

Abstract
The key roles of toll-like receptors (TLRs) as mediators of the detection and responses of immune cells to invading pathogens are well known. There are at least 13 mammalian TLRs which are integral membrane proteins with a leucine-rich extracellular domain and a cytoplasmic domain similar to that of the interleukin-1 receptor which initiates downstream signaling through kinases to activate transcription factors such as AP-1 and NFκB. TLRs are activated in glial cells (microglia, astrocytes and oligodendrocytes) and lymphocytes that infiltrate the nervous system in response to inflammation caused by infectious agents, tissue injury or autoimmune conditions. By inducing the production of pro-inflammatory cytokines and cell adhesion molecules in immune cells, TLRs may indirectly damage neurons in conditions such as ischemic stroke and multiple sclerosis. Recent findings suggest that neurons also express a subset of TLRs and that their activation promotes neuronal degeneration in experimental models of stroke and Alzheimer’s disease. TLRs may also play roles in regulating the processes of neurogenesis and neurite outgrowth, suggesting roles in neuronal plasticity. A better understanding of the molecular and cellular biology of TLRs in the normal and diseases nervous system, may lead to novel approaches for preventing neuronal degeneration and promoting recovery of function in an array of neurodegenerative conditions.
Keywords: Toll-like receptor, neurodegeneration, Alzheimer’s disease, stroke, multiple sclerosis
1. Introduction
Toll-like receptors (TLRs) are transmembrane pattern-recognition receptors (PRRs) that initiate signals in response to diverse pathogen-associated molecular patterns (PAMPs) 1. The involvement of Toll receptors in innate immunity was first described in Drosophila in 1988 by Hashimoto and colleagues 2. Following the description of Drosophila Toll in host defense against fungal infection, a mammalian homologue was identified 3 which recognizes lipopolysaccharides (LPS), a major cell wall component of gram-negative bacteria 4. Subsequently, a family of proteins structurally related to Drosophila Toll was identified, collectively referred to as TLRs. Depending on their arrangement as either homo- or heterodimers, each TLR complex recognizes distinct PAMPs derived from various microorganisms, including bacteria, viruses, protozoa and fungi 1.
TLRs are expressed in a variety of mammalian immune-related cell types such as B cells 5, mast cells 6, NK cells 7, regulatory T cells 8, macrophages, monocytes, dendritic cells 9, neutrophils 10, basophils 11 as well as non-immune cells such as epithelial 11 and endothelial cells 12. TLRs are also present in the brain where, until recently, their expression was believed to be limited to microglia 13, astrocytes 14, and oligodendrocytes 15. Recent findings, however, suggest that neurons express at least some TLRs 16. While TLRs mediate immunity in Drosophila 17, they were initially identified based on their role in establishing embryonic dorso-ventral polarity during body axis development 18. This implies a wider range of functionality than purely innate immunity. Indeed, TLRs have been implicated in several non-immune processes, such as bone metabolism 19, neurogenesis 20 and brain development 21, 22.
Until recently, TLRs have been examined predominantly for their contribution to immune-related disorders. However, cumulative evidence suggests that TLRs not only contribute to pathophysiology, but also play a vital role in facilitating neurodegenerative conditions. This review summarizes our current knowledge of the role of TLRs in the pathogenesis of brain disorders such as ischemic stroke, Alzheimer’s disease and multiple sclerosis, as well as the therapeutic potential of TLR intervention in such diseases.
2. Toll-Like Receptors
TLRs are major PRRs that have a central role in the initiation of innate immunity against invading microbial pathogens. These single membrane spanning proteins bear a leucine-rich extracellular domain, through which they recognize PAMP, and a cytoplasmic Toll/IL-1 Receptor (TIR) domain similar to that of the interleukin-1 receptor (IL-1R), which initiates downstream signaling 1. Each TLR by itself or in combination with other TLRs recognize distinct PAMPs that include lipids, lipoproteins, nucleic acids and proteins. TLRs are ubiquitous, present both in immune and non-immune cells, and their expression is rapidly altered in response to pathogens, cytokines and environmental stressors 23. Members of the TLR family and its signaling pathway components are alternatively spliced, producing proteins that have the potential to functionally alter inflammatory outcomes 24. Thus far alternative splicing has been demonstrated only for TLR4, but it is plausible that other TLR family members are alternatively expressed, as well as their signaling partners.
Thus far, 11 human and 13 mouse TLRs have been identified. TLRs rely on receptor dimerization to achieve specificity in agonist recognition. Although most TLRs appear to form homodimers, TLR2 only forms heterodimers with TLR1 and 6 as well as with CD36. TLRs may be segregated into groups based on the specific PAMPs they recognize. For example, TLR1, 2, 4 and 6 recognize lipids. TLR4 predominantly recognizes lipopolysaccharides (LPS) from gram-negative bacteria. TLR2 dimerizes with TLR1 to recognize triacylated lipopeptides from bacteria, such as Pam3Csk4, or with TLR6 to respond to a variety of PAMPs including peptidoglycan, diacylated lipopeptides such as Pam2CSK4, lipopolysaccharides of gram positive bacteria, fungal zymosan and mucoplasma lipopeptides. TLR10, expressed only in humans is also thought to heterodimerize with TLR2 and TLR1 23. The second class of TLRs includes TLR5 and TLR11, which are activated in response to protein ligation. TLR5 is mainly expressed in the intestine where it senses bacterial flagellin protein 25. TLR11 recognizes an unknown ligand of uropathogenic bacteria and a profiling-like molecule of the protozoan parasite Toxoplasma Gondii 23. TLR3, 7, 8 and 9 comprise the third group in the TLR family, and are localized intracellularly, where they are ideally positioned for activation by nucleic acids of bacterial and viral origin. TLR3 is activated in response to double stranded RNA (dsRNA) of viral origin. Human TLR8 and its murine orthologue, TLR7, recognize imidazoquinoline and viral ssRNA. TLR9 recognizes unmethylated CpG dinucleotides found in bacteria as well as viral genomes.
In addition to the numerous exogenous ligands that activate the different TLRs, endogenous TLR ligands have been identified in recent years. Endogenous TLR ligands include low molecular weight hyaluronic acid (HA), fibrinogen, fibronectin, beta defensins and heparin sulphate proteoglycans 26. During tissue injury or proteolysis, extracellular matrix components undergo cleavage, gaining the ability to act as TLR ligands, thus initiating TLR signaling. For example, high molecular weight HA is cleaved to low molecular weight HA, which subsequently binds TLR2 or 4 and activates TLR signaling cascades. Heat shock proteins, released from stressed cells, may also activate TLR4 27. In this way innate immune inflammatory responses may be activated without the presence of invading pathogens 28.
3. Toll-Like Receptor Signaling
Functional TLR signal transduction is complex and relies on receptor dimerization as well as the presence of accessory proteins and co-receptors, which regulate the signaling pathways initiated by each receptor. After recognition of PAMPs, TLRs activate the signaling components which results in appropriate immune responses required for host defense. The cytoplasmic region of TLRs shares a stretch of Toll/IL-1 receptor (TIR) domain, which mediates homo- and heterophilic interactions between TLRs and TIR-containing adapters. TLRs recruit a set of adaptor proteins with TIR domains by homophilic interaction of their TIR domains. The signaling pathways activated by TLRs are broadly classified into myeloid differentiation factor 88 (MyD88)-dependent and independent pathways as MyD88 is the universal adapter protein recruited by all TLRs except TLR3 1.
Upon receptor activation and interaction with MyD88, one or more TIR-containing adapter proteins TIRAP/Mal (TIR-domain-containing adapter/MyD88 adaptor-like), TICAM1/TRIF (TIR-domain-containing adaptor molecule 1/TIR-domain-containing adaptor-inducing interferon-γ) and TRAM (TRIF-related adaptor molecule) are recruited along with IL-1R-associated kinases (IRAK)-1, 2, 3, 4 or (M) (see figure 1). Once phosphorylated, IRAKs dissociate from MyD88 and interact with TNF receptor-associated factor 6 (TRAF6). TRAF6 forms a complex with Ubc13 and Uev1A to promote the synthesis of lysine 63-linked polyubiquitin chains, which in turn activate transforming growth factor β activated kinase 1 (TAK1), a mitogen-activated protein kinase kinase kinase (MAPKKK) 29. TAK1, in combination with an activator subunit TAB1, TAB2 or TAB3, activates two downstream pathways involving the IKK complex and the MAPK family (ERK, JNK or p38). The IKK complex, composed of the catalytic subunits IKKα and IKKβ and a regulatory subunit IKKγ, catalyzes the phosphorylation of IκB proteins 30. This phosphorylation leads to the degradation of IκBs and the subsequent nuclear translocation of the transcription factor nuclear factor kappa B (NFκB). Members of the MAPK family phosphorylate and activate the transcription factor activator protein 1 (AP-1). Activation of the transcription factors NFκB and AP-1 results in expression of pro-inflammatory cytokines such as interleukin (IL)-6, IL-1 and tumor necrosis factor (TNF)-α.
Figure 1.

TLR mediated signaling. (a) The Myd88 mediated pathway is shared by all the TLRs with the exception of TLR3. MyD88 recruits TRAF6 and members of the IRAK family. TRAF6, along with Uev1A and Ubc13 activates the TAK1 complex by a K63 linked ubiquitination. The TAK1 complex then activates the IKK complex that consists of IKKα, IKKβ and IKKγ, that further catalyzes IκB proteins phosphorylation. This in turn facilitates IκB proteins degradation by a proteasome-dependent manner, which allows NFκB translocation to the nucleus. In parallel to activating the IKK complex, TAK1 activates the MAPK pathway which culminates in AP-1 activation. The combination of AP-1 and NFκB controls inflammatory responses mediated by inflammatory cytokines. (b) The MyD88 independent pathway initiates when TRIF associates with TRAF3 which binds to TBK1 and IKKε. This binding culminates in IRF3 phosphorylation that facilitates IRF3 dimerization and translocation into the nucleus and transcription regulation. TRIF can also interact with TRAF6 which along with RIP1 mediates NFκB activation. TRIF can also induce IRF2 translocation to the nucleus and transcription through the PI3K-AKT pathway.
