

Summary
A link exists between endoplasmic reticulum (ER) biogenesis and the unfolded protein response (UPR), a complex set of signaling mechanisms triggered by increased demands on the protein folding capacity of the ER. The UPR transcriptional activator X-box binding protein 1 (XBP1) regulates the expression of proteins that function throughout the secretory pathway and is necessary for development of an expansive ER network. We previously demonstrated that overexpression of XBP1(S), the active form of XBP1 generated by UPR-mediated splicing of Xbp1 mRNA, augments the activity of the cytidine diphosphocholine (CDP-choline) pathway for biosynthesis of phosphatidylcholine (PtdCho) and induces ER biogenesis. Another UPR transcriptional activator, activating transcription factor 6α (ATF6α), primarily regulates expression of ER resident proteins involved in the maturation and degradation of ER client proteins. Here, we demonstrate that enforced expression of a constitutively active form of ATF6α drives ER expansion and can do so in the absence of XBP1(S). Overexpression of active ATF6α induces PtdCho biosynthesis and modulates the CDP-choline pathway differently than does enforced expression of XBP1(S). These data indicate that ATF6α and XBP1(S) have the ability to regulate lipid biosynthesis and ER expansion by mechanisms that are at least partially distinct. These studies reveal further complexity in the potential relationships between UPR pathways, lipid production and ER biogenesis.
Keywords: Endoplasmic reticulum biogenesis, Unfolded protein response, ATF6α, XBP1, Lipid biosynthesis
Introduction
All proteins that traffic through the secretory pathway begin their journey when they are translocated as nascent polypeptides into the endoplasmic reticulum (ER). The ER, a network of membrane-bound tubular and sac-like structures, provides an environment that is conducive for the folding and assembly of proteins into correct conformations that are functional and suitable for transport to their final destination (van Anken and Braakman, 2005). When the load of client proteins exceeds ER capacity, a set of inter-organelle signaling pathways responds to the stress by coordinately regulating translation and a broad program of gene transcription. This unfolded protein response (UPR) slows the flow of nascent polypeptides into the ER and increases the supply of ER resident folding assistants, thereby rebalancing protein load with folding capacity in the ER (Ron and Walter, 2007; Schroder and Kaufman, 2005). The UPR-regulated transcription factor X-box binding protein 1 (XBP1) has recently been linked to ER biogenesis (Shaffer et al., 2004; Sriburi et al., 2007; Sriburi et al., 2004), indicating that the UPR can enhance ER capacity by expanding its size.
Xbp1 is required for development of specialized secretory cell types such as antibody-secreting plasma cells (Iwakoshi et al., 2003; Reimold et al., 2001) and pancreatic acinar cells (Lee et al., 2005) that are characterized by expansive networks of rough ER. The UPR regulates XBP1 through a novel mechanism of mRNA splicing initiated by IRE1 (inositol-requiring mutant, first identified in yeast; also known as ERN1), an ER transmembrane kinase/endoribonuclease (Tirasophon et al., 1998; Wang et al., 1998). Upon activation, IRE1 excises a 26 nt sequence from Xbp1 mRNA. Ligation of the resulting 5′ and 3′ fragments yields a transcript that encodes XBP1(S), a basic leucine zipper (bZIP) protein with a strong transactivation domain (Calfon et al., 2002; Shen et al., 2001; Yoshida et al., 2001). Overexpression studies have demonstrated that XBP1(S) is sufficient to trigger expansion of rough ER (Sriburi et al., 2004), and this correlates with increased phospholipid biosynthesis and the expression of many proteins that function in the secretory pathway (Shaffer et al., 2004; Sriburi et al., 2007). These findings fit well with the essential role of Xbp1 in professional secretory cells; however, it remains unclear whether the ability to modulate ER abundance is unique to XBP1(S) or might also be a property of other UPR-regulated factors.
In addition to IRE1, two other ER transmembrane proteins, PKR-like ER kinase (PERK; EIF2AK3) (Harding et al., 1999; Shi et al., 1998) and activating transcription factor 6 (ATF6) (Haze et al., 2001; Haze et al., 1999), serve as proximal transducers of UPR pathways. When activated, PERK phosphorylates the α subunit of eukaryotic initiation factor 2 (eIF-2α; EIF2S1). This event efficiently impedes the formation of translation initiation complexes, providing a means for rapid repression of protein synthesis in response to ER stress (Harding et al., 2000; Scheuner et al., 2001). However, these conditions favor translation of activating transcription factor 4 (ATF4) owing to the presence of regulatory small open reading frames in the 5′ untranslated region of its mRNA (Lu et al., 2004; Vattem and Wek, 2004). ATF4 activates genes involved in the synthesis and transport of amino acids, the response to oxidative stress and apoptosis induced by chronic ER stress (Harding et al., 2003). The PERK pathway is required for the proper development and function of specialized secretory cells in the pancreas and skeletal system (Harding et al., 2001; Scheuner et al., 2005; Zhang et al., 2002; Zhang et al., 2006), but it has not been implicated in ER biogenesis.
The two isoforms of ATF6, α and β, each have an N-terminal cytosolic domain that includes a bZIP region and a transactivation domain (Haze et al., 2001; Haze et al., 1999). Onset of ER stress causes transport of ATF6 from the ER to the Golgi. The site 1 (MBTPS1) and site 2 (MBTPS2) proteases then cleave ATF6, releasing its cytosolic domain from the membrane to move into the nucleus and function as a transcriptional activator (Ye et al., 2000). Although ATF6α and β both have the ability to transactivate ER stress-responsive promoters (Haze et al., 2001; Haze et al., 1999), only ATF6α is essential for induction of certain UPR target genes (Wu et al., 2007; Yamamoto et al., 2007). Many of the genes regulated by ATF6α encode ER resident molecular chaperones, folding enzymes and factors involved in ER-associated degradation (ERAD) of misfolded proteins (Adachi et al., 2008; Wu et al., 2007; Yamamoto et al., 2007), indicating that ATF6α plays a major role in enhancing the capacity of the ER to properly deal with client proteins. Therefore, we hypothesized that ATF6α might also regulate ER abundance.
Here, we report that ATF6α, but neither ATF6β nor ATF4, has the ability to trigger expansion of the ER. This ATF6α-induced ER expansion can occur in the absence of XBP1 and involves induction of phospholipid biosynthesis by mechanisms that are at least partially distinct from those previously characterized for XBP1(S) (Sriburi et al., 2007; Sriburi et al., 2004). These data suggest that both the ATF6α and IRE1/XBP1 pathways of the UPR can be utilized to modulate lipid biosynthesis, ER biogenesis and the capacity of the secretory pathway.
Results
Expression of HA-tagged UPR transcription factors in CHO cells
To investigate the effects of ATF6α on the ER, we first established a system to transiently express hemagglutinin (HA)-tagged UPR transcriptional activators in Chinese hamster ovary (CHO) cells. We expressed the N-terminal 1-373 amino acids (aa) of ATF6α, termed ATF6α(1-373), which localizes to the nucleus and constitutively activates the transcription of many UPR target genes (Wang et al., 2000; Yoshida et al., 2000). In parallel, we transiently expressed an ATF6α mutant (KNR to TAA at aa 315-317), termed ATF6α(1-373)m1, that lacks transactivation activity (Wang et al., 2000). CHO cells were also transfected with constructs encoding ATF6β(1-393) (N-terminal domain, aa 1-393), XBP1(S) and ATF4. All of these constructs encode a bicistronic mRNA allowing for co-expression of the HA-tagged factors and enhanced green fluorescent protein (EGFP). As expected, immunofluorescence revealed efficient nuclear localization of ATF6α(1-373), whereas ATF6α(1-373)m1 was distributed throughout the cytoplasm (Fig. 1A). ATF6β(1-393), XBP1(S) and ATF4 primarily localized to the nucleus (Fig. 1A). Co-transfection of a UPR element (UPRE)-luciferase reporter construct revealed the transactivation activity of the transiently expressed HA-tagged factors. As expected (Haze et al., 2001; Lee et al., 2002; Wang et al., 2000; Yoshida et al., 2001), the UPRE-driven luciferase reporter was strongly induced by ATF6α(1-373), ATF6β(1-393) and XBP1(S) (Fig. 1B). By contrast, neither ATF6α(1-373)m1 nor ATF4 induced UPRE-mediated transcription (Fig. 1B).
Fig. 1.

Expression of HA-tagged UPR transcription factors in CHO cells. (A) CHO cells were transiently transfected with a vector expressing EGFP alone or vectors expressing EGFP and HA-tagged ATF6α(1-373), ATF6α(1-373)m1, ATF6β(1-393), XBP1(S) or ATF4. At 40 hours post-transfection, cells were fixed, stained with anti-HA monoclonal antibody and DAPI, and examined by fluorescence microscopy. (a) EGFP visualized with a FITC filter; (b) HA-tagged proteins visualized with a TRITC filter; (c) nuclei of cells stained with DAPI; (d) merged FITC, TRITC and DAPI images. Scale bars: 20 μm. (B) CHO cells were co-transfected with a reporter plasmid containing the firefly luciferase gene under the control of 5×ATF6 binding sites, a plasmid containing a lacZ gene under control of the CMV promoter and increasing amounts of the expression vectors as indicated. The total amount of expression vector DNA was maintained at a constant level by adding vector control DNA as necessary. At 40 hours post-transfection, cells were harvested and the relative ratio of firefly luciferase to β-galactosidase activity in each cell lysate was determined. The mean ± s.d. of three independent experiments is plotted.
