

Abstract
Lysosomes filled with glycogen are a major pathologic feature of Pompe disease, a fatal myopathy and cardiomyopathy caused by a deficiency of the glycogen-degrading lysosomal enzyme, acid α-glucosidase (GAA). To facilitate studies germane to this genetic disorder, we developed two in vitro Pompe models: myotubes derived from cultured primary myoblasts isolated from Pompe (GAA KO) mice, and myotubes derived from primary myoblasts of the same genotype that had been transduced with cyclin-dependent kinase 4 (CDK4). This latter model is endowed with extended proliferative capacity. Both models showed extremely large alkalinized, glycogen-filled lysosomes as well as impaired trafficking to lysosomes. Although both Pompe tissue culture models were derived from fast muscles and were fast myosin positive, they strongly resemble slow fibers in terms of their pathologic phenotype and their response to therapy with recombinant human GAA (rhGAA). Autophagic buildup, a hallmark of Pompe disease in fast muscle fibers, was absent, but basal autophagy was functional. To evaluate substrate deprivation as a strategy to prevent the accumulation of lysosomal glycogen, we knocked down Atg7, a gene essential for autophagosome formation, via siRNA, but we observed no effect on the extent of glycogen accumulation, thus confirming our recent observation in autophagy deficient Pompe mice [1] that macroautophagy is not the major route of glycogen transport to lysosomes. The in vitro Pompe models should be useful in addressing fundamental questions regarding the pathway of glycogen to the lysosomes and testing panels of small molecules that could affect glycogen biosynthesis or speed delivery of the replacement enzyme to affected lysosomes.
Keywords: autophagy, lysosomes, myotubes, Pompe disease
Introduction
Pompe disease is caused by the absence or deficiency of acid α-glucosidase (GAA), the enzyme responsible for the hydrolysis of glycogen to glucose in lysosomes. Without GAA, glycogen delivered to the lysosome cannot exit as glucose and therefore accumulates. Although glycogen accumulates in the lysosomes of many tissues of both Pompe patients and a knockout mouse model (GAA KO), the major consequences are borne by cardiac and skeletal muscle. With a severe deficiency, there is heart failure in infancy; when some enzyme is present, skeletal and diaphragmatic muscle weakness predominates [2]. Enzyme replacement therapy (ERT) with recombinant human enzyme (rhGAA) significantly improves cardiac function and markedly reduces early infant mortality caused by heart failure. Skeletal muscle has proven to be a more difficult target [3, 4]. The accumulation of glycogen in both humans and GAA KO is not limited to the lysosomes in skeletal muscle, but is also found in autophagic vacuoles containing cytoplasmic degradation products [5, 6]. In Pompe mice, these autophagic inclusions are present in fast glycolytic (Type II) muscle fibers but not in slow oxidative (Type I) muscle fibers [7], and their presence correlates with the failure of fast fibers to respond to ERT with rhGAA [8, 9].
In infants treated with rhGAA, early therapy improves the clinical outcome [10]. Early treatment with rhGAA is likewise advantageous for the mouse model [11, 12]. Therefore, improved strategies to block the earliest accumulation of glycogen in lysosomes or to reverse the established pathology are needed. The search for novel therapies could be greatly benefitted by an improved in vitro model of the disease. Current systems, such as human fibroblasts and myoblasts, as well as myoblasts derived from the GAA KO mice, are primitive models for the in vivo condition. To facilitate our studies of the pathogenesis and therapy of the disorder, we have developed and characterized two in vitro systems which display a later stage of muscle cell differentiation: multi-nucleated myotubes. One system was derived from GAA KO primary myoblasts and the other from GAA KO primary myoblasts that had been transduced with CDK4.
Materials and methods
Primary Myoblast and Myotube Culture
Satellite cells were cultured by plating individual muscle fibers from mice as previously described [13]. Briefly, gastrocnemius muscles from 1-2 month old GAA KO, WT, and muscle-specific autophagy-deficient GAA KO mice [1] were incubated in 0.2% collagenase (Sigma-Aldrich, St. Louis, MO) for 1 h at 37°C. Individual fibers were released by gentle pipeting of partially digested muscle bundles. Up to 10 fibers were plated in each well of a Cell-Tak™ (BD Pharmingen, San Diego, CA) coated 2-chambered cover glass or collagen coated 6 well plates and incubated in plating medium [10% horse serum (HS) in DMEM (Invitrogen, Carlsbad, CA)] for 4 days. After the satellite cells migrated from fibers, the medium was changed to proliferating medium [20% fetal bovine serum (FBS) and 10% HS in DMEM]; the satellite cells were incubated for an additional 3-4 days until the myoblasts became nearly confluent. Then the medium was changed to differentiation medium (2% HS in DMEM) and the cells were kept at 37°C for several weeks. Myotubes began to form approximately 1 week after plating while still in proliferation medium. To evaluate the reversibility of lysosomal swelling, rhGAA (0.5μM or 5μM; Genzyme Corp., Framingham, MA) was added to the culture medium and incubated with WT or GAA KO myotubes for 4 days.
Immunofluoresence Microscopy and PAS Staining
Cultured myotubes were fixed with 2% paraformaldehyde (PFA) for 30 minutes at 37°C. After several washes with PBS, myotubes were incubated with blocking reagent (MOM kit; Vector Laboratories, Burlingame, CA) for 1 h at room temperature. Myotubes were then incubated with primary antibodies overnight at 4°C, washed with PBS, incubated with secondary antibody for 2 h, and washed again with PBS before examination by confocal microscopy (Zeiss LSM 510, confocal images are single planes, 63 × 1.4 NA). Antibodies were as follows: rat anti-mouse Lysosomal-associated membrane protein 1 (LAMP1; 1:500; BD Pharmingen), mouse anti-skeletal fast myosin (1:500; Sigma-Aldrich), goat anti-mouse cathepsin D and goat anti-mouse cathepsin B (1:500; Sigma-Aldrich), mouse anti-GFP (1:500; Clontech, Mountain View, CA), mouse anti α-tubulin (1:500; Sigma-Aldrich), rabbit anti-bovine cation-independent mannose-6-phosphate receptor (CI-MPR) (1:15000; a gift from Dr. Stuart Kornfeld, Washington University School of Medicine, St. Louis, MO, USA). Alexa Fluor-conjugated antibodies (1:500, Molecular Probes, Eugene, OR) were used as secondary antibodies. For the Periodic Acid-Schiff (PAS) staining, myotubes were fixed with 5% formaldehyde in 95% ethanol for 5 min at room temperature. The PAS Kit (Sigma-Aldrich) was used according to the manufacturer's instructions.
pH Measurement in Live Fibers and Cultured Myotubes
Isolated live fibers were obtained from gastrocnemius muscles of 3 month old WT and GAA KO mice. They were incubated in 0.2% collagenase and individual fibers were obtained as described above. Isolated fibers were incubated in DMEM with 5% HS followed by staining with acridine orange (AO) [14, 15] or LysoSensor [16] (Molecular Probes). Fibers or live cultured myotubes were incubated with AO at 2.5μg/ml for 10 minutes. LysoSensor staining was performed by incubation with DND-152 (pKa=7.5) or DND-189 (pKa=5.2) at 1μM for 60 minutes. Fibers or myotubes were washed several times with medium and then analyzed on a Zeiss 510 confocal microscope (AO emission was collected with a 500-560 nm band pass filter (green) and 590-660 nm band pass filter (red)).
