

Abstract
Recent studies of calorie restriction in several organisms demonstrate an increase in mitochondrial activity that is associated with the salutary effects of this dietary restriction regimen. In this Essay, I speculate on how an increase in mitochondrial activity might provide benefit and discuss how diet, mitochondria, and sirtuins might interact in a pathway to slow aging and associated diseases.
The earliest molecular theory of aging proposed that reactive oxygen species (ROS) damage macromolecules progressively over time, leading to a gradual decline in cellular function (Harman, 1956). ROS are produced as by-products of electron transport by the cells’ power plants for energy, mitochondria, during the generation of ATP by respiration (Wallace, 2005). Thus, it has also been supposed that mitochondria play a uniquely important role in aging. Indeed, many constituents of mitochondria, such as the mitochondrial genome and components of the electron transport chain, are damaged with aging, consistent with their proximity to the site of ROS production. Furthermore, individuals with mitochondrial genetic diseases (Wallace, 2005) or mice that generate frequent mutations in mitochondrial DNA (Kujoth et al., 2005; Trifunovic et al., 2004, 2005) display phenotypes that resemble premature aging.
By this reckoning, it was first suggested that calorie restriction (CR), a dietary regimen that is known to extend life span in rodents and other organisms, exerts its salutary effects by slowing carbohydrate use, respiration, and the rate of damage produced by ROS. However, several recent findings have prompted a re-evaluation of this perhaps naive idea. First, a more careful examination of the physiology of CR has demonstrated that this regimen does not slow respiration but in several examples actually upregulates mitochondrial function (see below). Second, CR appears to be a regulated rather than a passive process and requires regulatory proteins, such as the sirtuins, to exert its effects (Guarente, 2006). This family of antiaging proteins, related to the yeast SIR2 and its mammalian ortholog SIRT1, comprises NAD-dependent protein deacetylases (Imai et al., 2000; Landry et al., 2000) that increase life span in yeast, the worm Caenorhabditis elegans, and the fruit fly Drosophila. Three of the seven mammalian sirtuins (SIRT3, 4, and 5) are targeted to mitochondria, and SIRT1 itself is a regulator of mitochondrial biogenesis (Guarente, 2006). Other regulatory proteins involved in metabolism, such as the TOR and FOXO proteins (Kaeberlein et al., 2005a; Kenyon, 2005), may also play important roles in CR, but their connection to mitochondria is less clear.
These findings lead to a picture in which CR can exert a positive effect on mitochondria, boosting mitochondrial activity and hence providing at least some of the salutary effects of CR. In this Essay, I will review examples in which CR and sirtuins upregulate the activity of mitochondria in different organisms. Moreover, I will present several speculative models to explain how upregulation of mitochondrial biogenesis may confer antiaging effects on cells and organisms.
Mitochondria and CR-Induced Longevity
A regimen of moderate CR in yeast extends replicative life span of mother cells and increases respiration by funneling more pyruvate to the mitochondria for metabolism to CO2 at the expense of fermentation (Lin et al., 2002) (see Figure 1). Importantly, blocking respiration by deleting the gene for cytochrome c1 prevents this CR-driven extension in life span. Moreover, increasing respiration in yeast constitutively through the enforced expression of the HAP4 transcriptional driver of mitochondrial biogenesis results in a long life span under normal caloric conditions that is not extended further by CR. The additional respiration during CR increases the activity of yeast SIR2 and thereby extends life span.
Figure 1. Calorie Restriction Pathways in Different Species.
