

Abstract
Overexpression of prostacyclin synthase (PGIS) decreases lung tumor multiplicity in chemical and cigarette-smoke induced murine lung cancer models. Prostacyclin signals through a single G-protein coupled receptor (IP), which signals through cAMP. To determine the role of this receptor in lung cancer chemoprevention by prostacyclin, PGIS-overexpressing mice were crossed to mice that lack the IP receptor (IP (−/−)). Carcinogen-induced lung tumor incidence was similar in IP(+/+), IP(+/−) and IP(−/−) mice, and overexpression of PGIS gave equal protection in all three groups, indicating that the protective effects of prostacyclin are not mediated through activation of IP. Since prostacyclin can activate members of the PPAR family of nuclear receptors, we examined the role of PPARγ in prostacyclin’s protection against lung tumorigenesis. Iloprost, a stable prostacyclin analog, activated PPARγ in non-transformed bronchial epithelial cells and in a subset of human non-small cell lung cancer cell lines (NSCLC). Iloprost-impregnated chow fed to wild-type mice resulted in elevated lung macrophages and decreased lung tumor formation. Transgenic animals with lung specific PPARγ-overexpression also developed fewer lung tumors. This reduction was not enhanced by administration of supplemental iloprost. These studies indicate that PPARγ is a critical target for prostacyclin mediated lung cancer chemoprevention, and may also have therapeutic activity.
Introduction
In the United States, lung cancer continues to be the leading cause of cancer death in both men and women, and worldwide the lung cancer epidemic will result in millions of cases yearly(1). While tobacco abstinence and smoking cessation are the critical first steps in reducing lung cancer rates, the majority of US lung cancers are diagnosed in former smokers, and to date no effective chemopreventive agents have been discovered. The large at-risk population (current and former smokers) and poor 5-year lung cancer survival rates (2) underscore the need for a better understanding of chemopreventive mechanisms and effective agents.
Prostaglandins play an important role in lung tumorigenesis and prostaglandin manipulation has been investigated for lung cancer chemoprevention. Cyclooxygenase (COX) inhibition decreases levels of prostaglandins, and large epidemiologic surveys have shown fewer lung cancers in ‘frequent aspirin users’ (3). Human trials evaluating COX inhibition and lung cancer chemoprevention are currently being conducted, but conflicting data in murine studies evaluating the role of non-specific COX or selective COX-2 inhibition makes interpretation of these results difficult. Studies using non-selective COX inhibitors have shown inhibition of lung tumorigenesis in mice (4, 5). However, mice receiving celecoxib (a selective COX-2 inhibitor) showed reduced pulmonary inflammation, but no differences in tumor multiplicity, and an actual increase in tumor size after exposure to an initiator-promoter model of lung tumorigenesis (6). In addition, studies showing excess cardiovascular events associated with chronic COX-2 inhibitor use raise concerns about long-term safety(7, 8). One potential cause for these effects may be lower levels of COX-2 mediated production of prostacyclin which has been shown to have atheroprotective properties in female mice(9). Additionally, a recent report showed that dysfunctional prostacyclin receptor (IP) mediated signaling (defective adenyl cyclase activation) promoted increased platelet aggregation and a predisposition to atherothrombosis (10).
COX activity generates numerous downstream mediators (including prostaglandins, prostacyclin, and thromboxanes) that can have pro-tumorigenic or anti-tumorigenic effects(11). Our laboratory has focused on manipulating prostaglandin production distal to the COX enzymes. Prostacyclin (PGI2) is a downstream metabolite of COX activity with anti-inflammatory and platelet inhibitory properties (12). It has also been shown to prevent metastases (13) and inhibit the growth of established micrometastases (14). Previously, we showed that selective pulmonary overexpression of prostacyclin synthase (PGIS) increases pulmonary prostacyclin and prevents lung tumor formation in response to either chemical carcinogenesis or exposure to tobacco smoke in mice (15, 16).
