

Abstract
Chromosome region 1q21.1 contains extensive and complex low-copy repeats, and copy number variants (CNVs) in this region have recently been reported in association with congenital heart defects1, developmental delay2,3, schizophrenia and related psychoses4,5. We describe 21 probands with the 1q21.1 microdeletion and 15 probands with the 1q21.1 microduplication. These CNVs were inherited in most of the cases in which parental studies were available. Consistent and statistically significant features of microcephaly and macrocephaly were found in individuals with micro-deletion and microduplication, respectively. Notably, a paralog of the HYDIN gene located on 16q22.2 and implicated in autosomal recessive hydrocephalus6 was inserted into the 1q21.1 region during the evolution of Homo sapiens7; we found this locus to be deleted or duplicated in the individuals we studied, making it a probable candidate for the head size abnormalities observed. We propose that recurrent reciprocal microdeletions and microduplications within 1q21.1 represent previously unknown genomic disorders characterized by abnormal head size along with a spectrum of developmental delay, neuropsychiatric abnormalities, dysmorphic features and congenital anomalies. These phenotypes are subject to incomplete penetrance and variable expressivity.
Our clinical cytogenetics laboratory has examined 16,557 samples from affected individuals by targeted array comparative genomic hybridization (array CGH) over 4 years. Samples were received for a wide range of referring diagnoses, including mental retardation, autism and/or congenital anomalies. During this period we have identified 27 probands with microdeletions (clinical information was available for 21 of these cases; Table 1) and 17 probands with microduplications (clinical information was available for 15 of these cases; Table 2) involving 1q21.1. All cases were found to have loss or gain corresponding to BAC clones (RP11-337C18, RP11-533N14 and RP11-102F23) within the distal 1q21.1 region, similar to gains or losses previously reported in a small number of cases1–3 but distinct from the deletion region implicated in thrombocytopenia absent radius (TAR) syndrome8 (Fig. 1a).
Table 1. Molecular and clinical findings in individuals with distal lq21.1 microdeletion.
| Age at diagnosis (years) | Growth | FOC Z score | Dysmorphic features | Developmental delay | Clinical features | Father | Mother | |
|---|---|---|---|---|---|---|---|---|
| Class I deletion (distal 1q21.1 region only, ∼1 Mb) | ||||||||
| 1 | 0.5 | Short stature | −2.34 | + | − | IUGR, GERD; 11 pairs of ribs | Del 1q21.1 | Normal |
| 1FATHER | Normal | n.d. | + | − | Depression, anxiety; rheumatoid arthritis | n.d. | n.d. | |
| 2 | 9 | Normal | −3.8 | + | + | Ankyloglossia; behavioral problems; hallucinations; sleep disturbance | Del 1q21.1 | Normal |
| 2FATHER | Normal | −2.34 | − | + (Speech delay) | Scoliosis | n.d. | n.d. | |
| 3 | 11 | Normal | −1.23 | + | + | SNHL, ankyloglossia; strabismus; dry eyes and mouth; behavioral problems | n.d. | n.d. |
| 4 | 0.2 | Short stature | −3.5 | + | + | IUGR; chorioretinal and iris coloboma, lens subluxation, microphthalmia; laryngomalacia VUR grade 4/5, hydronephrosis; postaxial polydactyly | Normal | Del 1q21.1 |
| 5 | 1 | Normal | −3.3 | + | − | None | Normal | Normal |
| 6 | 1 | FTT | −0.84 | − | + | Chiari I malformation | Normal | Del 1q21.1 |
| 7 | 0.1 | FTT | −4.9 | + | n.d. | Congenital hydrocephalus, agenesis corpus callosum | n.d. | n.d. |
| 8 | 0.8 | Normal | −2.06 | − | − | Trigonocephaly | Del 1q21.1 | Normal |
| 8FATHER | Normal | −3.6 | − | − | − | n.d. | n.d. | |
| 9 | 12 | Normal | −0.05 | − | + | Autism spectrum behaviors; two- to three-toe syndactyly | Normal | Normal |
| 10 | 8 | Normal | −3.5 | + | + | Seizures; feeding difficulties; precocious puberty | Del 1q21.1 | Normal |
| 10FATHER | Normal | −1.56 | − | − | None | n.d. | n.d. | |
| 11 | 7 | Normal | −2.34 | + | + | Cryptorchidism; postaxial polydactyly | Normal | Del 1q21.1 |
| 11MOTHER | Normal | −1.89 | − | − | Multiple sclerosis | n.d. | n.d. | |
| 12 | 2 | FTT | −2.9 | + | + | Hemangioma | Del 1q21.1 | Normal |
| 12FATHER | Normal | n.d. | − | Learning disability | Congenital cataract; ADHD | n.d. | n.d. | |