Analysis of Vesicular Trafficking
Myotubes were incubated overnight at 37°C with a fluid-phase endocytic marker, TMR-conjugated dextran (10,000 kD, 1mg/ml; Molecular Probes). The cells were washed and observed by confocal microscopy at 2 and 48 hours following removal of the TMR-dextran. In a separate set of experiments for the quantitative analysis of fluid phase endocytosis, myotubes were incubated overnight with Alexa Fluor 546-conjugated dextran (which is brighter and more photo stable than TMR-dextran; 10,000 kD, 1 mg/ml, Molecular Probes) and imaged on the confocal microscope at 3 h and 48 h after removal of the dextran. The distribution of the fluorescent signal from both Alexa Fluor 546 and TMR dextran in the myotubes was similar. Using Image J [17], myotubes were outlined and the distribution of the pixel values (0-255) of 8-13 myotubes was averaged. A background value measured outside the cells was subtracted from the mean. For analysis of the fluorescent intensity of individual lysosomes, vesicular structures - either visible because they contained Alexa Fluor 546 or identified in differential interference contrast (DIC) images that accompanied each fluorescent image - were circled, and the total fluorescent signal was calculated from the mean pixel value of the area outlined. A background value, measured outside any vesicular structure was subtracted from the mean.
TMR-conjugated dextran was also utilized for direct microinjection into the sarcoplasm of myotubes using the Eppendorf FemtoJet Microinjection System (Pi; 260 mmHg, Pc; 50 mmHg, Ti; 0.6 second) on a Leica DMR1 fluorescent microscope. Media in the dishes was replaced with fresh media and injected myotubes were imaged on the confocal microscope after 24 or 72 hours at 37°C.
To analyze receptor-mediated endocytosis, WT and GAA KO myotubes were incubated overnight in the presence of Alexa Fluor 546 rhGAA (prepared as described in [9]), washed for 2 hours, and examined by confocal microscopy.
Isolation of GAA KO Myoblasts with Extended Lifespan
Satellite cells from GAA KO gastrocnemius muscle fibers were transduced with a retrovirus housing a CDK4 gene, which is involved in the regulation of cell cycle and senescence [18]. The transduction was performed as described previously [19]. Briefly, we repeated the transduction twice daily for five days and then selected for cells resistant to G418 (Sigma-Aldrich). The cells were called CS1.
Transfection of CS1 Cells with GFP-LC3
CS1 cells were transiently transfected with pEGFP-LC3 using the FuGENE 6 reagents (Roche Applied Science, Indianapolis, IN) according to the manufacturer's instructions. Transfection was performed on myoblasts (passage 15) prior to differentiation into myotubes; myotubes were fixed, stained with anti-GFP and anti-LAMP1, and imaged by confocal microscopy.
Treatment of CS1 Cells with Atg7 siRNA
CS1 cells (passage 30) were plated at 2.0 × 104/ml in proliferation medium. On day 3 of culture the cells were switched to differentiation medium and transfected with Atg7 siRNA or control siRNA (Dharmacon, Lafayette, CO) using DharmaFECT 3 (Dharmacon) according to the manufacturer's instructions. Myotubes were fixed with 2% PFA 6 days later and then stained with PAS or LAMP1.
Western Blotting
Lysates from cultured myotubes were prepared in RIPA buffer containing PBS pH 7.4, 1% NP40, 0.5% sodium deoxycholate, 1% SDS, and the following proteinase inhibitors: 4mM Pefabloc SC, 10μg/ml aprotinin, 10μg/ml leupeptin (Roche Diagnostics), and 5μg/ml E-64 (Calbiochem, San Diego, CA). The lysates were separated by SDS-PAGE according to standard procedure. Blots were incubated with anti-LC3 antibody (Sigma-Aldrich) in Odyssey Buffer (LI-COR Biosciences, Lincoln, NE) followed by incubation with Alexa Fluor 680-conjugated secondary antibody (Molecular Probes). Blots were scanned on an infrared imager (LI-COR Biosciences).
Animal care and experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
Results
In Vitro Pompe Model: Myotubes Cultured from Primary Myoblasts
To establish a culture of highly differentiated Pompe muscle cells, we began by isolating myoblasts from satellite cells that had migrated from single muscle fibers [13]. These primary myoblasts derived from fast (Type II) gastrocnemius muscle of young WT and GAA KO mice grew well in culture, and after about one week they began to fuse into myotubes. The myotubes survived in culture up to four weeks before they detached from the culture dish. Experiments were carried out on two- to four- week old myotubes.
We characterized these GAA KO myotubes with respect to their lysosomal morphology. Staining for glycogen (PAS) and for late endosomes/ lysosomes (LAMP1) revealed PAS positive/ Lamp1 positive vesicular structures (Fig. 1A and B). Following formation of the myotubes, these structures increased dramatically in size, in some cases reaching diameters of 10 -15 microns (Fig. 1B-D). The average cross-sectional area of the largest LAMP1 positive vesicles in GAA KO myotubes was 30 μm2 as compared to 2.8 μm2 in the WT myotubes. These large LAMP1 positive vesicles were identified as lysosomes because they were negative for CI-MPR (a negative marker for lysosomes; not shown). Both GAA KO and WT myotubes showed fast myosin positivity, consistent with their fast muscle origin [20] (Fig. 1E).
Fig. 1.
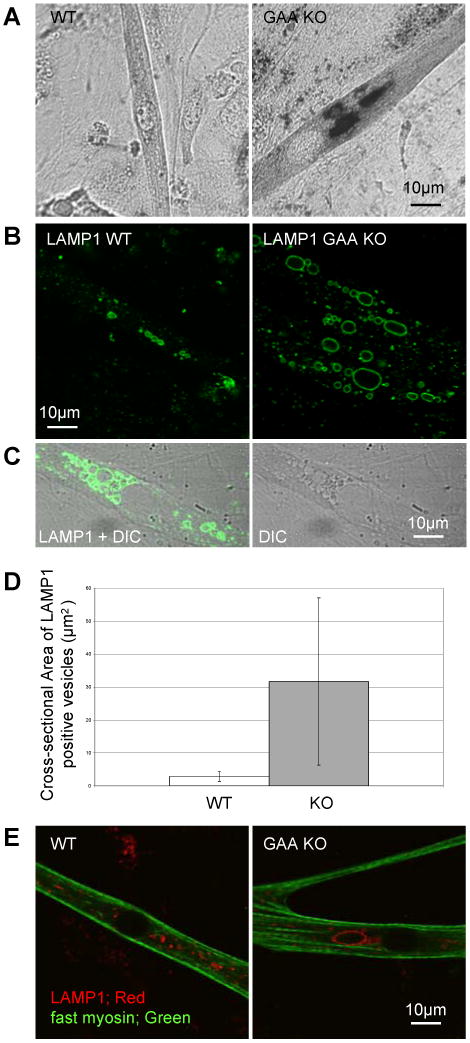
Characterization of lysosomes in myotubes cultured from GAA KO primary mouse myoblasts. (A) PAS stained WT and GAA KO myotubes. (B) LAMP1 stained myotubes cultured from WT and GAA KO primary myoblasts. (C) LAMP1 staining of GAA KO myotubes in DIC image where vacuoles are often recognized as lucent structures. (D) Myotubes were randomly selected from 4 week old WT and GAA KO cultures and areas of largest LAMP1 stained vacuoles in these myotubes were measured (n=16 for WT myotubes and n=40 for GAA KO myotubes, p=0.0003). (E) Fast myosin immunostained myotubes cultured from WT and GAA KO primary myoblasts.