Next, we transfected these expression vectors into CHO cells and then isolated EGFP-fluorescent cells by fluorescence-activated cell sorting (FACS). Immunoblot analysis of the sorted cells revealed similar levels of EGFP protein in each population, indicating comparable transfection efficiency and EGFP expression for each of the vectors (Fig. 2A). However, the respective HA-tagged UPR factors were present in variable amounts, with XBP1(S) and ATF4 at the lowest and highest level, respectively (Fig. 2A). The truncated forms of ATF6α and β were detected in similar quantities (Fig. 2A). The identities of the expressed UPR factors, except ATF4, were confirmed by immunoblotting with antibodies specific to each protein (Fig. 2B). In keeping with previous reports (Okada et al., 2002), we found significantly higher levels of ER chaperones and folding enzymes in the ATF6α(1-373)-expressing CHO cells, including GRP94 (HSP90B1), GRP78 (BiP; HSPA5), calreticulin, ERp72 (PDIA4) and protein disulfide isomerase (PDI; P4HB) (Fig. 2C). By contrast, these ER proteins were not upregulated in CHO cells expressing either ATF6α(1-373)m1 or the other UPR factors (Fig. 2C). Two components of the ER translocon, TRAPα (SSR1) and SEC61β, appeared to be slightly increased in the XBP1(S)-expressing cells (Fig. 2C), a finding consistent with other studies (Shaffer et al., 2004; Sriburi et al., 2007). Finally, the UPR transcription factor CHOP (DDIT3) was significantly expressed in the ATF6α(1-373)-expressing cells, whereas cells expressing the other UPR factors exhibited much lower levels of CHOP. Together, these data demonstrate that the HA-tagged UPR transcription factors were expressed and exhibited appropriate activity on known UPR target genes in the transfected CHO cells.
Fig. 2.

Immunoblot analysis of FACS-isolated CHO cells expressing HA-tagged UPR transcription factors. CHO cells were transfected with the indicated expression vectors. At 40 hours post-transfection, EGFP+ cells were collected by FACS. Cell lysates were prepared and analyzed by immunoblotting using antibodies against (A) the HA epitope and EGFP proteins, (B) actin, ATF6α, ATF6β and XBP1(S) proteins, (C) the KDEL sequence (GRP94 and GRP78) and the CHOP, calnexin (CNX), calreticulin (CRT), ERp72, protein disulfide isomerase (PDI), ribophorin, TRAPα and SEC61β proteins. The same lysates were used for all immunoblots.
Electron microscopy analysis of the ER in CHO cells expressing HA-tagged UPR transcription factors
The capacity of ATF6α to modulate the structure and abundance of the ER was then assessed using the CHO cell system for transient overexpression of the various UPR transcription factor constructs. To directly assess ER structure, transmission electron microscopy (TEM) analysis was performed on EGFP+ cells isolated by FACS. Remarkably, the majority of the ATF6α(1-373)-expressing cells exhibited profound alterations in ER morphology (Fig. 3A). The rough ER, as defined by the ribosomes studded along the ER membrane (Fig. 3A, lower right panel), in the ATF6α(1-373)-expressing cells was enlarged and exhibited a distended morphology (Fig. 3A, lower panels). This enlargement of the ER was not evident in cells expressing either ATF6α(1-373)m1 or any of the other UPR transcription factor constructs, including XBP1(S) (Fig. 3B). Consistent with these data, fluorescence microscopy analysis of CHO cells transiently co-expressing an ER-localized red fluorescent protein (DsRed2-ER) and ATF6α(1-373) revealed a mesh-like ER network that appeared more highly developed and prominent than that observed in cells co-expressing DsRed2-ER and ATF6α(1-373)m1 (supplementary material Fig. S1).
Fig. 3.

Electron microscopy analysis of the ER in CHO cells expressing HA-tagged UPR-transcription factors. CHO cells were transfected with the indicated expression vectors. At 40 hours post-transfection, EGFP+ cells were collected by FACS and then examined by TEM. (A) Vector alone- or ATF6α(1-373)-expressing cells at 4600× (left) and 25,000× (right). (B) ATF6α(1-373)m1-, ATF6β(1-393)-, XBP1(S)- and ATF4-expressing cells at 25000×. Arrows point to representative ER. For ATF6α(1-373), 19/20 cells examined by TEM exhibited ER expansion; for vector alone, ATF6α(1-373)m1, ATF6β(1-393) and ATF4, 0/20 cells and for XBP1(S), 0/50 cells, examined by EM exhibited ER expansion. Scale bars: 2 μm in 4600× frames and 500 nm in 25000× frames.
Since XBP1(S) has been shown to induce expansion of the rough ER in NIH-3T3 mouse fibroblast cells (Sriburi et al., 2004), we transfected our constructs encoding HA-tagged ATF6α(1-373) and XBP1(S) into NIH-3T3 cells and examined FACS-isolated EGFP+ cells by TEM. In NIH-3T3 cells, both ATF6α(1-373) and XBP1(S) induced expansion of the rough ER (supplementary material Fig. S2). Another study found evidence for ER expansion in a CHO cell line engineered to overexpress XBP1(S) (Tigges and Fussenegger, 2006); thus, the effects of XBP1(S) on the ER might vary among cell lines or be influenced by its expression level. In addition to CHO and NIH-3T3 cells, we also observed ER expansion upon overexpression of ATF6α(1-373) in two human cell lines (293 and HeLa) (supplementary material Figs S3 and S4). These data demonstrate that ATF6α(1-373), like XBP1(S), has the capacity to drive expansion of the rough ER. Furthermore, modulation of ER structure and abundance by ATF6α(1-373) is dependent on its ability to function as a transcriptional activator and is not cell type specific.
Analysis of XBP1 in ATF6α-induced expansion of the ER
Previous studies have shown that ATF6α(1-373) can strongly induce expression of Xbp1 mRNA (Yoshida et al., 2000). These transcripts can be modified by UPR-mediated splicing to encode XBP1(S), a factor that can direct ER biogenesis. Therefore, we reasoned that ER expansion in cells overexpressing ATF6α(1-373) might be due to induction of XBP1(S). To investigate this possibility, we first assessed the status of Xbp1 mRNA in CHO cells expressing ATF6α(1-373). Based on the sequence of a partial cDNA for hamster Xbp1 (supplementary material Fig. S5), primers were designed to amplify products derived from unspliced (306 bp product) and UPR-spliced (279 bp product) Xbp1 mRNA. Using these primers in RT-PCR, we observed that the Xbp1 mRNA in CHO cells under normal conditions was mostly unspliced (Fig. 4A, lane 1). As expected, treatment of cells with tunicamycin, an inhibitor of N-linked glycosylation that potently induces the UPR, led to an apparent increase in Xbp1 mRNA and robust UPR-mediated splicing (Fig. 4A, lane 2). Analysis of FACS-isolated ATF6α(1-373)-expressing cells revealed an apparent increase in Xbp1 mRNA as compared with both empty vector- and ATF6α(1-373)m1-expressing cells (Fig. 4A, lanes 3-5). Most importantly, the vast majority of Xbp1 transcripts in ATF6α(1-373)-expressing cells had not been modified by UPR-mediated splicing (Fig. 4A, lane 4), indicating that induction of XBP1(S) was not responsible for ER expansion in these cells. We then transfected Xbp1–/– mouse embryo fibroblasts (MEFs) with the ATF6α(1-373) expression vector and examined FACS-isolated EGFP+ cells by TEM. ATF6α(1-373)-expressing Xbp1–/– cells exhibited conspicuous enlargement and distension of the ER (Fig. 4B). These data demonstrate that ATF6α(1-373) can drive ER expansion by XBP1-independent mechanisms.
Fig. 4.

Analysis of XBP1 in ATF6α(1-373)-induced ER expansion. (A) CHO cells were transfected with the indicated expression vectors. At 40 hours post-transfection, EGFP+ cells were collected by FACS. As controls, CHO cells were left untreated (–) or treated with tunicamycin (Tm) for 6 hours and harvested. Total RNA was isolated and equivalent amounts of RNA (200 ng) from each sample were analyzed by RT-PCR using a primer set that amplifies hamster Xbp1 mRNA. Unspliced (Un) and UPR-spliced (S) Xbp1 transcripts yield 306 bp and 279 bp PCR products, respectively. (B) Xbp1–/– MEFs were nucleofected with the indicated expression vectors. At 40 hours post-transfection, EGFP+ cells were collected by FACS and then examined by TEM at the indicated magnifications. Arrows point to representative ER. For vector alone, 2/20 cells, and for ATF6α(1-373), 18/20 cells, examined by TEM exhibited ER expansion. Scale bars: 2 μm in 7900× frames and 500 nm in 19,000× frames.