| 13 | 6 | Normal | −0.77 | + | − | Aggressive behavior; ADHD; seizure disorder | n.d. | n.d. |
| 14 | 11 | Normal | −3.1 | + | + | Clubfeet; ankyloglossia; ADHD | Normal | Del 1q21.1 |
| 14MOTHER | Normal | −3.5 | − | Learning disability | ADHD | n.d. | n.d. | |
| 14SIBLING | 31 | Normal | −2.33 | − | Learning disability | Antisocial behavior | Normal | Del 1q21.1 |
| 15 | 1 | Normal | −1.17 | − | + | GERD; inguinal hernia | n.d. | n.d. |
| Class II deletion (distal 1q21.1 region and TAR region, ∼2 Mb) | ||||||||
| 16 | 4 | Normal | −4.3 | − | + | None | Del 1q21.1 | Normal |
| 17 | 5 | Short stature | −3.7 | + | + | Gestation complicated by maternal diabetes; cleft palate, preaxial polydactyly; SNHL; bicuspid aortic valve; congenital cystic adenomatoid malformation of the lung; pelvic kidney | Del 1q21.1 | Normal |
| 17FATHER | Normal | −1.88 | − | − | None | n.d. | n.d. | |
| 18 | 3 | Normal | −2.05 | + | + | Plagiocephaly; strabismus; MRI: brain asymmetry, enlargement of the lateral and III ventricles, increased subarachnoid space | n.d. | Normal |
| 19 | 2 | FTT | −2.34 | − | + | Frequent infections; pectus excavatum; small kidneys; hypospadias | Normal | Del 1q21.1 |
| 19MOTHER | Normal | −2.7 | − | − | None | n.d. | n.d. | |
| 20 | 6 | FTT | −3 | + | + | None | Del 1q21.1 | Normal |
| 21 | 2 | FTT | −2.4 | + | + (Speech delay) | Polyhydramnios; severe feeding difficulties, chronic vomiting | Normal | Normal |
ADHD, attention deficit hyperactivity disorder; FOC, frontal occipital circumference; FTT, failure to thrive; GERD, gastroesophageal reflux disease; IUGR, intrauterine growth retardation; SNHL, sensorineural hearing loss; VUR, vesicoureteral reflux; n.d., not determined.
Table 2. Molecular and clinical findings in individuals with distal 1q21.1 microduplication.
| Age at diagnosis (years) | Growth | FOC Z score | Dysmorphic features | Developmental delay | Clinical features | Father | Mother | |
|---|---|---|---|---|---|---|---|---|
| Class I duplication (distal 1q21.1 region only, ∼1 Mb) | ||||||||
| 22 | 1 | Normal | +2.7 | − | − | Complex congenital heart defect | Normal | Dup 1q21.1 |
| 22MOTHER | Normal | −1.55 | − | − | None | − | − | |
| 23 | 1 | FTT | +1.3 | + | + | Hypotonia; eczema | Normal | Dup 1q21.1 |
| 24 | 5 | Normal | 0 | − | + (Speech delay) | Seizures | Normal | Dup 1q21.1 |
| 25 | 1 | Normal | +3.1 | + | + | Esotropia; VI and VII nerve paresis; hypotonia | Normal | Dup 1q21.1 |
| 25MOTHER | Normal | +3 | − | Learning disability | Cataract; glaucoma; depression; anxiety | n.d. | n.d. | |
| 26 | 0.3 | Normal | 0.47 | − | − | Lower limb hypertonicity | Normal | Dup 1q21.1 |
| 27 | 1.5 | Normal | 0.62 | + | + | Chiari malformation; hemihypertrophy | Dup 1q21.1 | Normal |
| 27FATHER | Normal | +3 | − | − | None | n.d. | n.d. | |
| 28 | 3 | Normal | +5.2 | + | + | Dysphagia, GERD; Lower limb hypertonicity; toe-walking | n.d. | n.d. |
| 29 | 0.5 | FTT | −2.03 | + | Mild | GERD; lower limb hypertonicity | n.d. | n.d. |
| 30 | 5 | Normal | 0.64 | + | + | Cryptorchidism; hypotonia; seizures | Normal | Normal |
| 31 | 1 | Normal | −0.39 | − | + | Arthrogryposis; fifth-finger clinodactyly; Psoriasis; right-sided hyperpigmentation | Dup 1q21.1 | Normal |
| 31FATHER | Normal | −1.22 | − | − | None | n.d. | n.d. | |
| 32 | 7 | Normal | +2.6 | + | + | Scoliosis; hypospadias; Raynaud's phenomenon; Autism spectrum disorder | n.d. | n.d. |
| 33 | 0.5 | Normal | +3 | + | + | Esotropia; hydrocephalus; advanced bone age | Normal | Dup 1q21.1 |
| 33MOTHER | Normal | +2.06 | − | − | Normal | n.d. | n.d. | |
| 33SIBLING | 2 | Normal | +2.4 | − | Mild | None | Normal | Dup 1q21.1 |
| 34 | 16 | FTT | −2.3 | + | + | Autism; spastic diplegia | n.d. | n.d. |
| 35 | 4 | Normal | 0 | − | + | Autism | n.d. | n.d. |
| Complex TAR syndrome deletion with distal 1q21.1 duplication | ||||||||
| 36 | 5 | Normal | +2.4 | + | + | ADHD | Normal | Dup 1q21.1 |
| 36MOTHER | Normal | −2.9 | + | Learning disability | Congenital hip dysplasia; clubfeet | n.d. | n.d. | |
| 36SIBLING 1 | 8 | Normal | −1.06 | + | + (Speech delay) | Frequent otitis media | Normal | Dup 1q21.1 |
| 36SIBLING 2 | 4 | Normal | +1.65 | + | + (Speech delay) | None | Normal | Dup 1q21.1 |
ADHD, attention deficit hyperactivity disorder; FOC, frontal occipital circumference; FTT, failure to thrive; GERD, gastroesophageal reflux disease.
