

Abstract
Purpose
Frizzled-5 (Fzd5) is expressed in the developing retina of multiple species and appears to play species-specific roles during eye development. The present study analyzed the effects of tissue-specific deletion of Fzd5 on mammalian eye development.
Methods
To generate Fzd5 conditional knockout (CKO) mice, Fzd5+/− mice carrying the Six3-Cre transgene were crossed with Fzd5LoxP/LoxP mice. To determine which cell lineages in the eye had Cre recombinase activity, Six3-Cre transgenic mice were crossed with ROSA-26 reporter mice, and lacZ activity was assayed. Histologic analysis, immunofluorescence, and TUNEL labeling were performed from embryonic day (E)12.5 to postnatal stages to analyze vascularization, cell proliferation, retinal organization, and apoptosis.
Results
On conditional disruption of Fzd5 specifically in the retina, but not in vitreous hyaloid vasculature (VHV), an abnormal accumulation consisting of pericytes and endothelial cells was observed in the vitreous as early as E12.5. The abundant retrolental cells persisted into postnatal stages and appeared as a pigmented intravitreal mass. In Fzd5 CKO mice there was failure of normal apoptosis of the VHV, and cells in the persistent VHV were maintained in the cell cycle up to postnatal day 23. Moreover, morphogenesis of the retina adjacent to the vasculature was disrupted, leading to retinal folds, detachment, and abnormal lamination. This phenotype is similar to that of human eye disease persistent hyperplastic primary vitreous (PHPV).
Conclusions
Selective loss of Fzd5 in the retina results in PHPV and retinal defects through an apparently cell-nonautonomous effect, revealing a potential requirement for retina-derived signals in regulating the development of the VHV.
The Wnt genes encode a large family of secreted glycoproteins that elicit cellular responses by binding with membrane receptors, including the Frizzled (Fzd) and low-density lipoprotein-related receptors 5/6. Ten Fzd family members have been identified in humans and mice. Fzd5 is expressed in the developing retina in zebrafish, Xenopus laevis, chick, and mouse, which suggests a specific role for this receptor in regulating eye development. In zebrafish, Fzd5 mediates non-canonical signaling and promotes eye field formation.1 In X. laevis, Fzd5 activates the Wnt/β-catenin (canonical) pathway in the retina and controls the neural potential of retinal progenitors by regulating Sox2 expression.2 In mouse, Fzd5 is highly expressed in developing retina from embryonic day (E)8.5 to postnatal stages3,4 but is not required for early eye formation or for regulating neural potential of retinal progenitors,5 suggesting species-specific roles for Fzd5 in eye development. However, disruption of the Fzd5 gene results in embryonic lethality at approximately E10.56; thus, the role of Fzd5 at later stages of mammalian eye development is unknown.
During eye development a transient vascular system called the primary vitreous hyaloid vascular system (HVS) is formed. This vascular system provides nutrition to the developing lens and retina before the intraretinal vasculature is formed. Formation of the intraretinal vasculature begins with the entry of the hyaloid artery into the primitive vitreous through the fetal fissure, which then branches in the vitreous to form the vasa hyaloids propria and the tunica vasculosa lentis.7 In mouse, the formation of HVS starts at E10.5 and is fully formed by E13.5. Because the HVS is a transient capillary network, it undergoes regression through apoptosis at postnatal stages, which ensures the mature vitreous is avascular. Most of the branches of HVS regress by postnatal day (P)10, and the vitreous is completely avascular by P16.8–10 Failure of HVS regression causes the persistence of this fetal vasculature at postnatal stages and the formation of a retrolental mass.
Disruption of several genes, including Arf, can cause hyperplasia of the HVS even earlier, at embryonic stages, with a subsequent failure of regression.11 This results in abnormal retrolental tissue that can give rise to cataract, secondary glaucoma, retinal folding, dysplasia, and retinal detachment,7,9,12 similar to the congenital human eye disease persistent hyperplastic primary vitreous (PHPV). In human patients the mechanisms underlying this disease remain unclear.
To investigate the function of Fzd5 at later stages of mammalian eye development, we conditionally disrupted the Fzd5 gene specifically in the mouse retina. In the absence of retinal Fzd5, we observed hyperplastic hyaloid vitreous vasculature at embryonic stages in the mouse eye. This hyperplastic vitreous vasculature became heavily pigmented and failed to regress at postnatal stages. Retinal abnormalities such as retinal folds, detachment, and disrupted retina lamination occurred in the retina adjacent to the hyperplastic vitreous vasculature. This phenotype is reminiscent of the human eye disease PHPV. Given that Fzd5 was inactivated in the retina but not in the vitreous vasculature tissue, the persistent hyperplastic vitreous vasculature was likely caused by a cell-nonautonomous mechanism. Our data suggest that Fzd5 regulates the expression of retina-derived signals that influence hyaloid vitreous vasculature development in mammals.
Methods
Mice and Genotyping
Mice carrying a null allele of the Fzd5 gene (Fzd5+/−) were provided by Makoto Taketo.6 Mice with floxed Fzd5 alleles (Fzd5LoxP/LoxP) were provided by Hans Clevers.13 Six3-Cre transgenic mice were provided by Yasuhide Furuta14 (Cre line 69). Six3-Cre females heterozygous for the Fzd5 null allele (Fzd5+/−;Six3-Cre) were crossed with Fzd5LoxP/LoxP males to generate conditional Fzd5 mutant embryos (Fzd5LoxP/−;Six3-Cre). Littermates carrying a wild-type allele of Fzd5 were used as controls.