A single receptor for PGI2 has been described (IP/PGIR), which is a member of the G-protein coupled receptor family, containing 7 membrane-spanning domains (17). PGI2 binding to the IP receptor activates Gs and stimulates adenylyl cyclase, leading to increases in intracellular cAMP. PGI2 analogs, such as iloprost, have been shown to bind the IP receptor and increase cAMP levels (18). Studies using IP receptor knockout mice suggest that activation of the receptor mediates inflammation, particularly edema and pain, vascular homeostasis, and thrombosis prevention (for review, see Narumiya et al. 1999(19)) Reports have also shown that PGI2 and its analogs are ligands for peroxisomal proliferator-activated receptors (PPARs), which are members of the nuclear receptor superfamily of regulated transcription factors (20). The purpose of this study was to determine the contribution of IP versus PPARs in mediating the anti-tumorigenic effects of PGI2. Our experiments demonstrate that prostacyclin mediated lung cancer chemoprevention is independent of the IP receptor, and may be secondary to activation of PPARγ. Manipulation of prostaglandin production distal to COX, and/or direct activation of PPARγ, may represent attractive lung cancer chemopreventive strategies.
Materials and Methods
Development of transgenic PGIS overexpressors/IP receptor knockouts
As previously described, PGIS-transgenic mice were developed using a construct consisting of the human surfactant protein-C (SP-C) promoter and full-length rat PGIS cDNA(21). The SP-C promoter allows targeted expression to alveolar and airway epithelial cells(22). Transgenic mice were genotyped by performing PCR on genomic DNA isolated from tails as previously described (21). IP receptor knockout mice were the generous gift of S. Narumiya and were originally developed in the C57BL/6 background (23). The IP KO trait was successfully transferred from the C57BL/6 background to the FVB/N background by completing 10 generations of backcrosses. FVB/N IP KO mice were then bred with PGIS overexpressors to create the experimental animals (PGIS OE-IP KO mice). Animals were bred at the Denver VA Animal Facility. All procedures were performed with IACUC approved protocols at the University of Colorado Health Sciences Center and the Denver Veterans Affairs Medical Center.
Development of PPARγ overexpressing animals
Animals with lung specific PPARγ overexpression were created to examine the role of PPARγ in lung tumorigenesis. The 1.5kb human wt PPARγ1 cDNA was ligated into a construct containing the human SP-C promoter and the SV40 small T intron and poly A region (a generous gift of Dr. Jeffrey Whitsett). Transgenic mice were made at the University of Cincinnati transgenic facility as previously described (21). Individual founders in the FVB strain were propagated and genotyped by PCR using primers for the small T intron to indicate a PPARγ over-expressing transgenic mouse. Animals were bred at the University of Colorado Health Sciences Center animal facility.
Type II cell isolation and cAMP Determination
Murine type II alveolar cells were isolated from IP KO and WT animals to confirm that IP membrane receptor activity had been abolished. Lungs were isolated and perfused with 0.9% saline via the pulmonary artery until thorough blanching had occurred. The type II cells were isolated as previously reported(24). Briefly, the cell suspension was added to antibody plates (CD 45 and CD 32) that had been pre-washed × 2 with DMEM/HEPES and were incubated for 2 hours at 37°C in 5% CO2. Following incubation, media was gently removed from plates and centrifuged (8 minutes, 130×g). Freshly isolated Type II cells from a single mouse lung were resuspended in media containing 10% FCS with 100 μM IBMX (isobutylmethylxanthine, Sigma, St. Louis, MO) +/− 5 μM iloprost. The cells were incubated at 37ºC for 45 min, and then washed 3× quickly with cold phosphate buffered saline. The cell pellet was then lysed in cold 0.1M HCL. Cellular cAMP content was measured using the direct cyclic AMP kit from Assay Designs (Ann Arbor, MI). Results are reported as pmol cAMP/mg protein.
Immunoblotting
Freshly isolated Type II were lysed in cold RIPA buffer (10mM TrisHCl, 150 mM NaCl, 1% NaDOC, 1% Triton X-100, 0.1% SDS) containing protease inhibitors, centrifuged at 10,000×g for 5 minutes, and the supernatant saved. Protein was quantitated using Bio-Rad Protein Assay reagent (Bio-Rad). Protein samples were run on precast 4–20% Gradient Tris-HCl gels (Bio Rad). Antibody PPARγ was from Cayman (Ann Arbor MI). Blots were visualized by enhanced chemiluminescence, and changes in expression normalized to β-actin expression were determined by densitometry using at least three independent experiments.