Most of the TLRs seem to be absolutely dependent on the expression of MyD88 for all of their functions, whereas TLR3 and TLR4 are capable of signaling through a MyD88-independent pathway (see figure 1). Both TLR3 and TLR4 are unique from other TLRs by their ability to activate Interferon Regulatory Factor 3 (IRF3). Following TLR4 activation, a MyD88-independent pathway can be activated when TRIF is recruited in concert with TRAM. This culminates in MAPK signaling, activation of the transcription factors NFκB and IRF3 and production of type-I interferon (IFN) production. TRIF-dependent signaling following TLR3 activation acts through recruitment of the IKKs TBK1 and IKKε which activate IRF3 31. Alternatively, TLR3 may activate IRF2 through TRIF-dependent activation of phosphatidylinositol 3-kinase and AKT 32. Exceptionally, MyD88-dependent signaling of TLR7, TLR8, and TLR9 can also induce type I IFN production 1.
4. Toll-Like Receptors in the Nervous System
TLRs have traditionally been considered receptors expressed solely on antigen presenting cells of the immune system such as B cells, dendritic cells, monocytes and macrophages, where they mediate innate immunity. It is increasingly clear, however, that nearly all cells within the body express TLRs, including those within the CNS. This section will focus on the role of TLRs in different brain cell types such as microglial cells, astrocytes, oligodendrocytes and neurons.
Microglia
Microglial cells are bone marrow-derived macrophage-like cells that are resident within the CNS. Constituting about 10% of the adult CNS, they mediate neuronal immune interactions under both physiological and pathological conditions 33. A large body of evidence concluded that microglial cells express a wide repertoire of TLRs in addition to their adapter proteins, required for functional downstream TLR signaling. Microglia express a wide array of TLR mRNA in both mice 13 and humans 15. A recent study by Jack and colleagues showed that TLR1-9 are expressed in microglia, whereas TLR10 is not 34. A similar pattern of expression was shown by Bsibsi and colleagues. 15. Interestingly, a large variation in expression of TLRs 2, 3 and 4 is noted between different human donors 15. Upon activation, TLR mRNA and protein expression is increased in microglia. For example, Kielian and colleagues 35 showed that upon stimulation with TLR ligands such as LPS, peptidoglycan (PGN) or the gram-negative bacteria S. aureus, the expression of TLR2 mRNA and protein and MyD88 protein levels were elevated. Microglial cells are the key defense against invading pathogens within the CNS, and it is not surprising, therefore, that activation of these cells, either by a single type of ligand or a combination of ligands, leads to secretion of a milieu of cytokines and chemokines. For example, TLR4 stimulation on human microglia with LPS results in secretion of the proinflammatory cytokines TNF-α and IL-6 36, 37 as well as the anti-inflammatory cytokine IL-10 38. Further, human microglia are stimulated in response to the TLR 3 ligand poly I:C to produce IL-12, TNFα, IL-6, chemokine (C-X-C motif) ligand 10 (CXCL-10), IL-10 and IFNβ 34, 39. Stimulation of TLR2 results predominantly in the secretion of IL-6 and IL-10 34. In addition, stress conditions such as hypoxia enhance TLR expression in microglia 40, 41.
Although there is convergence in the signaling cascades initiated by different TLRs, there exists large diversity in the functional responses to TLR activation. In a recent study, Zuiderwijk-Sick and colleagues 42 found that microglia differentiated with macrophage colony stimulating factor express higher amounts of TLR8 encoding mRNA and protein compared to cells differentiated with granulocyte macrophage colony stimulating factor. As a result, these cells secrete higher amounts of proinflammatory cytokines in response to TLR8 activation. This opens the possibility that in response to different stimuli, microglial cells are capable of pathogen-specific responses. A similar phenomenon occurs following microglial activation of CD4 T cells 43. TLR3-activated glial cells induce TH1 polarization and IFNγ secretion of CD4 T cells without affecting their proliferation. In-contrast, TLR2 and 4 activated glial cells decrease glial Major Histocompatibility complex class II expression and depress CD4 T-Cell proliferation.
TLR signaling in microglia may have a role in cell death and survival following inflammatory activation. TLR4, but not TLR2, ligation sensitizes microglia to apoptosis via autocrine/paracrine IFNβ production 44. In addition, Lehnardt and colleagues 45 recently described a cooperation between TLR2 and caspase-8 in group B streptococcus (GBS) induced apoptosis. GBS overactivation of microglia cells induced apoptosis that was TLR2-MyD88 but not nitric oxide (NO) or caspase-3 mediated. In a similar manner, Arvalli and colleagues 46 showed that inhibition of TLR2 signaling blocked apoptosis of microglial cells incubated with herpes-simplex virus (HSV)-1. These studies suggest a paradigm in which auto-regulation of the innate immune system exists in the CNS which helps to prevent excessive inflammation during pathogen infection. In contrast, a recent study suggested a possible neuroprotective role for TLR4 signaling in microglia, as LPS was shown be responsible for induction of NFκB independent/ JNK-mediated GDNF gene expression 47.
Astrocytes
Similar to microglial cells astrocytes exhibit a wide expression of these receptors. mRNAs for TLRs 1-7, 9 and 10, are detectable in human primary astrocytes cultures shown to be microglia-free using astrocyte-specific GFAP staining.34 Murine astrocytes express TLR1-6 mRNA, and produce pro-inflammatory cytokines when stimulated by their respective ligands.14 However, these data still require further confirmation using single-cell PCR to exclude contaminating microglia as a source of TLR mRNA.
Several cytokines and chemokines are reportedly produced following TLR activation in astrocytes. Both cytokines and TLR agonists induce expression of chemokine (C-C motif) ligands (CCL)2, CCL3, CCL5, ICAM-1 and vascular cell adhesion molecule-1 (VCAM-1). In addition, LPS as well as Poly I:C induce stronger expression of TLRs 4 and 3 respectively. Moreover, LPS and Poly I:C induce production of IL-6, TNF-α, IFN- α4, IFN-β and iNOS that is even stronger than induced by cytokine activated astrocytes 48. Poly I:C activation also induces production of CXCL-10 34. When astrocytes are activated with dsRNA and LPS in parallel, they secrete IL-1α, IL-1β, IL-6, TNF-α, GM-CSF, LTβ and TGF-β3 cytokines while MIF secretion is inhibited. This type of activation has no effect on IL-2, IL-3, IL-4, IL-5, IL-10, TGF-β1, TGF-β2, TNFβ and IFN-γ secretion 49. However, this study did not exclude the possibility of microglial contamination of the cultures, implying that at least some of these cytokines may be microglial derived. Recently, statins were found to be involved in enhancing TLR4-mediated cytokine expression; Rho proteins mediate negative feedback inhibition of TLR4 signaling 50. Endogenous ligands are also responsible for TLR activation in astrocytes. TLR2 activation with soluble CD14 results in CXCL8, IL-6 and IL-12p40 cytokine production. Interestingly, in contrast to LPS-treated astrocytes, this type of activation does not induce secretion of the pro-inflammatory cytokines TNF-α and IL-1β 51.
Human astrocytes constitutively express high levels of TLR3 and TLR adaptor molecules such as MyD88, TIRAP/Mal and TICAM-1/TRIF 52, although the TRIF branch of TLR4 signaling is inactive 49. Consistent with this, stimulation of human astrocytes with pro-inflammatory cytokines or TLR agonists induces a selective upregulation of TLR3, implicating a key role for MyD88-independent signaling in astrocytes 53. Further, upon TLR3 activation, a collection of mediators produced by astrocytes themselves contribute to astrocyte growth inhibition, endothelial cell growth inhibition and survival of neurons in organotypic brain slice cultures 53. This suggests that astrocytes express TLR3 in response to neuroinflammation, which may mediate neuroprotection when activated.
While TLR expression and signaling in astrocytes has been reported by several labs, potential microglial contamination of astrocytic cultures can taint these results. Indeed, some labs have been unable to show TLR mRNA expression in human astrocytes. 54 It is therefore a necessity to provide stronger evidence for specificity in astrocyte responses to TLR activation. Since even 1% of contaminating glial cells can affect results obtained by PCR, this can be achieved by using single-cell PCR or alternatively through separation of astrocytes using magnetic beads followed by measurement of protein levels.
Oligodendrocytes
Compared to other cell types, very little is known regarding the expression and function of TLRs in oligodendrocytes. The first report on TLRs in these cells was made by Bsibi and colleagues 15 showing the predominant expression of TLR2 and 3. Evidence for TLR2 expression on oligodendrocytes was confirmed by Lehnardt and colleagues 55. While the exact role of TLR2 in oligodendrocytes is unknown, evidence suggests that activation of this receptor is involved in CNS repair. Oligodendrocyte precursor cells transplanted into adult rat retina and activated with the TLR2 ligand zymosan induce inflammation-mediated remyelination 56. This provides a possible neuroprotective role for TLR2 mediated signaling by oligodendrocytes by enhancing myelination of neurons in the CNS. Another report indicated the involvement of TLR signaling in oligodendrocytes during damage repair after spinal cord injury 57. TLR2 knock-out mice or mice with a loss of function mutation on TLR4 exhibit reduced myelination following spinal cord injury, suggesting that the presence of these receptors is necessary for proper remyelination. In contrast, a recent study revealed differential effects of intraspinal microinjection of TLR4 (LPS) and TLR2 (zymosan) ligands, and suggested that TLR2 activation is not always neuroprotective 58. Following LPS injection, oligodendrocyte progenitor cell (OPC) proliferation was significantly augmented, even exceeding lysolethicin, a positive regulator of OPC proliferation. In contrast, zymosan injection induced complete oligodendrocyte loss without OPC proliferation. In this model, zymosan also evoked delayed lesion expansion and demyelination of intact myelinated axons around the lesions. The seeming contradiction between the effects of LPS and zymosan on oligodendrocytes in different models is yet to be settled, and more research is necessary on the expression and functionality of TLRs in oligodendrocytes.