PCR was used to identify the genotypes of the embryos and mice, as previously reported.6,13,14 Primers complementary to neomycin (pn5b, CTA AAG CGC ATG CTC CAG ACT) and to Fzd5 downstream of the stop codon (sj2, CCT TTA GCA AAG AGT CCT AAC) were used to genotype the floxed Fzd5 allele, generating a 700-bp PCR product. The wild-type allele was determined using primers of f5x (AGA GGAGGC CTT ATA GA CG) and sj2 generating a 250-bp PCR product. The Fzd5 null allele was determined using primers complementary to neomycin (Neo, GCG CAT GCT CCA GAC TG) and Fzd5 (Fzd5, GAT GCG GAA GAG TGA CAC GA). The Cre gene was identified by using primers Cre159 (TCG ATG CAA CGA GTG ATG AG) and Cre160 (TTC GGC TAT ACG TAA CAG GG).
LacZ Activity
Six3-Cre homozygous transgenic mice were crossed with ROSA-26 reporter mice. The tissue was fixed in 4% paraformaldehyde in PBS for 25 minutes at room temperature, washed in PBS, saturated in 25% sucrose, and embedded in optimum cutting temperature compound (Tissue-Tek; Sakura Finetek, Torrance, CA). LacZ activity was analyzed on cryostat sections (16 μm) at E14.5 and P0 (n = 3 for each age) using X-gal substrate (USB Corporation, Cleveland, OH).
Histologic Analysis
Embryo and eye tissue at ages E12.5, E14.5, E17.5, P0, P8, P10, and P23 were fixed in 4% paraformaldehyde in PBS for 2 hours at room temperature or overnight at 4°C, saturated in 25% sucrose, and embedded as described. Cryostat sections (16 μm) were stained with hematoxylin (Fisher Scientific, Pittsburgh, PA) and eosin (Fisher Scientific). Sections crossing the central area of the eye, where eye and lens sizes were greatest, were used to measure the greatest anteroposterior diameter of the eye and the thickness and diameter of the lens. Thickness of cornea and retina were also measured on these sections. Student's t-test was used for statistics analysis.
In Situ RNA hybridization
In situ RNA hybridization was performed as previously described15 on serial cryostat sections of embryos at E13 through the central region of the eye. Digoxigenin-labeled antisense Fzd5 riboprobe (1 μg/mL) was used for the procedure. To ensure reproducibility, in situ hybridizations were repeated on both eyes from three wild-type embryos.
Immunofluorescence and Apoptosis Analysis
Cryostat sections were processed for immunofluorescence staining using the following primary antibodies: rat anti–PECAM-1 (1:75; BD PharMingen, San Diego, CA), goat anti–Brn3b (1:50; Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti–proliferating cell nuclear antigen (PCNA; 1:500; DAKO, Carpinteria, CA), rabbit anti–NG2 chondroitin sulfate proteoglycan (1:500; Chemicon, Temecula, CA), mouse anti-Tuj1 (1:1000; Covance, Princeton, NJ), and rabbit anti–Otx2 (1:1500; Chemicon). For PCNA labeling, antigen retrieval with citric acid and sodium citrate buffer (2 mM citric acid, 9 mM sodium citrate) was used. For visualization, sections were incubated with Alexa 568- or Alexa 488 – conjugated secondary antibodies (1:1000 – 1:3000; Molecular Probes, Eugene, OR) and were counterstained with 4,6-diamidino-2-phenylindole (DAPI; Roche, Basel, Switzerland). Cell apoptosis was monitored with a cell detection kit (In Situ Cell Death Detection Kit, Fluorescein; Roche) according to the manufacturer's instructions. Images were captured using a microscope (E800; Nikon, Tokyo, Japan) equipped with an RT slider digital camera (Diagnostic Instruments, Sterling Heights, MI). Throughout this study mice were treated in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Results
Abnormal Retrolental Tissue and Retina Dysplasia at Embryonic Stages Caused by Loss of Fzd5
To investigate Fzd5 function during later stages of mouse eye development, Six3-Cre–mediated recombination was used to generate Fzd5 conditional knockout mice. Six3-Cre mice were crossed with ROSA-26 reporter mice to determine which cell lineages exhibit successful Cre-mediated recombination. X-gal staining at E14.5 and P0 (n = 3 for each age) showed that β-galactosidase activity is specifically restricted to the neuro-retina and optic nerve (Figs. 1A, 1B, and data not shown) but does not occur in the hyaloid vitreous vasculature tissue and the lens (Fig. 1B). In the original report describing the generation of the Six3-Cre lines, lacZ activity is detected in the vitreous vasculature in a cross with the ROSA-26 reporter.14 However, this was for the transgenic line Cre49 (which has been discontinued), whereas the line used here was Cre69, which does not show activity in the vitreous vasculature (Fig. 1B, arrow; Yas Furuta, personal communication, August 7, 2008). Furthermore, in situ hybridization analysis showed that Fzd5 is expressed in the retina and optic stalk (Fig. 1C) but not in cells of the vitreous vasculature (Fig. 1D, arrow).
FIGURE 1.
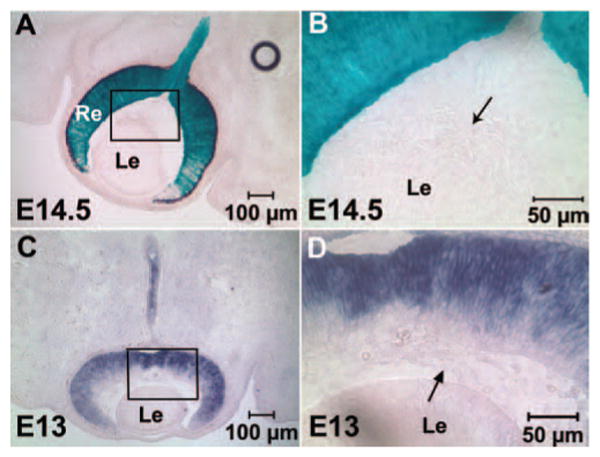
Six3-Cre activity and Fzd5 expression are present in the retina and optic stalk but not in the hyaloid vitreous vasculature. (A, B) Six3-Cre/Six3-Cre transgenic mice were crossed with ROSA-26 reporter mice to analyze which cell lineages in the eye express Cre activity by X-gal staining on cryostat sections of the embryonic eye at E14.5. (B) Inset in (A) at higher magnification. (C, D) Fzd5 in situ hybridization on cryostat sections at E13. (D) Inset in (C) at higher magnification. The primary hyaloid vitreous vasculature was negative for X-gal staining and Fzd5 expression (B, D, arrows; n = 3 for each). Re, retina; Le, lens.