Effect of ATF6α(1-373) on cellular lipids
In previous studies of XBP1(S)-induced ER biogenesis, we used retroviral transduction to enforce expression of this transcription factor in NIH-3T3 cells (Sriburi et al., 2007; Sriburi et al., 2004). Retroviral transduction is highly efficient (>90%) for NIH-3T3 cells, making it possible to perform analysis on bulk populations of transduced cells. Thus, we reasoned that this approach might facilitate further investigation of ATF6α(1-373)-mediated effects. We verified that expression of ATF6α(1-373) by retroviral transduction of NIH-3T3 cells results in enlargement and distension of the ER (supplementary material Fig. S6A) as we had observed in the transient transfection studies (Fig. 3; supplementary material Fig. S2). Importantly, UPR-mediated splicing of Xbp1 mRNA was not upregulated in ATF6α(1-373)-transduced cells (supplementary material Fig. S6B). These data prompted us to use retroviral transduction of NIH-3T3 cells as an experimental system to examine the effect of ATF6α(1-373) on the abundance of cellular lipids. The polar phospholipids phosphatidylcholine (PtdCho) and phosphatidylethanolamine (PtdEtn) increased by 50-60% in ATF6α(1-373)-transduced cells, whereas the level of sphingomyelin was unchanged (Fig. 5A). By contrast, there was a small increase in cholesterol and a decrease in cholesterol ester (Fig. 5A). The substantial increase in PtdCho and PtdEtn, which are major components of intracellular membranes, corroborates the observed ER expansion in ATF6α(1-373)-transduced cells.
Fig. 5.
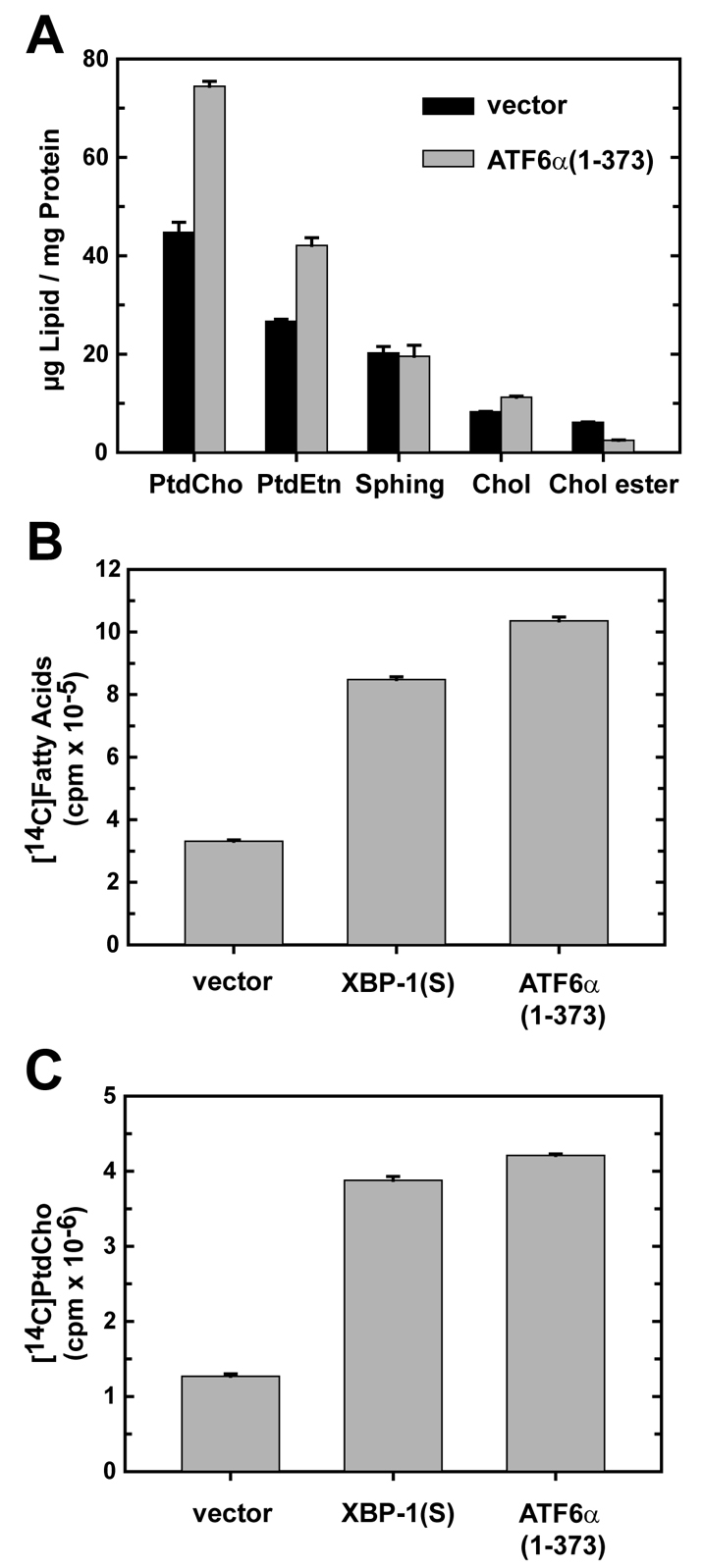
Analysis of lipid abundance and lipid biosynthesis in NIH-3T3 cells transduced with ATF6α(1-373). (A) NIH-3T3 cells were transduced with empty vector (black bars) or ATF6α(1-373) (gray bars) retroviruses and assessed at 48 hours post-transduction. Total amounts of phosphatidylcholine (PtdCho), phosphatidylethanolamine (PtdEtn), cholesterol (Chol), sphingolipid (Sphing) and cholesterol ester (Chol ester) were determined by flame ionization and normalized to total cellular protein. The results are the mean ± s.d. of triplicate determinations and are representative of two independent experiments. (B,C) NIH-3T3 cells were transduced with the indicated retroviruses. At 48 hours post-transduction, cells were metabolically labeled with [14C]acetate for 2 hours. The incorporation of radiolabel into total fatty acids (B) and PtdCho (C) was determined as described in Materials and Methods and normalized to 107 cells. The data are the mean ± s.d. of triplicate determinations and are representative of two independent experiments.
We then determined whether the increase in phospholipids in ATF6α(1-373)-transduced cells correlated with upregulation of de novo lipid biosynthesis. Fatty acids are necessary for phospholipid production; thus, we performed metabolic labeling studies with radiolabeled acetate and measured both fatty acid and phospholipid biosynthesis. For comparison, XBP1(S)-transduced cells were assessed in parallel. In both XBP1(S)- and ATF6α(1-373)-transduced cells, fatty acid biosynthesis increased significantly (∼three- to 3.5-fold) (Fig. 5B). Synthesis of PtdCho, the most abundant phospholipid in cellular membranes, was strongly induced (∼fourfold) upon enforced expression of either ATF6α(1-373) or XBP1(S) (Fig. 5C). Together, these data demonstrate that ATF6α(1-373), like XBP1(S), can trigger events that augment the biosynthesis and abundance of PtdCho.
Effect of ATF6α(1-373) on the CDP-choline pathway for PtdCho biosynthesis
As a key component of cellular membranes, PtdCho plays a crucial role in ER biogenesis (Sriburi et al., 2007). Therefore, we investigated the underlying mechanism for increased production of PtdCho in ATF6α(1-373)-transduced cells. In mammalian cells, the cytidine diphosphocholine (CDP-choline) pathway, also known as the Kennedy pathway, is primarily responsible for PtdCho biosynthesis (Lykidis and Jackowski, 2001). First, choline kinase (CK) phosphorylates choline in the presence of ATP, producing phosphocholine. Second, in the presence of phosphocholine cytidylyltransferase (CTP), choline cytidylyltransferase (CCT) utilizes phosphocholine to produce CDP-choline, and this is considered to be the rate-limiting step in the CDP-choline pathway (Jackowski and Fagone, 2005). Finally, either choline phosphotransferase (CPT1; CHPT1) or choline/ethanolamine phosphotransferase (CEPT1) transfers the phosphocholine moiety of CDP-choline to diacylglycerol (DAG), yielding PtdCho. Here, we consider the activities of CPT1 (Henneberry et al., 2000) and CEPT1 (Henneberry and McMaster, 1999) collectively as CPT activity. In ATF6α(1-373)-transduced cells, CK activity increased ∼threefold (Fig. 6A), CCT activity was unchanged (Fig. 6B), and CPT activity was elevated ∼fourfold (Fig. 6C). Interestingly, our previous work showed that XBP1(S)-transduced cells exhibit no change in CK activity, a small increase in CCT activity and a large increase in CPT activity (Sriburi et al., 2004). These data indicate that ATF6α(1-373) and XBP1(S) have the capacity to differentially modulate enzymatic activities of the CDP-choline pathway, thereby enhancing PtdCho biosynthesis by distinct mechanisms.
Fig. 6.