We then assessed biochemical properties of the GAA KO myotubes by determining the degree of acidity of their lysosomes using pH sensitive metachromatic dyes. Experiments with AO, which shows red fluorescence in an acidic environment and green fluorescence in a neutral environment, demonstrated gross abnormalities in the condition of the large vesicles. The small lysosomes of WT myotubes (Fig. 2A) showed red fluorescence. By contrast, the large GAA KO lysosomes showed green fluorescence (Fig. 2A and B), indicating that the lumen of these expanded structures is not acidic. It should be noted that small acidic vesicles exhibiting red fluorescence are also present just above or beneath the vacuolar membrane of these large lysosomes as well as elsewhere in the cytoplasm.
Fig. 2.
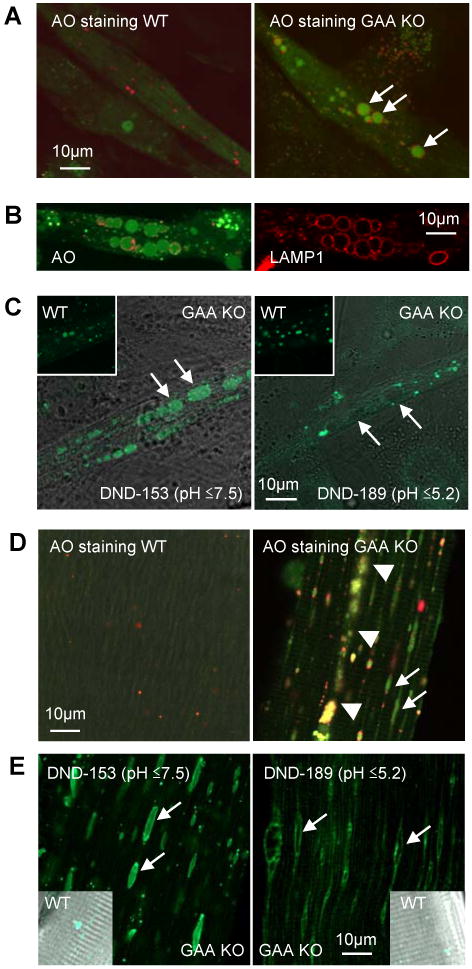
Analysis of pH of expanded lysosomes. (A) AO stained live cultured myotubes derived form WT and GAA KO primary myoblasts. AO shows red fluorescence (650 nm) in an acidic environment and green fluorescence (526 nm) in a neutral environment. Expanded lysosomes in GAA KO myotubes show green fluorescence (arrows). Small structures clustered around the expanded alkalinized lysosomes are acidic vesicles. (B) Live imaging of GAA KO myotubes loaded with AO (left panel) followed by fixation and LAMP1 staining of the same myotubes. (C) LysoSensor DND-153 (pH ≤7.5) fluoresces green in large lysosomes of GAA KO (left panel) but LysoSensor DND-189 (pH ≤5.2) does not (right panel). Green fluorescence is seen in WT with both dyes. (D) AO stained live fibers isolated from white gastrocnemius muscle (predominantly fast Type II) of WT and GAA KO mice. Arrows show alkalinized lysosomes (green fluorescence) in GAA KO fibers. In WT fibers, small red vesicles (normal sized lysosomes) are scattered. Arrowheads point to area of autophagic buildup. (E) Live GAA KO myofibers stained with LysoSensors. LysoSensor DND-153 (pH≤7.5) fluoresces green in large lysosomes (arrows) but the lysosomes do not stain with LysoSensor DND-189 (pH ≤5.2). The lysosomes in the WT fibers stain with both LysoSensors.
To evaluate the pH of the large lysosomes more precisely, we used two LysoSensors: DND-189, which fluoresces in an acidic environment (pH≤ 5.2), and DND-153, which fluoresces in both an acidic and in a neutral environment (pH≤ 7.5). The lumen of the swollen lysosomes stained with DND-153 but not with DND-189, establishing the pH range of these vacuoles to lie between 5.2 and 7.5 (Fig. 2C).
To see if this profound lysosomal abnormality mirrors that in muscle fibers, we checked lysosomal pH in live cultured single muscle fibers of GAA KO mice. Green fluorescence was observed in the lysosomes of these fibers treated with AO, and experiments with DND-189 and DND-153 placed the lysosomal pH within the 5.2-7.5 range (Fig. 2D and E). Furthermore, immunocytochemistry showed that enlarged lysosomes in cultured GAA KO myotubes did not contain cathepsins B or D (Fig. 3), two classical lysosomal enzymes. These enzymes were present in the lumen of WT lysosomes and in the smaller vesicles adjacent to the lysosomal membrane in GAA KO myotubes.
Fig. 3.
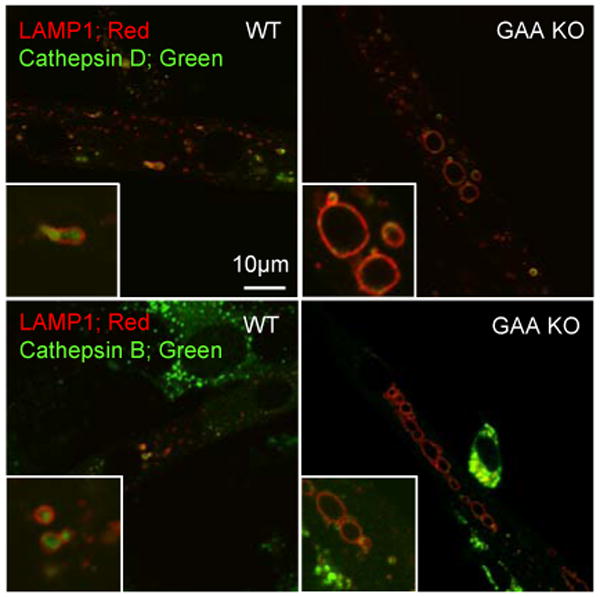
Analysis of myotubes for cathepsin D and B. WT and GAA KO myotubes were doubly immunostained for LAMP1 and either cathepsin D or B. In WT myotubes, the majority of the LAMP1 positive vacuoles contain cathepsin D. No cathepsin D staining is observed in expanded GAA KO lysosomes. A similar result is evident for cathepsin B. Small structures clustered around the expanded lysosomes (similar to those shown in Fig. 2) are cathepsin D and B positive.
