113c. [DOI] [PubMed] [Google Scholar]
- 26.Parks LW, Schlenk F. The stability and hydrolysis of S-adenosylmethionine; isolation of S-ribosylmethionine. J Biol Chem. 1958;230:295–305. [PubMed] [Google Scholar]
- 27.Goedecke K, Pignot M, Goody RS, Scheidig AJ, Weinhold E. Structure of the N6-adenine DNA methyltransferase M.TaqI in complex with DNA and a cofactor analog. Nat Struct Biol. 2001;8:121–5. doi: 10.1038/84104. [DOI] [PubMed] [Google Scholar]
- 28.Demirci H, Gregory ST, Dahlberg AE, Jogl G. Crystal structure of the Thermus thermophilus 16 S rRNA methyltransferase RsmC in complex with cofactor and substrate guanosine. J Biol Chem. 2008;283:26548–56. doi: 10.1074/jbc.M804005200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schluckebier G, Zhong P, Stewart KD, Kavanaugh TJ, Abad-Zapatero C. The 2.2 A structure of the rRNA methyltransferase ErmC ’ and its complexes with cofactor and cofactor analogs: Implications for the reaction mechanism. J Mol Biol. 1999;289:277–291. doi: 10.1006/jmbi.1999.2788. [DOI] [PubMed] [Google Scholar]
- 30.Maravic G, Bujnicki JM, Flogel M. Mutational analysis of basic residues in the N-terminus of the rRNA : m(6)A methyltransferase ErmC ’. Folia Microbiol (Praha) 2004;49:3–7. doi: 10.1007/BF02931637. [DOI] [PubMed] [Google Scholar]
- 31.O’Farrell HC, Xu Z, Culver GM, Rife JP. Sequence and structural evolution of the KsgA/Dim1 methyltransferase family. BMC Res Notes. 2008;1:108. doi: 10.1186/1756-0500-1-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.March PE, Lerner CG, Ahnn J, Cui X, Inouye M. The Escherichia coli Ras-like protein (Era) has GTPase activity and is essential for cell growth. Oncogene. 1988;2:539–44. [PubMed] [Google Scholar]
- 33.Sayed A, Matsuyama S, Inouye M. Era, an essential Escherichia coli small G-protein, binds to the 30S ribosomal subunit. Biochem Biophys Res Commun. 1999;264:51–4. doi: 10.1006/bbrc.1999.1471. [DOI] [PubMed] [Google Scholar]
- 34.Sharma MR, Barat C, Wilson DN, Booth TM, Kawazoe M, Hori-Takemoto C, Shirouzu M, Yokoyama S, Fucini P, Agrawal RK. Interaction of Era with the 30S ribosomal subunit implications for 30S subunit assembly. Mol Cell. 2005;18:319–29. doi: 10.1016/j.molcel.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 35.Maravic G, Feder M, Pongor S, Flogel M, Bujnicki JM. Mutational analysis defines the roles of conserved amino acid residues in the predicted catalytic pocket of the rRNA : m(6)A methyltransferase ErmC ’. J Mol Biol. 2003;332:99–109. doi: 10.1016/s0022-2836(03)00863-5. [DOI] [PubMed] [Google Scholar]
- 36.Feder M, Purta E, Koscinski L, Cubrilo S, Vlahovicek GM, Bujinicki JM. Virtual screening and experimental verification to identify potential inhibitors of the ErmC methyltransferase responsible for bacterial resistance against macrolide antibiotics. Chemmedchem. 2008;3:316–322. doi: 10.1002/cmdc.200700201. [DOI] [PubMed] [Google Scholar]
- 37.Schubert HL, Phillips JD, Hill CP. Structures along the catalytic pathway of PrmC/HemK, an N-5-glutamine AdoMet-dependent methyltransferase. Biochemistry. 2003;42:5592–5599. doi: 10.1021/bi034026p. [DOI] [PubMed] [Google Scholar]
- 38.Demirci H, Gregory ST, Dahlberg AE, Jogl G. Multiple-site trimethylation of ribosomal protein L11 by the PrmA methyltransferase. Structure. 2008;16:1059–1066. doi: 10.1016/j.str.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Denoya C, Dubnau D. Mono- and dimethylating activities and kinetic studies of the ermC 23 S rRNA methyltransferase. J Biol Chem. 1989;264:2615–24. [PubMed] [Google Scholar]
- 40.Koyama Y, Hoshino T, Tomizuka N, Furukawa K. Genetic transformation of the extreme thermophile Thermus thermophilus and of other Thermus spp. J Bacteriol. 1986;166:338–40. doi: 10.1128/jb.166.1.338-340.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thompson J, Dahlberg AE. Testing the conservation of the translational machinery over evolution in diverse environments: assaying Thermus thermophilus ribosomes and initiation factors in a coupled transcription-translation system from Escherichia coli. Nucleic Acids Res. 2004;32:5954–5961. doi: 10.1093/nar/gkh925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hendrickson WA, Horton JR, LeMaster DM. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): a vehicle for direct determination of three-dimensional structure. EMBO J. 1990;9:1665–72. doi: 10.1002/j.1460-2075.1990.tb08287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 44.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cohen SX, Morris RJ, Fernandez FJ, Ben Jelloul M, Kakaris M, Parthasarathy V, Lamzin VS, Kleywegt GJ, Perrakis A. Towards complete validated models in the next generation of ARP/wARP. Acta Crystallogr D. 2004;60:2222–2229. doi: 10.1107/S0907444904027556. [DOI] [PubMed] [Google Scholar]
- 47.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jogl G, Tao X, Xu YW, Tong L. COMO: a program for combined molecular replacement. Acta Crystallogr D. 2001;57:1127–1134. doi: 10.1107/s0907444901006783. [DOI] [PubMed] [Google Scholar]
- 49.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 50.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 51.Bailey S. The CCP4 Suite - programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 52.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. Procheck - a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 53.Clamp M, Cuff J, Searle SM, Barton GJ. The Jalview Java alignment editor. Bioinformatics. 2004;20:426–427. doi: 10.1093/bioinformatics/btg430. [DOI] [PubMed] [Google Scholar]
- 54.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 55.Shatsky M, Nussinov R, Wolfson HJ. A method for simultaneous alignment of multiple protein structures. Proteins Struct Funct Bioinformat. 2004;56:143–156. doi: 10.1002/prot.10628. [DOI] [PubMed] [Google Scholar]
- 56.Shatsky M, Nussinov R, Wolfson HJ. Optimization of multiple-sequence alignment based on multiple-structure alignment. Proteins Struct Funct Bioinformat. 2006;62:209–217. doi: 10.1002/prot.20665. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.