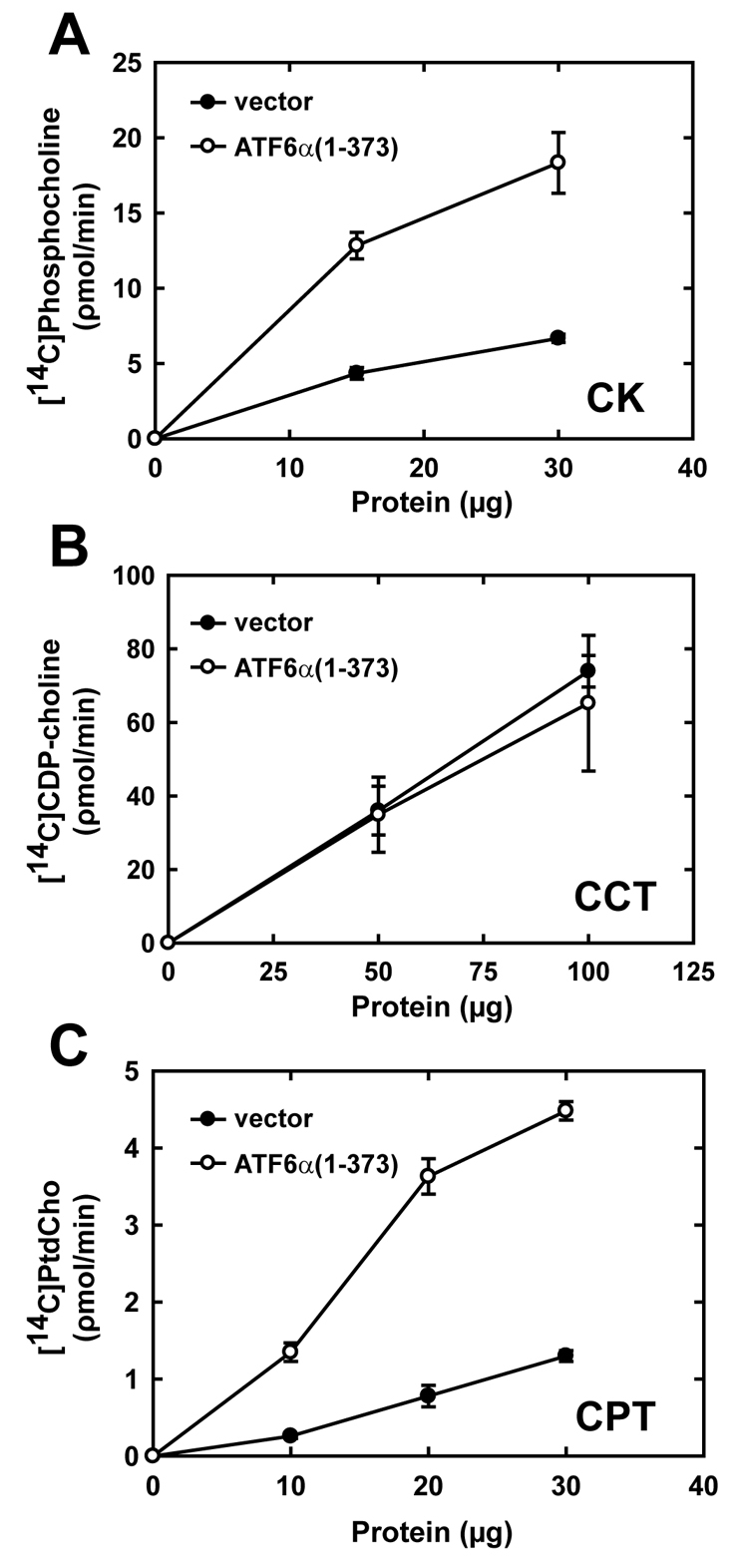
Enzymatic activities in the CDP-choline pathway of PtdCho synthesis in NIH-3T3 fibroblasts transduced with ATF6α(1-373). The relative enzymatic activities of CK, CCT and CPT were determined using lysates or microsomes prepared from NIH-3T3 cells harvested 48 hours post-transduction with empty vector (black circle) and ATF6α(1-373) (white circle) retroviruses. The rates of production of phosphocholine, CDP-choline and PtdCho were compared as a function of total protein in each assay and are plotted as the mean ± s.d. obtained for each protein concentration. (A) Data for CK are averaged from quadruplicate determinations and are representative of two independent experiments. (B) Data for CCT are averaged from six determinations obtained in two independent experiments. (C) Data for CPT are averaged from triplicate determinations and are representative of two independent experiments.
Quantitative analysis of gene expression revealed a substantial elevation in mRNA for both the α and β isoforms of CK in ATF6α(1-373)-transduced cells (Fig. 7), suggesting that increased expression accounts for the increased CK activity in this system. In contrast to ATF6α(1-373), enforced expression of XBP1(S) had no effect on CK expression (supplementary material Table S1). The levels of mRNA for CCT, CPT1 and CEPT1 were unchanged in ATF6α(1-373)-transduced cells (Fig. 7), analogous to previous findings for the XBP1(S) system (Sriburi et al., 2004). Thus, the large increase in CPT activity that occurs in both ATF6α(1-373)- and XBP1(S)-transduced cells must be regulated by post-transcriptional control of CPT1 and/or CEPT1.
Fig. 7.
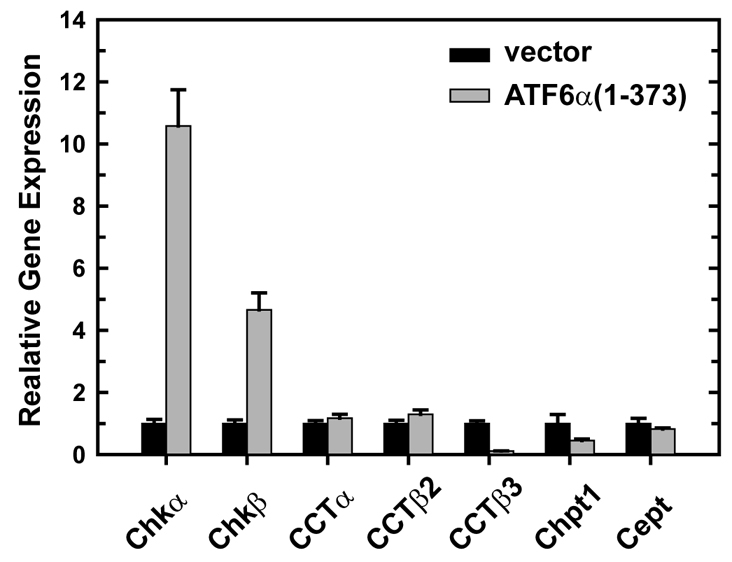
Expression of CDP-choline pathway enzymes in NIH-3T3 fibroblasts transduced with ATF6α(1-373). NIH-3T3 cells were transduced with empty vector (black bars) or ATF6α(1-373) (gray bars) retroviruses and harvested at 48 hours post-transduction. Total RNA was prepared and the relative levels of expression of the CK α and β isoforms (Chkα and Chkβ), the CCT isoforms α, β2 and β3, and Chpt1 and Cept1 were measured by quantitative real-time PCR using gene-specific primers and probes. The amount of target RNA was normalized to endogenous Gapdh as a reference. The mean ± s.e.m. of triplicate determinations is plotted and is representative of two independent experiments.
Effect of ATF6α(1-373) on the secretory pathway
When cells are exposed to pharmacological agents that disrupt protein folding in the ER, ATF6α primarily upregulates expression of ER quality-control proteins including chaperones, folding enzymes and ERAD components (Adachi et al., 2008; Wu et al., 2007; Yamamoto et al., 2007). Therefore, we asked whether a similar pattern of gene expression occurred upon enforced expression of ATF6α(1-373) in NIH-3T3 cells. Microarray analyses revealed that the expression of many secretory pathway genes (∼60 genes) was enhanced in ATF6α(1-373)-transduced cells, with the largest subgroup being genes for ER chaperones and folding enzymes (Table 1). In addition, there was increased expression of genes involved in the targeting and translocation of nascent polypeptides into the ER (four genes), genes implicated in various aspects of transport within the secretory pathway (16 genes) and genes linked to ERAD (four genes) in cells expressing ATF6α(1-373) (Table 1). We reasoned that these changes in gene expression were consistent with expansion of the ER and an increase in secretory capacity. Our previous studies showed that enforced expression of XBP1(S) in NIH-3T3 cells results in elevated expression of ∼120 secretory pathway genes (Sriburi et al., 2007). By comparison with the ATF6α(1-373) microarray data, it is notable that XBP1(S)-transduced cells exhibit induction of much larger groups of genes involved in targeting and translocation (19 genes) and in transport within the secretory pathway (40 genes), as well increased expression of ER chaperones, folding enzymes and ERAD components (supplementary material Table S2). Thus, we predicted that enforced expression of XBP1(S) would augment secretory capacity to a greater extent than ATF6α(1-373). To explore the effects of ATF6α(1-373) and XBP1(S) on secretory output, we enforced expression of these factors in NIH-3T3 cells stably expressing secreted alkaline phosphatase (SEAP) and monitored secretion of active SEAP using a chemiluminescence assay. Enforced expression of either ATF6α(1-373) or XBP1(S) enhanced secretion of SEAP, with XBP1(S) having the greatest effect (Fig. 8A). To ensure that these effects were not clone specific, we performed the experiment using two separate clones of NIH-3T3 cells expressing SEAP (3T3-SEAP.4 and 3T3-SEAP.9). ATF6α(1-373) and XBP1(S) augmented secretion of SEAP by both clones by ∼fourfold and sevenfold, respectively (Fig. 8B). These data demonstrate that active ATF6α and XBP1(S) both have the ability to drive the expansion of a functional ER. Moreover, ATF6α(1-373) facilitates enhanced production of secretory cargo, but to a lesser extent than does XBP1(S), which appears to serve as a master regulator of the entire secretory pathway.
Table 1.