Neurons
During the past 4 years, evidence for the neuronal expression of TLRs has increased, suggesting a role for this receptor family in neurons during physiological as well as pathological conditions. Human NTera2 (NT-2) cells, a transformed teratoma forming cell line with a neuronal-like phenotype, express mRNA for TLR1, 2, 3, and 4 59. We have recently shown the expression of the mRNA for TLRs1-9 as well as protein levels of TLR 2, 3 and 4 in rat primary neuronal cultures 16. Another recent report showed TLR1-8 mRNA expression in neurons in vivo 60. In addition, TLR2 and 6 proteins were shown to be expressed in murine neurons in vivo under physiological conditions. TLR 2, 4, 6, 7 and 8 protein expression is induced following infection in a parasitic model of neurocysticercosis 60. Wadachi and colleagues found that both human and rat sensory nociceptive neurons co-express TLR4 and CD14, supporting the hypothesis that LPS might activate these neurons via TLR4/CD14 complex 61. Another recent study provided evidence that neurons from both the central and peripheral nervous systems express TLR3 and that it is concentrated at the growth cones of neurons 62.
In addition to its role in brain diseases (see below), it is now known that neuronal TLR activation plays a role in development. The TLR3 ligand Poly I:C induces collapse of growth cones and irreversibly inhibits neurite extension of cortical and hippocampal neurons following their extraction from E14 embryos, independently of NFκB, an effect not apparent in TLR3 knock out mice. Our recent study found that treatment of cultured embryonic cortical neurospheres with a TLR3 ligand significantly reduced proliferating (BrdU-labeled) cells and neurosphere formation, whereas NPC from TLR3-deficient embryos formed greater numbers of neurospheres compared to neurospheres from wild-type embryos. Numbers of proliferating cells, as assessed by phospho histone H3 (PH3) and proliferating cell nuclear antigen (PCNA) labeling, were also increased in the developing cortex of TLR3-deficient mice compared to wild-type mice in vivo. Our findings revealed a novel role for TLR3 in the negative regulation of NPC proliferation in the developing brain (Lathia et al., 2008, submitted). Similarly, the TLR8 agonist, R848, inhibits neurite outgrowth and induces neuronal apoptosis in mouse cortical cultures 22. A distinct difference is apparent between the effects of TLR activation in differentiated neurons and neuronal progenitor cells. Recently published work by Rolls and colleagues described the effects of TLR2 and 4 on neuronal differentiation 20. Both TLR2 and TLR4 are expressed on adult neural stem/progenitor cells; however a dichotomy exists between TLR2 and 4 in regard to neuronal differentiation. While TLR2 deficiency impairs hippocampal neurogenesis, the absence of TLR4 results in enhanced proliferation and neuronal differentiation [44]. In addition, TLR2 and TLR4 both were shown to modulate the self renewal and cell fate of neuronal progenitors. These processes are mediated by MyD88 and NFκB signaling pathways. However, TLR signaling through NFκB is specific to neuronal progenitor cells, and the signaling mediators of TLR activity in differentiated neurons remain unknown. An issue disregarded by many groups studying neuronal damage in neurodegenerative disorders is the effect of different TLR ligands on neurons both in vitro and in vivo. The inability of neurons to respond to the classical TLR ligands such as LPS (TLR4) or Pam3CSK4 (TLR2), questions the functionality of TLRs in these cells. It is possible that neuronal TLRs respond to endogenous ligands but not pathogen-derived ligands. Structural studies of different neuronal TLRs may provide an answer for this question.
5. Toll-Like Receptors in Neurodegeneration
Although TLRs are traditionally considered to respond to invading pathogens, they can be activated in the absence of microbial infection 63 and regulate neurogenesis 20. Studies examining inflammatory markers in normal brain aging have also suggested a dynamic regulation of TLRs with advancing age. Aging is associated with increased secretion of pro-inflammatory cytokines, whereas levels of the anti-inflammatory cytokines decrease 64, 65. Further, aged human brains exhibit upregulated transcription of pro-inflammatory genes 66. Perhaps not surprisingly, TLR gene transcription also changes profoundly with age 67, suggesting these receptors may participate in both normal aging and age-related disease. In this section we discuss the role of TLRs in brain diseases such as ischemic stroke, Alzheimer’s disease, multiple sclerosis and in other neurodegenerative conditions.
5.1 Role of TLRs in ischemic stroke
The ability of TLRs to mediate inflammatory responses in immune cells suggests their involvement in inflammatory responses and ischemia-induced neuronal damage. Expression studies confirm that cerebral ischemia results in the upregulation of mRNA for TLR2, TLR4 and TLR9 in mouse CNS 68. Further, mice lacking TLR2 or TLR4 exhibit reduced infarct size compared to wild-type mice following cerebral focal ischemia injury 68, 69. Inflammatory responses play a crucial role in cerebral ischemic injuries; however the complex array of mechanisms and the precise role of TLRs in mediating neuronal damage remain vague.
The first direct study identifying a role for TLR4 in ischemia/reperfusion injury following middle cerebral artery occlusion (MCAO) utilized TLR4 mutant mice 69. In these mice, the intracellular region of TLR4 bears a point mutation rendering it unresponsive to LPS 70. TLR4 mutant mice exhibit improved neurological behavior and reduced edema as well as reduced levels of pro-inflammatory cytokine secretion to the serum such as TNF-α and IL-6 following MCAO. Similar results were reported in a model of global cerebral ischemia/reperfusion (I/R) model and using TLR4 mutant mice 71. Global cerebral I/R elevated TLR4 expression, resulting in activation of NF-κB pathway and release of the pro-inflammatory cytokines TNF-α and IL-6. TLR4 mutant mice, however, exhibited decreased neuronal damage and apoptosis and a concomitant reduced secretion of pro-inflammatory cytokines. Further, instead of NF-κB, the AKT-GSK3-β survival pathway is activated in TLR4 mutant mice, suggesting its possible role in the beneficial outcome following ischemic stroke in TLR4 deficient animals. Likewise, a TLR4 mutation confers protection against MCAO and retinal ganglion cell axotomy 72. Following MCAO, loss of TLR4 function is associated with reduced expression of p38, JNK and ERK1/2 and iNOS in damaged neurons, suggesting the necessity of TLR4 in MCAO injury.
Another recent study by Caso and colleagues 73 showed that TLR4-deficient mice had lower infarct volumes and better outcomes in neurological and behavioral tests. Mice that lacked TLR4 had minor expression of stroke-induced IRF-1, inducible nitric oxide synthase, and cyclooxygenase-2 (COX-2), mediators implicated in brain damage. The levels of IFN-β and of the lipid peroxidation marker malondialdehyde were also lower in brains from TLR4-deficient mice than in those from control mice. In addition, Caso and colleagues found that the expression of matrix metalloproteinase-9, which is induced and mediates brain damage, was also reduced in TLR4 deficient mice after experimental stroke Caso et al., 2007. Similar to these studies in TLR4 deficient mice, Lehnardt and colleagues 45, 74 found that TLR2 deficient mice develop a decreased CNS injury compared to wild type mice in a model of focal cerebral ischemia. TLR2 mRNA is up-regulated in wild type mice during cerebral ischemia and they found that in ischemic brains, TLR2 protein is expressed in infiltrating lesion-associated microglia 24 to 72 hours post inchemia. Lehnardt and colleagues concluded that TLR2 in microglia propagates stroke-induced CNS injury 45, 74. In addition to inducing cytokine production by intrinsic glial cells, recent findings suggest that following a stroke TLRs upregulates the expression of adhesion molecules that facilitate the infiltration of lymphocytes into the ischemic brain region which likely contributes to neuronal damage 16. Taken together, these studies indicate TLR4 signaling modulates the severity of ischemia-induced neuronal damage and suggests TLR4 as a target of stroke therapy.
The specific role of neuronal TLRs during and following ischemic stroke recently began to unravel when Tang and colleagues 16 showed the pivotal role of TLR2 and 4 in ischemic brain injury. Neurons were found to express a broad range of TLRs (1-9) under physiological conditions. However, as early as one hour after initiation of I/R in vivo, neurons induce a high expression of TLR2 and 4. This suggests that neurons are the first cells to respond to ischemia by expressing TLR2 and 4, while 24 to 72 hours following ischemic injury, the expression of TLR2 is shifted to infiltrating microglia 16, 74. Importantly, cortical neuronal cultures from TLR2 and 4 deficient mice show increased survival after glucose deprivation. This suggests that the mechanisms mediating ischemic damage, and its prevention in TLR2 and 4 mutant mice, are not only facilitated by inflammation induced by astrocytes, microglia, macrophages or infiltrating immune cells, but also by the early activation of TLRs in neurons themselves.
Currently, relatively little is known about the mechanisms of TLRs in brain stroke, and many basic questions must be answered before a broad picture will emerge. Thus far, studies on TLRs in ischemic brain stroke have mainly focused on ischemic damage in TLR4 and to a lesser extent, TLR2, mutant mice. While this approach has provided a first glimpse into the relevance of TLR signaling in ischemic brain stroke, it has not enabled an understanding of the role of TLR signaling in specific cell types. This issue is of high importance due to the involvement of many different cells in the pathology of ischemic brain stroke, e.g: neurons, astrocytes, microglial, endothelial cells and invading immune cells (see figure 2). One potential strategy to approach the issue of neuronal TLR signaling is through knocking down neuronal TLRs by using a neuron-specific promoter. This is likely to provide a clearer answer to the question whether TLR signaling in the neurons themselves plays a detrimental role in neuronal damage following ischemic brain stroke.
Figure 2.