Carcinogenesis protocol
FVB/N mice (PGIS OE-IP KO, PPARγ OE and wild-type controls) 8–12 weeks of age were maintained on a standard, anti-oxidant free laboratory chow (Lab Diet™, PMI Nutrition International, St. Louis, Missouri, USA) and given food and water ad libitum. They were kept on cedar-free bedding with a 12-hour light/dark cycle in climate controlled animal facilities. Animal were subjected to a single intraperitoneal injection of ethyl carbamate (Sigma Chemical Co., St. Louis, Missouri, USA) at a dose of 1mg/g mouse weight dissolved in normal saline. Animals were sacrificed 20 weeks later via pentobarbital overdose. Lungs were removed and tumors counted, dissected from surrounding tissue and their diameters measured using a dissection microscope (5× magnification).
Iloprost Chemoprevention
To determine if the chemoprevention observed in the transgenic PGIS overexpressor could be replicated with an oral prostacyclin analogue, animals were fed anti-oxidant free chow (AIN-76A, Test Diet, Richmond, IN) containing 3% ground iloprost tablets (a gift of Schering AG, Berlin, Germany). Iloprost clathrate tablets contain 0.38 mg iloprost per 65 mg tablet. The iloprost chow was started one week prior to the urethane treatment and animals were sacrificed 20 weeks after urethane exposure. For experiments with the PPARγ-OE mice, some animals were also started on iloprost chow 5 weeks after urethane administration.
Iloprost activation of PPAR response element
RL-65 cells, a normal, immortalized rat lung epithelial cell line (25), were obtained from ATCC. Lung cancer cell lines (A549, H661) were obtained from the University of Colorado Cancer Center. Cells were transiently transfected using lipofectamine with constructs encoding a PPARγ response element (PPARγ-RE) plasmid or PPARδ response element plasmid (PPARδ-RE) (26), each linked to a luciferase reporter, along with a plasmid encoding β-galactosidase (βgal) under the control of the CMV promoter to normalize for transfection efficiency. Following transfection, cells were incubated overnight, and then stimulated with 10μM iloprost or vehicle control (0.1% DMSO) for an additional 24 h. In a second set of experiments, following transfection, cells were stimulated with 10 μM iloprost, 10 μM of the PPARγ inhibitor T0070907 (Cayman Chemical Co., Ann Arbor, MI), or both agents. Cells were harvested, washed, and resuspended in Luciferase Reporter Lysis Buffer (Promega). Cell lysates were centrifuged and analysed for luciferase and β-gal activities. PPAR-RE activity was normalized to β-gal and presented as relative light units per milliunit of β-gal.
Macrophage immunohistochemistry
Lungs from iloprost- and control chow-fed animals were evaluated for the presence of macrophages to determine if iloprost altered the number of inflammatory cells. For immunohistochemistry, whole lungs were formalin-fixed and paraffin-embedded. Paraffin sections were deparaffinized in xylene, rehydrated through a graded ethanol series, and underwent antigen retrieval (Antigen unmasking solution; Vector) by heating for 20 min at 125°C in a decloaking chamber (Biocare). Sections were then blocked in 3% goat serum and exposed to rabbit anti-F4/80 (1:100) overnight at 4°C. Antigen:antibody complexes were visualized using kits from Vector Laboratories and sections lightly counterstained with hematoxylin. Sections were visualized using an Olympus light microscope equipped with SPOT software.
Statistical analysis
Data are presented as means ± SEM. Differences between groups were identified using the Student’s unpaired t-test. One-way analysis of variance (ANOVA) was used to compare more than two groups and post-hoc Newman-Keuls test were used to identify differences between groups. Under all circumstances, P < 0.05 was considered to be significant.
Results
Confirmation of PGIS OE/IP KO mouse genotype and phenotype
To examine whether the IP receptor is required for the protective effect of overexpression of PGIS, we created a mouse which overexpressed PGIS and lacked the IP receptor, in a congenic FVB/N strain. To transfer the IP KO strain from the C57BL/6 to the FVB/N background, 10 generations of backcrosses were completed. To confirm that the IP KO animals lacked functional cell membrane IP activity, we isolated type II cells from IP KO and wild-type mice. These type II cells were then exposed to exogenous iloprost, and cell lysates were examined for changes in cAMP levels (Figure 1A). Type II cells from wild-type mice showed a 3-fold increase in cAMP levels in response to iloprost, whereas IP KO mice failed to show any significant increase, consistent with a lack of a functional IP receptor.
Figure 1. IP receptor status does not effect PGIS-mediated lung tumor prevention.