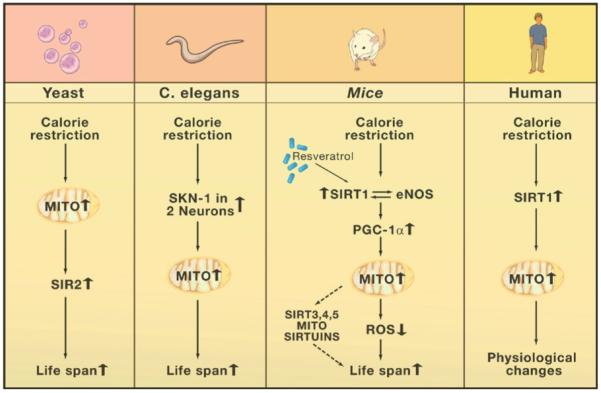
In yeast, SIR2 has been implicated downstream of mitochondrial changes in response to calorie restriction (CR), whereas in mammals the SIR2 ortholog SIRT1 has been implicated upstream of mitochondrial changes. In C. elegans, sirtuins have not been implicated in dietary restriction to date. The pathway in mice shows that the increase in mitochondrial number and activity may work via the mitochondrial sirtuins SIRT3, 4, and 5 or by reducing reactive oxygen species (ROS). The drug Resveratrol and CR may increase SIRT1 activity, which is part of an autoregulatory feedback loop that includes the enzyme endothelial nitric oxide synthase (eNOS).
In C. elegans, CR was recently shown to require two sensory neurons, termed ASI neurons, in the head of the roundworm (Bishop and Guarente, 2007). Signals emanating from these two cells trigger an increase in respiration, which, again, proved to be essential for CR-induced life span extension, as two different inhibitors of electron transport prevented this effect (see Figure 1). The response to CR in C. elegans requires two transcription factors, SKN-1 (Bishop and Guarente, 2007) and PHA-4 (Panowski et al., 2007), but so far sirtuins have not been implicated.
It appears that the degree and timing of energy limitation may influence the relationship between mitochondrial function and longevity. In yeast, a CR regimen using one tenth as much glucose as the moderate CR regimen, described above, was shown to extend yeast life span by a mechanism that did not require mitochondria or SIR2 (Kaeberlein et al., 2005b). In C. elegans, an RNAi-imposed reduction in mitochondrial function during development surprisingly extends life span, but in adult worms such a reduction in mitochondrial function has no effect (Dillin et al., 2002; Rea et al., 2007).
A priori, one cannot be certain which of the experimental conditions in the lower organisms is relevant to the standard CR protocol in mammals (30%-40% reduction of ad libitum feeding). However, recent findings have begun to link these mechanisms to mammalian CR. First, mice lacking SIRT1 are unable to mount the increase in foraging activity typical of this diet (Chen et al., 2005), suggesting a role of this sirtuin in the brain during CR. Moreover, transgenic mice that over-express SIRT1 resemble CR mice in physiological parameters (decreased blood glucose, insulin, fat, and cholesterol) and behavioral assays (improved rotarod performance and delayed mating) (Bordone et al., 2007).
Second, CR increases respiration as well as mitochondrial number per cell in mice (Nisoli et al., 2005) (see Figure 1). These increases require the endothelial nitric oxide synthase (eNOS); nitric oxide is known to be capable of activating the SIRT1 promoter in cultured cells. These findings fit with the fact that the NAD/NADH ratio and SIRT1 protein levels are increased in several rodent tissues during CR (Cohen et al., 2004; Guarente, 2006). Interestingly, SIRT1 also deacetylates and activates the eNOS enzyme (Mattagajasingh et al., 2007), indicating that a positive feedback mechanism between SIRT1 and eNOS may reset the levels of this sirtuin during CR.
Further molecular insight into the increase in mitochondrial function in CR is suggested by the relationship between SIRT1 and PGC-1α, a transcriptional coactivator of nuclear genes encoding mitochondrial proteins. SIRT1 was shown to deacetylate PGC-1α at several lysine residues and thereby increase its ability to transcriptionally activate target genes (Rodgers et al., 2005; Gerhart-Hines et al., 2007). Thus, the increase in SIRT1 during CR should result in mitochondrial biogenesis in tissues such as muscle and white fat. Indeed, feeding mice the putative SIRT1 activator resveratrol upregulates mitochondrial number in muscle (Lagouge et al., 2006). This compound also triggers numerous salutary effects to counteract a high caloric diet, including improved physical activity and longer average life span (Baur et al., 2006; Lagouge et al., 2006).