Figure 1.
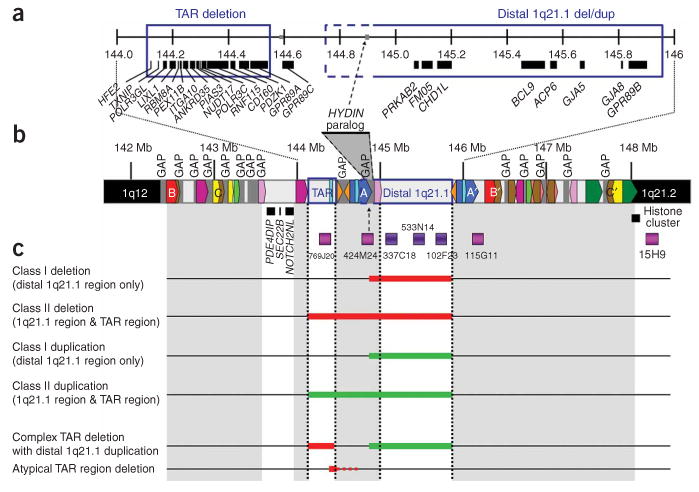
Schematic representation of chromosome 1q21.1 based on the March 2006 freeze of the reference human genome sequence (NCBI build 36.1) and summary of molecular findings. (a) An enlargement of the region between 144 and 146 Mb. Known genes are indicated by black bars. (b) The region from 142 to 148 Mb. The minimal regions for the TAR syndrome deletion and the distal 1q21.1 deletion/duplication are shown with blue boxes. Array-CGH clones are shown in dark violet, and additional FISH clones used during the course of the study are shown in magenta. Gaps in the reference sequence are indicated by gray breaks in the chromosome bar. The putative location of the HYDIN paralog is indicated by a gray triangle bounded by dashed lines. Regions of segmental duplication are depicted by colored boxes. A pair of large, directly oriented intrachromosomal repeats flanks the distal 1q21.1 region (blue blocks labeled A and A′). In addition, there is another pair of directly oriented repeats flanking the first ∼4 Mb of the sub-band and encompassing both the TAR region and the distal 1q21.1 region (red blocks labeled B and B′). Finally, there is a pair of blocks flanking a large portion of the sub-band 1q21.1 (yellow blocks labeled C and C′), in opposite orientation. Several other blocks are present, but gaps in the reference sequence preclude further detailed characterization at this time. (c) Overview of the deletions and duplications in affected individuals described in this report. Thick red horizontal lines indicate minimally deleted regions, whereas thick green horizontal lines indicate minimally duplicated regions. The complex deletion/duplication rearrangement in individual 36 is depicted with red and green bars, and the single case with atypical TAR region deletion is indicated with a solid red and dashed red line (see text for details).
We examined the genomic structure of the 1q21.1 region to determine the relationships among the various low-copy repeats (LCRs) (Fig. 1b). As previously indicated2, this region contains numerous LCRs that could mediate genomic rearrangements. The reference sequence used in our analysis (Human Genome Build 36.1, from the University of California Santa Cruz (UCSC) genome browser) contains 15 gaps within sub-band 1q21.1, many of which are adjacent to LCRs, thus limiting the analysis, and revision of the reference sequence for this region is likely to alter the overall structure of the LCRs.