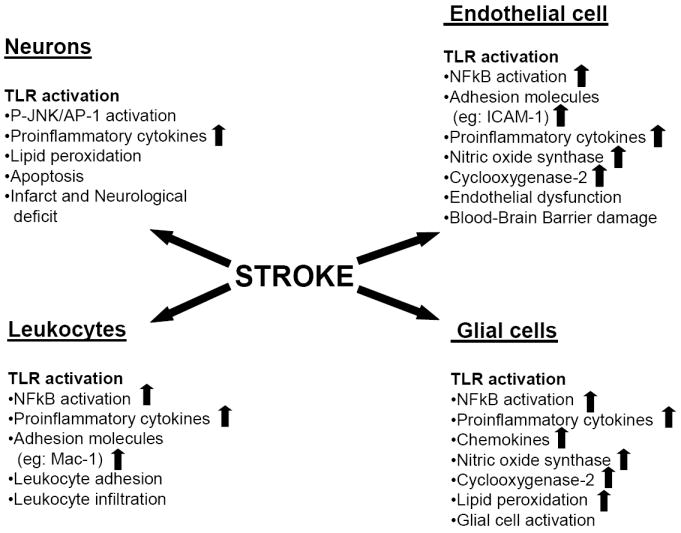
The roles of TLR signaling in glial cells, endothelial cells, leukocytes and neuronal cells during the pathogenesis of ischemic stroke. During ischemic stroke, TLRs are activated and induce NFκB activation in glial cells, endothelial cells and infiltrating lymphocytes. In neurons, the JNK/AP-1 pathway is activated. This culminates in proinflammatory cytokines and chemokines secretion, nitric oxide synthase and cox-2 expression induction. Endothelial cells dysfunction causes blood-brain-barrier permeability which promotes further lipid peroxidation, apoptosis of neurons, infarct and neurological deficits as the final outcome.
5.2 TLRs in Alzheimer’s disease
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by gradual onset and advancement of memory loss and other cognitive deficits. Definitive diagnosis of AD is based on the presence of extracellular amyloid plaques comprised of neurotoxic amyloid β-peptide (Aβ) which is generated by proteolysis of the β-amyloid precursor protein (APP), and intracellular neurofibrillary tangles composed of hyperphosphorylated insoluble forms of tau protein 75. Genetic factors that either cause or predispose to AD include mutation in APP and presenilins 1 and 2 (which cause early-onset autosomal dominant inherited AD) and polymorphisms in apolipoprotein E (ApoE4 increases the risk of AD). Activation of the innate immune response by reactive glia is a consistent pathological event in AD. Neuroinflammation in AD brain is concentrated at sites of Aβ plaques, which exhibit increased levels of pro-inflammatory cytokines, complement components and proteases 76, 77. Aβ plaques are surrounded and infiltrated by activated astrocytes and microglia, which are believed to be the major source of local inflammatory components 76.
Neuroinflammation is proposed to play a major role AD pathogenesis, as long term treatment with non-steroidal anti-inflammatory drugs reduces AD risk and may delay disease progression 78, 79. Further, TLR expression is upregulated in the AD brain. A screening of TLRs in murine models of AD revealed an upregulation of TLR2 and TLR7 transcription levels compared to wild-type controls 80. TLR2 and 4 expression is increased in the brain of AD patients 81. Further, multiple TLR genes (1-8) are expressed in microglia in postmortem tissue from AD patients, with varying levels of expression 15. The increased expression of TLRs in AD positions them as potential players in neurodegenerative mechanisms and disease progression.
The TLR4 gene has emerged as a candidate susceptibility gene for AD. A common missense polymorphism occurs at the TLR4 gene locus resulting from an adenine to guanine substitution 896 nucleotides downstream of the transcription start site. This substitution causes the replacement of glycine for aspartic acid at amino acid 299 (Asp299Gly), and alters the structure of the extracellular domain of TLR4. This mutation attenuates TLR4 signaling in response to LPS and diminishes the ability to induce inflammation 82. Accordingly, this polymorphism is associated with decreased cardiovascular disease and successful aging 83. The Asp299Gly polymorphism was associated with a decreased risk of late-onset AD in an Italian population cohort, independent of the susceptibility gene APOE ε4 84. However, the exact mechanism by which the Asp299Gly polymorphism mediates neuroprotection and decreases risk for late-onset AD remains to be elucidated.
In AD brain, activated glia expressing high levels of TLR4 and TLR2 surround Aβ plaques 67, 81. The close association between Aβ plaques and reactive astrocytes and microglia has led to the assertion that these cells contribute to plaque formation 85. APP transgenic mice, which progressively accumulate Aβ deposits, exhibit significant increases in TLR4 mRNA expression compared to wildtype littermates 81. Young APP transgenic mice treated for 12 weeks with LPS (a TLR4 ligand) exhibit high numbers of activated microglia and astrocytes throughout the neocortex and hippocampus. Further, following LPS treatment there was significant accumulation of intraneuronal Aβ, and these cells were in close proximity to activated microglia 86.
We have examined the role of TLR4 in AD using primary neuronal cultures from TLR4 mutant mice 87. Aβ damages neurons by causing membrane-associated oxidative stress and the production of the lipid peroxidation product 4-hydroxynonenal (HNE). We found that TLR4 expression increases during exposure to Aβ and the lipid peroxidation product HNE. Further, JNK and caspase-3 activity levels are augmented in neurons exposed to Aβ and HNE. Selective elimination of TLR4 function significantly suppresses the abilities of Aβ and HNE to induce activation of JNK and caspase-3 87, suggesting that TLR4 expression increases neuronal vulnerability to Aβ-induced damage. Consistent with this, we found that levels of TLR4 are decreased in AD brain tissue samples compared to control subjects. Taken together, this suggests that neurons expressing TLR4 have increased sensitivity to Aβ and are vulnerable to degeneration in AD.
In addition to epidemiological studies that suggest mutations in TLR4 decrease susceptibility to neurodegeneration, other data indicate that activation of TLR4 is required for clearance of Aβ in AD. For example, DiCarlo and colleagues 88 showed that in both young and old APP/presenilin-1 double mutant transgenic mice, an acute intrahippocampal injection of LPS significantly decreases Aβ deposition, dependent upon microglial activation 89. Mice carrying double mutations in mouse APP and human PS1 with exon 9 deletion, as well as a point mutation in TLR4, exhibit augmented Aβ deposition in both the neocortex and hippocampus 90. Cultured microglial cells from these mice were unresponsive to TLR4 ligands, while activation of TLR4 in cultured microglial cells from wild type littermates induces phagocytosis of Aβ 90. In addition to TLR4, activation of other TLRs may also contribute to Aβ clearance. Activation of TLR2 by peptidoglycan (PGN) initiates Aβ uptake by the microglial cell line, BV-2 90, 91. TLR 9 activation by CpG also results in significant clearance of Aβ from cultured mouse microglia 90, 92.
Whereas TLRs are activated by exogenous pathogens, mounting evidence indicates that Aβ itself activates TLRs and mediates microglial activation. Aβ stimulates microglia, as assessed by nitric oxide and TNF-α production, at similar levels to LPS (TLR4), Pam (TLR2) and CpG (TLR9) 93. Walter and colleagues recently showed that in mouse microglial cultures aggregated Aβ stimulates production of nitrite and TNF-α. However, Aβ stimulation of microglial cultures from mice bearing a point mutation in TLR4 was significantly diminished, suggesting that microglial activation by Aβ at least in part requires functional TLR4 81. Consistent with this, TLR4 mutant AD mice also exhibit diminished TNF-α, IL-1beta, IL-10 and IL-7 compared to TLR4 wildtype AD mice, suggesting that the upregulation of these cytokines by activated microglia in AD is dependent upon TLR4 signaling 94. Aβ sensitizes microglia to stimulation by LPS 95, and administration of Aβ together with LPS increases activation of TLR4, leading to increased release of nitric oxide and TNF-α 93. Costimulation of TLRs with Aβ and Pam3Cys enhances TLR2 stimulation. However, the enhancement of TLR signaling by Aβ is not homogenous, as Aβ depresses TLR9 activation by CpG 93. This may be due to the location of the TLR9 receptor in the endoplasmic reticulum and/or different signaling mechanisms of TLR receptors 96, 97. Further, activation of TLR9 by CpG can have anti-inflammatory activities 98.
The mechanism by which TLR activation results in Aβ clearance is unclear. The human G-protein-coupled formyl peptide receptor-like 1 (FPRL1) is essential for human macrophage phagocytosis of Aβ 92. Its mouse homologue, mFPR2, has recently been identified as important in microglial clearance of Aβ following TLR activation in murine models of Alzheimer’s disease. The phagocytosis of Aβ protein by both TLR2 and TLR9 is dependent upon activation of mFPR2 in cultured mouse microglia. Activation of both TLR2 and TLR9 initiates p38 MAPK signaling, expression of mFPR2 and subsequent Aβ internalization 91, 92. Consistent with this, Aβ clearance following TLR4 activation is blocked by the G-protein inhibitor pertussis toxin 90, suggesting that TLR4-induced Aβ phagocytosis occurs through a similar signaling pathway and requires mFPR2. Therefore, cell surface expression of mFPR2 may serve as a “sensor” of Aβ in TLR-activated microglia 91, 92.
At present, it still remains to be determined if the activation of TLRs by Aβ contributes to and/or inhibits AD progression. Contrasting data exist. on the precise role of TLRs in Aβ deposition. Therefore there may be a balance of TLR activation in which mild activation is beneficial, promoting Aβ uptake and breakdown. However, excessive activation of TLRs on microglia may lead to the accumulation of cytotoxic compounds such as reactive oxygen species, cytokines, complements and proteases causing damage and eventual neuronal loss 76. TLR signaling pathways are a potential therapeutic target in AD; however more work remains to delineate the complex interaction of TLRs in Aβ deposition and clearance and its precise role in AD development.
5.3 TLRs in Multiple sclerosis
Multiple sclerosis (MS) is a chronic inflammatory and demyelinating disease of the CNS. It is characterized by recurrent neurological dysfunction, interrupted with periods of functional recovery. Pathologically, MS is characterized by multiple demyelinating lesions within the CNS which frequently demonstrate axonal transection 99. The etiology of MS is unknown, although it is hypothesized to be an immune-mediated disease where autoreactive T cells enter the CNS and drive a proinflammatory reaction resulting in tissue injury, possibly subsequent to infection 100, 101. MS lesions within the CNS are associated with neuroinflammation and are surrounded by infiltrating T-lymphocytes, monocytes and macrophages, as well as activated microglia and reactive astrocytes 102. The most common mouse model of MS, Experimental Autoimmune Encephalomyelitis (EAE), is induced by immunizing mice with adjuvant and myelin components of the CNS, such as myelin basic protein, proteolipid protein or myelin oligodendrocyte glycoprotein. Development of EAE is dependent upon activation of pro-inflammatory CD4 Th1 cells by antigen presenting cells, resulting in leukocyte infiltration into the CNS, expression of inflammatory genes, microglial activation, astrogliosis and demyelinating lesions 103-105.