To investigate the roles of Fzd5 during mouse eye development, the ocular morphology of Fzd5 conditional knockout mice was analyzed at E12.5, E14.5, and E17.5 by hematoxylin and eosin staining. At E12.5 (n = 3) and E14.5 (n = 3), abnormally abundant retrolental mass tissue was observed in the vitreous body of Fzd5LoxP/−;Six3-Cre embryos, possibly representing hyaloid vitreous vasculature (Figs. 2B, 2D, arrowheads). In the Fzd5LoxP/−;Six3-Cre embryos at E14.5, the retina was 51% thicker and the cornea was 120% thicker than in controls, respectively (P < 0.01). The lens diameter was also reduced by 17% in the Fzd5LoxP/−;Six3-Cre embryos (seven eyes analyzed) compared with control (six eyes analyzed), but this was variable and not highly significant (P = 0.037). In addition, the lamination of the neuroblasts in the retinal area that was adjacent to the abnormal retrolental mass tissue was disrupted (Fig. 2D, white arrow). This disruption was not observed in the retinal area distant from the retrolental mass tissue. At E17.5 (n = 2), abnormal retrolental mass tissue persisted in the vitreous and the posterior surface of the lens (Fig. 2F, arrowhead), and in some instances this mass tissue extended into the optic nerve. The disorganization of the retina adjacent to the vitreous mass tissue was more obvious at this stage (Fig. 2F, white arrow). Sagittal sections at E14.5 did not reveal defects in optic fissure closure (data not shown). These results suggest that loss of Fzd5 in the retina results in the excess formation of retrolental mass tissue in the vitreous through a cell-nonautonomous mechanism because neither Fzd5 expression nor Cre recombinase activity was detectable in the vitreous and lens.
FIGURE 2.
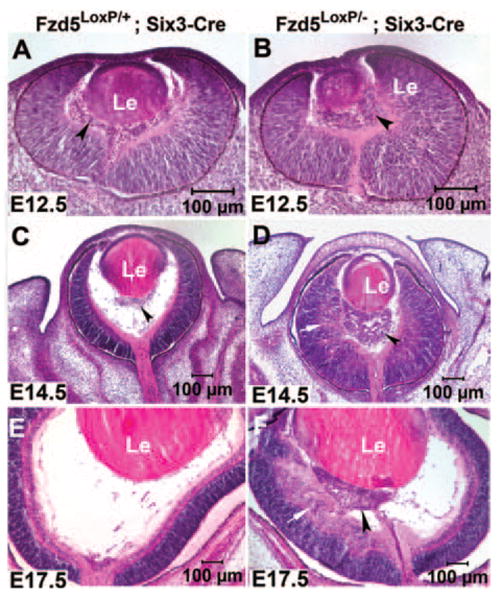
Abnormal abundant retrolental tissue was observed at embryonic stages in eyes of Fzd5LoxP/−;Six3-Cre mice. Hematoxylin and eosin staining of cryostat sections of Fzd5LoxP/+;Six3-Cre mice (A, C, E) and Fzd5LoxP/−;Six3-Cre mice (B, D, F) at E12.5 (n = 3), E14.5 (n = 3), and E17.5 (n = 2), respectively. (A, C, arrowheads) Vitreous vasculature. (B, D, F, arrowheads) Abnormal abundant retrolental tissue. (D, F, arrows) Disorganization of the inner retina adjacent to the abnormal retrolental tissue.
Development of Retrolental Mass Tissue in Fzd5LoxP/−;Six3-Cre Mice into Pigmented Hyperplastic Primary Vitreous Vasculature
To determine the identity of the retrolental mass tissue observed in Fzd5LoxP/−;Six3Cre mice, immunofluorescence staining with specific cell markers was performed. We hypothesized that the abnormal retrolental mass tissue observed in Fzd5LoxP/−;Six3Cre mice at embryonic stages was primary vitreous vasculature. To confirm this, the endothelial cell marker PECAM and the pericyte marker NG2 were used for immunofluorescence staining at E12.5 and E14.5. In control Fzd5LoxP/+;Six3-Cre eyes, PECAM-positive and NG2-positive cells were found at the posterior surface of the lens, in the vitreous body, at the inner surface of central retina, and in the optic nerve head (Figs. 3B, 3F, arrowheads) as part of the normal primary vitreous vasculature. In Fzd5LoxP/−;Six3-Cre mice there was an accumulation of PECAM-positive endothelial cells and NG2-positive pericytes at E12.5 (n = 3) that became more pronounced at E14.5 (n = 2; Figs. 3D, 3H, arrowheads). Abundant PECAM-positive cells were also found in the abnormal retrolental mass tissue in the Fzd5LoxP/−;Six3-Cre eyes at P0 (n = 4; Fig. 3L, arrowhead) and formed vessel-like structures (data not shown), suggesting that the mass tissue in the vitreous of Fzd5LoxP/−;Six3-Cre mice was hyperplastic vitreous vasculature.
FIGURE 3.