We carried out FISH analyses to refine the breakpoint regions. BAC clone RP11-769J20 was used to examine whether CNVs extend through the TAR syndrome region. BAC clones RP11-115G11 (which maps within the telomeric repeat region but in a region of relatively unique sequence) and RP11-15H9 (which maps to sub-band 1q21.2, distal to the telomeric repeats and the cluster of histone genes) were used to determine the telomeric extent of the CNVs (summarized in Fig. 1c).
Combined array-CGH and FISH results indicated that deletions cluster into two classes. Class I deletions are defined as involving only the distal 1q21.1 region, with telomeric breakpoints mapping within clone RP11-115G1. Class II deletions are defined as larger deletions including both the TAR syndrome and distal 1q21.1 regions, with telomeric breakpoints mapping within RP11-115G1 or further within the telomeric LCR (Fig. 1c).
Among microduplication cases, 16 of 17 involved only the distal 1q21.1 region (class I). In one case (clinical information not available), the microduplication extended from the TAR region through the distal 1q21.1 region (class II). In all microduplication cases, clone RP11-115G11 showed three signals whereas RP11-15H9 showed two signals, indicating that the breakpoints were located within or distal to RP11-115G11. One individual (subject 36) with microduplication of the distal 1q21.1 region was found to have an intriguing complex rearrangement involving microdeletion of the TAR region, which was inherited from the mother and also present in two similarly affected siblings. Both parental samples were available for analysis in 17 of 21 microdeletion cases and 12 of 15 microduplication cases, and the CNVs were inherited from a parent in most cases (Tables 1 and 2). There were three de novo microdeletions and one de novo microduplication.
Certain facial dysmorphic features such as frontal bossing, deep-set eyes and bulbous nose in individuals with 1q21.1 microdeletion (Fig. 2), and frontal bossing and hypertelorism in individuals with 1q21.1 microduplication (Fig. 3) seem to be common. Otherwise, no distinctive pattern of facial dysmorphic features was noted. A wide range of congenital anomalies was present, although without a clearly apparent pattern (Tables 1 and 2). Developmental delay and/or learning disabilities were reported in most cases. Finally, behavioral abnormalities were frequently observed, including attention deficit hyperactivity disorder (ADHD), autism, anxiety/depression, antisocial behavior, aggression and even hallucinations. It should be noted that many of the affected individuals are young and may not yet have manifested atypical behaviors at the time of examination. Furthermore, clinically relevant abnormalities including congenital cataract, learning disabilities, ADHD, depression/anxiety and antisocial behavior were present in some but not all parents carrying the 1q21.1 CNVs (Tables 1 and 2), and these parents had not previously been suspected of having a chromosomal abnormality.
Figure 2.
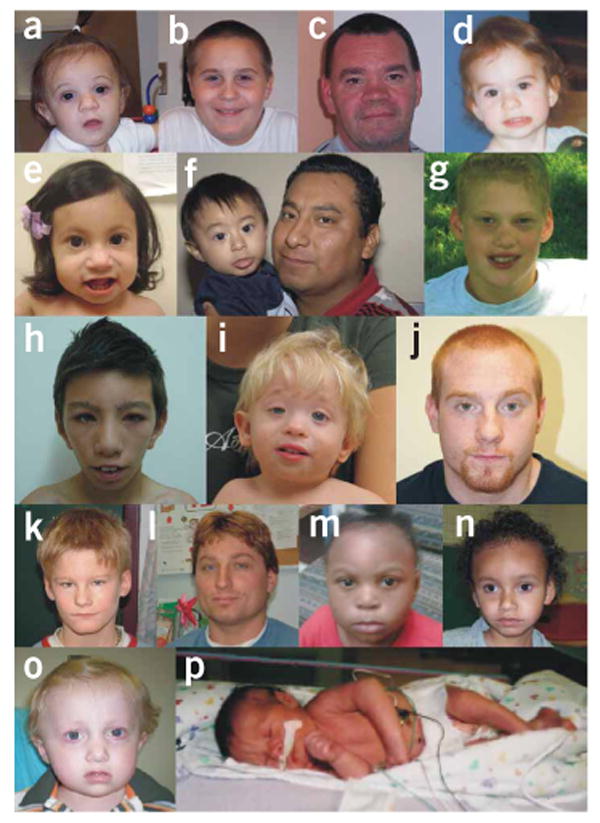
Facial appearance of individuals with the 1q21.1 microdeletion. (a) Subject 1. (b) Subject 2. (c) Subject 2FATHER. (d) Subject 3. (e) Subject 5. (f) Subjects 8 and 8FATHER. (g) Subject 9. (h) Subject 11. (i) Subject 12. (j) Subject 12FATHER. (k) Subject 14. (l) Subject 14SIBLING. (m) Subject 15. (n) Subject 18. (o) Subject 19. (p) Subject 7. Frontal bossing, deep-set eyes and bulbous nose were frequently present.
