

Abstract
The tumor suppression function of p53 is mostly conferred by its transactivation activity, which is inactivated by p53 mutations in ~50% of human cancers. In cancers harboring wild type p53, the p53 transactivation activity may be compromised by other mechanisms. Identifying the mechanisms by which wild type p53 transactivation activity can be abrogated may provide insights into the molecular etiology of cancers harboring wild type p53. In this report, we show that BCCIP, a BRCA2 and CDKN1A-interacting protein, is required for the transactivation activity of wild type p53. In p53 wild type cells, BCCIP knock down by RNA interference diminishes the transactivation activity of p53 without reducing the p53 protein level, inhibits the binding of p53 to the promoters of p53 target genes p21 and HDM2, and reduces the tetrameric formation of p53. These data demonstrate a critical role of BCCIP in maintaining the transactivation activity of wild type p53 and further suggest down-regulation of BCCIP as a novel mechanism to impair the p53 function in cells harboring wild type p53.
The tumor suppressor gene p53 is a transcription factor that can be activated by a variety of stress signals, including DNA damage (1–3). Upon activation, p53 forms a homotetramer that binds to specific DNA sequences in the promoter of p53-regulated genes (4–6), which leads to the transcription activation of these target genes. These p53 target genes subsequently regulate cell cycle progression, cell death, DNA repair, and DNA replication to maintain genomic stability and to prevent tumorigenesis.
Mutations in p53 are found in ~50% of all human cancers, and inactivation of p53 leads to cancer predisposition in animal models (7). A key element for the tumor suppressor function of p53 is its transactivation activity (5, 6). Cancer-bearing p53 mutations are often defective in its transcription activity (5, 8, 9), and mice expressing transactivation-deficient p53 are pre-disposed to cancer (5, 9). In cancers harboring wild type p53, the p53 tumor suppression activity may be circumvented by other genetic alternations that impair the transcription activity. For example, overexpression of mouse double minute 2 gene (MDM2),2 or its human homologue (HMD2), promotes the degradation of wild type p53, thus inhibiting the transcription activity of p53 (10). In some cancer types, such as breast cancer, p53 mutation is detected in only ~20% of the total cases. Therefore, identification of alternative mechanisms by which p53 transactivation function is impaired may provide further insights into the molecular etiology of the human cancers harboring wild type p53.
BCCIPα is a BRCA2 and CDKN1A (p21, Cip1, and Waf1)-interacting protein, which has also been named Tok-1α (11, 12). A second isoform, BCCIPβ, sharing an N-terminal acidic domain and a central conserved domain but with a distinct C-terminal domain, also interacts with BRCA2 and p21 (13, 14). Previous studies have suggested a functional role of BCCIP in homologous recombinational repair and G1/S cell cycle control (12–15). Expression of BCCIP is down-regulated in kidney cancer (16). More than 70% of astrocytic brain tumors lack BCCIP expression, and the lack of BCCIP expression is correlated with the aggressiveness of astrocytic brain tumors.3 In this report, we found that down-regulation of BCCIP overrides the transcriptional activity of wild type p53, suggesting a novel mechanism by which the p53 transactivation function can be compromised. It has significant implication for the understanding of tumorigenesis in cancers harboring wild type p53.
EXPERIMENTAL PROCEDURES
Cell Culture
HCT116 and COS7 cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen) with 10% fetal bovine serum (Hyclone, Logan, UT), 20 mM glutamine, and 1% penicillin/streptomycin (Invitrogen). Plasmids were transfected into cells using the Geneporter transfection kit (Gene Therapy Systems Inc., San Diego, CA).
Antibodies and Western Blot
Rabbit anti-BCCIPα/β antibodies were reported previously (11). Anti-HDM2, anti-GST, and anti-p53 (No. 1801) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-p53 (Ab-6) was purchased from Calbiochem (La Jolla, CA), and anti-β-actin antibody was from Sigma. Protein extracts were prepared from cells lysed with 50 mM HEPES, pH 7.6, 250 mM NaCl, 5 mM EDTA, 0.1% Nonidet P-40. Proteins were separated by SDS-PAGE, and Western blotting was performed as described (11).