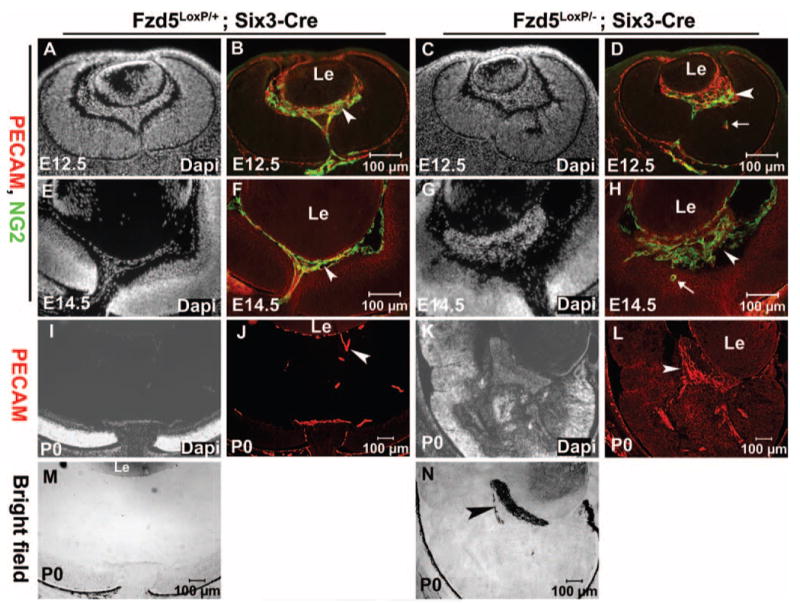
Retrolental mass tissue in the eyes of Fzd5LoxP/−;Six3-Cre mice is primary vitreous vasculature and is heavily pigmented. (A–H) Immunofluorescence staining of endothelial cell marker PECAM (red) and NG2 (green) on cryostat sections of Fzd5LoxP/+;Six3-Cre (A, B, E, F) and Fzd5LoxP/−;Six3-Cre (C, D, G, H) eyes at E12.5 (n = 3) and E14.5 (n = 2). (B, D, F, H) PECAM and NG2 double-immunofluorescence staining. (A, C, E, G) Corresponding DAPI staining. (D, H, arrows) Ectopic PECAM and NG2-positive cells in the retina. (I– L) Immunofluorescence staining of PECAM on cryostat section of Fzd5LoxP/+;Six3-Cre (J) and Fzd5LoxP/−;Six3-Cre (L) at P0 (n = 4). White arrowheads: PECAM and NG2-positive vitreous vasculature. (I, K) Corresponding DAPI staining. (M, N) Bright-field images at P0. (N, black arrowhead) Pigmented hyperplastic vitreous vasculature.
One possibility was that Fzd5 mutant cells derived from the neural retina or optic nerve migrate to the vitreous to generate the phenotype. However, because endothelial cells and pericytes are derived from the periocular mesenchyme, do not express Fzd5, and are not present in the retina at E12.5 and E14.5 (Figs. 3B, 3F), these excess endothelial cells and pericytes in the vitreous of Fzd5LoxP/−;Six3-Cre mice were unlikely to have migrated from the retina. In fact, the opposite was observed, with occasional invasion of PECAM-positive endothelial cells and NG2-positive pericytes into Fzd5LoxP/−;Six3-Cre retina (Figs. 3D, 3H, arrows). Furthermore, we confirmed that at the stages between E14.5 and P0, the retrolental cells did not express neural progenitor markers (Pax6, Chx10), markers of retinal neurons (TUJ1, Brn3b), or retinal pigment epithelium (Otx2; see Supplementary Fig. S1, online at http://www.iovs.org/cgi/content/full/49/12/5561/DC1, and data not shown), further suggesting that the excess cells were not derived from the neural retina or retinal pigment epithelium. Bright-field imaging indicated that the retrolental mass tissue was heavily pigmented by P0 (Fig. 3N, arrowhead); hence, it remains possible that neural crest-derived melanocytes migrate to the vitreous or that cells from the retinal pigment epithelium migrate into the mass at late embryonic stages. Other cell types may be present in the mass at this stage, including cells derived from the retina sclera or optic nerve. We conclude that initially the abnormal retrolental mass tissue in the Fzd5LoxP/−;Six3-Cre eye is hyperplastic primary vitreous vasculature, though additional cell types may ultimately contribute to the phenotype.
Persistence of Retrolental Hyperplastic Vitreous Vasculature at Postnatal Stages
Normally, the primary vitreous vasculature is a transient blood vessel system that regresses through apoptosis at postnatal stages to ensure that the mature vitreous is avascular.8–10 However, the hyperplastic vitreous vasculature in the Fzd5LoxP/−;Six3-Cre eye persisted as a pigmented mass tissue at postnatal stages P0 (n = 7), P10 (n = 4), and even P23 (n = 6); Figs. 4B, 4E, 4H). This hyperplastic vitreous vasculature was occasionally attached to the lens or to the inner surface of the retina (Figs. 4B, 4E). In addition, the eye size in Fzd5LoxP/−;Six3-Cre mice was reduced by 24% (P < 0.05) compared with control littermates at P0. Morphogenesis of the retina adjacent to the hyperplastic vitreous vasculature was disrupted, leading usually to multiple abnormalities such as partial retinal detachment and occasionally to complete detachment, retinal folds, and abnormal retinal lamination, especially in the inner retinas and rosette structures (data not shown; Figs. 4C, 4E, 4F, 4H, 4I). The retina distant from the hyperplastic vitreous vasculature appeared morphologically normal (Figs. 4E, 4H, and data not shown), suggesting that retinal dysplasia is a secondary effect caused by the interaction between the hyperplastic vitreous vasculature and the adjacent retina and is not directly caused by the loss of Fzd5 in the retina.
FIGURE 4.