In Vitro Pompe Model: Myotubes Cultured from CDK4-transduced Primary Myoblasts
Generation of Pompe myotubes from primary myoblasts was limited by the proliferative capacity of these cells. The primary myoblasts fell into senescence after only one or two passages in culture. This feature made the system cumbersome, requiring re-isolation of primary myoblasts for every experiment. We sought therefore to streamline our model system.
It had previously been shown that human myoblasts transduced with CDK4-containing retroviruses yielded myoblasts with an extended lifespan [19]. For this purpose, we isolated a population of CDK4- transduced GAA KO myoblasts derived from satellite cells of fast gastrocnemius muscle. These myoblasts (CS1 cells) could undergo multiple passages in culture while retaining their potential to terminally differentiate into multi-nucleated myotubes. At this writing, we have confirmed that CS1 myoblasts that have undergone 80 passages in culture can still form myotubes. The CS1-derived myotubes were morphologically indistinguishable from those derived from primary myoblasts; they were fast myosin positive (not shown); and they accumulated glycogen in large lysosomes (Fig. 4). We concluded therefore that we could utilize these in vitro Pompe models interchangeably.
Fig. 4.
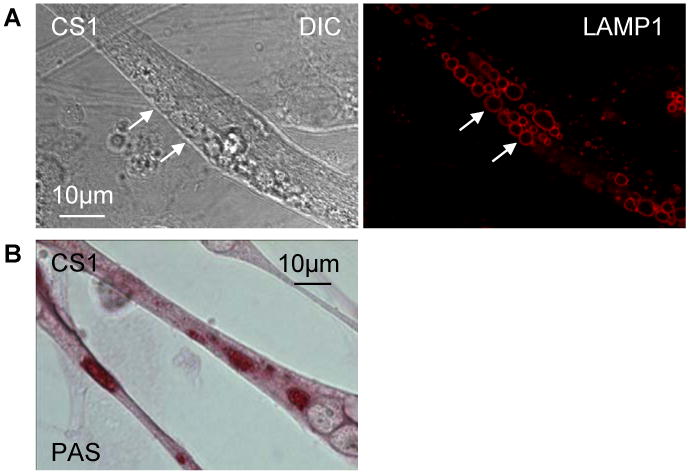
Characterization of lysosomes in myotubes cultured from CDK4 transduced GAA KO primary myoblasts. (A) As in myotubes derived from GAA KO primary myoblasts, enlarged vesicles (arrows) are observed in CDK4 transduced cells (left, DIC). These vesicles are positive for LAMP1 (right, red). (B) PAS staining for glycogen is positive in expanded lysosomes.
Examination of Endocytic Trafficking in an In Vitro Pompe Model
We next studied vesicular trafficking in the myotubes derived from primary myoblasts. Fluid phase endocytosis was evaluated using Tetramethylrhodamine (TMR)-labeled dextran in WT and GAA KO myotubes. Dextran-filled lysosomes were observed at 2 hours following removal of the labeled dextran from the medium in the WT myotubes. In the GAA KO myotubes, TMR dextran was detected in small and large lysosomes, but the intensity in the large lysosomes was much lower (Fig. 5A). The lysosomal origin of these structures was confirmed by LAMP1 staining of the same myotubes which were fixed after imaging. After 48 hours, however, the GAA KO cultures showed an increase in signal intensity in the large lysosomes, but no such increase was observed in the WT lysosomes (Fig. 5B), suggesting that fluid phase endocytosis is sluggish in the Pompe model.
Fig. 5.
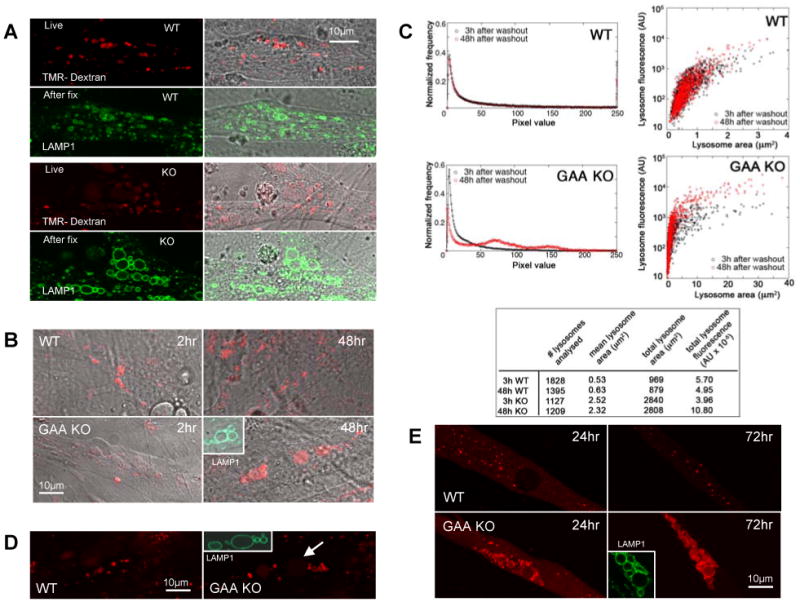
Examination of trafficking in cultured myotubes. (A) WT and GAA KO cultured myotubes incubated with 1mg/ml TMR-dextran (red) overnight followed by a two-hour wash in medium lacking labeled dextran. Small lysosomes (LAMP1 positive green vacuoles) in WT are positive for dextran but enlarged lysosomes in GAA KO show weak staining. (B) GAA KO expanded lysosomes (LAMP1-positive structures) show strong staining for dextran 48 hours after removal of labeled dextran. (C) Quantification of fluid phase endocytosis in cultured myotubes loaded with Alexa Fluor 546-labeled dextran. Left panel: histograms of the pixel values of 8-13 myotubes show a shift in the distribution of the pixel values between 3h and 48 h in the GAA KO but not in the WT. Right panel: analysis of the fluorescent signal in the cross-section of individual lysosomes. WT lysosomes (the majority of which is ∼1 μm2) change very little in the fluorescence signal between 3h and 48 h after washout of the dye. In contrast, GAA KO lysosomes, especially the large ones (> 5 μm2), increased in fluorescence between 3h and 48 h after washout of the dye, indicating that trafficking of labeled dextran had continued after it had ceased in WT. The table shows that the total lysosomal area in KO myotubes is significantly larger than in WT and that the summed fluorescence of the KO lysosomes doubled between 3h and 48h, while there was a slight decrease in the WT lysosomes fluorescence. (D) WT and GAA KO myotubes were incubated in the presence of Alexa-Fluor 546 rhGAA (red) followed by a two-hour wash. Lysosomes of WT myotubes are positive. Small lysosomes but not the large lysosomes in GAA KO contain labeled rhGAA. (E) TMR-dextran was microinjected into myotubes. Twenty four hours after injection, dextran is localized in small vesicles in both WT and GAA KO; small structures clustered around the expanded lysosomes in the GAA KO (similar to those shown in Fig. 2 & 3) are dextran positive. Three days after injection, expanded vacuoles (LAMP1 positive) stained strongly for dextran in GAA KO myotubes and no background dextran is seen in the cytoplasm. The image of the LAMP1 staining (inset) was collected from the same myotubes that were analyzed in live cultures.