Figure 3.
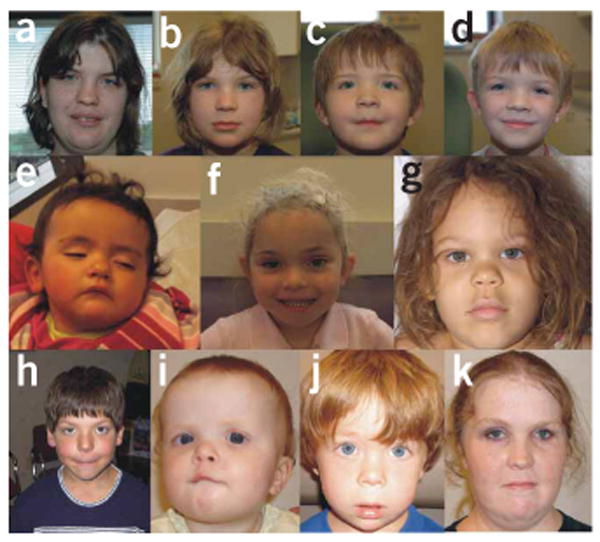
Facial appearance of individuals with the 1q21.1 microduplication. (a) Subject 36MOTHER. (b) Subject 36SIBLING 1. (c) Subject 36SIBLING 2. (d) Subject 36. (e) Subject 22. (f) Subject 27. (g) Subject 35. (h) Subject 32. (i) Subject 33. (j) Subject 33SIBLING. (k) Subject 33MOTHER. Hypertelorism and frontal bossing seem to be common in individuals with the microduplication.
Individuals with microdeletion and microduplication differed significantly in their mean head sizes (unpaired Student's t-test, P < 0.0001). The mean Z score for microdeletion cases (probands, parents and siblings carrying the microdeletion) was −2.53 (95% confidence interval (c.i.) = −2.96; −2.11), significantly different from the population mean, which is zero by definition (one sample t-test, P < 0.0001). Among these individuals 21 of 29 had a head circumference Z score less than −2. There was greater variation among microduplication cases, with 10 of 24 having head circumference Z scores greater than +2. Nevertheless, the mean Z score for microduplication cases (probands, parents and siblings carrying the microdeletion) was +0.95 (95% c.i. = 0.06; 1.83), which was statistically different from the population mean (one sample t-test, P < 0.05). Because of the possible effect of shared genetic background on head size among parents and siblings, we also carried out the statistical analyses on probands only, finding that the mean Z score for probands with the microdeletion was −2.55 (95% c.i. = −3.12; −1.98) and the mean Z score for probands with the microduplication was +1.15 (95% c.i. = 0.03; 2.28). Both the microdeletion and microduplication groups were still significantly different from each other (unpaired t-test, P < 0.0001) and from the population mean (deletion probands, one sample t-test, P < 0.0001; duplication probands, one sample t-test, P < 0.05). To rule out ascertainment bias for head size abnormalities within the referred population, which is enriched for individuals with clinically relevant phenotypes, we obtained head circumference measurements for 50 randomly selected individuals referred for array-CGH testing during the same time period in which the index cases were received. These control individuals had a much wider range of head circumference measurements than either the microdeletion or microduplication cases. The mean Z score among control individuals was −0.62 (95% c.i. = −1.33; 0.08), which was not statistically different from the population mean (one sample t-test, P = 0.083) (Fig. 4a). In addition, we found statistically significant differences between microdeletion cases and controls (unpaired t-test, P < 0.0001) and between microduplication cases and controls (unpaired t-test, P < 0.01), thus strengthening the finding that 1q21.1 CNVs are important in determining head size (Fig. 4a).
Figure 4.