Finally, some of these effects found in mice also have been observed in a six month human trial in which subjects were calorie restricted to a degree that resulted in the expected reductions in body weight and blood insulin (Civitarese et al., 2007). Most strikingly, muscle punch biopsies from these calorie-restricted individuals showed upregulation of SIRT1, eNOS, the mitochondrial protein TFAM, and mitochondrial number, compared to biopsies from control individuals on a normal diet (Figure 1). These findings suggest that a conserved pathway may operate in mammals during CR involving the activation of SIRT1 and eNOS that triggers an increase in mitochondrial activity.
Why Is Activation of Mitochondria Good?
Increased respiration in yeast and worms is required for CR-induced longevity, but the situation in mammals is only correlative. However, it is still worth considering possible mechanisms to explain how salutary effects might be caused by or associated with the increase in number of mitochondria, if only to spur new experiments. In yeast, Sir2p lies downstream of (that is, it responds to) increased respiration in the CR longevity pathway (Figure 1). It has been suggested that this sirtuin is activated by the respiration-triggered increase in the NAD/NADH ratio (Lin et al., 2004) and the induction of the PNC1 nicotinamidase (Anderson et al., 2003). In mammals on CR, it seems likely that sirtuins both trigger (SIRT1) and respond to (SIRT3, 4, 5) an increase in mitochondrial number and activity (Figure 1). I consider below five possible ways in which increased mitochondrial number and activity may be an integral part of a mechanism by which CR exerts antiaging effects in mammals.
A Mitochondrial Buffer to Aging
There is ample evidence that damage to mitochondria increases progressively with age (Wallace, 2005). This has been observed in the form of the accumulation of mutations in mitochondrial DNA and a decline in the activity of mitochondrial enzymes and components of the electron transport chain. In the case of aging skeletal muscle syncitia, for example, zones of metabolically inactive tissue have been observed, due to expansion of mitochondria that become damaged during aging (Bua et al., 2006). A large body of evidence links mitochondrial dysfunction with diabetes (Lowell and Shulman, 2005), although a recent report surprisingly showed that deleting the apoptosis inducing factor AIF1 in muscle or liver reduced mitochondrial function and had salutary effects on metabolic disease (Pospisilik et al., 2007).
A higher pool of functional mitochondria may ameliorate tissue damage simply by buffering cells (or the syncitia in muscle) against the gradual decline in the ability to produce energy as mitochondria become damaged during aging. One may well wonder why mitochondrial number is not normally set to a higher level during ad libitum feeding to forestall this decline. It is important to remember that aging occurs postreproductively and is nonadaptive. Thus, under normal conditions, mitochondrial number and function will only fall sway to selective pressure until the reproductive period has been completed. During subsequent aging, there is minimal selective pressure to maintain mitochondrial (or any other) robustness. By this logic, CR and perhaps other stressors may impose a new selective landscape in which robust somatic maintenance, rather than reproduction, is now at a premium and mitochondrial biogenesis favored.
Mitochondrial Regulation of Sirtuins
A second possibility is that the increase in mitochondrial function serves a regulatory function by changing the activity of regulatory proteins, exemplified by CR and Sir2p in yeast. Proteins capable of responding to metabolic changes in mitochondria that alter the NAD/NADH ratio include the three sirtuins located in that cellular compartment (SIRT3, 4, and 5). Both SIRT3 and SIRT4 have been shown to have important metabolic functions in mitochondria (Guarente, 2006). Importantly, these two sirtuins were also recently shown to be involved in a stress-resistance pathway (Yang et al., 2007). Cells that sustain DNA damage will acutely deplete their nuclear NAD pool because of the activation of the NAD-cleaving enzyme poly-ADP-ribose polymerase (PARP), which is involved in DNA repair. However, the mitochondrial pool of NAD is preserved in part through the translocation of the NAD synthetic enzyme Nampt to mitochondria, where reinforced synthesis of NAD, SIRT3, and SIRT4 appears to be required to prevent apoptosis.