There is a marked increase in TLR expression in MS lesions. Microglial cells from MS patients express TLRs 1-8, as detected by PCR 15. Moreover, while healthy white matter from MS patients does not contain TLRs, active lesions are associated with high expression of TLR3 and TLR4 on microglia and astrocytes. Examination of TLR3 and TLR4 localization revealed that early active MS lesions are associated with vesicular localization of TLR3 and TLR4 within microglia, located near blood vessels at the outer edges of lesions. In contrast, late active lesions also contain astrocytes bearing surface TLR3 and TLR4 15. This suggests that early lesions are characterized by microglia infiltration, while astrocytes are also active in later MS lesions. However, the precise role of TLR3 and TLR4 activation in these lesions is unknown.
Similar to human lesions, TLR expression is upregulated in the brain and spinal cord in animal models of MS. In EAE, TLR2 gene expression is increased within the barrier microvasculature and circumventricular organs within the brain as early as 10 days following myelin oligodendrocyte glycoprotein administration 105. Moreover, a striking upregulation of TLR2 mRNA is observed 3 weeks post immunization, during the symptomatic stages of disease. At this time, TLR2 mRNA is localized to microglia throughout the brain and spinal cord, but is absent from reactive astrocytes. Concurrent with TLR2 mRNA upregulation throughout the brain and spinal cord, there is also upregulation of NFκB, TNF-α and MCP-1 in TLR2 expressing regions. However, TLR2 deficient mice remain susceptible to EAE, suggesting that while TLR2 transcription is regulated during EAE progression, it is not required for disease development 106. Prinz and colleagues 106 recently expanded on this study to examine the expression of a wide range of TLRs within EAE spinal cord. At the initial stages of EAE, simultaneous with leukocyte infiltration of the CNS, there is an increase in TLR1, TLR2, TLR4, TLR6, TLR7, TLR8, TLR9 as well as MyD88 within the spinal cord. At late stages of EAE, TLR7 and TLR9 mRNA expression was further increased, suggesting signaling through these receptors is involved in late active lesions. Consistent with this, the spinal cord of TLR9 deficient mice exhibited smaller white matter lesions and the number of infiltrating leukocytes was depressed106. Therefore, TLR9 activation may be more significant in altering EAE progression than TLR2.
Murine models of MS confirm the necessity of select TLR signaling in lesion development. Mice deficient in MyD88, an adaptor molecule required for TLR signaling, are not susceptible to EAE following immunization with myelin oligodendrocyte glycoprotein 107, suggesting that MyD88-dependent pathways are essential for development of EAE. In contrast, the absence of TLR4 exacerbates EAE symptoms, concurrent with augmented proinflammatory cytokine expression in dendritic cells and serums, suggesting these receptors regulate disease progression 107. Although augmented peripheral immune proinflammatory cytokine production following EAE induction in TLR9 deficient mice suggests it also modulates disease severity 107, spinal cord white matter lesions in these mice are smaller and the number of infiltrating leukocytes is depressed 106. The seeming disparity in these data may result from differences in the immunization method used to induce EAE. While a detrimental effect of TLR activation is implied by these studies, Touil and colleagues 108 recently raised the possibility of TLRs as neuroprotective in EAE. Stimulation of TLR3 with poly I:C suppresses relapsing demyelination in EAE, associated with increased IFN-β production and CCL2 108. This suggests that while MyD88-dependent signaling through TLR4 and TLR9 may mediate disease progression, TLR3 activation may protect from disease.
While TLRs may contribute to the development of MS, exactly how they may modify the disease process within the CNS is unknown. One hypothesis asserts that in response to proinflammatory cytokines, microglia are capable of serving as antigen-presenting cells which can activate CD4+ T cells and facilitate neuroinflammation 109. Consistent with this, activation of both TLR3 and TLR4 in human microglia and astrocytes evokes the release of CXCL-10, which is an important chemoattractant for CD4+ T cells 34, 110. Therefore, TLR activation may be an essential step in converting microglia to antigen presenting cells and facilitate T cell infiltration of MS lesions. Alternatively, TLRs may induce production of proinflammatory cytokines and thereby inflict damage. In mixed glial cultures, activation of microglial TLR4 by LPS results in damage and death of both oligodendrocytes 111 and neurons 112. Moreover, intracerebral injection of LPS, a TLR4 agonist, in neonatal rats induces oligodendrocyte loss and hypomyelination, mediated in part by the proinflammatory cytokine IL-1β 111, 113, 114. Recently, Li and colleagues 114 showed that the proinflammatory cytokine, IL-23p19 is expressed in macrophages and microglia in human MS white matter lesions, and that its expression in human microglia is induced following activation of TLR2 and TLR4. Taken together, this suggests a role for microglia in mediating CNS damage through cytokine production.
Although TLRs are traditionally thought to solely recognize pathogen-associated molecular patterns and thereby protect the body from invasion of microbial pathogens, the expression of TLRs within MS suggests novel roles for these receptors in mediating neurological disorders. However more work remains to be done to delineate the precise roles and mechanisms by which TLRs contribute to MS lesion development, particularly in differentiating the contribution of microglia and astrocytes to progression and/or protection of neuronal damage. The severity of white matter lesions in MS may be modulated in part by the balance of specific TLR signaling in microglia and astrocytes. Moreover, the potential roles of TLR signaling in neurons to the neurodegenerative component of MS remains to be elucidated. Human MS tissue exhibits TLR expression in both microglia and astrocytes, and a similar pathology is induced in EAE. It is important to note, however, that EAE is induced with adjuvant containing TLR ligands, therefore complicating the interpretation of results indicating an involvement of TLRs in disease progression. Therefore, these data must be considered carefully, and the proposed mechanisms must be confirmed in studies on human MS tissue. Clearly, more work remains to delineate the potential mechanisms by which TLR signaling modulates disease progression and, further, how activation of multiple pathways alters neuroinflammation and CNS damage.
5.4 TLRs in other neurodegenerative disorders
Despite the emerging role of TLRs in stoke, AD and MS, very little is known regarding the function of these receptors in other neurodegenerative disorders. A recent screening of murine models of AD, Parkinson’s disease and amyotrophic lateral sclerosis (ALS) revealed dynamic changes in TLR expression 80. Expression levels for TLR2, TLR5 and CD14 are increased in Parkinson’s disease mice, and expression levels for TLR1, TLR 2, TLR 7, TLR 9 and CD14 are increased in ALS transgenic mice. In addition, upregulation of TLR3 is reported in ALS cerebellar Purkinje neurons and Bergmann glia 115. Upregulation of TLR mRNA is also observed in primary mixed glial cultures from Pick’s disease and olivopontocerebellar atrophy patients 15. The availability of mouse models of Parkinson’s disease and ALS offer the opportunity to critically evaluate the roles of TLRs in the disease processes.
6. Therapeutic approaches
Evidence is emerging that TLRs are not only activated in response to microbial infection, but are critically involved in mediating neurological dysfunction. The extensive involvement of TLRs in neurodegenerative disorders provides ample opportunity for promoting and/or inhibiting their signaling to intervene in disease progression. However, it may prove exceedingly difficult to achieve the correct balance and appropriate timing of such interventions. Proper targeting of TLRs will require extensive understanding of the pathways activated, cell-specific responses, and at what time during disease progression TLRs are active.
The targeting of TLR-regulated pathways will require therapies that are intricately tailored for individual neurodegenerative conditions. While some TLR-related neuronal damage is similar across neurological disorders, TLR expression modifications are varied across different disorders, and moreover, often there exists variability within patients of the same disease 15, 80. The Asp299Gly polymorphism is linked to late-onset AD 84, and this is supported by studies implicating TLR4 activation in Aβ plaque clearance 90. In contrast, while TLR4 signaling may contribute to MS development, this mutation does not appear to alter susceptibility to MS 116, 117. Therefore, targeted TLR-based therapies will require an understanding of both the contribution of TLRs to genetic predisposition as well as involvement in sporadic development.
Both human and animal studies implicating TLRs in neural degeneration suggest direct modulation of TLR signaling as an ideal therapy. For example, stimulation of TLR2, TLR4 or TLR9 on microglia increases Aβ clearance in AD models 84; stimulation of these TLRs in AD patients may reduce Aβ plaque load and increase survival. Inhibition of MyD88 and/or stimulation of TLR4 or TLR9 may slow disease progression in MS 106, 107. However, many of the current TLR agonists/antagonists are high molecular weight compounds that may not cross the blood-brain barrier. Therefore specific strategies are necessary to circumvent this barrier and allow administration of TLR treatments to the CNS. During ischemic stroke, however, the blood-brain barrier is not a limiting factor. If treatment can be given within the correct time, this breakdown may be taken advantage of and TLR2 or TLR4 antagonists may be applied to the CNS without limiting factors. Consistent with this, TLR2 and 4 knock out mice were shown to be more protected against ischemic brain stroke. In contrast to this, it was recently shown that activation of NFκB is directly related to hypoxia-inducible factor 1-α (HIF1-α) induction, which is implicated in a better outcome 118. Therefore, activating NFκB through TLR signaling may increase stroke survival and functional recovery.
A unique therapy for combating the detrimental effects of TLR activation may be through TLR-induced TLR-suppression. Tasaki and colleagues were the first to demonstrate that low dose systemic administration of LPS (TLR4 ligand) prevents ischemic brain damage following MCAO in spontaneously hypertensive rats 119. LPS pretreatment also successfully blocks MCAO-induced brain damage in a mouse stroke model 120. This TLR-induced tolerance is dependent upon a small inflammatory response prior to ischemic damage, and inhibits inflammatory reactions subsequent to MCAO, particularly suppression of the TNFα pathway, thereby conferring neuroprotection 120, 121. LPS pretreatment-induced neuroprotection may be mediated by a shift in TLR4-signaling from a MyD88-dependent to –independent pathway. Instead of producing the prototypical TNFα in response to LPS, LPS-tolerant macrophages produce IFNβ, a downstream product of the MyD88-independent pathway 122. Therefore, TLR4 agonist pretreatment may change the TLR4 signaling pathway and therefore provide an ideal target for stroke therapy in at-risk patient populations.