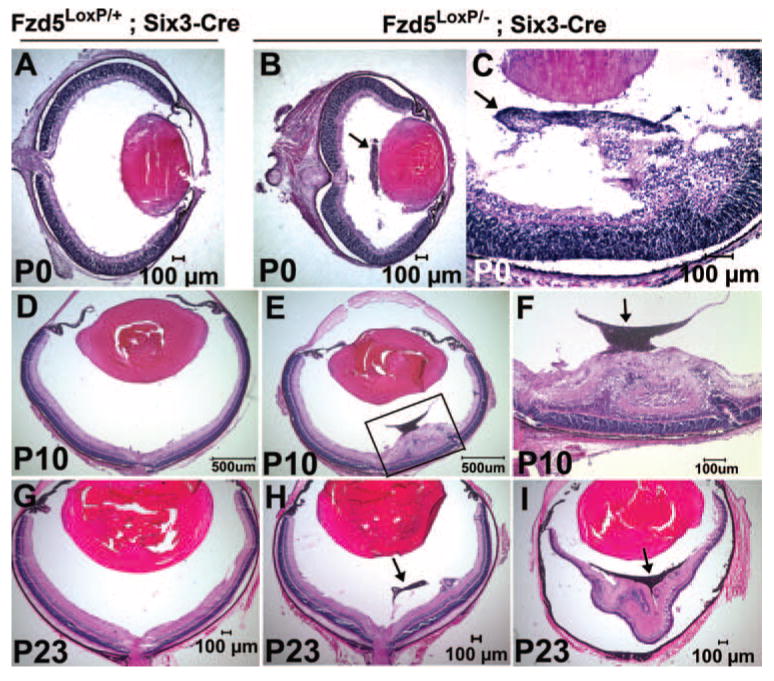
Abnormal retrolental tissue in Fzd5LoxP/–;Six3-Cre mice is persistent at postnatal stages and results in retinal abnormal lamination, folds, and detachment. (A, D, G) Hematoxylin and eosin staining of cryostat sections of Fzd5LoxP/+;Six3-Cre retina at the indicated ages (P0, n = 8; P10, n = 2; P23, n = 3). (B, C, E, H, I) Hematoxylin and eosin staining of Fzd5LoxP/–;Six3-Cre retina at the indicated ages. (P0, n = 7; P10, n = 4; P23, n = 6). (F) Insert in (E) at higher magnification. (B, C, F, H, I, arrows) Retrolental mass tissue.
Disorganization of Ganglion Cell Layer Adjacent to the Hyperplastic Vitreous Vasculature Showing an Increase in Cell Death
Given that morphogenesis of the retina adjacent to the hyperplastic vitreous vasculature was disrupted and the lamination of the ganglion cell layer (GCL) appeared to be most severely affected (Figs. 4C, 4E, 4F, 4H), we further analyzed the morphology of the GCL by performing immunostaining for the ganglion cell marker Brn-3b. The GCL was formed properly in control Fzd5LoxP/+;Six3-Cre mice (n = 2) (Supplementary Fig. S1E). However, in Fzd5LoxP/−;Six3-Cre mice (n = 4), the lamination of the GCL was severely disrupted and mixed with the neuroblast layer (Supplementary Fig. S1F).
The dysplasia of the retina adjacent to the hyperplastic vitreous vasculature suggested that cell survival of the dysplastic retina could be affected; therefore, TUNEL staining was performed at P0 to monitor cell apoptosis. In the retinas of Fzd5LoxP/+;Six3-Cre mice (n = 3), only a few TUNEL-positive cells were observed in the GCL (Figs. 5A–C). However, many TUNEL-positive cells were observed in the disorganized area of the GCL in Fzd5LoxP/−;Six3-Cre mice (n = 3) (Figs. 5D–F), which suggested that cell death was increased in disorganized areas of the retina.
FIGURE 5.
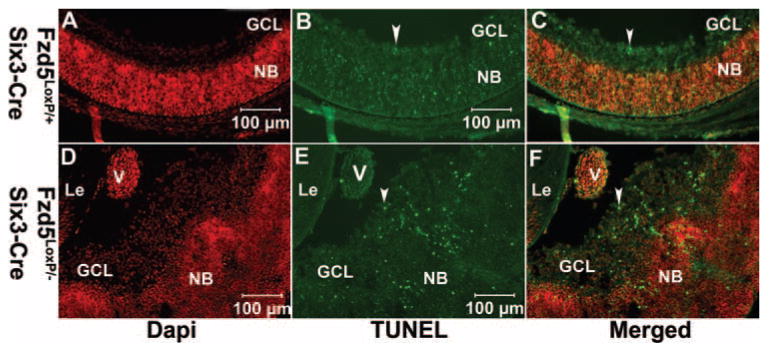
Increased cell death is observed in the disorganized retinal ganglion cell layer adjacent to the hyperplastic vitreous vasculature in Fzd5LoxP/–;Six3-Cre mice. (A– F) TUNEL analysis on cryostat sections of Fzd5LoxP/+;Six3-Cre (n = 3) and Fzd5LoxP/–;Six3-Cre (n = 3) mice eyes at P0. Fluorescence microscope images show DAPI (A, D, red), TUNEL (B, E, green), and the respective merged images (C, F). (B, C, E, F, arrowheads) TUNEL-positive cells. GCL, ganglion cell layer; NB, neuroblasts; V, hyperplastic vasculature.
Abnormal Apoptosis and Proliferation of the Hyperplastic Vitreous Vasculature Associated with Failure of Vascular Regression
The primary vitreous vasculature normally regresses through apoptosis beginning at P4 and peaking at P7 to P8, with all the vasculature regressed by P16.8–10 Inhibition of apoptosis can result in persistence of the vitreous vasculature.16–18 In Fzd5LoxP/–;Six3-Cre mice, the vitreous vasculature was persistent even at P23 (Figs. 4H, 4I). We therefore assessed apoptosis in the vitreous vasculature to determine whether it might be reduced. In control Fzd5LoxP/+;Six3-Cre mice, a few remaining vasculature cells were observed on the posterior surface of the lens and in the vitreous at P10 (Figs. 6A, 6B), and these were TUNEL positive (Figs. 6C, 6D), consistent with previously reported results.8,10 However, no TUNEL labeling was detectable in the hyperplastic vitreous vasculature cells of Fzd5LoxP/–; Six3-Cre mice at P10 (n = 4) and P23 (n = 3; Figs. 6G–I and data not shown). The hyperplastic vitreous vasculature in the Fzd5LoxP/–;Six3-Cre eye failed to regress; we tested the cell proliferation status of these cells by immunostaining for PCNA, which is expressed in all phases of the cell cycle.19 In control mice at P8 and P10, PCNA-positive cells were observed in the inner nuclear layer of the retina but not in the vitreous (Figs. 7A–C and data not shown). In contrast, in Fzd5LoxP/–;Six3-Cre eyes, the hyperplastic vitreous vasculature cells were PCNA positive (P8, n = 2; P10, n = 3 3; Figs. 7D–F and data not shown). Thus, persistence of the vitreous vasculature in Fzd5LoxP/–;Six3-Cre mice may be attributed to the failure of normal apoptosis and to ongoing proliferation.