Knock Down of BCCIP Expression by Short Hairpin RNA (shRNA)
Two vectors were used to express shRNA to knock down BCCIP expression. The plasmid pPUR/U6 (17) uses a puromycin resistance cassette, while the pSilencer2.1Hyg (Ambion Inc., Austin, TX) uses hygromycin B as the selection marker. The control vectors express either a scrambled sequence 5′-ACT ACC GTT GTT ATA GGT G-3′ (Ambion Inc.) in pSilencer2.1Hygomycin or a green fluorescent protein cDNA sequence 5′-GGT TAT GTA CAG GAA CGC A-3′ in pPUR/U6. To knock down both BCCIPα and BCCIPβ isoforms, several shRNA sequences targeted at the shared region of BCCIPα and BCCIPβ were used, including shRNA-αβ311 (5′-GTG TGA TTA AGC AAA CGG ATG-3′), shRNA-αβ633 (5′-GCC ATG TGG GAA GTG CTA C-3′), and shRNA-αβ730 (5′-GCT GCG TTA ATG TTT GCA AAT-3′). We found that application of a single shRNA can only cause ~50% down-regulation of BCCIP. To further knock down BCCIPα/β, we combined two shRNAs, αβ311 and αβ633, or αβ311 and αβ730, creating ~95% down-regulation of BCCIP. This is feasible because pPUR/U6 and pSilencer use different selection markers, puromycin and hygromycin B, respectively.
Radiation Survival Assays
The radiation sensitivities of HCT116 and HCT116 cells with BCCIP down-regulations were determined by colony formation assays. The numbers of cells to be plated for each radiation dosage group were determined by a pilot experiment in order to yield 50–150 surviving colonies/100-mm plate. Cells in each dosage group were plated in triplicate. After the appropriate number of cells were plated for 18 h, cells were irradiated with Cs-137 γ-rays (dose rate, 0.893 Gy/min), and colonies were grown for 12–14 days after which the colonies were fixed with methanol and stained with 1% crystal violet. The number of colonies was normalized to the number of cells plated to calculate the survival fraction. The experiments were repeated at least twice.
Luciferase Reporter Gene Assay
A p53 reporter plasmid p53-Luc was purchased from Stratagene (La Jolla, CA). This reporter plasmid p53-Luc confers p53-dependent expression of luciferase. MG15-luc (kindly provided by Dr. Bert Vogelstein) was a negative control with the mutation at p53 binding sites (18). WWP-luc (kindly provided by Dr. Bert Vogelstein) was a reporter gene with 2.4-kb p21WAF1 promoter region (19).
Chromatin Immunoprecipitation Assay to Detect the Binding of p53 with p21 and HDM2 Promoters
Chromatin immunoprecipitation assays were performed with the Chromatin Immunoprecipitation Assay kit (Upstate Biotechnology, Lake Placid, NY) according to the manufacturer’s instructions. Briefly, after cells were cross-linked and their chromatin sheared, extract from an equal number of cells was subjected to immunoprecipitation with agarose-conjugated monoclonal anti-p53 antibodies or agarose-conjugated control mouse IgG. The amounts of input and precipitated p53 in each sample were detected by immunoblotting with an anti-p53 polyclonal anti-body. After the chromatin immunoprecipitation assay, the precipitated p53-bound p21 or HDM2 promoter DNAs were detected by PCR and quantified with the DNA Engine Opticon™ 2 Real-time Detection System (M J Research Inc., Reno, NV). The primers for the p21 promoter were 5′-TCC ACC TTT CAC CAT TCC CCT ACC C-3′ (forward) and 5′-CAG CCC AAG GAC AAA ATA GCC ACC A-3′ (reverse). The primers for HDM2 promoter were 5′-CGG GAG GTC CGG ATG ATC GCA GG-3′ (forward) and 5′-GTC GGT GCT TAC CTG GAT CAG C-3′ (reverse).