The absence of a strong fluorescent signal in the large, glycogen-filled GAA KO lysosomes at 2 hours could, however, have been the effect of dilution of the fluorescence, assuming that all of the available dextran had reached the lysosomes. To eliminate this possibility we did quantitative analysis using Alexa Fluor 546-labeled dextran as a fluid phase marker. After overnight incubation with the labeled dextran, WT and GAA KO myotubes were washed and imaged following incubation in dextran-free medium for 3 hours. The cultures were then placed back in the incubator, and the same myotubes were imaged 48 hours after washout of the dye, using identical imaging parameters.
Images of 8-13 myotubes were used to create a mean pixel intensity distribution (PID) diagram of all the fluorescence. No change in the PID in the WT myotubes between 3 and 48 hours was registered (Fig. 5C, left top panel). In contrast, in the GAA KO there was a shift in PID from lower to higher intensity pixels (Fig. 5C, left bottom panel) after 48 hours, implicating a trafficking defect. Furthermore, we have also estimated the amount of Alexa Fluor 546 dextran in the cross-section of individual lysosomes (Fig. 5C). This showed that between 3 and 48 hours, individual WT lysosomes gained little to no fluorescence whereas large (≥ 5 μm2) GAA KO lysosomes did (Fig. 5C, right panels). During the interval between 3 and 48 hours, the WT lysosomes lost a small portion of their fluorescent signal, while the GAA KO lysosomes more than doubled their signal (Fig. 5C, Table). In some of the very large GAA KO lysosomes the signal increased about 10- fold. These results point very strongly towards a delay in the delivery of fluid phase endocytosed materials to the large, glycogen-filled lysosomes.
We have also analyzed receptor-mediated transport in WT and GAA KO myotubes. When these myotubes were treated with labeled rhGAA, the enzyme accumulated within 2 hours in small lysosomes of both cell types but not in the glycogen-swollen lysosomes in GAA KO myotubes (Fig. 5D). Since exogenously added rhGAA reaches the lysosome via mannose-6-phosphate (cation-independent) receptor mediated endocytosis [21], this experiment suggests retardation of the process in the GAA KO myotubes.
Finally, to assess the transfer of cytosolic content to the lysosomes, TMR-labeled dextran was microinjected into the cytosol of WT and GAA KO myotubes followed by incubation for 24 or 72 hours. In the WT, about the same amount of TMR dextran was observed in lysosomes after 24 and 72 hours, while the staining significantly increased in the GAA KO during that time period, showing slow movement of labeled dextran to its lysosomal destination (Fig. 5E).
Examination of Autophagy in In Vitro Pompe Models
Based on the autophagic abnormality observed in fast muscle fibers of GAA KO mice, we expected but did not find autophagic buildup in either model. We addressed, therefore, the question of whether constitutive autophagy was functional in our in vitro Pompe models. LC3-II, a specific autophagosomal marker [22] derived from the processing of LC3-I, was detectable by Western blotting in GAA KO myotubes (Fig. 6A). When CS1 cells were transfected with GFP-LC3, followed by fixation and immunostaining for LAMP1 (Fig. 6B and C), the lysosomal and autophagosomal markers co-localized, suggesting that fusion between lysosomes and autophagosomes had occurred. Cultured myotubes therefore have both glycogen accumulation and functional basal autophagy, but they lack autophagic buildup.
Fig. 6.
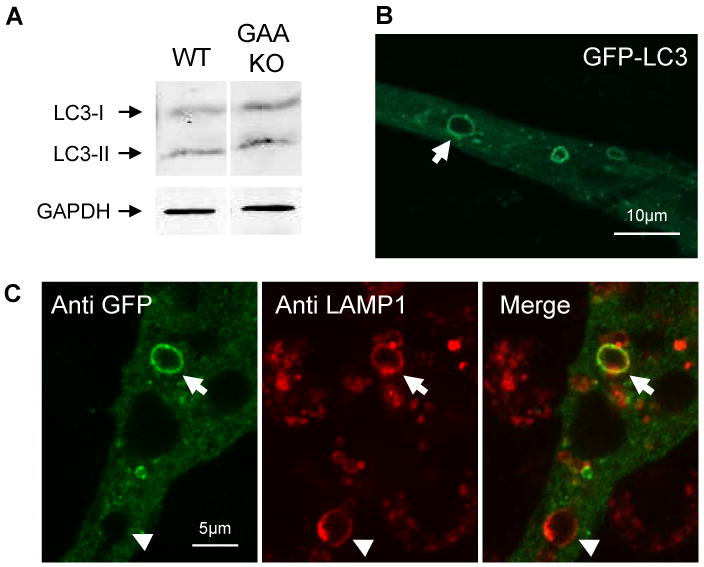
Analysis of autophagy in in vitro Pompe models. (A) Western blotting of lysates obtained from WT and GAA KO myotubes using LC3 antibodies. (B) Following transfection of CS1 cells with GFP-LC3, green fluorescence is observed in structures similar in appearance to expanded lysosomes. (C) Post-transfection immunostaining using anti-GFP and anti- LAMP1 reveals the presence of a doubly-labeled vacuole, an autophagolysosome (arrow). Note arrowhead which shows a vacuole that stains only for LAMP1.
Use of the In Vitro Pompe Model to Evaluate Therapeutic Approaches
We then turned to using these Pompe models for therapeutic studies. Substrate deprivation has proved an effective therapy in some lysosomal storage diseases [23, 24], and this route has been shown to be effective in Pompe mouse muscle cells as well [25]. Because autophagy has been the presumed mechanism of glycogen delivery to the lysosomes [26, 27], we anticipated that glycogen accumulation in GAA KO mice might be blocked by inactivation of autophagy. Our recent data show, to the contrary, that glycogen accumulation is unimpaired in the muscle of autophagy-deficient Pompe (Atg5-deficient GAA KO) mice [1]. We have now demonstrated that myotubes derived from these Atg5-deficient GAA KO mice accumulate the same amount of glycogen as myotubes derived from GAA KO mice (Fig. 7A-D).
Fig. 7.