In the steady-state condition of CR, any changes in the NAD/NADH ratio due to altered metabolic activity in mitochondria are likely to equilibrate to the other cellular compartments by shuttle systems that operate across the mitochondrial membrane. Thus, the non-mitochondrial sirtuins and indeed any other cellular proteins that bind to NAD may also be affected. In principle, it is also possible that changes in other small molecules due to altered mitochondrial activity may play important regulatory roles during CR.
Mitochondrial Regulation of ROS
ROS remain a likely cause of the decline in cellular and organismal vitality during aging. There is good evidence that species that are long-lived produce high levels of antioxidant enzymes and, moreover, that CR leads to a reduction in the production of ROS in a given organism (Weindruch and Walford, 1988). Furthermore, mice with a transgene targeting the antioxidant enzyme catalase to the mitochondrial matrix have a longer life span (Schriner et al., 2005). Although ROS can be generated by a variety of enzymes of oxidative metabolism, the electron transport chain is their most significant source. How might the biogenesis of mitochondria, described above, relate to the reduction of ROS observed during CR? It was first assumed that ROS production by the electron transport chain would be proportional to the respiration rate; however, recent evidence suggests that this view is too simplistic (Lopez-Lluch et al., 2006). In fact, most ROS are produced by complexes I (NADH dehydrogenase) and III (cytochrome b-c1 complex) in the electron transport chain (Barja, 2007; Pamplona and Barja, 2007), and this production actually appears to be enhanced by the stalling of electrons at these complexes (Kushnareva et al., 2002; Barros et al., 2004) (see Figure 2). For example, ischemia will stall electron transport due to lack of a terminal electron acceptor and cause ROS production upon oxygen reperfusion resulting in severe tissue damage.
Figure 2. Mitochondrial Biogenesis and Reactive Oxygen Species.
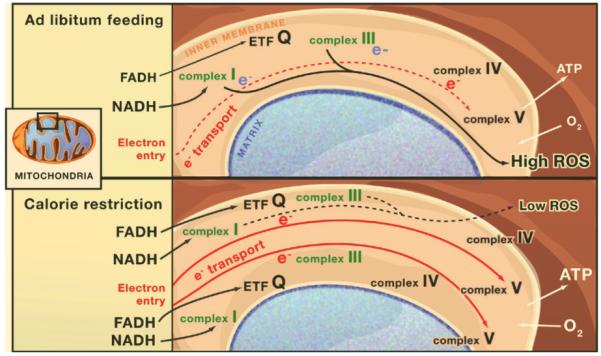
Shown is mitochondrial biogenesis during calorie restriction versus ad libitum feeding in mice and its proposed effects on reactive oxygen species (ROS). In the ad libitum case, the number of electron transport chains is low, and if the rate of entry of electrons (red e-) exceeds the slowest step of flow through the chain, stalling of electrons at mitochondrial complexes I and III (blue e-) and production of ROS will be favored. During calorie restriction, mitochondrial biogenesis increases the number of electron transport chains, thereby reducing the rate of electron entry per electron transport chain. Calorie restriction may also increase the fraction of electrons that bypass complex I by entering the electron transport chain via the electron transfer flavoprotein dehydrogenase (ETF). These effects may reduce the production of ROS during calorie restriction and hence mitigate cellular damage, aging, and disease.