Secretory pathway genes upregulated in ATF6α(1-373)-transduced NIH-3T3 fibroblasts
| Function/location | Gene |
|---|---|
| Targeting and translocation | |
| Signal sequence receptor | Ssr3* |
| Translocon | Sec63* |
| Signal peptidase | Sec11l1*, Sec11l3* |
| N-linked glycosylation | |
| Core oligosaccharide synthesis | Alg12* |
| Oligosaccharyltransferase | Ddost*, Rpn1*, Rpn2* |
| Protein folding | |
| Chaperones | Dnajb9§, Dnajb11‡, Dnajc10*, Fkbp2*, Fkbp7‡, Fkbp11†, Fkbp14*, Hspa5*, Hsp90b1†, Hyou1‡, Ppib* |
| Disulfide bond formation | Ero1b‡, Erp29*, Pdia3†, Pdia4§, Pdia6*, Txndc4*, Txndc11† |
| ER-associated degradation | Derl1†, Edem1‡, Herpud1‡, Syvn1§ |
| Vesicular trafficking and transport | |
| Anterograde transport (ER→Golgi) | |
| COPII vesicles | Sec23a†, Sec24d*, Sec31l1*, Yif1a* |
| Cargo receptors | Mcfd2* |
| SNAREs | Betl1* |
| Retrograde transport (ER←Golgi) | |
| COPI vesicles | Arfgap3*, Copz1* |
| Cargo receptors | Kdelr3* |
| Transport/recycling in the Golgi | Blzf1*, Vdp* |
| Exocytosis | |
| SNAREs | Stx5a* |
| Small GTPases | Rab3a* |
| Others | |
| ER proteins | Atp2a2*, Creb3*, Eif2ak3†, Ggcx*, Hmox1†, Ormdl3*, Rrbp1†, Rcn3*, Piga†, Sdf2l1§, Surf4*, Wfs1† |
| Golgi proteins | Golga3*, Golgb1*, Gcc1*, Gopc†, Rabac1* |
Affymetrix microarray analysis revealed that a large subset of genes encoding proteins that function in the secretory pathway were upregulated in ATF6α(1-373)-transduced NIH-3T3 fibroblasts (≥2-fold as compared with empty vector controls; P<0.05). These genes are grouped according to function and/or location. Fold increase in expression in ATF6α(1-373)-transduced versus empty vector-transduced cells: *≥2 to <5; †≥5 to <10; ‡≥10 to <20; §≥20
Fig. 8.
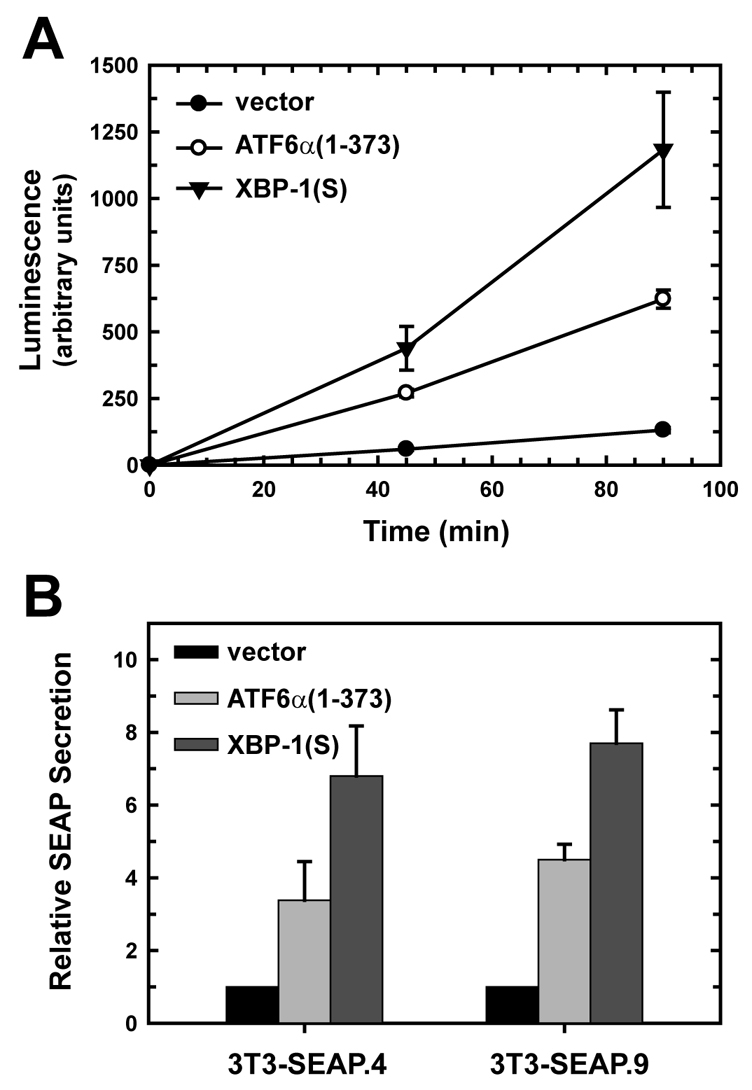
Effect of ATF6α(1-373) and XBP1(S) on the secretory activity of NIH-3T3 fibroblasts. (A) NIH-3T3 cells stably expressing secreted alkaline phosphatase (SEAP) were transduced with empty vector (black circle), ATF6α(1-373) (white circle) and XBP1(S) (triangle) retroviruses. At 48 hours post-transduction, cells were washed, shifted into fresh media and then cultured for 45 or 90 minutes. Culture supernatants harvested at each time were assessed for SEAP activity using a chemiluminescence assay, and luminescence readings were normalized to cell number. The mean ± s.d. of triplicate determinations is plotted and is representative of five independent experiments. (B) Two separate clones of SEAP-expressing NIH-3T3 cells (3T3-SEAP.4 and 3T3-SEAP.9) were transduced with empty vector (black bars), ATF6α(1-373) (light-gray bars) or XBP1(S) (dark-gray bars) retroviruses. SEAP secretion was assessed 48 hours post-transduction as described in A and calculated as the level relative to SEAP secretion by empty vector-transduced cells (set at 1). The mean ± s.d. is plotted (3T3-SEAP.4, n=3; 3T3-SEAP.9, n=2).
Discussion
Recent studies have uncovered links between the UPR transcriptional activator XBP1(S), lipid biosynthesis, ER biogenesis and proper development of specialized secretory cell types. Our current data demonstrate that the UPR transcription factor ATF6α also possesses the ability to drive lipid biosynthesis and expansion of the ER. By contrast, neither ATF6β nor ATF4 affected the morphology or size of the ER in our experimental system. Importantly, ATF6α-driven ER expansion was not accompanied by an increase in UPR-mediated splicing of Xbp1 mRNA. Although enforced expression of active ATF6α upregulates Xbp1 mRNA (Yoshida et al., 2000), it apparently does not elicit events that trigger increased IRE1 activity. These findings argue that the increased lipid biosynthesis and ER expansion that occur upon enforced expression of active ATF6α cannot be attributed to induction of XBP1(S). Furthermore, robust enlargement of the ER was observed when active ATF6α was overexpressed in Xbp1–/– MEFs. Therefore, ATF6α can induce XBP1(S)-independent expansion of the ER.
The process of ER expansion requires an increased supply of the lipids necessary for membrane biogenesis. Phospholipids, such as PtdCho and PtdEtn, are particularly enriched in ER membranes, and these were increased substantially in cells overexpressing active ATF6α. Metabolic labeling studies with radiolabeled acetate revealed that enforced expression of active ATF6α augmented the biosynthesis of both fatty acids and PtdCho. Moreover, these increases in de novo lipid biosynthesis were similar to those observed for cells overexpressing XBP1(S). It is noteworthy that in a previous study we observed a reduction in the incorporation of radiolabeled choline into PtdCho in ATF6α(1-373)-transduced NIH-3T3 cells (Sriburi et al., 2004), a finding that seems inconsistent with our current data. However, we have recently found that choline uptake is severely reduced in ATF6α(1-373)-transduced cells (data not shown), providing an explanation for the results of the earlier choline-labeling experiments. We speculate that this phenomenon might be related to the high CK activity present in ATF6α(1-373)-transduced cells, reasoning that the predicted increase in the pool of intracellular phosphocholine might feedback to slow the uptake of additional choline. Whatever the exact mechanism, it is clear that the choline supply is sufficient to support a ∼fourfold increase in PtdCho biosynthesis in ATF6α(1-373)-transduced cells.
The predominant means for PtdCho biosynthesis in mammalian cells is the CDP-choline pathway, and CCT, which converts phosphocholine to CDP-choline in the presence of CTP, is considered to be rate-limiting in this biosynthetic process (Jackowski and Fagone, 2005). Indeed, our previous work revealed that the elevated PtdCho biosynthesis in XBP1(S)-transduced NIH-3T3 cells can be attributed to increased CCT activity (Sriburi et al., 2007). Thus, it was surprising that the increase in PtdCho biosynthesis in cells overexpressing active ATF6α did not correlate with a measurable increase in CCT activity. However, CK activity, which is strongly upregulated in ATF6α(1-373)-transduced (Fig. 6A) but not XBP1(S)-transduced (Sriburi et al., 2004) cells, appears regulatory for the CDP-choline pathway in some systems (Kent, 2005). These findings, coupled with the fact that elevated CPT activity alone is not sufficient to augment PtdCho production (Sriburi et al., 2007; Wright et al., 2001), suggest that increased CK activity plays a key role in driving PtdCho biosynthesis in ATF6α(1-373)-transduced cells.