Binding of p53 with Its Consensus Binding Sequence Detected by a Biotin-Streptavidin Pulldown Assay
One palindrome oligonucleotide, containing biotin on the nucleotide at the 5′-position, was used in the pulldown assays. The sequence of the oligonucleotide was 5′-biotin-GGA CAT GCC CGG GCA TGT CC-3′. The sequence has been reported to be the strongest p53 binding consensus (20). Nuclear extracts were prepared as follows. Briefly, the cells were suspended in three packed cell volumes of hypotonic buffer (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol with protease inhibitors) and allowed to swell for 10 min on ice. The cells were homogenized and centrifuged for 30 min at 10,000 × g. The released nuclei were suspended in half the packed cell volume of low salt buffer (20 mM HEPES, pH 7.9, 20 mM KCl, 1.5 mM MgCl2, 0.1 mM EDTA, 25% glycerol, 0.2 mM dithiothreitol, and the mixture of protease inhibitors), followed by the dropwise addition of high salt buffer (20 mM HEPES, pH 7.9, 0.6 M KCl, 1.5 mM MgCl2, 25% glycerol, 0.2 mM dithiothreitol, and the mixture of protease inhibitors). The nuclear suspensions were extracted for 30 min at 4 °C with gentle agitation and centrifuged for 30 min at 14,000 × g. The supernatants (nuclear extracts) were stored at −80 °C in aliquots. One microgram of each double-stranded oligonucleotide was incubated with 300 μg of nuclear protein for 20 min at room temperature in binding buffer containing 12% glycerol, 12 mM HEPES, pH 7.9, 4 mM Tris. pH 7.9, 150 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, and 10 μg of poly(dI-dC) competitor. Following the incubation, 30 μl of streptavidin-agarose (Pierce Biotechnology) was added to the reaction and incubated at 4 °C for 4 h. Prior to this step, 300 μl of the original streptavidin-agarose bead preparation was preabsorbed with 500 μl of bovine serum albumin, 50 μg of poly(dI-dC), and 50 μg of sheared salmon sperm DNA for 30 min at 25 °C. The streptavidin-agarose beads were washed three times and resuspended in 300 μl of the binding buffer. The protein-DNA-streptavidin-agarose complex was washed three times with binding buffer and loaded onto an SDS gel. The bound proteins were separated on a 10% SDS-PAGE and then Western blotted with p53 antibody.
p53 Tetramer Formation
To test whether BCCIP promotes p53 tetramer formation in vitro, 1 μg of recombinant p53 was incubated for 30 min with 1 μg of recombinant BCCIP (50 mM Tris/HCl, pH 7.5, 50 mM NaCl, 4 mM EDTA, 100 ng/μl bovine serum albumin, 20% (v/v) glycerol) followed by addition of 10 mM diamide (Sigma) to cross-link interacting proteins as described previously by others (21). After 20 min, the incubation reaction mixtures were separated in a SDS-polyacrylamide gel (10%) and transferred to a nitrocellulose membrane. Immunodetection of p53 was performed by using p53 Ab6 antibody.
RESULTS
Lack of p21 and HDM2 Expressions in BCCIP Knockdown Cells
Our previous work showed that overexpression of either BCCIPα or BCCIPβ increases p21 level and partial knock down of BCCIPα or/and BCCIPβ by RNA interference reduces p21 mRNA levels (15). Furthermore, we showed that the induction of p21 by BCCIP overexpression is dependent on p53 and that the p53 transactivation activity is enhanced by BCCIP overexpression (15). To further identify the mechanism by which BCCIP regulates p21 expression, BCCIP expression was reduced by expression of shRNA targeted at several common regions of BCCIPα and BCCIPβ (Fig. 1A). Because a single shRNA can only reduce the level of BCCIP by ~50%, we applied two independent BCCIP shRNAs in HCT116 cells, BCCIP shRNA-αβ633 and shRNA-αβ311 (see “Experimental Procedures” for details). This resulted in >95% down-regulation of BCCIP. The cells were exposed to 3 or 10 Gy of γ-irradiation. At 1, 6, and 16 h after the irradiation, we observed down-regulation of p21 in BCCIP knockdown cells (Fig. 1B). A previous report has shown that partial knock down of BCCIP also results in reduced mRNA level of p21 (15). The expression of another p53 target gene, HDM2, is also inhibited in BCCIP knockdown cells. It is critical to point out that BCCIP knock down resulted in the down-regulation of p21 and HDM2 but p53 protein level was not reduced (Fig. 1B), suggesting that the transcription activity of p53 in BCCIP knockdown cells is impaired.
FIGURE 1. Reduced expressions of p21 and HDM2 in BCCIP knockdown cells.