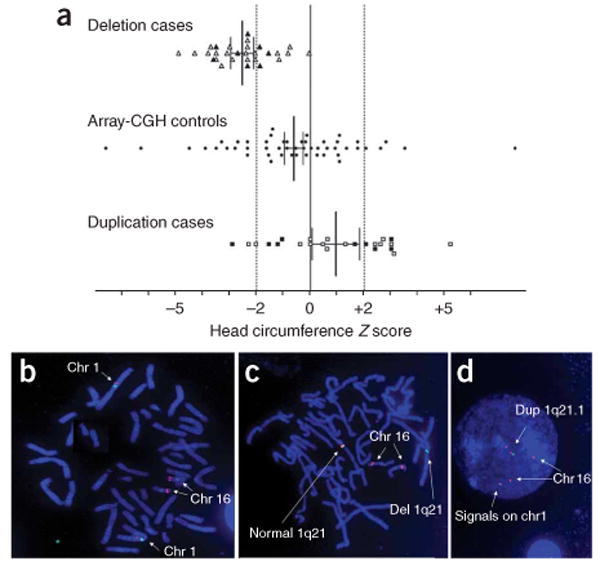
Microdeletions and microduplications of 1q21.1 are associated with head size abnormalities and include the 1q21.1 HYDIN paralog. (a) Head circumference measurements in individuals with microdeletion (triangles), controls (circles) and individuals with microduplication (squares) are plotted as age- and sex-matched Z scores. Parents carrying the CNVs are represented by filled symbols. Bars indicate mean and 95% confidence intervals. The vertical dashed lines mark the −2 and +2 Z scores. (b–d) FISH for HYDIN using BAC clone RP11-424M24 (red signals) and centromere probe for chromosome 1 (green signal). In the control sample (b), red signals are seen on both 16q22 and 1q21.1, confirming the previously reported intrachromosomal HYDIN duplication. In samples from affected individuals, the 1q21.1 HYDIN paralog is deleted in individuals with 1q21.1 microdeletion (c) and duplicated in individuals with 1q21.1 microduplication (d).
It was previously reported that a large interchromosomal duplication containing a paralog of the HYDIN gene on chromosome 16q22.2 was inserted into 1q21.1 during primate evolution, but this contig is not accounted for in the current build of the human genome7. Homozygous inactivating mutations of HYDIN cause hydrocephalus in mice6, probably as a result of impaired ciliary function of ependymal cells9. Notably, a conserved protein domain found several times in the HYDIN protein and designated as ASH (ASPM, SPD-2, Hydin) was identified at the N terminus of abnormal spindle-like microcephaly-associated protein (ASPM)10,11, which is implicated in control of cerebral cortex size12. Moreover, individuals with cytogenetically visible deletions on 16q22.2 that presumably involve the HYDIN gene have been reported to have microcephaly13–15.
The 1q21.1 HYDIN paralog is exclusively expressed in the brain6, and we suggest that it could be involved in the head size abnormalities seen in our affected individuals. Accordingly, the 1q21.1 HYDIN paralog is deleted or duplicated in all individuals with deletion or duplication of distal 1q21.1, respectively (Fig. 4b–d). These results also indicate that this locus maps in one of the two gap segments between the TAR syndrome region and the distal 1q21.1 region. Indeed, it was predicted on the basis of sequence data that the duplicated paralog extends into a contig (NT_034398) mapping to 1q21.1 (ref. 7). Given that the LCRs flanking the distal 1q21.1 region most likely mediate reciprocal deletion or duplication (Fig. 1b, blue blocks labeled A and A′), the most parsimonious placement of the HYDIN paralogous segment is within the gap immediately centromeric to the distal 1q21.1 region (Fig. 1a,b, gray triangle bounded by dashed lines). Consistent with this proposal, we have also identified a proband with an atypical deletion beginning approximately at nt 144253465 but not involving the distal 1q21.1 region, most likely mediated by a pair of small segmental duplications in this region (Fig. 1b, light blue blocks, and Fig. 1c). This atypical deletion does not include the HYDIN paralog by FISH (data not shown), and the head circumference of this individual is within the normal range. Our results are particularly of note in light of the recent finding of an association between 1q21.1 deletions and schizophrenia4,5, and the recognition of smaller head circumference and facial dysmorphic features among some schizophrenic individuals16–18.
The group of affected individuals described herein represents the largest collection of individuals with microdeletions or microduplications within chromosome 1q21.1 reported in the literature. We have clearly demonstrated a dosage effect of 1q21.1 copy number on head size. Most of the probands in our cohort presented with developmental delay, and many of them also manifested a range of behavioral abnormalities, nonspecific dysmorphic features and congenital anomalies. These findings are similar to the broad range of referring diagnoses among people undergoing clinical array-CGH testing, thus posing a challenge to the delineation of specific syndromic features other than head size abnormalities. We found only one individual with microdeletion who had a bicuspid aortic valve, so the initial report of an association between distal 1q21.1 deletions and congenital heart defects1 is probably a result of ascertainment bias.