Studies of Atf6α–/– cells have recently shown that ATF6α is primarily responsible for transcriptional induction of a cohort of ER proteins including chaperones, folding enzymes and ERAD components (Adachi et al., 2008; Wu et al., 2007; Yamamoto et al., 2007). By contrast, XBP1(S) targets some of these same genes as well as a variety of others that are involved at multiple steps in the secretory pathway, such as targeting and translocation of nascent polypeptides into the ER and vesicular transport (Lee et al., 2003; Shaffer et al., 2004). These gene expression profiles are in close agreement with our analysis of secretory pathway gene expression in ATF6α(1-373)- and XBP1(S)-transduced NIH-3T3 cells (Table 1 and supplementary material Table S2). Thus, ATF6α provides more `machinery' for the folding and disposal of proteins by the ER, whereas XBP1(S) directs a broader program that also includes `machinery' to boost production and transport of secretory cargo. Our observation that XBP1(S) augmented SEAP secretion to a greater extent than did active ATF6α (Fig. 8) supports this model. It will be particularly interesting to investigate whether ATF6α plays any roles in the development and function of professional secretory cells. In this regard, activation of ATF6α has been observed in B-lymphocytes differentiating into antibody-secreting cells (Gass et al., 2002; Gass et al., 2008).
A small group of lipid metabolism genes was upregulated in ATF6α(1-373)-transduced cells (supplementary material Table S1). For example, in addition to the choline kinase genes (Chkα and Chkβ), the genes encoding acetyl-coenzyme A carboxylase 2 (Acacb), a major regulator of fatty acid oxidation in mitochondria, and fatty acid synthase (Fasn), a key enzyme in fatty acid biosynthesis, were strongly induced in cells overexpressing active ATF6α. Interestingly, these genes were not upregulated in XBP1(S)-transduced cells (supplementary material Table S1), providing further evidence that ATF6α and XBP1(S) differ in their abilities to affect various aspects of lipid metabolism. Presently, we have no evidence that the Chkα/β, Acacb and Fasn genes are induced by pharmacological agents that trigger the UPR and induce known ATF6α target genes; thus, it seems unlikely that they are directly regulated by ATF6α. Rather, it seems that ATF6α has the ability to indirectly influence events that govern cellular lipid supplies. Further investigation of these issues is warranted and might shed light on the mechanisms by which UPR pathways are utilized to meet the demands of lipid metabolism as well as the demands of the secretory pathway.
Materials and Methods
Expression vectors
Both pCGNATF6α-(1-373) and pCGNATF6α-(1-373)m1 were generously provided by Ron Prywes (Columbia University, New York, NY). The plasmid pCGNATF6α-(1-373) encodes the N-terminal domain of human ATF6α (aa 1-373) with an HA epitope tag at the N-terminus. The plasmid pCGNATF6α-(1-373)m1 is the same as pCGNATF6α-(1-373) except that aa 315-317 were changed from KNR to TAA as described previously (Wang et al., 2000). To construct pCGNATF6α-(1-373)-IRES-EGFP and pCGNATF6α-(1-373)m1-IRES-EGFP, the NheI-NotI internal ribosome entry site (IRES)-EGFP fragment from pIRES2-EGFP (Clontech, Mountain View, CA) was treated with Klenow polymerase and inserted into pCGNATF6α-(1-373) and pCGNATF6α-(1-373)m1 vectors that had been digested with BamHI and treated with Klenow polymerase. pCGNATF6β-(1-393)-IRES-EGFP was constructed by amplifying a cDNA fragment (aa 1-393) of human ATF6β from tunicamycin-treated 293T cells by standard RT-PCR using a random primer and specific primers: hATF6β-F, 5′-TTTCCTTCTAGAATGGCGGACCTGATGCTG-3′; hATF6β-R, 5′-TTTCCTAGCGCTCAAGACCCTAACTTGAGCTC-3′. The ATF6β(1-393) cDNA fragment was digested with XbaI and Eco47III and inserted into pCGNATF6α-(1-373)-IRES-EGFP that had been digested with XbaI and Eco47III, yielding pCGNATF6β-(1-393)-IRES-EGFP.
pCGNXBP1-(S)-IRES-EGFP was constructed by amplifying a cDNA encoding UPR-spliced human XBP1 from pcDNA-XBP1(S) (Yoshida et al., 2001) by standard PCR using specific primers: hXBP1-s-F, 5′-TTTCCTTCTAGAATGGTGGTGGTGGCAGCC-3′; hXBP1-s-R, 5′-TTTCCTGTCGACTTAGACACTAATCAGCTGGG-3′. The XBP1(S) cDNA was digested with XbaI and HincII and inserted into pCGNATF6α-(1-373)-IRES-EGFP that had been digested with XbaI and Eco47III, yielding pCGNXBP1-(S)-IRES-EGFP. pCGNATF4-IRES-EGFP was constructed by amplifying a cDNA encoding mouse ATF4 from NIH-3T3 cells by standard RT-PCR using a random primer and specific primers: mATF4-F, 5′-TTTCCTTCTAGAATGAGCTTCCTGAACAGCG-3′; mATF4-R, 5′-TTTCCTAGCGCTCTACGGAACTCTCTTCTTCCC-3′. The ATF4 cDNA fragment was digested with XbaI and Eco47III and inserted into pCGNATF6α-(1-373)-IRES-EGFP that had been digested with XbaI and Eco47III, yielding pCGNATF4-IRES-EGFP. To construct the pCGN-IRES-EGFP control vector expressing only EGFP, the NheI-NotI IRES-EGFP fragment from pIRES2-EGFP (Clontech) was treated with Klenow polymerase and inserted into pCGNATF6α-(1-373) that had been digested with XbaI and BamHI and treated with Klenow polymerase. All RT-PCR was performed using the Reverse Transcription System (Promega, Madison, WI) and Expand High Fidelity PCR System (Roche Applied Science, Indianapolis, IN). The pcDNA3.1-SEAP vector was provided by Susan Baker (Loyola University Medical Center, Chicago, IL). The retroviral plasmids pBMN-I-GFP, pBMN-hATF6(373)-I-GFP (which encodes aa 1-373 of human ATF6α) and pBMN-hXBP1(S)-I-GFP [which encodes full-length human XBP1(S)] were described previously (Sriburi et al., 2004).
Cell culture, cell lines and retroviral transduction
Chinese hamster ovary (CHO) cells deficient in dihydrofolate reductase (DHFR) were grown in Minimal Essential Medium (MEM) Alpha (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Hyclone, Logan, UT), 10 μg/ml adenosine, 10 μg/ml deoxyadenosine and 10 μg/ml thymidine. Cell lines (NIH-3T3, 293, 293T and HeLa) were grown in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% FBS (Hyclone and Atlanta Biologicals, Atlanta, GA). NIH-3T3 fibroblasts stably expressing SEAP were generated by transfection of cells with the pcDNA3.1-SEAP vector and selection of single-cell clones in G418 (Invitrogen). To obtain Xbp1–/– MEFs, male and female Xbp1+/– mice (provided by Laurie H. Glimcher, Harvard Medical School, Boston, MA) were crossed. Embryos were dissected on day 14.5 and Xbp1–/– MEFs were prepared as described (Scheuner et al., 2001). MEFs were maintained in DMEM supplemented with 10% FBS, L-glutamine, 1% penicillin G/streptomycin, and additional essential and non-essential amino acids (Invitrogen) at 37°C in a 5% CO2 incubator. Genotypes of MEFs were determined by PCR amplification of genomic DNA using specific primers: mXBP764s, 5′-GTATGCGTGTGCGTGTGCGTTTTG-3′; mXBP980as, 5′-CATCCGGGCTTTCTTTCTATCTCG-3′; NEO887, 5′-CACCGGACAGGTCGGTCTTG-3′. Production of ecotropic retroviral particles and retroviral transduction of NIH-3T3 cells were described previously (Gunn et al., 2004).
Transfection, fluorescence microscopy and FACS
For fluorescence microscopy, CHO cells were plated on gelatin-coated glass cover slips in 60 mm dishes the day before transfection using Fugene6 (Roche) and plasmids as described previously (Back et al., 2006). Microscopy analysis of HA-tagged proteins and EGFP was performed as described (Back et al., 2006). Images were captured using a photomultiplier tube (PMT) detector of a LSM 510 META confocal microscope (Carl Zeiss, Thornwood, NY) equipped with a C-Apochromat 63×/1.2W correction objective lens. Laser lines used were 364 nm (Coherent Enterprise laser), 488 nm (argon laser) or 543 nm (helium neon 1 laser) to excite DAPI/Alexa Fluor 350, EGFP or TRITC/DsRed-ER, respectively. Fluorescence was detected using the following filters: 385-470 (DAPI), 505-550 (FITC) and long-pass 560 (TRITC). All data were processed using Photoshop (Adobe, Mountain View, CA). For FACS isolation of EGFP+ cells, cells were plated on 10 cm dishes the day before transfection. Transfections were performed using a mixture of Lipofectamine and Plus reagents (Invitrogen) and plasmids according to the manufacturer's instructions. At 40 hours post-transfection, culture medium was removed and cells were washed three times with PBS. The cells were harvested by trypsinization and filtered through nylon screens (45 μm; BD Biosciences, San Jose, CA) to obtain single-cell suspensions. EGFP+ cells were isolated using a FACSVantage dual laser flow cytometer (BD Biosciences). Xbp1–/– MEFs were nucleofected using Nucleofector technology (Amaxa, Gaithersburg, MD) as described previously (Back et al., 2006).