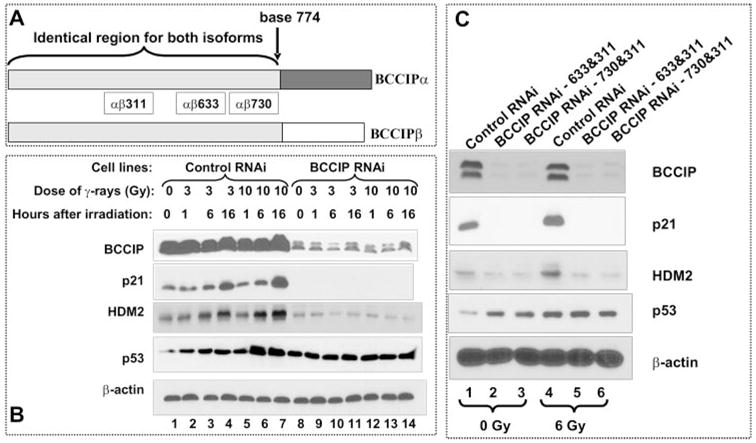
A, BCCIP isoforms and the common regions between BCCIPα and BCCIPβ targeted by RNA interference. B, HCT116 cells were transfected with vectors expressing shRNA-αβ311 and shRNA-αβ633. The control cells were transfected with pPUR/U6/GFPshRNA and pSilencer2.1Hyg Neg. After the cells were exposed to 3 or 10 Gy of γ-irradiation, the BCCIP, p21, HDM2, and p53 proteins levels were detected by Western blot at various times (1, 6, and 16 h) after the irradiation. Shown are the expressions of p53, p21, and HDM2 in control (lanes 1–7) and BCCIP knockdown cells (lanes 8 –14). C, HCT116 cells were transfected with vectors expressing shRNA-αβ311/shRNAαβ633 (lanes 2 and 5) and shRNA-αβ311/shRNA-αβ730 (lanes 3 and 6). Six hours after 6 Gy of irradiation, the levels of BCCIP, p21, HDM2, and p53 were detected by Western blot. β-actin was detected as loading control.
To confirm this, combination of BCCIP shRNA-αβ730 and shRNA-αβ311 was used to down-regulate BCCIP (Fig. 1C). Again in these BCCIP knockdown cells, we observed significant down-regulation of p21 and HDM2 proteins (Fig. 1C), although p53 expression increased. These data strongly suggest that, in the absence of BCCIP, the expression of p53 downstream target genes p21 and HDM2 is severely impaired, suggesting an abrogation of p53 transcription activity in BCCIP knockdown cells. In this report, we focus on the mechanism by which BCCIP down-regulation abolishes p21 and HDM2 expression. The mechanism by which BCCIP knock down induces p53 is not the focus of this report but will be discussed (see “Discussion”).
Inhibition of p53 Transactivation Activity in BCCIP Knockdown Cells
Because the expression of p21 and HDM2 is regulated by p53, we hypothesized that the transactivation activity of p53 is impaired in the BCCIP knockdown cells. To test this, a plasmid (p53-luc) containing 13 copies of the p53 binding consensus sequence upstream of a luciferase reporter gene was transfected into cells. As shown in Fig. 2A, the reporter activity was induced by γ-irradiation in control cells. However, the p53 reporter activity was significantly reduced in BCCIP knockdown cells, suggesting a lack of p53 protein transcription activity. To confirm this, the expression of another luciferase reporter (WWP-Luc) that is regulated by a 2.4-kb promoter DNA sequence of the p21 gene was transfected into the HCT116 cells. As shown in Fig. 2B, the p21 promoter activity was also severely inhibited in the absence of BCCIP. The activity of a control reporter (pGal4-Luc) that is driven by the Gal4 promoter and independent of p53 was not altered in BCCIP knockdown cells (data not shown). These data strongly suggest that the p53 transactivation activity is impaired in BCCIP knockdown cells, even though p53 protein level is increased (Fig. 1, B and C).
FIGURE 2. Inhibition of p53 transactivation activity by BCCIP knock down.

The p53-luc reporter plasmid contains 13 copies of the p53 binding consensus sequence upstream of the luciferase gene. The p21 promoter reporter plasmid (WWP-Luc) contains 2.4 kb of p21 promoter sequences. These reporter plasmids were individually transfected into control and two BCCIP knockdown HCT116 cells. Forty-two hours after transfection, cells were irradiated with 3 Gy of γ-irradiation. Luciferase activities were measured 6 h after irradiation. Panel A shows the transactivation activity of p53 toward the p53-luc reporter, and panel B shows the transcription activity of p53 toward the p21 promoter. As a control, GAL4-luc, a luciferase reporter driven by GAL4-responsive elements, was not inhibited in BCCIP knockdown cells (data not shown).