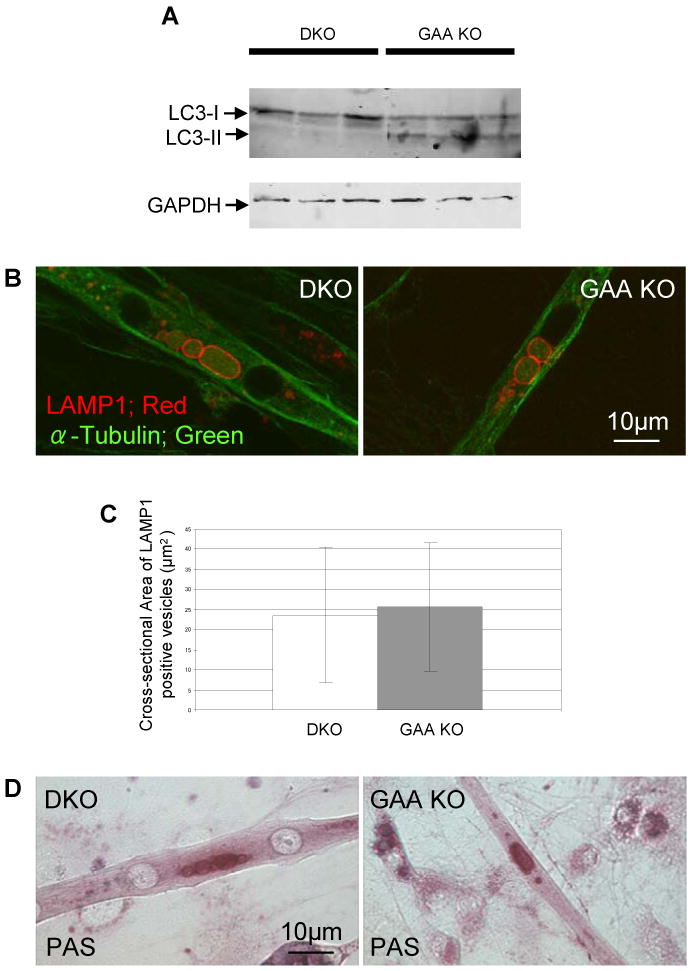
Lysosomes in autophagy deficient myotubes. Myotubes were prepared from primary myoblasts of skeletal muscle-specific autophagy-deficient GAA KO mice (DKO) [1]. (A) Absence of autophagy is confirmed by western blotting using LC3 antibody. (B) Enlarged lysosomes are shown by LAMP1 staining of DKO myotubes. (C) The difference between the size of the LAMP-I positive vacuoles in DKO and GAA KO mice is statistically insignificant (n=36 myotubes for GAA KO and n=41 myotubes for DKO; p =0.6). (D) Glycogen accumulation in DKO myotubes is shown by PAS staining.
To extend this finding, we suppressed another key autophagic gene, Atg7 [28, 29], this time in an in vitro Pompe model, CS1 cells. As expected, Atg7 siRNA in GAA KO myotubes blocked the conversion of LC3-I to LC3-II (Fig. 8A), but there was a negligible effect on the accumulation of glycogen in the lysosomes (Fig. 8B-D). Therefore, these findings strongly reinforce that macroautophagy is not the major pathway of glycogen delivery to muscle cell lysosomes. The concordance of these in vivo and in vitro observations further validates these tissue culture models of Pompe disease.
Fig. 8.
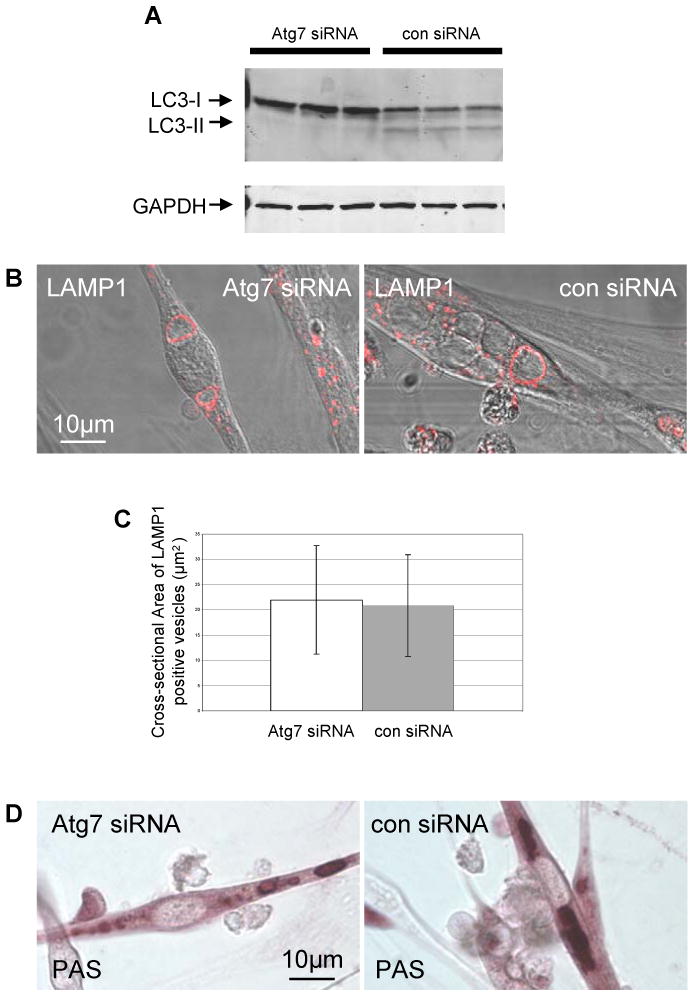
Inhibition of macroautophagy with Atg7 siRNA in GAA KO myotubes. (A) GAA KO myoblasts (CS1 cells) were treated with Atg7 siRNA or negative control siRNA. Macroautophagy is successfully inhibited by Atg7 siRNA as shown by the absence of LC3-II signal in western blotting. (B) Atg7 siRNA treated cells contain large LAMP1 positive lysosomes. (C) The difference between the area of swollen lysosomes in the control and Atg7 siRNA treated cells is statistically insignificant (n=30 for control siRNA and n=35 for Atg7 siRNA; p=0.66). (D) Glycogen accumulation in Atg7 siRNA treated CS1 cells is confirmed by PAS staining.
To determine the feasibility of rescuing cells that had accumulated lysosomal glycogen in this in vitro model, myotubes were incubated with unlabelled rhGAA, which reaches lysosomes via mannose 6-phosphate-mediated endocytosis. Following incubation with unlabelled rhGAA for four days, the diseased myotubes exhibited a clear dose-dependent reduction in lysosome size. At high doses of the therapeutic enzyme the lysosomes were reduced to WT size (Fig. 9A and B). This therapeutic responsiveness to exogenously provided replacement enzyme thus parallels the clearance of glycogen from lysosomes observed in the slow fibers in GAA KO mice treated with rhGAA [8].
Fig. 9.