Transmission electron microscopy
FACS-isolated EGFP+ cells were fixed by immersion in 2.5% glutaraldehyde in 0.1 M Sorensen buffer, post-fixed in 1% osmium tetroxide, and stained in 3% uranyl acetate. The cells were dehydrated in ethanol and embedded in Epon. Ultra-thin sections were post-stained with uranyl acetate and lead citrate and examined using a Philips CM100 electron microscope at 60 kV. Images were recorded digitally with a Kodak 1.6 Megaplus camera system operated using AMT software (Advanced Microscopy Techniques, Danvers, MA). Data were processed using Photoshop.
UPRE-luciferase reporter assays
CHO cells were plated on 60-mm dishes the day before transfection. Transfections were performed using Fugene6 (Roche Applied Science), expression plasmids, 5×UPRE-luciferase reporter plasmid (1.6 μg) and pCMV-β-galactosidase (0.2 μg). The total amount of effector plasmid DNAs was maintained at a constant level by adding control plasmid pCGN-IRES-EGFP as necessary. The 5×UPRE-luciferase reporter plasmid (formerly known as p5XATF6GL3) containing the firefly luciferase gene under the control of five ATF6 binding sites was described previously (Wang et al., 2000; Lee et al., 2002). Reporter assays were performed as described (Lee et al., 2002). Firefly luciferase activity values were normalized by β-galactosidase activity values that reflect transfection efficiency.
Immunoblot analysis
Cell lysates were prepared from EGFP+ sorted CHO cells using Nonidet P40 lysis buffer (1% NP40, 50 mM Tris-Cl pH 7.5, 150 mM NaCl, 0.05% SDS, 0.5 mM Na-vanadate, 100 mM NaF, 50 mM β-glycerophosphate, 1 mM PMSF) supplemented with the Complete Protease Inhibitor Cocktail (Roche) as described (Back et al., 2006). Immunoblot analyses were performed as described previously (Back et al., 2006) using anti-EGFP monoclonal antibody (BD Biosciences), anti-actin monoclonal antibody (MP Biomedicals, Solon, OH), anti-ATF6α monoclonal antibody (Imgenex, San Diego, CA), anti-HA monoclonal antibody and anti-XBP1 and anti-CHOP polyclonal antibodies (Santa Cruz, Santa Cruz, CA), anti-KDEL monoclonal antibody and anti-CNX, anti-CRT, anti-ERp72 and anti-PDI polyclonal antibodies (Stressgen, Ann Arbor, MI). Rabbit polyclonal antibodies against translocon-associated protein α (TRAPα) and SEC61β were described previously (Rutkowski et al., 2007). Rabbit polyclonal ATF6β antiserum against the sequence corresponding to amino acids 9-20 (EIADPTRFFTDNC) of murine ATF6β was described previously (Wu et al., 2007). Rabbit polyclonal antibody against ribophorin was a gift from Vishwanath R. Lingappa (UCSF, San Francisco, CA).
RT-PCR analysis of hamster Xbp1 mRNA splicing
Total RNA was prepared from CHO cells using Trizol (Invitrogen) as specified by the manufacturer. Unspliced and UPR-spliced Xbp1 mRNAs were then detected by standard RT-PCR using oligo(dT) primers to synthesize cDNA followed by PCR with primers specific for hamster Xbp1: haXBP1-F, 5′-CTCGCTTGGGAATGGATGTG-3′; haXBP1-R, 5′-GGTAGACCTCTGGGAGTTC-3′. Primers were designed based on the sequence of a partial cDNA for hamster Xbp1 (supplementary material Fig. S5). PCR products (306 bp from unspliced Xbp1 mRNA and 279 bp from UPR-spliced Xbp1 mRNA) were separated by electrophoresis on 3% agarose gels and visualized by ethidium bromide staining. The partial cDNA for hamster Xbp1 was obtained using total RNA isolated from untreated or tunicamycin-treated (10 μg/ml, 6 hours) CHO cells. Several pairs of primers corresponding to mouse and human Xbp1 mRNA sequence were used in RT-PCR. Among them, a human forward primer (5′-AGCTGGAACAGCAAGTGG-3′) and a mouse reverse primer (5′-CTAGAGGCTTGGTGTATAC-3′) amplified products of the expected sizes (533 bp and 507 bp). The product derived from the unspliced form of hamster Xbp1 mRNA (533 bp) was predominant when total RNA from untreated cells was used, whereas the product derived from the UPR-spliced form of hamster Xbp1 mRNA (507 bp) was predominant when total RNA from tunicamycin-treated cells was used. The amplified cDNAs corresponding to unspliced and UPR-spliced hamster Xbp1 mRNA were subcloned into pGEM-T Easy (Promega) and sequenced.
Phospholipid analysis
Cells (2×107) were pelleted by centrifugation, flash frozen and stored at –80°C until analysis. Lipids were isolated, fractionated by thin-layer chromatography (TLC), and then detected and quantified by flame ionization using an Iatroscan instrument (Iatron Laboratories, Tokyo, Japan) as previously described (Sriburi et al., 2004).
Analysis of fatty acid and PtdCho biosynthesis
NIH-3T3 cells were transduced with retroviral vectors. At 48 hours post-transduction, cells were cultured in DMEM containing 3 μCi/ml [methyl-14C]acetic acid (55 mCi/mmol; American Radiolabeled Chemicals, St Louis, MO) for 2 hours at 37°C. Labeled cells were then harvested by trypsinization and pelleted by centrifugation. Lipids were extracted from the samples as previously described (Fagone et al., 2007; Sriburi et al., 2004). The level of fatty acid biosynthesis was assessed using scintillation spectroscopy to measure incorporation of radioactive acetate into the lipid fraction. The level of PtdCho biosynthesis was assessed by resolving the lipids by TLC along with authentic standards to identify PtdCho. The plate was developed in a solvent system of chloroform:methanol:acetic acid:water (50:27:7:1, v/v) to resolve PtdCho from other lipid species. Signals from radiolabeled PtdCho were detected using a Typhoon 8600 Variable Mode Imager (GE Healthcare, Piscataway, NJ) and quantified using ImageQuant software (GE Healthcare).
Analysis of CK, CCT and CPT enzymatic activity
Pelleted frozen cells (2×107) were used for assays of CK and CCT enzymatic activity as described previously (Sriburi et al., 2004). CPT activity was assessed as described previously (Sriburi et al., 2004) using microsomes prepared from pelleted frozen cells (2×107).
Analysis of gene expression by real-time PCR
Quantitative real-time PCR was performed as described previously using gene-specific primers and probes for the CCT α, β2 and β3 isoforms (Sriburi et al., 2004), the CK α and β isoforms, and Chpt1 and Cept1 (Fagone et al., 2007).
Microarray analysis
Gene expression in NIH-3T3 cells transduced with empty vector and ATF6α(1-373) retroviruses was analyzed by microarray as described previously (Sriburi et al., 2007) using Affymetrix GeneChip (Affymetrix, Santa Clara, CA) mouse genome arrays MOE430A or MOE430V2 at the Hartwell Center for Bioinformatics and Biotechnology, St Jude Children's Research Hospital, Memphis, TN. For empty vector- and ATF6α(1-373)-transduced cells, a total of four separate experiments were performed. The statistical analyses were performed on data sets from all of the experiments that were independently processed and hybridized. The statistical significance of ATF6α(1-373) overexpression was determined using a two-tailed unpaired t-test with the confidence intervals set at 95% and data with P-values ≤0.05 considered significant.
Analysis of SEAP secretion by NIH-3T3 cells
NIH-3T3 fibroblasts stably expressing SEAP were transduced with retroviral vectors. At 48 hours post-transduction, cells were washed, shifted into fresh media and then cultured for various times. Culture supernatants were harvested and assessed for SEAP activity using the Great EscAPe SEAP Reporter System (Clontech) chemiluminescence assay.
Supplementary Material
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/122/10/1636/DC1
We thank John McNulty and Linda Fox of the Loyola University Imaging Facility, Patricia Simms of the Loyola University Flow Cytometry Facility, Jina Wang and Daren Hemingway of St Jude Children's Research Hospital and the University of Michigan M.I.L. (Microscope and Image Analysis Lab) and Flow Cytometry Core Facility for expert technical support; and LeeTerry Moore for laboratory assistance. We gratefully acknowledge Kezhong Zhang for Xbp1+/+ and Xbp1–/– MEFs. This work was supported by NIH grants GM61970 (J.W.B.), T32AI007508 (H.B.), GM45737 (S.J.), DK042394 (R.J.K.), HL052173 (R.J.K.) and HL057346 (R.J.K.). This work was also supported by a Cancer Center (CORE) Support Grant CA21765 (to St Jude Children's Research Hospital) and the American Lebanese Syrian Associated Charities. R.J.K. is an investigator of the Howard Hughes Medical Institute. Deposited in PMC for release after 12 months.