FIGURE 6.
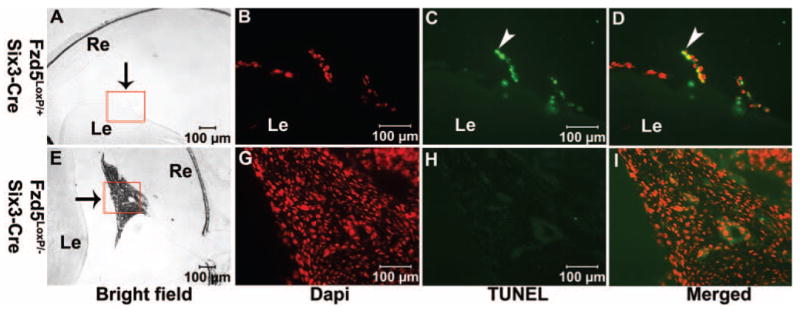
Cell apoptosis in the hyperplastic vitreous vasculature in Fzd5LoxP/–;Six3-Cre mice is inhibited. (A–I) TUNEL staining on cryostat sections of Fzd5LoxP/+;Six3-Cre (A–D; n = 2) and Fzd5LoxP/–;Six3-Cre (E–I; n = 4) mice eyes at age P10. (A, E) Bright-field images. (B–D, G–I) Inserts are shown at higher magnification. (A, E, arrows) Vitreous vasculature. (B, G) DAPI staining (red). (C, H) TUNEL staining (green). (D, I) The respective merged images. (C, D, arrowheads) TUNEL-positive cell.
FIGURE 7.
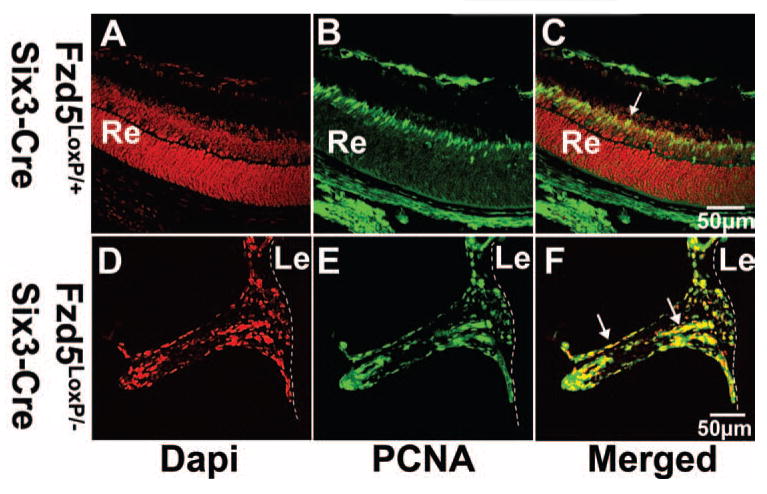
Persistent hyperplastic vitreous vasculature cells are maintained in the cell cycle. (A–C, D–F) Immunofluorescence staining of PCNA on cryostat sections of Fzd5LoxP/+;Six3-Cre (n = 2) and Fzd5LoxP/−;Six3-Cre (n = 2) mice eyes at age P8. (A–C) Apoptosis of vitreous vasculature usually peaks at P7 to P8; therefore, retina rather than vitreous vasculature of Fzd5LoxP/+;Six3-Cre mice was used for positive control of PCNA immunofluorescence staining. Fluorescence microscope images show DAPI (A, D, red), PCNA (B, E, green), and the respective merged images (C, F). (C, F, arrows) PCNA-positive cells. Dashed lines indicate the posterior surface of the lens.
Discussion
Fzd5 is highly expressed in the developing mouse retina from E8.5 to postnatal stages.3,4 We show that selective loss of Fzd5 in the retina affects the normal development of the hyaloid vitreous vasculature and suggest that this occurs through a cell-nonautonomous mechanism. Loss of Fzd5 in the retina causes abnormal accumulation of the hyaloid vitreous vasculature at embryonic stages and persistence of a retrolental mass after birth. Notably, these ocular defects closely resemble those observed in human PHPV.
PHPV is a congenital human eye disease that occurs in infancy and childhood. It is characterized by the presence of pigmented retrolental mass tissue caused by persistent hyperplastic primary vitreous vasculature in the vitreous body. This retrolental mass tissue attaches to the inner neuroretina or the posterior surface of the lens, or both, resulting in various ocular abnormalities, such as retinal folds, retinal detachment, abnormal retinal lamination, and lens destruction.7,12,20 These ocular abnormalities lead to vision loss and even blindness in humans, but the pathogenesis of the disease is unclear.
Several mouse models have shown early hyperplasia and persistence of the HVS. For example, the Arf tumor-suppressor gene is normally expressed in perivascular cells of the HVS from E12.5 through P5 and constrains their proliferation by blocking Pdgfrb-dependent signals.21 Arf mutant mice show abnormal early proliferation, with the appearance of a retrolental mass as early as E14.5 that persists through postnatal stages.11 TGF-β knockout mice also exhibit overgrowth of the vitreous vasculature at embryonic stages because of the loss of lens-derived TGF-β, which normally inhibits the proliferation of vasculature cells.22 In addition, alterations in retinoic acid signaling can affect the migration of periocular mesenchyme cells that contribute to the vitreous vasculature, causing an accumulation of cells in the vitreous.23 In each of these cases affecting cell proliferation or cell migration, HVS hyperplasia occurs at embryonic stages, as in the PHPV phenotype caused by conditional disruption of Fzd5 in the retina.