Defective p53 Binding with Its Targeting Promoter Sequences in BCCIP Knockdown Cells
p53 transactivates its downstream target genes by binding with their promoter sequences (19). To further understand the mechanism by which BCCIP knock down inhibits the p53 transactivation activity, we hypothesized that the binding of p53 to its target DNA sequences is hindered in BCCIP knockdown cells. To test this, we used chromatin immunoprecipitation assay to measure the binding of p53 to the p21 and HDM2 promoters. After the p53 protein was precipitated, the p53-associated p21 and HDM2 promoter DNAs were detected by PCR. As shown in Fig. 3A, the binding of p53 with p21 and HDM2 promoter DNA was enhanced after γ-irradiation in control cells. However, the amounts of p53-co-precipitated p21 and HDM2 promoter DNA were significantly reduced in BCCIP knockdown cells (Fig. 3A), although no less amount of p53 proteins were precipitated in BCCIP knockdown cells than the control (Fig. 3B). Non-specific IgG did not precipitate p53 protein nor did p21 and HDM2 promoters (data not shown). Based on real-time PCR quantification, the binding of p53 with p21 and HDM2 promoters was reduced by 4–5-folds in irradiated BCCIP knockdown cells (Fig. 3, C and D).
FIGURE 3. Reduced binding of p53 with p21 and HDM2 promoters in BCCIP knockdown cells.

Six hours after irradiation (IR) of control and BCCIP knockdown cells, chromosome immunoprecipitation assay was performed (see “Experimental Procedures”). Panel A shows the amount of p21 and HDM2 promoter DNA co-precipitated with p53 as detected by PCR. Panel B shows the amount of input and precipitated p53 proteins in the chromatin immunoprecipitation assay. Panels C and D show the relative amounts of p21 (C) and HDM2 (D) promoters as quantified by real-time PCR. Panel E shows a reduced binding of p53 with its binding consensus DNA in BCCIP knockdown cells. Six hours after the control and BCCIP knockdown cells were irradiated, nuclear protein extracts were incubated with streptavidin-agarose and biotin-conjugated double-stranded oligonucleotides of the p53 binding consensus sequence. After washing, the agarose-bound proteins were separated on a 10% SDS-PAGE and then detected by Western blot with p53 antibody.
To confirm that the wild type p53 failed to bind with the consensus sequence in BCCIP knockdown cells, biotin-labeled p53 consensus (20) double-stranded DNA was incubated with cell extracts. The cellular proteins bound with these sequences were precipitated by streptavidin resins and detected by anti-p53 blot (see “Experimental Procedures” for details). As shown in Fig. 3E, binding of p53 protein with this DNA sequence is increased by irradiation in control cells but significantly less p53 protein was co-precipitated with the consensus p53 binding DNA in BCCIP knockdown cells data confirm an inhibition of p53 binding with its target sequence in BCCIP knockdown cells, suggesting that the reduction of p53 transcription activity in BCCIP knockdown due to a role of BCCIP in regulating p53 binding to DNA sequences. However, BCCIP appears unable to bind with p21 or HDM2 promoter DNA (data not shown).
Down-regulation of BCCIP in Wild Type p53 Cells Radiation Resistance
Lack of p53 transactivation associated with radiation resistance and cell cycle defects. We have previously shown that BCCIP down-regulation abrogates DNA damage-induced G1/S checkpoint activation in p53 wild type HT1080 cells (14). To that BCCIP defect confers radiation resistance, the expression in HCT116 was knocked down by BCCIP-and radiation sensitivities were measured by colony formation assay (see “Experimental Procedures”). As shown 4, down-regulation of BCCIP in HCT116 cells confers radiation resistance to the same level as that of HCT116(p53–) cells. These data, in combination with previously results (14), further support a role of BCCIP in p53-related functions.
BCCIP Promotes p53 Tetramer Formation
Having shown that BCCIP is required for the binding of p53 with the promoters of its target genes, we further investigated the potential mechanisms underlining this observation. Several factors, including Ser-15 phosphorylation in the N-terminal transactivation domain (23) and p53 acetylation in the p53 C-terminal domain (24), have been shown to regulate p53 transactivation activity. However, we did not find significant alterations of Ser-15 phosphorylation or p53 acetylation in BCCIP knockdown cells (data not shown). In addition, p53 remains in the nucleus in BCCIP knockdown cells (data not shown). Because it is the tetrameric form of p53 that binds with its target DNA sequences (4), we tested whether BCCIP is required for the formation of tetrameric p53. After incubation of recombinant p53 protein with purified BCCIPα and BCCIPβ, the protein solution was cross-linked by diamide (21) and analyzed by SDS-PAGE. As shown in Fig. 5, incubation of p53 with either BCCIPα or BCCIPβ significantly increases the formation of tetrameric p53 protein. Therefore, we suggest that BCCIP is required for the formation of p53 tetramer, which is the transcriptionally active conformation of p53. These data suggest that BCCIP may promote p53 transcription activity by facilitating the formation of p53 tetramers, which then bind to promoter DNA sequences to activate target gene transcription. Although BCCIP is required for the p53 tetramer formation, we did not observe a cross-link between p53 and BCCIP (Fig. 5), suggesting that BCCIP may promote p53 tetramer formation without a stable interaction between them.