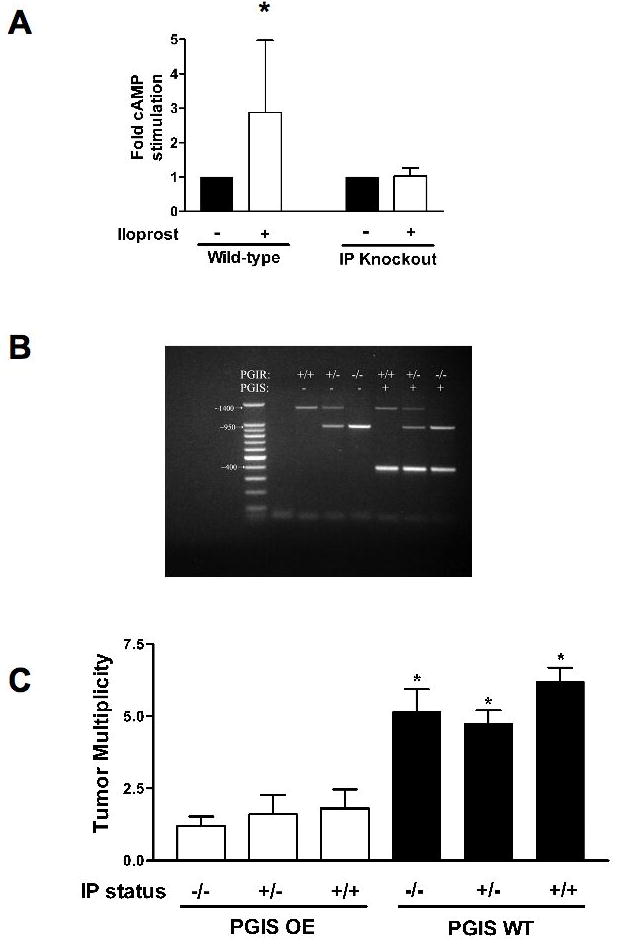
A. Intracellular cAMP content was measured in Type II cells isolated from wild-type or IP receptor knockout mice that were stimulated with iloprost (white bars) or vehicle control (black bars). cAMP content was only increased in wild-type mice stimulated with iloprost. *P<0.05 vs Control. B. Agarose gel electrophoresis of the PCR products for PGIS transgene (400bp band) and IP receptor knockout (PGIR; 1400bp wild-type band and/or 950bp knockout band). C. The indicated genetic mice were injected with 1mg/g mouse weight ethyl carbamate. Twenty weeks later, mice were sacrificed and lung tumors counted. PGIS mice developed significantly fewer lung tumors regardless of IP receptor status. *P<0.05
PGIS overexpression-mediated prevention of lung tumor formation is independent of membrane IP receptor
PGIS OE/IP KO mice were generated by breeding heterozygote PGIS animals with IP KO animals. PCR-based genotyping of the resultant animals is illustrated in Figure 1B with the expected genetically modified progeny. The indicated groups of mice were subjected to a single ethyl carbamate injection to initiate lung tumorigenesis. Animals were sacrificed after 20 weeks and tumor incidence and multiplicity determined. IP null animals did not show a difference in tumor multiplicity compared to wild-type (5.1 ± 0.8 vs. 6.2 ± 0.48, p>0.05) or IP heterozygotes (5.1 ± 0.8 vs. 4.7 ± 0.45, p>0.05) (Figure 1C). Overexpression of PGIS decreased tumor number in wild-type, IP(−/−) and IP(+/−) mice to the same extent, and these were all significantly less than PGIS wild-type animals. There were no differences in tumor incidence between the various experimental groups (data not shown). Tumor histologies were reviewed to confirm they represented pulmonary adenomas.
Prostacyclin analogs activate PPARγin lung epithelial cells and NSCLC cell lines
Since the chemoprotective effects of PGI2 were independent of IP status, we sought to identify other targets which mediated the anti-tumorigenic effect seen in PGIS-OE mice. Several studies have shown that PGI2 and its analogs can activate members of the PPAR family of nuclear receptors(27, 28). To examine the ability of prostacyclin analogs to engage PPARs, RL65 cells (a non-transformed rat lung epithelial cell line) were transiently transfected with a reporter constructs encoding three copies of a consensus PPARγ or PPARδ (26) response elements linked to firefly luciferase, and then stimulated with iloprost. Iloprost exposure increased PPARγ activity 3-fold (p<0.05); but no concomitant increase in PPARδ-RE activity was observed (Figure 2A). We further examined the ability of iloprost to activate PPARγ in NSCLC. In two adenocarcinoma cell lines, A549 and H661, iloprost increased PPAR-RE activity by approximately 2-fold. Furthermore, a specific PPARγ ligand binding antagonist, T0070907, completely blocked this stimulation (Figure 2B). These data suggest that prostacyclin and iloprost may mediate inhibition of lung tumorigenesis through activation of PPARγ. We next examined levels of IP expression in the NSCLC cell lines by RT-PCR. Both cell lines had undetectable levels of IP (data not shown), indicating that the activation of PPARγ was independent of IP expression.