Electron stalling occurs when the rate of entry of electrons into complex I of the electron transport chain exceeds their rate of transit through the slowest step of the chain. For example, if electron transit through complex IV were limiting, rapid entry of electrons into the electron transport chain could trigger electron stalling at complexes I and III (Figure 2). Ironically, the old idea that CR reduces carbohydrate use as fuel for the TCA cycle could explain a reduction in ROS production because NADH production and the rate of entry of electrons into the electron transport chain would be reduced. But this idea has been discredited because respiration does not decrease during CR.
The biogenesis of mitochondria during CR offers another possible explanation. The increase in electron transport chain components should restrain ROS production because it will increase the number of entry points for electrons into the electron transport chain, thereby lowering the rate of entry into the chain and reducing the probability of electrons becoming stalled at complexes I and III (Figure 2). Further studies on mitochondrial bioenergetics should resolve whether such an effect is observed in tissues from animals on CR. The model discussed in this section does not require any additional regulatory components to translate the upregulation of mitochondria into beneficial effects.
Mitochondrial Reprogramming to Fat Oxidation
CR changes the way in which different available energy sources in food are handled by the body. In caloric excess, carbohydrates are used for energy and fat is stored, presumably because it has a higher energy content per gram. However, during CR all energy sources are used, notably fat and amino acids, and the animal minimizes carbohydrate use in peripheral tissues to maintain adequate blood glucose levels for the brain (Weindruch and Walford, 1988). Therefore, in energy-producing tissues like muscle, there is a shift toward the β-oxidation of fatty acids to derive energy during CR, a process that occurs in mitochondria.
Unlike carbohydrate catabolism, in which most of the reducing power generated for electron transport is in the form of NADH, β-oxidation of fatty acids produces FADH at a molar equivalence of NADH. FADH must enter the electron transport chain not via complex I but via the electron transfer flavoprotein dehydrogenase (ETF) (coupled to acyl-CoA dehydrogenase) (Figure 2). ETF passes electrons to ubiquinone, which then donates them to complex III and thence onward down the chain. Therefore, use of fat as an energy source increases the frequency with which electrons enter the electron transport chain by bypassing complex I. Given that complex I is one of the primary stations of ROS production, this simple metabolic shift from carbohydrate to fat utilization may reduce ROS production.
But what about the increase in mitochondrial biogenesis observed during CR? When electrons enter the electron transport chain at complex I, their transport results in the synthesis of three molecules of ATP by oxidative phosphorylation. However, when electrons enter at ETF only two ATP molecules are made because the proton-pumping complex I has been bypassed. Thus, it is possible that the observed mitochondrial biogenesis during CR compensates for the reduction in ATP production per reducing equivalent when fat is used as an energy source. This model would suggest that it is not an increase in respiration or even mitochondrial biogenesis per se that is beneficial during CR but rather the partial bypass of complex I in the production of ATP. In this regard, it is interesting to note that the human CR trial did not record any increase in respiration but actually showed a decrease in whole body energy expenditure (Civitarese et al., 2007), even though there was an increase in mitochondrial components.
As for model three, this idea is appealing for its simplicity but bears the caveat of placing great emphasis on the importance of ROS in aging. It is interesting to note that one of the most effective drugs for diabetes, metformin, can act as a partial inhibitor of complex I (Owen et al., 2000). Much of the benefit of this drug in treating metabolic disease has been attributed to the resulting activation of AMP-dependent protein kinase (AMPK) (Zhou et al., 2001), which helps to reduce gluconeogenesis in liver and to drive the oxidation of fat in muscle. However, it is clearly possible that the inhibition of complex I per se can help to reduce cellular damage and to foster long-term health benefits in metformin-treated individuals. The activation of AMPK may still serve an important regulatory role by inducing fatty-acid oxidation to ensure the maintenance of energy production.