Subsequently, the HVS normally regresses during the early postnatal period through apoptosis. Disruption of cell death regulators (Bax, Bak) or genes normally required for macrophages to trigger apoptosis of vascular endothelial cells of the HVS prevents vascular regression.8,16–18,24 In some cases, defects in the formation of the retinal vasculature can lead to abnormal persistence of the HVS, presumably to accommodate hypoxic demand from the developing retina. This is observed with a loss of function of Frizzled-4, norrin, VEGF isoform, and collagen XVIII.10,25–27 Although failure of regression was observed in the conditional Fzd5 knockout, this was likely secondary to the early hyperplasia and loss of normal organization of the HVS.
In our mouse model, Fzd5 was specifically removed from the retina but not from the lens or vitreous vasculature tissue. Therefore, the PHPV phenotype observed in Fzd5 conditional knockout mice is likely caused by altered signals from the retina in the absence of Fzd5. Although retinal dysplasia was observed in the retina adjacent to the hyperplastic vitreous vasculature, regions of the retina that were distant from the hyperplastic vitreous vasculature appeared to be morphologically normal. Therefore, the retinal dysplasia phenotype seems to be a secondary defect caused by the hyperplastic vitreous vasculature, which is a common feature of PHPV.
We found that retinal Fzd5 expression is required for preventing abnormal accumulation of the vitreous vasculature, but the mechanism underlying this apparently nonautonomous effect remains unclear. One possibility is that retinal Fzd5 may be involved in regulating the expression of secreted proangiogenesis factors such as VEGF or antiangiogenesis factors such as placental growth factor, which participate in governing the formation, regression, or both of the vitreous vasculature.27–29 A second plausible mechanism is that Fzd5 signaling in the retina may affect the periocular mesenchyme, which is initially formed by cells originating from neural crest and mesoderm30–32 and provides multiple cell lineages that contribute to the vitreous vasculature.32,33 Further studies will shed light on the exact cellular and molecular mechanisms governing vitreous vasculature formation and regression and on how this is altered in the absence of retinal Fzd5.
Acknowledgments
The authors thank Makoto Taketo, Hans Clevers, and Yasuhide Furuta for generously providing the mice carrying a null allele of the Fzd5 gene, the mice with floxed Fzd5 alleles (Fzd5LoxP/LoxP), and Six3-Cre transgenic mice, respectively.
Supported by National Institutes of Health Grant EY14954 (SF, MV) and by an unrestricted grant from Research to Prevent Blindness, Inc., to the Department of Ophthalmology.
Footnotes
Disclosure: J. Zhang, None; S. Fuhrmann, None; M.L. Vetter, None
References
- 1.Cavodeassi F, Carreira-Barbosa F, Young RM, et al. Early stages of zebrafish eye formation require the coordinated activity of Wnt11, Fz5, and the Wnt/beta-catenin pathway. Neuron. 2005;47:43–56. doi: 10.1016/j.neuron.2005.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Raay TJ, Moore KB, Iordanova I, et al. Frizzled 5 signaling governs the neural potential of progenitors in the developing Xenopus retina. Neuron. 2005;46:23–36. doi: 10.1016/j.neuron.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 3.Borello U, Buffa V, Sonnino C, Melchionna R, Vivarelli E, Cossu G. Differential expression of the Wnt putative receptors Frizzled during mouse somitogenesis. Mech Dev. 1999;89:173–177. doi: 10.1016/s0925-4773(99)00205-1. [DOI] [PubMed] [Google Scholar]
- 4.Kemp CR, Willems E, Wawrzak D, et al. Expression of Frizzled5, Frizzled7, and Frizzled10 during early mouse development and interactions with canonical Wnt signaling. Dev Dyn. 2007;236:2011–2019. doi: 10.1002/dvdy.21198. [DOI] [PubMed] [Google Scholar]
- 5.Burns C, Zhang J, Brown E, et al. Investigation of Frizzled-5 during embryonic neural development in mouse. Dev Dyn. 2008;237:1614–1626. doi: 10.1002/dvdy.21565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishikawa T, Tamai Y, Zorn AM, et al. Mouse Wnt receptor gene Fzd5 is essential for yolk sac and placental angiogenesis. Development. 2001;128:25–33. doi: 10.1242/dev.128.1.25. [DOI] [PubMed] [Google Scholar]
- 7.Goldberg MF. Persistent fetal vasculature (PFV): an integrated interpretation of signs and symptoms associated with persistent hyperplastic primary vitreous (PHPV): LIV Edward Jackson Memorial Lecture. Am J Ophthalmol. 1997;124:587–626. doi: 10.1016/s0002-9394(14)70899-2. [DOI] [PubMed] [Google Scholar]
- 8.Reichel MB, Ali RR, D'Esposito F, et al. High frequency of persistent hyperplastic primary vitreous and cataracts in p53-deficient mice. Cell Death Differ. 1998;5:156–162. doi: 10.1038/sj.cdd.4400326. [DOI] [PubMed] [Google Scholar]
- 9.Ito M, Yoshioka M. Regression of the hyaloid vessels and pupillary membrane of the mouse. Anat Embryol. 1999;200:403–411. doi: 10.1007/s004290050289. [DOI] [PubMed] [Google Scholar]