CNVs within 1q21.1 can be found in apparently normal individuals (as was also observed for unaffected carriers of the TAR syndrome microdeletion8) or associated with psychiatric phenotypes such as schizophrenia4,5. The presence of parents carrying 1q21.1 CNVs but not manifesting clinically evident phenotypes raises the issue of whether 1q21.1 microdeletions and microduplications are benign CNVs or are pathogenic variants with incomplete penetrance. Indeed, CNVs within distal 1q21.1 were previously reported in normal individuals in population screens of genomic variation4,19 but only at a frequency of ∼0.02% in two large control populations in studies of schizophrenia4,5. Moreover, while this manuscript was under review, Mefford et al. reported a series of 4,737 controls in which no 1q21.1 microdeletions and only one microduplication case were found20. Furthermore, no 1q21.1 CNVs were detected in 550 normal parental cases analyzed in our laboratory. Therefore, these CNVs occur at a higher frequency among individuals referred for clinical array-CGH testing and are unlikely to be benign.
It is clear that an important consideration in genetic counseling for these syndromes is the potential for reduced penetrance and variable expressivity, which has also been reported for well-characterized syndromes such as 22q11.2 microdeletion, where phenotypically mild deletion carriers have escaped clinical recognition until they had children with more severe manifestations21. Findings such as these and the recently described 16p13.11 and 15q13.3 microdeletion syndromes22,23 raise difficult questions in the context of genetic counseling for newly diagnosed cases and particularly for prenatal diagnosis.
In conclusion, our results demonstrate that recurrent reciprocal microdeletions and microduplications within 1q21.1 represent novel genomic disorders consisting of microcephaly or macrocephaly, respectively, and can manifest with a range of developmental delay, neuropsychiatric abnormalities, dysmorphic features and a variety of other congenital anomalies. These features seem to be subject to incomplete penetrance and variable expressivity. We propose that the 1q21.1 HYDIN paralog (which was deleted or duplicated in all of our cases and in several deletion cases reported by Mefford et al.20) is dosage sensitive and is important in determining head size. Other genes within the deleted or duplicated region could mediate other phenotypes associated with 1q21.1 CNVs, perhaps in association with other genetic or genomic variants.
Methods
Cases and controls
Clinical information was available for 21 microdeletion and 15 microduplication cases. Clinical evaluations of parents carrying the CNVs were also available in 9 of 21 microdeletion cases and in 6 of 15 microduplication cases. Clinical evaluations consisted of a comprehensive medical history, family history, growth measurements and dysmorphology examination. Pictures of the affected individuals were obtained after informed consent, and consent for publication was also obtained. Fifty controls were randomly selected from 16,557 array-CGH cases referred from Texas Children's Hospital during the same time period.
Array CGH and FISH analyses
Array-CGH analysis was carried out as described previously on DNA obtained from peripheral blood samples from the affected individuals24. Phytohemagglutinin-stimulated peripheral blood lymphoblast cultures from the affected individuals and their parents were used for metaphase and interphase FISH analyses using standard protocols.
1q21.1 LCR structure analysis
We examined 1q21.1 segmental duplications downloaded from the UCSC genome browser, Segmental Duplication track (Human Genome Build 36.1). Segmental duplications within the 1q21.1 region were assembled into a table along with their matching segments. Segmental duplications matching only outside the 1q21.1 region were excluded for the purpose of this analysis. Because of the large number of segments, we restricted the analysis to ten large blocks. We identified groups of related sequences and mapped these regions to obtain a composite view of linked segmental duplications.
Head circumference analysis
Head circumferences were measured using standard techniques. Age- and sex-matched percentiles were obtained using Abase, a PalmOS-based calculator25, and converted into Z scores. Each of the three groups being studied (microdeletion cases, microduplication cases and controls) was compared to zero (the population mean) using a one sample t-test and against each other using an unpaired two-tailed t-test with Welch's correction for unequal variances. Analysis and graphing was carried out in GraphPad Prism 5.
Footnotes
Author Contributions: N.B.-P. coordinated clinical data collection. Clinical information was provided by N.B.-P., J.S.B., F.S., J.B., C.A.B., B.G., B.L., M.S., J.S., A.P., T.L., G.K., S.L.-S., C.W., E.R.R., T.A.G., G.L.A., T.H., T.R., S.A., M.T.G., J.W.I., E.O., B.N., S.S.R., P.I.B., D.K.G., S.N., A.D.G., S.M.B., C.-T.F., A.S. and W.D.W. Array-CGH and FISH analysis were carried out by A.P., C.A.B., T.S., S.R.L., S.-H.L.K., P.S. and S.-W.C. Data interpretation, critical revisions and writing of the manuscript were carried out by N.B.-P., J.S.B. and A.P. J.R.L., P.S. and S.W.C. contributed to the writing of the manuscript. A.P. provided supervision and oversaw manuscript preparation and revision.
Competing Interests Statement: The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturegenetics/.