In contrast to most TLRs, TLR3 signals through the MyD88-independent pathway, and may provide a unique therapeutic target for neurological disease. Cultured adult human astrocytes predominantly express TLR4, however stimulation with the pro-inflammatory cytokines TNF-α, IL-1β, IFN-g, IL-12, IL-4 or IL-6 results in significant expression of TLR3 mRNA 53. Gene expression profiling of these cells indicates that in response to TLR3 activation, the expression of neuroprotective mediators is induced. Among these are TGF-β2, which protects against demyelination from viral infection 123, ciliary neurotrophic factor and leukemia inhibitory factor, both of which support remyelination 124, 125 as well as several anti-inflammatory cytokines such as IL-9, IL-10 and IL-11. In addition, media from TLR3-activated astrocytes promotes neuronal survival in organotypic brain slices. Consistent with this, TLR3 stimulation suppresses relapsing demyelination in EAE 108. These data suggest astrocytic TLR3 expression promotes neuronal survival and protection against demyelination, and combined with their presence in late active lesions 15, implicate TLR3 bearing astrocytes as a neuroprotective and repair response to neuronal damage in MS. Therefore, stimulation of TLR3 may reduce neuronal damage in MS; however further studies delineating the exact role of TLR3 in actual multiple sclerosis models, such as EAE, are necessary.
Targeting TLRs in neurological disease will not be without difficulties. One potential obstacle to targeting TLR signaling in disease is that virtually all cells in the body express TLRs. If chronic administration of TLR agonists is necessary, overstimulation of the immune system may occur, which limits dosage capability as well as frequency of application 126. CNS-specific isoforms of TLR agonists which possess high selectivity could prevent such overstimulation of peripheral immune responses. In addition, partial agonists may be useful in preventing overstimulation of TLRs in the same tissues. Another potential hurdle to TLR-directed therapeutics is cross-talk between receptor subtypes. TLR agonists that activate the MyD88-dependent pathway can induce upregulation of effectors shared by other TLRs, evoking a higher efficacy of receptor activation. Alternatively, activation of one TLR pathway can induce tolerance to stimulation of other TLRs sharing that pathway 127. Therefore, the consequences of targeted TLR stimulation on similar signaling pathways must be carefully considered. In this regard, an indirect but perhaps more specific targeting scheme may be to direct antagonism to the adaptor, accessory and/or negative regulatory molecules exclusive to individual TLRs.
7. Conclusion
It has become evident in recent years that TLR expression and functionality extends well beyond the boundaries of immune cells, as increasing numbers of cells appear to express and respond to TLRs and their ligands. Among the most interesting non-immune tissues that appear to express TLRs are cells within the CNS. All types of CNS residing cells, namely astrocytes, microglia, oligodendrocytes and neurons, express different subsets of TLRs in both murine models and human cells. It is not surprising, therefore, that TLRs play such important and central roles in neurodegenerative processes.
TLRs are intimately involved in several neurodegenerative disorders including AD, MS and ischemic brain stroke, which imply a broad functionality of these receptors in neurological disease. Thus far, the majority of studies performed on these pathologies have either correlated TLR expression with disease progression and pathology, or used transgenic murine models to counteract the pathological effect of the disease. However, one outstanding question that remains to be answered is the role of neuronal TLR signaling. There exist two main approaches have not been extensively undertaken, but may provide crucial answers to the question of TLRs in neuronal tissue. (1) Cell specific knock-down of specific TLRs, or (2) the effect of specific TLR ligands on neurons both in vitro and in vivo These approaches would allow the role of TLRs as well as the signaling pathways initiated by TLR activation to be elucidated. For example, since TLR 2 and 4 KO mice show beneficial outcome following ischemic brain stroke, follow-up studies with knock-down of TLR4 in neurons will provide an excellent answer for the role of TLR 4 in neurons during ischemic stroke. In addition, no apparent effect of TLR ligands for TLRs 2-9 is observed on neuronal morphology or viability in post-mitotic cortical neurons (following 9 days in culture) (E. Okun, unpublished observations). The only exception is R848, which induces neuronal toxicity at high doses (100 μM) through TLR7 22. Further, unlike other CNS-resident cells, TLR activation in neurons does not lead to NF-κB activation. Therefore, two interesting possibilities emerge: (1) Neuronal TLRs are expressed as different splice variants that perhaps block signaling or (2) non-canonical, e.g.: non-NFkB signaling occurs through them. Both possibilities are interesting and much more research is yet to be done in this field. We believe that TLRs, one of the ancient defense systems of our cells, will soon emerge as an outstanding target for intervention in our relentless efforts in finding cures for neurodegenerative disorders.
Abbreviations
- TLR
Toll-like receptor
- AD
Alzheimer’s Disease
- MS
Multiple sclerosis
- ALS
Amyotrophic lateral sclerosis
- PD
Parkinson’s Disease
- I/R
Ischemia / Reperfusion
- NFκB
Nuclear factor kappa B
- PAMP
pathogen-associated molecular patterns
- PRR
pattern-recognition receptors
- NK
Natural killer
- TIR
Toll/IL-1 Receptor
- HA
Hyaluronic acid
- MyD88
myeloid differentiation factor 88
- EAE
Experimental Autoimmune Encephalomyelitis
- MCAO
middle cerebral artery occlusion
- IRAK
IL-1R-associated kinases
- PGN
peptidoglycan
- GBS
group B streptococcus
- OPC
oligodendrocyte progenitor cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Hashimoto C, et al. The Toll gene of Drosophila, required for dorsal-ventral embryonic polarity, appears to encode a transmembrane protein. Cell. 1988;52:269–279. doi: 10.1016/0092-8674(88)90516-8. [DOI] [PubMed] [Google Scholar]
- 3.Medzhitov R, et al. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 4.Hoshino K, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752. [PubMed] [Google Scholar]
- 5.Gerondakis S, et al. Regulating B-cell activation and survival in response to TLR signals. Immunol Cell Biol. 2007;85:471–475. doi: 10.1038/sj.icb.7100097. [DOI] [PubMed] [Google Scholar]
- 6.Iwamura C, Nakayama T. Toll-like receptors in the respiratory system: their roles in inflammation. Curr Allergy Asthma Rep. 2008;8:7–13. doi: 10.1007/s11882-008-0003-0. [DOI] [PubMed] [Google Scholar]
- 7.Eriksson M, et al. TLRs mediate IFN-gamma production by human uterine NK cells in endometrium. J Immunol. 2006;176:6219–6224. doi: 10.4049/jimmunol.176.10.6219. [DOI] [PubMed] [Google Scholar]
- 8.Sutmuller R, et al. Regulatory T cells and toll-like receptors: regulating the regulators. Ann Rheum Dis. 2007;66(Suppl 3):iii91–95. doi: 10.1136/ard.2007.078535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaisho T, Akira S. Toll-like receptor function and signaling. J Allergy Clin Immunol. 2006;117:979–987. doi: 10.1016/j.jaci.2006.02.023. quiz 988. [DOI] [PubMed] [Google Scholar]
- 10.Sabroe I, Whyte MK. Toll-like receptor (TLR)-based networks regulate neutrophilic inflammation in respiratory disease. Biochem Soc Trans. 2007;35:1492–1495. doi: 10.1042/BST0351492. [DOI] [PubMed] [Google Scholar]
- 11.Yoshimoto T, Nakanishi K. Roles of IL-18 in basophils and mast cells. Allergol Int. 2006;55:105–113. doi: 10.2332/allergolint.55.105. [DOI] [PubMed] [Google Scholar]
- 12.Gibson FC, 3rd, et al. Engagement of specific innate immune signaling pathways during Porphyromonas gingivalis induced chronic inflammation and atherosclerosis. Front Biosci. 2008;13:2041–2059. doi: 10.2741/2822. [DOI] [PubMed] [Google Scholar]
- 13.Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- 14.Bowman CC, et al. Cultured astrocytes express toll-like receptors for bacterial products. Glia. 2003;43:281–291. doi: 10.1002/glia.10256. [DOI] [PubMed] [Google Scholar]
- 15.Bsibsi M, et al. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 16.Tang SC, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lemaitre B, et al. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 18.Belvin MP, Anderson KV. A conserved signaling pathway: the Drosophila toll-dorsal pathway. Annu Rev Cell Dev Biol. 1996;12:393–416. doi: 10.1146/annurev.cellbio.12.1.393. [DOI] [PubMed] [Google Scholar]
- 19.Bar-Shavit Z. Taking a toll on the bones: regulation of bone metabolism by innate immune regulators. Autoimmunity. 2008;41:195–203. doi: 10.1080/08916930701694469. [DOI] [PubMed] [Google Scholar]
- 20.Rolls A, et al. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol. 2007;9:1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- 21.Ma Y, et al. TLR8: an innate immune receptor in brain, neurons and axons. Cell Cycle. 2007;6:2859–2868. doi: 10.4161/cc.6.23.5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma Y, et al. Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. J Cell Biol. 2006;175:209–215. doi: 10.1083/jcb.200606016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akira S, et al. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 24.Wells CA, et al. Alternate transcription of the Toll-like receptor signaling cascade. Genome Biol. 2006;7:R10. doi: 10.1186/gb-2006-7-2-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uematsu S, et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat Immunol. 2008 doi: 10.1038/ni.1622. [DOI] [PubMed] [Google Scholar]