FIGURE 5. Promotion of p53 tetramer formation by BCCIP.

Purified p53 (1μg) was incubated with an equal amount of BCCIPα or BCCIPβ, cross-linked with diamide, and analyzed with anti-p53 Western blot. Indicated are the monomeric and tetrameric forms of p53.
BCCIP Weakly Interacts with the p53
We next addressed the potential mechanisms by which BCCIP may regulate p53 tetramerization. Because acetylation and phosphorylation of p53 (modification that may regulate p53 transcription activity) were not altered in BCCIP knockdown cells (data not shown), we focused on whether BCCIP may interact with p53, although we anticipated that this interaction, if any, would be transient or weak. Recombinant GST-tagged BCCIPα, BCCIPβ, and GST (negative control) were incubated with His-tagged p53 recombinant protein. We found that pull down of GST-BCCIPα and GST-BCCIPβ fusion protein (but not GST alone) with glutathione beads co-precipitates with p53 (Fig. 6A), suggesting a direct interaction between BCCIP and p53. We also expressed Myc-tagged p53 in HCT116 (p53−/−) cells (Myc-UBC9 was transfected as a control). After the Myc-p53 and Myc-UBC9 were precipitated by anti-Myc resin, we detected a small portion of endogenous BCCIPα and BCCIPβ co-precipitated with p53 (Fig. 6B). In addition, when Myc-p53 and Myc-UBC9 were co-expressed with FLAG-tagged BCCIPα and BCCIPβ, we found that a small amount of p53 was co-precipitated with FLAG-BCCIP, but not UBC9 (Fig. 6C). These data suggest an interaction between the full-length p53 and BCCIP. As shown in Fig. 5, diamide, which cross-links protein by forming a disulfate link, did not cause cross-link between p53 and BCCIP (Fig. 5), and we were not able to precipitate endogenous p53 with BCCIP (data not shown). Thus, the interaction between full-length p53 and BCCIP is likely to be transient or weak.
FIGURE 6. Interaction between BCCIP and p53.

A, in vitro binding between p53 and BCCIP. 1 μg of purified GST, GST-BCCIPα, or GST-BCCIPβ was incubated with 1 μg of His-p53. After precipitation with glutathione-agarose, the bound proteins were resolved in SDS-PAGE and stained. Shown are the amounts of precipitated GST, GST-BCCIPα, GST-BCCIPβ, and His-p53 proteins. B, co-precipitation of endogenous BCCIP with Myc-p53. Myc-p53 protein was expressed in HCT116 (p53−/−) cells and precipitated with anti-Myc antibodies. Myc-UBC9 was used as a control. The co-precipitated BCCIP was detected by Western blot (top panel). p53-negative HCT116 cells were used to avoid potential interference of untagged p53 on the co-precipitation. C, co-precipitation of p53 with FLAG-BCCIP. Myc-p53 and FLAG-BCCIP were co-expressed in COS7 cells. Myc-UBC9 was used as a negative control. The protein extracts were precipitated with mouse anti-FLAG beads. Precipitated proteins were detected by rabbit anti-Myc (top panel) and anti-FLAG (bottom panel).
DISCUSSION
In this report, we show that BCCIP weakly associates with p53. Without BCCIP, wild type p53 fails to form tetramers (Fig. 5), cannot bind with its target promoter sequences (Fig. 3), and is defective in transactivation function (Fig. 2). Furthermore, down-regulation of BCCIP confers resistance of HCT116 cells to radiation damage to the same level as p53 defect (Fig. 4). Thus, BCCIP defects override the wild type p53 transactivation activity, suggesting a critical role of BCCIP in maintaining the wild type p53 transactivation function and p53 tumor suppression activity.
FIGURE 4. BCCIP knock down confers resistance to radiation damage.

The radiation survival curves of isogenic HCT116 cells with different p53 and BCCIP backgrounds are shown (for details see “Results”).