References
- 1.Christiansen J, et al. Chromosome 1q21.1 contiguous gene deletion is associated with congenital heart disease. Circ Res. 2004;94:1429–1435. doi: 10.1161/01.RES.0000130528.72330.5c. [DOI] [PubMed] [Google Scholar]
- 2.Sharp AJ, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 3.Shaffer LG, et al. Targeted genomic microarray analysis for identification of chromosome abnormalities in 1500 consecutive clinical cases. J Pediatr. 2006;149:98–102. doi: 10.1016/j.jpeds.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Stefansson H, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stone JL, et al. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davy BE, Robinson ML. Congenital hydrocephalus in hy3 mice is caused by a frameshift mutation in Hydin, a large novel gene. Hum Mol Genet. 2003;12:1163–1170. doi: 10.1093/hmg/ddg122. [DOI] [PubMed] [Google Scholar]
- 7.Doggett NA, et al. A 360-kb interchromosomal duplication of the human HYDIN locus. Genomics. 2006;88:762–771. doi: 10.1016/j.ygeno.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 8.Klopocki E, et al. Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am J Hum Genet. 2007;80:232–240. doi: 10.1086/510919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lechtreck KF, Delmotte P, Robinson ML, Sanderson MJ, Witman GB. Mutations in Hydin impair ciliary motility in mice. J Cell Biol. 2008;180:633–643. doi: 10.1083/jcb.200710162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bond J, et al. Protein-truncating mutations in ASPM cause variable reduction in brain size. Am J Hum Genet. 2003;73:1170–1177. doi: 10.1086/379085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ponting CP. A novel domain suggests a ciliary function for ASPM, a brain size determining gene. Bioinformatics. 2006;22:1031–1035. doi: 10.1093/bioinformatics/btl022. [DOI] [PubMed] [Google Scholar]
- 12.Bond J, et al. ASPM is a major determinant of cerebral cortical size. Nat Genet. 2002;32:316–320. doi: 10.1038/ng995. [DOI] [PubMed] [Google Scholar]
- 13.Callen DF, et al. High resolution mapping of interstitial long arm deletions of chromosome 16: relationship to phenotype. J Med Genet. 1993;30:828–832. doi: 10.1136/jmg.30.10.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujiwara M, Yoshimoto T, Morita Y, Kamada M. Interstitial deletion of chromosome 16q: 16q22 is critical for 16q- syndrome. Am J Med Genet. 1992;43:561–564. doi: 10.1002/ajmg.1320430311. [DOI] [PubMed] [Google Scholar]
- 15.Natt E, Magenis RE, Zimmer J, Mansouri A, Scherer G. Regional assignment of the human loci for uvomorulin (UVO) and chymotrypsinogen B (CTRB) with the help of two overlapping deletions on the long arm of chromosome 16. Cytogenet Cell Genet. 1989;50:145–148. doi: 10.1159/000132745. [DOI] [PubMed] [Google Scholar]
- 16.McNeil TF, Cantor-Graae E, Nordstrom LG, Rosenlund T. Head circumference in ‘preschizophrenic’ and control neonates. Br J Psychiatry. 1993;162:517–523. doi: 10.1192/bjp.162.4.517. [DOI] [PubMed] [Google Scholar]
- 17.Ward KE, Friedman L, Wise A, Schulz SC. Meta-analysis of brain and cranial size in schizophrenia. Schizophr Res. 1996;22:197–213. doi: 10.1016/s0920-9964(96)00076-x. [DOI] [PubMed] [Google Scholar]
- 18.Kelly BD, Lane A, Agartz I, Henriksson KM, McNeil TF. Craniofacial dysmorphology in Swedish schizophrenia patients. Acta Psychiatr Scand. 2005;111:202–207. doi: 10.1111/j.1600-0447.2004.00473.x. [DOI] [PubMed] [Google Scholar]
- 19.Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mefford HC, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson DI, Goodship JA, Burn J, Cross IE, Scambler PJ. Deletions within chromosome 22q11 in familial congenital heart disease. Lancet. 1992;340:573–575. doi: 10.1016/0140-6736(92)92107-q. [DOI] [PubMed] [Google Scholar]
- 22.Hannes FD, et al. Recurrent reciprocal deletions and duplications of 16p13.11: the deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J Med Genet. 2008 June 11; doi: 10.1136/jmg.2007.055202. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharp AJ, et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet. 2008;40:322–328. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheung SW, et al. Development and validation of a CGH microarray for clinical cytogenetic diagnosis. Genet Med. 2005;7:422–432. doi: 10.1097/01.gim.0000170992.63691.32. [DOI] [PubMed] [Google Scholar]
- 25.Zankl A, Molinari L. ABase—a tool for the rapid assessment of anthropometric measurements on handheld computers. Am J Med Genet A. 2003;121A:146–150. doi: 10.1002/ajmg.a.20185. [DOI] [PubMed] [Google Scholar]