- 26.Pandey S, Agrawal DK. Immunobiology of Toll-like receptors: emerging trends. Immunol Cell Biol. 2006;84:333–341. doi: 10.1111/j.1440-1711.2006.01444.x. [DOI] [PubMed] [Google Scholar]
- 27.Lehnardt S, et al. A vicious cycle involving release of heat shock protein 60 from injured cells and activation of toll-like receptor 4 mediates neurodegeneration in the CNS. J Neurosci. 2008;28:2320–2331. doi: 10.1523/JNEUROSCI.4760-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimada M, et al. Hyaluronan fragments generated by sperm-secreted hyaluronidase stimulate cytokine/chemokine production via the TLR2 and TLR4 pathway in cumulus cells of ovulated COCs, which may enhance fertilization. Development. 2008;135:2001–2011. doi: 10.1242/dev.020461. [DOI] [PubMed] [Google Scholar]
- 29.Wang C, et al. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 30.Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 31.Arancibia SA, et al. Toll-like receptors are key participants in innate immune responses. Biol Res. 2007;40:97–112. doi: 10.4067/s0716-97602007000200001. [DOI] [PubMed] [Google Scholar]
- 32.Sarkar SN, et al. Novel roles of TLR3 tyrosine phosphorylation and PI3 kinase in double-stranded RNA signaling. Nat Struct Mol Biol. 2004;11:1060–1067. doi: 10.1038/nsmb847. [DOI] [PubMed] [Google Scholar]
- 33.Pessac B, et al. Microglia: origin and development. Bull Acad Natl Med. 2001;185:337–346. discussion 346-337. [PubMed] [Google Scholar]
- 34.Jack CS, et al. TLR signaling tailors innate immune responses in human microglia and astrocytes. J Immunol. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- 35.Kielian T, et al. Toll-like receptor 2 (TLR2) is pivotal for recognition of S. aureus peptidoglycan but not intact bacteria by microglia. Glia. 2005;49:567–576. doi: 10.1002/glia.20144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan A, et al. Phagocytosis of apoptotic inflammatory cells by microglia and its therapeutic implications: termination of CNS autoimmune inflammation and modulation by interferon-beta. Glia. 2003;43:231–242. doi: 10.1002/glia.10258. [DOI] [PubMed] [Google Scholar]
- 37.Becher B, et al. Regulation of CD14 expression on human adult central nervous system-derived microglia. J Neurosci Res. 1996;45:375–381. doi: 10.1002/(SICI)1097-4547(19960815)45:4<375::AID-JNR6>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 38.Williams K, et al. IL-10 production by adult human derived microglial cells. Neurochem Int. 1996;29:55–64. doi: 10.1016/0197-0186(95)00138-7. [DOI] [PubMed] [Google Scholar]
- 39.Alexopoulou L, et al. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 40.Cassiani-Ingoni R, et al. Cytoplasmic translocation of Olig2 in adult glial progenitors marks the generation of reactive astrocytes following autoimmune inflammation. Exp Neurol. 2006;201:349–358. doi: 10.1016/j.expneurol.2006.04.030. [DOI] [PubMed] [Google Scholar]
- 41.Ock J, et al. Regulation of Toll-like receptor 4 expression and its signaling by hypoxia in cultured microglia. J Neurosci Res. 2007;85:1989–1995. doi: 10.1002/jnr.21322. [DOI] [PubMed] [Google Scholar]
- 42.Zuiderwijk-Sick EA, et al. Differentiation of primary adult microglia alters their response to TLR8-mediated activation but not their capacity as APC. Glia. 2007;55:1589–1600. doi: 10.1002/glia.20572. [DOI] [PubMed] [Google Scholar]
- 43.Jack CS, et al. Th1 polarization of CD4+ T cells by Toll-like receptor 3-activated human microglia. J Neuropathol Exp Neurol. 2007;66:848–859. doi: 10.1097/nen.0b013e3181492a7. [DOI] [PubMed] [Google Scholar]
- 44.Jung DY, et al. TLR4, but not TLR2, signals autoregulatory apoptosis of cultured microglia: a critical role of IFN-beta as a decision maker. J Immunol. 2005;174:6467–6476. doi: 10.4049/jimmunol.174.10.6467. [DOI] [PubMed] [Google Scholar]
- 45.Lehnardt S, et al. TLR2 and caspase-8 are essential for group B Streptococcus-induced apoptosis in microglia. J Immunol. 2007;179:6134–6143. doi: 10.4049/jimmunol.179.9.6134. [DOI] [PubMed] [Google Scholar]
- 46.Aravalli RN, et al. Inhibition of toll-like receptor signaling in primary murine microglia. J Neuroimmune Pharmacol. 2008;3:5–11. doi: 10.1007/s11481-007-9097-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanaka T, et al. NF-kappaB independent signaling pathway is responsible for LPS-induced GDNF gene expression in primary rat glial cultures. Neurosci Lett. 2008;431:262–267. doi: 10.1016/j.neulet.2007.11.051. [DOI] [PubMed] [Google Scholar]
- 48.Carpentier PA, et al. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia. 2005;49:360–374. doi: 10.1002/glia.20117. [DOI] [PubMed] [Google Scholar]
- Krasowska-Zoladek A, et al. Kinetics of inflammatory response of astrocytes induced by TLR 3 and TLR4 ligation. J Neurosci Res. 2007;85:205–212. doi: 10.1002/jnr.21088. [DOI] [PubMed] [Google Scholar]
- 50.Konat GW, et al. Statins enhance toll-like receptor 4-mediated cytokine gene expression in astrocytes: implication of Rho proteins in negative feedback regulation. J Neurosci Res. 2008;86:603–609. doi: 10.1002/jnr.21509. [DOI] [PubMed] [Google Scholar]
- 51.Bsibsi M, et al. Identification of soluble CD14 as an endogenous agonist for Toll-like receptor 2 on human astrocytes by genome-scale functional screening of glial cell derived proteins. Glia. 2007;55:473–482. doi: 10.1002/glia.20473. [DOI] [PubMed] [Google Scholar]
- 52.Farina C, et al. Preferential expression and function of Toll-like receptor 3 in human astrocytes. J Neuroimmunol. 2005;159:12–19. doi: 10.1016/j.jneuroim.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 53.Bsibsi M, et al. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia. 2006;53:688–695. doi: 10.1002/glia.20328. [DOI] [PubMed] [Google Scholar]
- 54.Aravalli RN, et al. Toll-like receptors in defense and damage of the central nervous system. J Neuroimmune Pharmacol. 2007;2:297–312. doi: 10.1007/s11481-007-9071-5. [DOI] [PubMed] [Google Scholar]
- 55.Lehnardt S, et al. A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J Immunol. 2006;177:583–592. doi: 10.4049/jimmunol.177.1.583. [DOI] [PubMed] [Google Scholar]
- 56.Setzu A, et al. Inflammation stimulates myelination by transplanted oligodendrocyte precursor cells. Glia. 2006;54:297–303. doi: 10.1002/glia.20371. [DOI] [PubMed] [Google Scholar]
- 57.Kigerl KA, et al. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J Neurochem. 2007;102:37–50. doi: 10.1111/j.1471-4159.2007.04524.x. [DOI] [PubMed] [Google Scholar]
- 58.Schonberg DL, et al. Oligodendrocyte generation is differentially influenced by toll-like receptor (TLR) 2 and TLR4-mediated intraspinal macrophage activation. J Neuropathol Exp Neurol. 2007;66:1124–1135. doi: 10.1097/nen.0b013e31815c2530. [DOI] [PubMed] [Google Scholar]
- 59.Prehaud C, et al. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J Virol. 2005;79:12893–12904. doi: 10.1128/JVI.79.20.12893-12904.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mishra BB, et al. Expression and distribution of Toll-like receptors in the brain during murine neurocysticercosis. J Neuroimmunol. 2006;181:46–56. doi: 10.1016/j.jneuroim.2006.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wadachi R, Hargreaves KM. Trigeminal nociceptors express TLR-4 and CD14: a mechanism for pain due to infection. J Dent Res. 2006;85:49–53. doi: 10.1177/154405910608500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cameron JS, et al. Toll-like receptor 3 is a potent negative regulator of axonal growth in mammals. J Neurosci. 2007;27:13033–13041. doi: 10.1523/JNEUROSCI.4290-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Z, Schluesener HJ. Mammalian toll-like receptors: from endogenous ligands to tissue regeneration. Cell Mol Life Sci. 2006;63:2901–2907. doi: 10.1007/s00018-006-6189-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Godbout JP, Johnson RW. Interleukin-6 in the aging brain. J Neuroimmunol. 2004;147:141–144. doi: 10.1016/j.jneuroim.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 65.Ye SM, Johnson RW. An age-related decline in interleukin-10 may contribute to the increased expression of interleukin-6 in brain of aged mice. Neuroimmunomodulation. 2001;9:183–192. doi: 10.1159/000049025. [DOI] [PubMed] [Google Scholar]
- 66.Lu T, et al. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 67.Letiembre M, et al. Innate immune receptor expression in normal brain aging. Neuroscience. 2007;146:248–254. doi: 10.1016/j.neuroscience.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 68.Ziegler G, et al. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun. 2007;359:574–579. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]
- 69.Cao CX, et al. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem Biophys Res Commun. 2007;353:509–514. doi: 10.1016/j.bbrc.2006.12.057. [DOI] [PubMed] [Google Scholar]
- 70.Poltorak A, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 71.Hua F, et al. Activation of Toll-like receptor 4 signaling contributes to hippocampal neuronal death following global cerebral ischemia/reperfusion. J Neuroimmunol. 2007;190:101–111. doi: 10.1016/j.jneuroim.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kilic U, et al. TLR-4 deficiency protects against focal cerebral ischemia and axotomy-induced neurodegeneration. Neurobiol Dis. 2008;31:33–40. doi: 10.1016/j.nbd.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 73.Caso JR, et al. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- 74.Lehnardt S, et al. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J Neuroimmunol. 2007;190:28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 75.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Akiyama H, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McGeer PL, et al. Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J Alzheimers Dis. 2006;9:271–276. doi: 10.3233/jad-2006-9s330. [DOI] [PubMed] [Google Scholar]