It has been reported that a reductase, Ref1/APE1, transiently interacts with p53 and is required for p53 tetramer formation (21). Similar to APE1/Ref1 and p53 interaction, the interaction between BCCIP and p53 is likely transient and weak, as diamide cannot cross-link purified APE1 with p53 in vitro yet it promotes the tetramer formation (21). The function of Ref1/APE in promoting p53 activity is dependent on the APE1 reductase activity mediated by critical cysteine residues (21). We also tested whether BCCIP may act as a reductase to promote p53 tetramer formation. Mutation of cysteine residues 153, 157, 213, and 216 in the common region between BCCIPα and BCCIPβ did not affect the role of BCCIP in promoting p53 tetramerization (data not shown). Therefore, it is likely that BCCIP may regulate p53 tetramer formation by a distinct mechanism, such as by serving as a p53 chaperone. This prediction is subject to further investigation.
Although our data suggest that BCCIP may regulate p53 transcription activity by promoting tetramer formation, the potential contribution of alternative mechanisms cannot be ruled out. First, Gottifredi et al. (25) have shown that replication blockage by hydroxyurea results in binding of p53 to the stalled replication sites, which sequesters p53 away from the promoter, thus limiting the transcription activity of p53 (25, 26). When there are a large number of stalled replication forks, p53 binding to stalled replication forks may lead to down-regulation of its transcription activity (25, 26). Because BCCIP is involved in homologous recombinational repair (13), it is possible that the BCCIP defect produces an excessive level of stalled replication forks that then sequester p53 away from the promoters. Second, a nonspecific DNA binding domain is located in the p53 C terminus (27). According to one model, this C-terminal domain may bind to DNA non-specifically and then slide along the linear DNA to locate the specific binding motif (28). It is possible that the interaction of BCCIP with the p53 affects the initiation of the nonspecific DNA binding of p53, which affects the localization of p53 to the consensus binding motif. Third, it has been reported that overexpression of BRCA2 inhibits p53 transcription activity (22). Because BCCIP interacts with BRCA2, it is possible that BCCIP may regulate p53 transcription activity via BRCA2. In addition, although our data suggest that lack of p53 transcription activity significantly contributes to the down-regulation of p21, we cannot exclude that alternative mechanisms may also contribute to the down-regulation of p21 in BCCIP knockdown cells, as BCCIP directly interacts with p21 and basal level of p21 expression can still be observed in some-p53 deficient cells.
One consistent observation is that down-regulation of BCCIP increases p53 protein level, although this increased p53 does not confer enhanced transactivation activity due to the failure of p53 to bind with target promoters. Although the mechanism by which BCCIP knock down increases p53 is not the focus of this report and remains to be determined in the future, two explanations are plausible. First, because of the lack of p53 transactivation activity, HDM2 expression is significantly inhibited. This may provide a feedback mechanism to increase p53 protein level. Second, it has been reported that defect of BRCA2 results in elevated p53 protein level. Because BCCIP interacts with BRCA2 and BCCIP is involved in recombinational repair (13), it is possible that lack of BCCIP results in accumulation of endogenous DNA damage, which in turn may stabilize the p53 protein. Nevertheless, the elevated p53 protein level in BCCIP knockdown cells does not confer transactivation activity of p53 as demonstrated in our report.
A previous report has shown a role of BCCIP in homologous recombinational DNA repair (13), which implies that down-regulation of BCCIP would sensitize cells to radiation. On the other hand, defective BCCIP abrogates the p53 function and thus may confer resistance to DNA damage. As shown in Fig. 4, the overall outcome of these apparently opposite effects is radiation resistance in HCT116 cells. This has potential implications in tumor progression and radiation therapy: the reduced homologous recombinational repair combined with resistance to DNA damage would promote further genomic instability and tumor progression, while the resistance to radiation in BCCIP down-regulated cells may imply a poor outcome of radiation therapy for cancers with defective BCCIP but wild type p53.
We have previously reported that expression of BCCIP is down-regulated in kidney cancer (16). The expression of BCCIP is absent in >70% of astrocytic brain tumor, and the lack of BCCIP expression is correlated with the aggressiveness of astrocytic brain tumors,3 suggesting an etiological role of BCCIP in cancer. Data shown in this report have demonstrated that the transcriptional activity of wild type p53 can be compromised due to BCCIP down-regulation. Thus, BCCIP defect may contribute to tumorigenesis by overruling the p53 transactivation activity in cancers harboring wild type p53.
Acknowledgments
We thank Jingmei Liu (UMDNJ) for critical reading of the manuscript and Dr. Bert Vogelstein (The Johns Hopkins University) for providing the HCT116 cells and p53 target gene expression reporters.