Recycling of Damaged Mitochondria
Cells have the cytosolic ubiquitin/proteasome system and mitochondrial proteases to degrade damaged or unwanted proteins and to replace them with newly synthesized ones (Varshavsky, 2005). Cells can also degrade damaged structures as large as organelles by a process called autophagy (Scherz-Shouval and Elazar, 2007). This process transports damaged organelles, for example mitochondria, to lysosomes where they are degraded and their contents recycled. Obviously, if mitochondria were degraded in this fashion, new synthesis would be required to maintain energy homeostasis. In this regard, it is possible that the increase in mitochondrial biogenesis induced by CR is a consequence of increased organelle turnover, and any increase in net mitochondrial synthesis is but a corollary. An increase in mitochondrial turnover might be beneficial for cells, given that they are among the most damaged structures during aging. It would not matter according to this model whether ROS or other agents generate the damage; the important outcome would be better maintenance of young mitochondria in aging cells. Consistent with this idea, autophagy has been reported to be required for the CR extension of life span in C. elegans (Jia and Levine, 2007).
Importantly, a recent paper showed that SIRT1 deacetylates and activates several proteins of the autophagic machinery (Lee et al., 2007). Cells from SIRT1-deficient mice are defective in autophagy, and SIRT1-deficient mice have damaged organelles and display early postnatal lethality like mice defective in autophagy. Thus, the idea that salutary effects of CR spring from the activation of autophagy and the clearance of defective mitochondria is attractive.
However, several questions arise. First, can the autophagic machinery selectively recognize and destroy damaged mitochondria? Second, does CR actually induce autophagy in mammals? One might expect the answer to be yes, given that one consequence of autophagy is to provide a source of carbon and energy, and induction of autophagy during acute starvation is clearly observed. However, long-term CR is a steady-state process and does not impose the same degree of energy limitation as starvation, so whether it triggers autophagy is still an open question. In conclusion, the interesting possibility that autophagy of damaged mitochondria plays an important role in mammalian CR awaits further experimentation.
Conclusion
Mitochondria have long been proposed to play an important role in aging. Recent genetic findings in lower organisms have pinpointed sirtuins as antiaging genes, and at least four of the seven mammalian sirtuin homologs have mitochondria-associated functions. CR is perhaps the most robust intervention that extends mammalian life span and has been associated with an increase in SIRT1 levels in several tissues and a corresponding increase in mitochondrial components. Here, I have presented several models for how this increase in mitochondria may have the effect of slowing aging and disease. Some of the models rely on an important role of ROS in the aging process, whereas others do not. It is hoped that the next few years will see a further convergence of genetic pathways with mitochondrial function, which will provide a comprehensive view of aging and antiaging mechanisms and will also explain how CR works. It seems likely that we are on the right track of acquiring this understanding, and that it will involve mechanisms rich in new and old ideas about aging and how to counteract it.
REFERENCES
- Anderson R, Bitterman K, Wood J, Medvedik O, Sinclair D. Nature. 2003;423:181–185. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barja G. Rejuvenation Res. 2007;10:215–224. doi: 10.1089/rej.2006.0516. [DOI] [PubMed] [Google Scholar]
- Barros MH, Bandy B, Tahara EB, Kowaltowski AJ. J. Biol. Chem. 2004;279:49883–49888. doi: 10.1074/jbc.M408918200. [DOI] [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, et al. Nature. 2006;444:280–281. [Google Scholar]
- Bishop NA, Guarente L. Nature. 2007;447:545–549. doi: 10.1038/nature05904. [DOI] [PubMed] [Google Scholar]
- Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, Steele AD, Crowe H, Marmor S, Luo J, et al. Aging Cell. 2007;6:759–767. doi: 10.1111/j.1474-9726.2007.00335.x. [DOI] [PubMed] [Google Scholar]