- 10.Fukai N, Eklund L, Marneros AG, et al. Lack of collagen XVIII/endostatin results in eye abnormalities. EMBO J. 2002;21:1535–1544. doi: 10.1093/emboj/21.7.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKeller RN, Fowler JL, Cunningham JJ, et al. The Arf tumor suppressor gene promotes hyaloid vascular regression during mouse eye development. Proc Natl Acad Sci U S A. 2002;99:3848–3853. doi: 10.1073/pnas.052484199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haddad R, Font RL, Reeser F. Persistent hyperplastic primary vitreous: a clinicopathologic study of 62 cases and review of the literature. Surv Ophthalmol. 1978;23:123–134. doi: 10.1016/0039-6257(78)90091-7. [DOI] [PubMed] [Google Scholar]
- 13.van Es JH, Jay P, Gregorieff A, et al. Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat Cell Biol. 2005;7:381–386. doi: 10.1038/ncb1240. [DOI] [PubMed] [Google Scholar]
- 14.Furuta Y, Lagutin O, Hogan BL, Oliver GC. Retina- and ventral forebrain-specific Cre recombinase activity in transgenic mice. Genesis. 2000;26:130–132. [PubMed] [Google Scholar]
- 15.Kanekar S, Perron M, Dorsky R, et al. Xath5 participates in a network of bHLH genes in the developing Xenopus retina. Neuron. 1997;19:981–994. doi: 10.1016/s0896-6273(00)80391-8. [DOI] [PubMed] [Google Scholar]
- 16.Hahn P, Lindsten T, Tolentino M, Thompson CB, Bennett J, Du-naief JL. Persistent fetal ocular vasculature in mice deficient in bax and bak. Arch Ophthalmol. 2005;123:797–802. doi: 10.1001/archopht.123.6.797. [DOI] [PubMed] [Google Scholar]
- 17.Lang RA, Bishop JM. Macrophages are required for cell death and tissue remodeling in the developing mouse eye. Cell. 1993;74:453–462. doi: 10.1016/0092-8674(93)80047-i. [DOI] [PubMed] [Google Scholar]
- 18.Lobov IB, Rao S, Carroll TJ, et al. WNT7b mediates macrophage-induced programmed cell death in patterning of the vasculature. Nature. 2005;437:417–421. doi: 10.1038/nature03928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barton KM, Levine EM. Expression patterns and cell cycle profiles of PCNA, MCM6, cyclin D1, cyclin A2, cyclin B1, and phosphorylated histone H3 in the developing mouse retina. Dev Dyn. 2008;237:672–682. doi: 10.1002/dvdy.21449. [DOI] [PubMed] [Google Scholar]
- 20.Pollard ZF. Persistent hyperplastic primary vitreous: diagnosis, treatment and results. Trans Am Ophthalmol Soc. 1997;95:487–549. [PMC free article] [PubMed] [Google Scholar]
- 21.Thornton JD, Swanson DJ, Mary MN, et al. Persistent hyperplastic primary vitreous due to somatic mosaic deletion of the arf tumor suppressor. Invest Ophthalmol Vis Sci. 2007;48:491–499. doi: 10.1167/iovs.06-0765. [DOI] [PubMed] [Google Scholar]
- 22.Saika S, Saika S, Liu CY, et al. TGFβ2 in corneal morphogenesis during mouse embryonic development. Dev Biol. 2001;240:419–432. doi: 10.1006/dbio.2001.0480. [DOI] [PubMed] [Google Scholar]
- 23.Ozeki H, Shirai S, Ikeda K, Ogura Y. Critical period for retinoic acid-induced developmental abnormalities of the vitreous in mouse fetuses. Exp Eye Res. 1999;68:223–228. doi: 10.1006/exer.1998.0591. [DOI] [PubMed] [Google Scholar]
- 24.Kato M, Patel MS, Levasseur R, et al. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol. 2002;157:303–314. doi: 10.1083/jcb.200201089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Q, Wang Y, Dabdoub A, et al. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116:883–895. doi: 10.1016/s0092-8674(04)00216-8. [DOI] [PubMed] [Google Scholar]
- 26.Richter M, Gottanka J, May CA, Welge-Lussen U, Berger W, Lutjen-Drecoll E. Retinal vasculature changes in Norrie disease mice. Invest Ophthalmol Vis Sci. 1998;39:2450–2457. [PubMed] [Google Scholar]
- 27.Stalmans I, Ng YS, Rohan R, et al. Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J Clin Invest. 2002;109:327–336. doi: 10.1172/JCI14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rutland CS, Mitchell CA, Nasir M, Konerding MA, Drexler HC. Microphthalmia, persistent hyperplastic hyaloid vasculature and lens anomalies following overexpression of VEGF-A188 from the alphaA-crystallin promoter. Mol Vis. 2007;13:47–56. [PMC free article] [PubMed] [Google Scholar]
- 29.Feeney SA, Simpson DA, Gardiner TA, Boyle C, Jamison P, Stitt AW. Role of vascular endothelial growth factor and placental growth factors during retinal vascular development and hyaloid regression. Invest Ophthalmol Vis Sci. 2003;44:839–847. doi: 10.1167/iovs.02-0040. [DOI] [PubMed] [Google Scholar]
- 30.Johnston MC, Noden DM, Hazelton RD, Coulombre JL, Coulombre AJ. Origins of avian ocular and periocular tissues. Exp Eye Res. 1979;29:27–43. doi: 10.1016/0014-4835(79)90164-7. [DOI] [PubMed] [Google Scholar]
- 31.Le Lievre CS, Le Douarin NM. Mesenchymal derivatives of the neural crest: analysis of chimaeric quail and chick embryos. J Embryol Exp Morphol. 1975;34:125–154. [PubMed] [Google Scholar]
- 32.Gage PJ, Rhoades W, Prucka SK, Hjalt T. Fate maps of neural crest and mesoderm in the mammalian eye. Invest Ophthalmol Vis Sci. 2005;46:4200–4208. doi: 10.1167/iovs.05-0691. [DOI] [PubMed] [Google Scholar]
- 33.Kanakubo S, Nomura T, Yamamura K, Miyazaki J, Tamai M, Osumi N. Abnormal migration and distribution of neural crest cells in Pax6 heterozygous mutant eye, a model for human eye diseases. Genes Cells. 2006;11:919–933. doi: 10.1111/j.1365-2443.2006.00992.x. [DOI] [PubMed] [Google Scholar]