- 78.Stewart WF, et al. Risk of Alzheimer’s disease and duration of NSAID use. Neurology. 1997;48:626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- 79.in t’ Veld BA, et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med. 2001;345:1515–1521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- 80.Letiembre M, et al. Screening of innate immune receptors in neurodegenerative diseases: A similar pattern. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 81.Walter S, et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol Biochem. 2007;20:947–956. doi: 10.1159/000110455. [DOI] [PubMed] [Google Scholar]
- 82.Arbour NC, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–191. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- 83.Balistreri CR, et al. Role of Toll-like receptor 4 in acute myocardial infarction and longevity. Jama. 2004;292:2339–2340. doi: 10.1001/jama.292.19.2339. [DOI] [PubMed] [Google Scholar]
- 84.Minoretti P, et al. Effect of the functional toll-like receptor 4 Asp299Gly polymorphism on susceptibility to late-onset Alzheimer’s disease. Neurosci Lett. 2006;391:147–149. doi: 10.1016/j.neulet.2005.08.047. [DOI] [PubMed] [Google Scholar]
- 85.Mackenzie IR, et al. Role of microglia in senile plaque formation. Neurobiol Aging. 1995;16:797–804. doi: 10.1016/0197-4580(95)00092-s. [DOI] [PubMed] [Google Scholar]
- 86.Sheng JG, et al. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol Dis. 2003;14:133–145. doi: 10.1016/s0969-9961(03)00069-x. [DOI] [PubMed] [Google Scholar]
- 87.Tang SC, et al. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp Neurol. 2008 doi: 10.1016/j.expneurol.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.DiCarlo G, et al. Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol Aging. 2001;22:1007–1012. doi: 10.1016/s0197-4580(01)00292-5. [DOI] [PubMed] [Google Scholar]
- 89.Herber DL, et al. Microglial activation is required for Abeta clearance after intracranial injection of lipopolysaccharide in APP transgenic mice. J Neuroimmune Pharmacol. 2007;2:222–231. doi: 10.1007/s11481-007-9069-z. [DOI] [PubMed] [Google Scholar]
- 90.Tahara K, et al. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain. 2006;129:3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen K, et al. Activation of Toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid beta peptide. J Biol Chem. 2006;281:3651–3659. doi: 10.1074/jbc.M508125200. [DOI] [PubMed] [Google Scholar]
- 92.Iribarren P, et al. CpG-containing oligodeoxynucleotide promotes microglial cell uptake of amyloid beta 1-42 peptide by up-regulating the expression of the G-protein- coupled receptor mFPR2. Faseb J. 2005;19:2032–2034. doi: 10.1096/fj.05-4578fje. [DOI] [PubMed] [Google Scholar]
- 93.Lotz M, et al. Amyloid beta peptide 1-40 enhances the action of Toll-like receptor-2 and -4 agonists but antagonizes Toll-like receptor-9-induced inflammation in primary mouse microglial cell cultures. J Neurochem. 2005;94:289–298. doi: 10.1111/j.1471-4159.2005.03188.x. [DOI] [PubMed] [Google Scholar]
- Jin JJ, et al. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease. J Neuroinflammation. 2008;5:23. doi: 10.1186/1742-2094-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gasic-Milenkovic J, et al. Beta-amyloid peptide potentiates inflammatory responses induced by lipopolysaccharide, interferon -gamma and ‘advanced glycation endproducts’ in a murine microglia cell line. Eur J Neurosci. 2003;17:813–821. doi: 10.1046/j.1460-9568.2003.02506.x. [DOI] [PubMed] [Google Scholar]
- 96.Latz E, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. 2004;5:190–198. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- 97.Wagner H. The immunobiology of the TLR9 subfamily. Trends Immunol. 2004;25:381–386. doi: 10.1016/j.it.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 98.Krieg AM. CpG motifs: the active ingredient in bacterial extracts? Nat Med. 2003;9:831–835. doi: 10.1038/nm0703-831. [DOI] [PubMed] [Google Scholar]
- 99.Trapp BD. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 100.Grigoriadis N, Hadjigeorgiou GM. Virus-mediated autoimmunity in Multiple Sclerosis. J Autoimmune Dis. 2006;3:1. doi: 10.1186/1740-2557-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kurtzke JF. Epidemiologic evidence for multiple sclerosis as an infection. Clin Microbiol Rev. 1993;6:382–427. doi: 10.1128/cmr.6.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Prat A, Antel J. Pathogenesis of multiple sclerosis. Curr Opin Neurol. 2005;18:225–230. doi: 10.1097/01.wco.0000169737.99040.31. [DOI] [PubMed] [Google Scholar]
- 103.Johns TG, et al. Myelin oligodendrocyte glycoprotein induces a demyelinating encephalomyelitis resembling multiple sclerosis. J Immunol. 1995;154:5536–5541. [PubMed] [Google Scholar]
- 104.Swanborg RH. Experimental autoimmune encephalomyelitis in rodents as a model for human demyelinating disease. Clin Immunol Immunopathol. 1995;77:4–13. doi: 10.1016/0090-1229(95)90130-2. [DOI] [PubMed] [Google Scholar]
- 105.Zekki H, et al. The clinical course of experimental autoimmune encephalomyelitis is associated with a profound and sustained transcriptional activation of the genes encoding toll-like receptor 2 and CD14 in the mouse CNS. Brain Pathol. 2002;12:308–319. doi: 10.1111/j.1750-3639.2002.tb00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Prinz M, et al. Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J Clin Invest. 2006;116:456–464. doi: 10.1172/JCI26078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marta M, et al. Unexpected regulatory roles of TLR4 and TLR9 in experimental autoimmune encephalomyelitis. Eur J Immunol. 2008;38:565–575. doi: 10.1002/eji.200737187. [DOI] [PubMed] [Google Scholar]
- 108.Touil T, et al. Cutting Edge: TLR3 stimulation suppresses experimental autoimmune encephalomyelitis by inducing endogenous IFN-beta. J Immunol. 2006;177:7505–7509. doi: 10.4049/jimmunol.177.11.7505. [DOI] [PubMed] [Google Scholar]
- 109.Olson JK, et al. Direct activation of innate and antigen-presenting functions of microglia following infection with Theiler’s virus. J Virol. 2001;75:9780–9789. doi: 10.1128/JVI.75.20.9780-9789.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pisegna S, et al. p38 MAPK activation controls the TLR3-mediated upregulation of cytotoxicity and cytokine production in human NK cells. Blood. 2004;104:4157–4164. doi: 10.1182/blood-2004-05-1860. [DOI] [PubMed] [Google Scholar]
- 111.Lehnardt S, et al. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002;22:2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lehnardt S, et al. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pang Y, et al. Disturbance of oligodendrocyte development, hypomyelination and white matter injury in the neonatal rat brain after intracerebral injection of lipopolysaccharide. Brain Res Dev Brain Res. 2003;140:205–214. doi: 10.1016/s0165-3806(02)00606-5. [DOI] [PubMed] [Google Scholar]
- 114.Li Y, et al. Increased IL-23p19 expression in multiple sclerosis lesions and its induction in microglia. Brain. 2007;130:490–501. doi: 10.1093/brain/awl273. [DOI] [PubMed] [Google Scholar]
- 115.Jackson AC, et al. Expression of Toll-like receptor 3 in the human cerebellar cortex in rabies, herpes simplex encephalitis, and other neurological diseases. J Neurovirol. 2006;12:229–234. doi: 10.1080/13550280600848399. [DOI] [PubMed] [Google Scholar]
- 116.Kroner A, et al. Impact of the Asp299Gly polymorphism in the toll-like receptor 4 (tlr-4) gene on disease course of multiple sclerosis. J Neuroimmunol. 2005;165:161–165. doi: 10.1016/j.jneuroim.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 117.Reindl M, et al. Mutations in the gene for toll-like receptor 4 and multiple sclerosis. Tissue Antigens. 2003;61:85–88. doi: 10.1034/j.1399-0039.2003.610108.x. [DOI] [PubMed] [Google Scholar]
- 118.Rius J, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tasaki K, et al. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res. 1997;748:267–270. doi: 10.1016/s0006-8993(96)01383-2. [DOI] [PubMed] [Google Scholar]
- 120.Rosenzweig HL, et al. Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke. 2004;35:2576–2581. doi: 10.1161/01.STR.0000143450.04438.ae. [DOI] [PubMed] [Google Scholar]
- 121.Rosenzweig HL, et al. Endotoxin preconditioning protects against the cytotoxic effects of TNFalpha after stroke: a novel role for TNFalpha in LPS-ischemic tolerance. J Cereb Blood Flow Metab. 2007;27:1663–1674. doi: 10.1038/sj.jcbfm.9600464. [DOI] [PubMed] [Google Scholar]
- 122.Broad A, et al. Toll-like receptor interactions: tolerance of MyD88-dependent cytokines but enhancement of MyD88-independent interferon-beta production. Immunology. 2007;120:103–111. doi: 10.1111/j.1365-2567.2006.02485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Drescher KM, et al. TGF-beta 2 reduces demyelination, virus antigen expression, and macrophage recruitment in a viral model of multiple sclerosis. J Immunol. 2000;164:3207–3213. doi: 10.4049/jimmunol.164.6.3207. [DOI] [PubMed] [Google Scholar]
- 124.Stankoff B, et al. Ciliary neurotrophic factor (CNTF) enhances myelin formation: a novel role for CNTF and CNTF-related molecules. J Neurosci. 2002;22:9221–9227. doi: 10.1523/JNEUROSCI.22-21-09221.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Linker RA, et al. CNTF is a major protective factor in demyelinating CNS disease: a neurotrophic cytokine as modulator in neuroinflammation. Nat Med. 2002;8:620–624. doi: 10.1038/nm0602-620. [DOI] [PubMed] [Google Scholar]
- 126.Parkinson T. The future of toll-like receptor therapeutics. Curr Opin Mol Ther. 2008;10:21–31. [PubMed] [Google Scholar]
- 127.Bagchi A, et al. MyD88-dependent and MyD88-independent pathways in synergy, priming, and tolerance between TLR agonists. J Immunol. 2007;178:1164–1171. doi: 10.4049/jimmunol.178.2.1164. [DOI] [PubMed] [Google Scholar]