- Bua E, Johnson J, Herbst A, Delong B, McKenzie D, Salamat S, Aiken JM. Am. J. Hum. Genet. 2006;79:469–480. doi: 10.1086/507132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Steele AD, Lindquist S, Guarente L. Science. 2005;310:1641. doi: 10.1126/science.1118357. [DOI] [PubMed] [Google Scholar]
- Civitarese AE, Carling S, Heilbronn LK, Hulver MH, Ukropcova B, Deutsch WA, Smith SR, Ravussin E. PLoS Med. 2007;4:e76. doi: 10.1371/journal.pmed.0040076. 10.1371/journal.pmed.0040076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Nature. 2006;444:868–874. doi: 10.1038/nature05486. [DOI] [PubMed] [Google Scholar]
- Harman D. J. Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Jia K, Levine B. Autophagy. 2007;3:597–599. doi: 10.4161/auto.4989. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, Powers R, Steffen K, Westman E, Hu D, Dang N, Kerr E, Kirkland K, Fields S, Kennedy BK. Science. 2005a;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, Hu D, Kerr EO, Tsuchiya M, Westman EA, Dang N, Fields S, Kennedy BK. PLoS Genet. 2005b;1:e69. doi: 10.1371/journal.pgen.0010069. 10.1371/journal.pgen.0010069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, et al. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Kushnareva Y, Murphy AN, Andreyev A. Biochem. J. 2002;368:545–553. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, et al. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R. Proc. Natl. Acad. Sci. USA. 2000;97:5807–5811. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IH, Cao L, Mostoslavsky R, Lombard D, Liu J, Bruns N, Tsokos M, Alt FW, Finkel T. Proc. Natl. Acad. Sci. USA. 2007 doi: 10.1073/pnas.0712145105. in press. 10.1073/PNAS.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SJ, Ford E, Haigis M, Liszt G, Guarente L. Genes Dev. 2004;18:12–16. doi: 10.1101/gad.1164804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, Fink GR, Guarente L. Nature. 2002;418:344–348. doi: 10.1038/nature00829. [DOI] [PubMed] [Google Scholar]
- Lopez-Lluch G, Hunt N, Jones B, Zhun M, Jamieson H, Hilmer S, Cascajo MV, Allard J, Ingram DK, Navas P, de Cabo R. Proc. Natl. Acad. Sci. USA. 2006;103:1768–1773. doi: 10.1073/pnas.0510452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell BB, Shulman GI. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- Mattagajasingh I, Kim CS, Nagvi A, Yamamori T, Hoffman TA, Jung SB, DeRicco J, Kasuno K, Irani K. Proc. Natl. Acad. Sci. USA. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, et al. Science. 2005;310:314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- Owen MR, Doran E, Halestrap AP. Biochem. J. 2000;348:607–614. [PMC free article] [PubMed] [Google Scholar]
- Pamplona R, Barja G. Ageing Res. Rev. 2007;6:189–210. doi: 10.1016/j.arr.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillen A. Nature. 2007;447:550–555. doi: 10.1038/nature05837. [DOI] [PubMed] [Google Scholar]
- Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, et al. Cell. 2007;131:476–491. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- Rea SL, Ventura N, Johnson TE. PLoS Biol. 2007;5:e259. doi: 10.1371/journal.pbio.0050259. 10.1371/journal. pbio.0050259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Scherz-Shouval R, Elazar Z. Trends Cell Biol. 2007;17:422–427. doi: 10.1016/j.tcb.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Schriner S, Linford N, Martin GM, Treuting P, Ogburgn C, Emond M, Coskun P, Ladiges W, Wolf N, Van Remmen H, et al. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink J, Rovio A, Bruder C, Bohlooly Y, Gidlof S, Oldfors A, Wibom R, et al. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Hansson A, Wredenber A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson NG. Proc. Natl. Acad. Sci. USA. 2005;102:17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. Trends Biochem. Sci. 2005;30:283–286. doi: 10.1016/j.tibs.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Annu. Rev. Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weindruch R, Walford RL. The Retardation of Aging and Disease by Dietary Restriction. Charles C Thomas Publishing, LTD; Springfield, IL: 1988. [Google Scholar]
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, et al. Cell. 2007;130:432–442. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. J. Clin. Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]