Footnotes
This work was supported by National Institutes of Health Grants ES08353 and CA115488 and by U. S. Army Medical Research and Materiel Command Grants DAMD17-02-1-0515 and DAMD17-03-1-0317. The costs of publication of this article were defrayed in part by the payment of page charges.
The abbreviations used are: MDM2, mouse double minute 2 gene; HDM2, human homologue of MDM2; BCCIP, BRCA2 and CDKN1A-interacting protein; GST, glutathione S-transferase; shRNA, short hairpin RNA; Gy, gray.
J. Liu, H. Lu, H. Ohgaki, A. Merlo, and Z. Shen, manuscript in preparation.
References
- 1.Vousden KH, Lu X. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 2.Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW. Science. 2000;287:1824–1827. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- 3.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- 4.Friedman PN, Chen X, Bargonetti J, Prives C. Proc Natl Acad Sci U S A. 1993;90:3319–3323. doi: 10.1073/pnas.90.8.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jimenez GS, Nister M, Stommel JM, Beeche M, Barcarse EA, Zhang XQ, O’Gorman S, Wahl GM. Nat Genet. 2000;26:37–43. doi: 10.1038/79152. [DOI] [PubMed] [Google Scholar]
- 6.Vogelstein B, Lane D, Levine AJ. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 7.Hollstein M, Sidransky D, Vogelstein B, Harris CC. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 8.Hollstein M, Rice K, Greenblatt MS, Soussi T, Fuchs R, Sorlie T, Hovig E, Smith-Sorensen B, Montesano R, Harris CC. Nucleic Acids Res. 1994;22:3551–3555. [PMC free article] [PubMed] [Google Scholar]
- 9.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 10.Bond GL, Hu W, Levine AJ. Curr Cancer Drug Targets. 2005;5:3–8. doi: 10.2174/1568009053332627. [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Yuan Y, Huan J, Shen Z. Oncogene. 2001;20:336–345. doi: 10.1038/sj.onc.1204098. [DOI] [PubMed] [Google Scholar]
- 12.Ono T, Kitaura H, Ugai H, Murata T, Yokoyama KK, Iguchi-Ariga SM, Ariga H. J Biol Chem. 2000;275:31145–31154. doi: 10.1074/jbc.M003031200. [DOI] [PubMed] [Google Scholar]
- 13.Lu H, Guo X, Meng X, Liu J, Allen C, Wray J, Nickoloff JA, Shen Z. Mol Cell Biol. 2005;25:1949–1957. doi: 10.1128/MCB.25.5.1949-1957.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meng X, Liu J, Shen Z. Cell Cycle. 2004;3:343–348. [PubMed] [Google Scholar]
- 15.Meng X, Lu H, Shen Z. Cell Cycle. 2004;3:1457–1462. doi: 10.4161/cc.3.11.1213. [DOI] [PubMed] [Google Scholar]
- 16.Meng X, Liu J, Shen Z. Gene. 2003;302:139–146. doi: 10.1016/s0378-1119(02)01098-3. [DOI] [PubMed] [Google Scholar]
- 17.Meng X, Yuan Y, Maestas A, Shen Z. J Biol Chem. 2004;279:6098–6105. doi: 10.1074/jbc.M306794200. [DOI] [PubMed] [Google Scholar]
- 18.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Nat Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 19.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 20.Funk WD, Pak DT, Karas RH, Wright WE, Shay JW. Mol Cell Biol. 1992;12:2866–2871. doi: 10.1128/mcb.12.6.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanson S, Kim E, Deppert W. Oncogene. 2005;24:1641–1647. doi: 10.1038/sj.onc.1208351. [DOI] [PubMed] [Google Scholar]
- 22.Marmorstein LY, Ouchi T, Aaronson SA. Proc Natl Acad Sci U S A. 1998;95:13869–13874. doi: 10.1073/pnas.95.23.13869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 24.Gu W, Roeder RG. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 25.Gottifredi V, Shieh S, Taya Y, Prives C. Proc Natl Acad Sci U S A. 2001;98:1036–1041. doi: 10.1073/pnas.021282898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takimoto R, El-Deiry WS. Proc Natl Acad Sci U S A. 2001;98:781–783. doi: 10.1073/pnas.98.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robles AI, Linke SP, Harris CC. Oncogene. 2002;21:6898–6907. doi: 10.1038/sj.onc.1205563. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Lagowski JP, Vanderbeek GE, Kulesz-Martin MF. Cancer Biol Ther. 2004;3:1102–1108. doi: 10.4161/cbt.3.11.1189. [DOI] [PubMed] [Google Scholar]