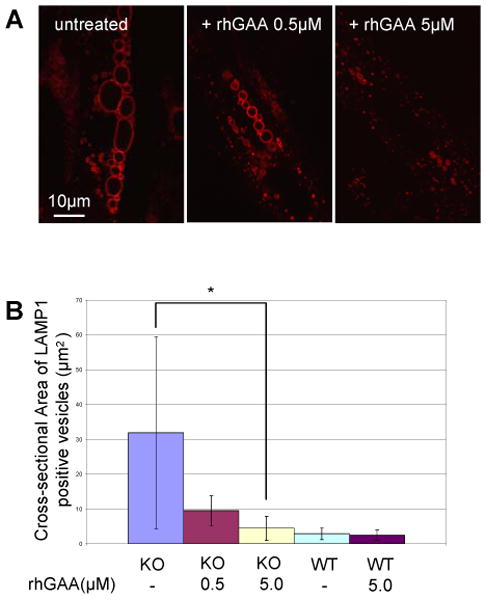
Effect of the therapeutic enzyme (rhGAA) in GAA KO myotubes. (A) GAA KO myotubes were treated with rhGAA. After 4 days of incubation, LAMP1 positive structures shrink in a dose dependent manner. (B) Graphical presentation of data in A. Myotubes analyzed: n=26 for GAA KO (control); n=23 for GAA KO myotubes treated with 0.5 uM rhGAA; n=19 for GAA KO myotubes treated with 5.0μM rhGAA; n=8 for WT myotubes; n=9 for WT myotubes treated with 5.0μM rhGAA. *p=0.00003.
Discussion
Earlier studies of myoblasts isolated from whole muscle biopsies of GAA KO mice revealed significant expansion of the endocytic vesicles and the presence of a subset of alkalinized vesicles (lysosomes or late endosomes) [7]. While the myoblasts derived in this fashion from WT and GAA KO mice grew well in culture, elimination of fibroblasts was a challenge, and the myotubes were short. Superior results were obtained when we altered our protocol and isolated myoblasts from satellite cells that had migrated from single muscle fibers [13]. The myotubes derived from these myoblasts show hugely expanded, glycogen-filled lysosomes (much larger than those in myoblasts) which are comparable with those observed in GAA KO muscle fibers, indicating that these myotube models replicate this hallmark of Pompe disease.
Furthermore, what we observed as a mild pH abnormality in myoblasts [7] became in the myotubes a severe acidification defect, as is found in live Pompe muscle fibers. Interestingly, however, small acidic vesicles (exhibiting red fluorescence) are also present just above or beneath the relatively alkalinized (exhibiting green fluorescence) vacuolar membrane in AO staining of GAA KO myotubes, suggesting the possibility of impaired fusion of endocytic vesicles with large alkalinized lysosomes. Impaired acidification in lysosomes might cause a variety of detrimental effects in muscle cells. Because many of the lysosomal enzymes show highest activity in an acidic environment, the functions of these enzymes might be impaired. The observed absence of cathepsins in expanded lysosomes suggests that lysosomal enzyme delivery or processing may be altered, but this remains to be elucidated.
In GAA KO myotubes, endocytic trafficking into the enlarged vacuoles was delayed. Although endocytosed dextrans finally accumulated in the enlarged lysosomes, vesicular fusion was remarkably slow. This may be related to the abnormal acidification since abnormal lysosomal pH may disturb fusion between lysosomes and endocytic vesicles, as has been shown in experiments on vesicular fusion using proton pump inhibitors [30, 31].
In both humans with Pompe disease and GAA KO mice there is extensive autophagic accumulation in the skeletal muscle fibers [7, 32]. In mice, this process is known to be limited to fast glycolytic fibers [7]. The regions of autophagic accumulation in fast fibers are most often located centrally and extend longitudinally so that eventually they may run almost the length of a fiber and almost half its width. The contractile apparatus appears disturbed, and the cells atrophy [9]. Such fast cells respond poorly to ERT. In contrast, GAA KO slow oxidative fibers lack autophagic buildup and respond well to ERT [8].
The in vitro generated Pompe myotubes, like muscle fibers of the GAA KO mice, contain enlarged glycogen-filled lysosomes with a profound acidification defect. Although these GAA KO myotubes are derived from fast fiber satellite cells and retain the property of fast fibers (as shown by fast myosin expression) their disease phenotype resembles the phenotype of slow fibers in that they lack autophagic buildup and respond well to therapy with rhGAA. It is possible that autophagic buildup requires a much longer time than the lifetime of myotube in culture to develop, but this point remains to be elucidated.
Although our in vitro model system did not replicate the autophagic buildup characteristic of fast live muscle fibers, nonetheless the system is certainly suitable for studies designed to decrease or prevent glycogen accumulation in the lysosomes. We have now clearly demonstrated both in vivo and in this in vitro system that inhibiting autophagy fails to prevent lysosomal glycogen accumulation. Other pathways, for example chaperon-mediated autophagy or microautophagy may offer a suitable target to block glycogen accumulation. The in vitro Pompe model systems should be useful in addressing fundamental questions regarding the pathway of glycogen to the lysosomes and testing panels of small molecules or other compounds that could affect glycogen biosynthesis or speed delivery of replacement enzyme to affected lysosomes.
Acknowledgments
We would like to thank Dr. Evelyn Ralston for help with the imaging procedures, and Lauren Shea and Victoria Hill for help in generating autophagy deficient Pompe mice. The retroviral vector pSRalphaMSV-CDK4-tkneo was given to us by Charles J Sherr M.D. Ph.D., a Howard Hughes Medical Institute Investigator at St. Jude Children's Research Hospital.
This research was supported by the Intramural Research Program of the NIH (National Institute of Arthritis and Musculoskeletal and Skin Diseases [NIAMS]). Dr. Takikita and Dr. Myerowitz were supported in part by a CRADA between the NIH and Genzyme Corporation.