References
- Adachi, Y., Yamamoto, K., Okada, T., Yoshida, H., Harada, A. and Mori, K. (2008). ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct. Funct. 33, 75-89. [DOI] [PubMed] [Google Scholar]
- Back, S. H., Lee, K., Vink, E. and Kaufman, R. J. (2006). Cytoplasmic IRE1alpha-mediated XBP1 mRNA splicing in the absence of nuclear processing and endoplasmic reticulum stress. J. Biol. Chem. 281, 18691-18706. [DOI] [PubMed] [Google Scholar]
- Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., Clark, S. G. and Ron, D. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92-96. [DOI] [PubMed] [Google Scholar]
- Fagone, P., Sriburi, R., Ward-Chapman, C., Frank, M., Wang, J., Gunter, C., Brewer, J. W. and Jackowski, S. (2007). Phospholipid biosynthesis program underlying membrane expansion during B-lymphocyte differentiation. J. Biol. Chem. 282, 7591-7605. [DOI] [PubMed] [Google Scholar]
- Gass, J. N., Gifford, N. M. and Brewer, J. W. (2002). Activation of an unfolded protein response during differentiation of antibody-secreting B cells. J. Biol. Chem. 277, 49047-49054. [DOI] [PubMed] [Google Scholar]
- Gass, J. N., Jiang, H. Y., Wek, R. C. and Brewer, J. W. (2008). The unfolded protein response of B-lymphocytes: PERK-independent development of antibody-secreting cells. Mol. Immunol. 45, 1035-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn, K. E., Gifford, N. M., Mori, K. and Brewer, J. W. (2004). A role for the unfolded protein response in optimizing antibody secretion. Mol. Immunol. 41, 919-927. [DOI] [PubMed] [Google Scholar]
- Harding, H. P., Zhang, Y. and Ron, D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271-274. [DOI] [PubMed] [Google Scholar]
- Harding, H. P., Zhang, Y., Bertolotti, A., Zeng, H. and Ron, D. (2000). Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 5, 897-904. [DOI] [PubMed] [Google Scholar]
- Harding, H. P., Zeng, H., Zhang, Y., Jungries, R., Chung, P., Plesken, H., Sabatini, D. D. and Ron, D. (2001). Diabetes mellitus and exocrine pancreatic dysfunction in Perk–/– mice reveals a role for translational control in secretory cell survival. Mol. Cell 7, 1153-1163. [DOI] [PubMed] [Google Scholar]
- Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., Sadri, N., Yun, C., Popko, B., Paules, R. et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619-633. [DOI] [PubMed] [Google Scholar]
- Haze, K., Yoshida, H., Yanagi, H., Yura, T. and Mori, K. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787-3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haze, K., Okada, T., Yoshida, H., Yanagi, H., Yura, T., Negishi, M. and Mori, K. (2001). Identification of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem. J. 355, 19-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneberry, A. L. and McMaster, C. R. (1999). Cloning and expression of a human choline/ethanolaminephosphotransferase: synthesis of phosphatidylcholine and phosphatidylethanolamine. Biochem. J. 339, 291-298. [PMC free article] [PubMed] [Google Scholar]
- Henneberry, A. L., Wistow, G. and McMaster, C. R. (2000). Cloning, genomic organization, and characterization of a human cholinephosphotransferase. J. Biol. Chem. 275, 29808-29815. [DOI] [PubMed] [Google Scholar]
- Iwakoshi, N. N., Lee, A. H., Vallabhajosyula, P., Otipoby, K. L., Rajewsky, K. and Glimcher, L. H. (2003). Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 4, 321-329. [DOI] [PubMed] [Google Scholar]
- Jackowski, S. and Fagone, P. (2005). CTP: phosphocholine cytidylyltransferase: paving the way from gene to membrane. J. Biol. Chem. 280, 853-856. [DOI] [PubMed] [Google Scholar]
- Kent, C. (2005). Regulatory enzymes of phosphatidylcholine biosynthesis: a personal perspective. Biochim. Biophys. Acta 1733, 53-66. [DOI] [PubMed] [Google Scholar]
- Lee, A. H., Iwakoshi, N. N. and Glimcher, L. H. (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448-7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, A. H., Chu, G. C., Iwakoshi, N. N. and Glimcher, L. H. (2005). XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 24, 4368-4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K., Tirasophon, W., Shen, X., Michalak, M., Prywes, R., Okada, T., Yoshida, H., Mori, K. and Kaufman, R. J. (2002). IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 16, 452-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, P. D., Harding, H. P. and Ron, D. (2004). Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 167, 27-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykidis, A. and Jackowski, S. (2001). Regulation of mammalian cell membrane biosynthesis. Prog. Nucleic Acid Res. Mol. Biol. 65, 361-393. [DOI] [PubMed] [Google Scholar]
- Okada, T., Yoshida, H., Akazawa, R., Negishi, M. and Mori, K. (2002). Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem. J. 366, 585-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimold, A. M., Iwakoshi, N. N., Manis, J., Vallabhajosyula, P., Szomolanyi-Tsuda, E., Gravallese, E. M., Friend, D., Grusby, M. J., Alt, F. and Glimcher, L. H. (2001). Plasma cell differentiation requires the transcription factor XBP-1. Nature 412, 300-307. [DOI] [PubMed] [Google Scholar]
- Ron, D. and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519-529. [DOI] [PubMed] [Google Scholar]
- Rutkowski, D. T., Kang, S. W., Goodman, A. G., Garrison, J. L., Taunton, J., Katze, M. G., Kaufman, R. J. and Hegde, R. S. (2007). The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol. Biol. Cell 18, 3681-3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner, D., Song, B., McEwen, E., Liu, C., Laybutt, R., Gillespie, P., Saunders, T., Bonner-Weir, S. and Kaufman, R. J. (2001). Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 7, 1165-1176. [DOI] [PubMed] [Google Scholar]
- Scheuner, D., Mierde, D. V., Song, B., Flamez, D., Creemers, J. W., Tsukamoto, K., Ribick, M., Schuit, F. C. and Kaufman, R. J. (2005). Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat. Med. 11, 757-764. [DOI] [PubMed] [Google Scholar]
- Schroder, M. and Kaufman, R. J. (2005). The mammalian unfolded protein response. Ann. Rev. Biochem. 74, 739-789. [DOI] [PubMed] [Google Scholar]
- Shaffer, A. L., Shapiro-Shelef, M., Iwakoshi, N. N., Lee, A. H., Qian, S. B., Zhao, H., Yu, X., Yang, L., Tan, B. K., Rosenwald, A. et al. (2004). XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 21, 81-93. [DOI] [PubMed] [Google Scholar]
- Shen, X., Ellis, R., Lee, K., Liu, C. Y., Yang, K., Solomon, A., Yoshida, H., Morimoto, R., Kurnit, D. M., Mori, K. et al. (2001). Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 107, 893-903. [DOI] [PubMed] [Google Scholar]
- Shi, Y., Vattem, K. M., Sood, R., An, J., Liang, J., Stramm, L. and Wek, R. C. (1998). Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 18, 7499-7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriburi, R., Jackowski, S., Mori, K. and Brewer, J. W. (2004). XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 167, 35-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriburi, R., Bommiasamy, H., Buldak, G. L., Robbins, G. R., Frank, M., Jackowski, S. and Brewer, J. W. (2007). Coordinate regulation of phospholipid biosynthesis and secretory pathway gene expression in XBP-1(S)-induced endoplasmic reticulum biogenesis. J. Biol. Chem. 282, 7024-7034. [DOI] [PubMed] [Google Scholar]
- Tigges, M. and Fussenegger, M. (2006). Xbp1-based engineering of secretory capacity enhances the productivity of Chinese hamster ovary cells. Metab. Eng. 8, 264-272. [DOI] [PubMed] [Google Scholar]
- Tirasophon, W., Welihinda, A. A. and Kaufman, R. J. (1998). A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812-1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Anken, E. and Braakman, I. (2005). Versatility of the endoplasmic reticulum protein folding factory. Crit. Rev. Biochem. Mol. Biol. 40, 191-228. [DOI] [PubMed] [Google Scholar]
- Vattem, K. M. and Wek, R. C. (2004). Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 101, 11269-11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. Z., Harding, H. P., Zhang, Y., Jolicoeur, E. M., Kuroda, M. and Ron, D. (1998). Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 17, 5708-5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y., Shen, J., Arenzana, N., Tirasophon, W., Kaufman, R. J. and Prywes, R. (2000). Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J. Biol. Chem. 275, 27013-27020. [DOI] [PubMed] [Google Scholar]
- Wright, M. M., Henneberry, A. L., Lagace, T. A., Ridgway, N. D. and McMaster, C. R. (2001). Uncoupling farnesol-induced apoptosis from its inhibition of phosphatidylcholine synthesis. J. Biol. Chem. 276, 25254-25261. [DOI] [PubMed] [Google Scholar]
- Wu, J., Rutkowski, D. T., Dubois, M., Swathirajan, J., Saunders, T., Wang, J., Song, B., Yau, G. D. and Kaufman, R. J. (2007). ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 13, 351-364. [DOI] [PubMed] [Google Scholar]
- Yamamoto, K., Sato, T., Matsui, T., Sato, M., Okada, T., Yoshida, H., Harada, A. and Mori, K. (2007). Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev. Cell 13, 365-376. [DOI] [PubMed] [Google Scholar]
- Ye, J., Rawson, R. B., Komuro, R., Chen, X., Dave, U. P., Prywes, R., Brown, M. S. and Goldstein, J. L. (2000). ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355-1364. [DOI] [PubMed] [Google Scholar]
- Yoshida, H., Okada, T., Haze, K., Yanagi, H., Yura, T., Negishi, M. and Mori, K. (2000). ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 20, 6755-6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, H., Matsui, T., Yamamoto, A., Okada, T. and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881-891. [DOI] [PubMed] [Google Scholar]
- Zhang, P., McGrath, B., Li, S., Frank, A., Zambito, F., Reinert, J., Gannon, M., Ma, K., McNaughton, K. and Cavener, D. R. (2002). The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell. Biol. 22, 3864-3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W., Feng, D., Li, Y., Iida, K., McGrath, B. and Cavener, D. R. (2006). PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab. 4, 491-497. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.