References
- 1.Raben N, Hill V, Shea L, Takikita S, Baum R, Mizushima N, Ralston E, Plotz P. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet. 2008;17:3897–3908. doi: 10.1093/hmg/ddn292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirschhorn R, Reuser AJ. Glycogen storage disease type II; acid alpha-glucosidase (acid maltase) deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited disease. McGraw-Hill; New York: 2001. pp. 3389–3420. [Google Scholar]
- 3.Fukuda T, Roberts A, Plotz P, Raben N. Acid alpha-glucosidase deficiency (Pompe disease) Curr Neurol Neurosci Rep. 2007;7:71–77. doi: 10.1007/s11910-007-0024-4. [DOI] [PubMed] [Google Scholar]
- 4.van der Ploeg A, Reuser A. Pompe's disease. Lancet. 2008;372:1342–1353. doi: 10.1016/S0140-6736(08)61555-X. [DOI] [PubMed] [Google Scholar]
- 5.Engel AG, Hirschhorn R, Huie ML. Acid Maltase Deficiency. In: Engel AG, Franzini-Armstrong C, editors. Myology. McGraw-Hill; New York: 2004. pp. 1559–1586. [Google Scholar]
- 6.Nishino I. Autophagic vacuolar myopathies. Curr Neurol Neurosci Rep. 2003;3:64–69. doi: 10.1007/s11910-003-0040-y. [DOI] [PubMed] [Google Scholar]
- 7.Fukuda T, Ewan L, Bauer M, Mattaliano R, Zaal K, Ralston E, Plotz P, Raben N. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann Neurol. 2006;59:700–708. doi: 10.1002/ana.20807. [DOI] [PubMed] [Google Scholar]
- 8.Raben N, Fukuda T, Gilbert A, de Jong D, Thurberg B, Mattaliano R, Meikle P, Hopwood J, Nagashima K, Nagaraju K, Plotz P. Replacing acid alpha-glucosidase in Pompe disease: recombinant and transgenic enzymes are equipotent, but neither completely clears glycogen from type II muscle fibers. Mol Ther. 2005;11:48–56. doi: 10.1016/j.ymthe.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 9.Fukuda T, Ahearn M, Roberts A, Mattaliano R, Zaal K, Ralston E, Plotz P, Raben N. Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease. Mol Ther. 2006;14:831–839. doi: 10.1016/j.ymthe.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thurberg B, Lynch Maloney C, Vaccaro C, Afonso K, Tsai A, Bossen E, Kishnani P, O'Callaghan M. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for Pompe disease. Lab Invest. 2006;86:1208–1220. doi: 10.1038/labinvest.3700484. [DOI] [PubMed] [Google Scholar]
- 11.Raben N, Lu N, Nagaraju K, Rivera Y, Lee A, Yan B, Byrne B, Meikle P, Umapathysivam K, Hopwood J, Plotz P. Conditional tissue-specific expression of the acid alpha-glucosidase (GAA) gene in the GAA knockout mice: implications for therapy. Hum Mol Genet. 2001;10:2039–2047. doi: 10.1093/hmg/10.19.2039. [DOI] [PubMed] [Google Scholar]
- 12.Raben N, Jatkar T, Lee A, Lu N, Dwivedi S, Nagaraju K, Plotz P. Glycogen stored in skeletal but not in cardiac muscle in acid alpha-glucosidase mutant (Pompe) mice is highly resistant to transgene-encoded human enzyme. Mol Ther. 2002;6:601–608. [PubMed] [Google Scholar]
- 13.Rosenblatt J, Lunt A, Parry D, Partridge T. Culturing satellite cells from living single muscle fiber explants. In Vitro Cell Dev Biol Anim. 1995;31:773–779. doi: 10.1007/BF02634119. [DOI] [PubMed] [Google Scholar]
- 14.Moriyama Y, Takano T, Ohkuma S. Acridine orange as a fluorescent probe for lysosomal proton pump. J Biochem. 1982;92:1333–1336. doi: 10.1093/oxfordjournals.jbchem.a134053. [DOI] [PubMed] [Google Scholar]
- 15.Busch G, Lang H, Lang F. Studies on the mechanism of swelling-induced lysosomal alkalinization in vascular smooth muscle cells. Pflugers Arch. 1996;431:690–696. doi: 10.1007/BF02253831. [DOI] [PubMed] [Google Scholar]
- 16.Lin H, Herman P, Kang J, Lakowicz J. Fluorescence lifetime characterization of novel low-pH probes. Anal Biochem. 2001;294:118–125. doi: 10.1006/abio.2001.5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rasband WS. U. S. National Institutes of Health; Bethesda, Maryland, USA: 19972008. ImageJ. http://rsb.info.nih.gov/ij/ [Google Scholar]
- 18.Zou X, Ray D, Aziyu A, Christov K, Boiko A, Gudkov A, Kiyokawa H. Cdk4 disruption renders primary mouse cells resistant to oncogenic transformation, leading to Arf/p53-independent senescence. Genes Dev. 2002;16:2923–2934. doi: 10.1101/gad.1033002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu C, Mouly V, Cooper R, Mamchaoui K, Bigot A, Shay J, Di Santo J, Butler-Browne G, Wright W. Cellular senescence in human myoblasts is overcome by human telomerase reverse transcriptase and cyclin-dependent kinase 4: consequences in aging muscle and therapeutic strategies for muscular dystrophies. Aging Cell. 2007;6:515–523. doi: 10.1111/j.1474-9726.2007.00306.x. [DOI] [PubMed] [Google Scholar]
- 20.Rosenblatt J, Parry D, Partridge T. Phenotype of adult mouse muscle myoblasts reflects their fiber type of origin. Differentiation. 1996;60:39–45. doi: 10.1046/j.1432-0436.1996.6010039.x. [DOI] [PubMed] [Google Scholar]
- 21.Van der Ploeg A, Kroos M, Willemsen R, Brons N, Reuser A. Intravenous administration of phosphorylated acid alpha-glucosidase leads to uptake of enzyme in heart and skeletal muscle of mice. J Clin Invest. 1991;87:513–518. doi: 10.1172/JCI115025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Platt F, Neises G, Reinkensmeier G, Townsend M, Perry V, Proia R, Winchester B, Dwek R, Butters T. Prevention of lysosomal storage in Tay-Sachs mice treated with N-butyldeoxynojirimycin. Science. 1997;276:428–431. doi: 10.1126/science.276.5311.428. [DOI] [PubMed] [Google Scholar]
- 24.Jeyakumar M, Dwek R, Butters T, Platt F. Storage solutions: treating lysosomal disorders of the brain. Nat Rev Neurosci. 2005;6:713–725. doi: 10.1038/nrn1725. [DOI] [PubMed] [Google Scholar]
- 25.Douillard-Guilloux G, Raben N, Takikita S, Batista L, Caillaud C, Richard E. Modulation of glycogen synthesis by RNA interference: towards a new therapeutic approach for glycogenosis type II. Hum Mol Genet. 2008 doi: 10.1093/hmg/ddn290. In press. [DOI] [PubMed] [Google Scholar]
- 26.Schiaffino S, Hanzlíková V. Autophagic degradation of glycogen in skeletal muscles of the newborn rat. J Cell Biol. 1972;52:41–51. doi: 10.1083/jcb.52.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kondomerkos D, Kalamidas S, Kotoulas O, Hann A. Glycogen autophagy in the liver and heart of newborn rats. The effects of glucagon, adrenalin or rapamycin. Histol Histopathol. 2005;20:689–696. doi: 10.14670/HH-20.689. [DOI] [PubMed] [Google Scholar]
- 28.Levine B, Klionsky D. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 29.Yoshimori T. Autophagy: a regulated bulk degradation process inside cells. Biochem Biophys Res Commun. 2004;313:453–458. doi: 10.1016/j.bbrc.2003.07.023. [DOI] [PubMed] [Google Scholar]
- 30.Mousavi S, Kjeken R, Berg T, Seglen P, Berg T, Brech A. Effects of inhibitors of the vacuolar proton pump on hepatic heterophagy and autophagy. Biochim Biophys Acta. 2001;1510:243–257. doi: 10.1016/s0005-2736(00)00354-0. [DOI] [PubMed] [Google Scholar]
- 31.Weisz O. Organelle acidification and disease. Traffic. 2003;4:57–64. doi: 10.1034/j.1600-0854.2003.40201.x. [DOI] [PubMed] [Google Scholar]
- 32.Raben N, Takikita S, Pittis M, Bembi B, Marie S, Roberts A, Page L, Kishnani P, Schoser B, Chien Y, Ralston E, Nagaraju K, Plotz P. Deconstructing Pompe disease by analyzing single muscle fibers: to see a world in a grain of sand…. Autophagy. 2007;3:546–552. doi: 10.4161/auto.4591. [DOI] [PubMed] [Google Scholar]