Figure 2. Iloprost treatment drives PPARγ activity in vitro.
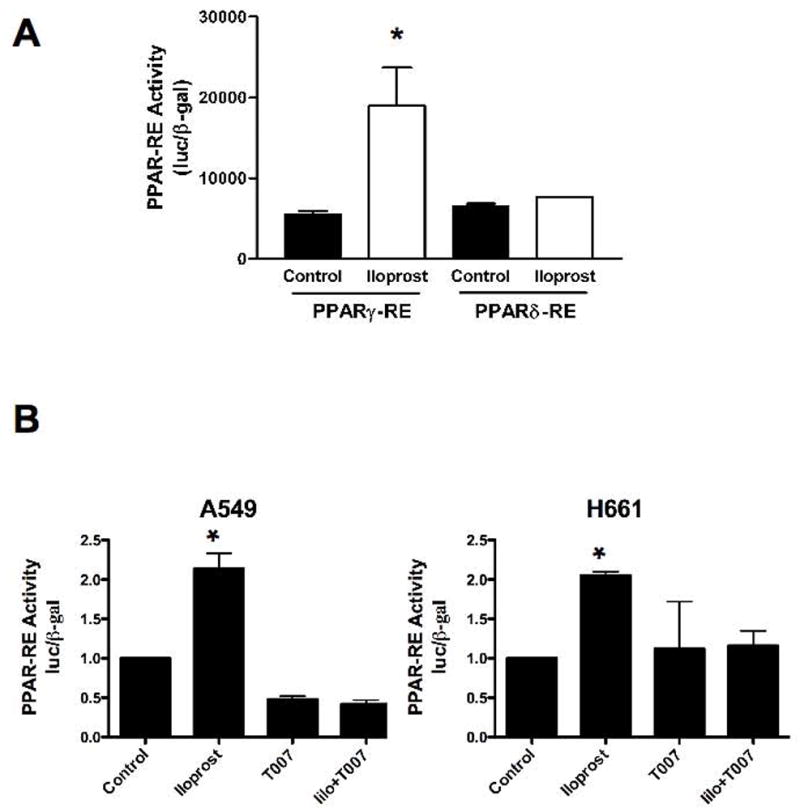
A. RL-65 cells were transiently transfected with a PPARγ-RE/luciferase or PPARδ-RE/luciferase construct along with a plasmid encoding β-galactosidase (β-gal) under the control of the CMV promoter to normalize for transfection efficiency. Following transfection, cells were incubated overnight, and then stimulated with 10μM iloprost or vehicle (Control) for an additional 24 hours. Cells were harvested and PPAR-RE activity normalized to β– gal was determined (*P<0.05 vs. Control). B. A549 and H661 cells were transiently transfected with the PPARγ-RE reporter construct along with a plasmid encoding β-gal. After an overnight incubation, cells were stimulated with 10 μM iloprost (Ilo), 10 μM of the PPARγ inhibitor T0070907, or both agents. Cells were harvested after 24 hours and PPARγ-RE normalized to β–gal was determined. (*P<0.05 vs. control and T007 treated cells)
PPARγ OE mice, with selective airway epithelial overexpression, developed fewer lung tumors
To directly test the role of PPARγ in chemoprevention of lung tumorigenesis, we created mice with targeted overexpression of PPARγ using the SPC promoter (PPARγ OE). Mice were genotyped using PCR (Fig. 3A), and propagated as heterozygotes in the FVB/N strain. Overexpression of PPARγ was verified by immunoblotting isolated Type II cells from wild-type and PPARγ OE mice with anti-PPARγ antibody. PPARγ OE mice showed a marked increase in PPARγ expression (Fig. 3B). PPARγ-OE mice did not display any phenotypic abnormalities, and had normal lung architecture when H&E stains were reviewed (Fig. 3C).
Figure 3. Characterization of PPARγ overexpressing transgenic mice.
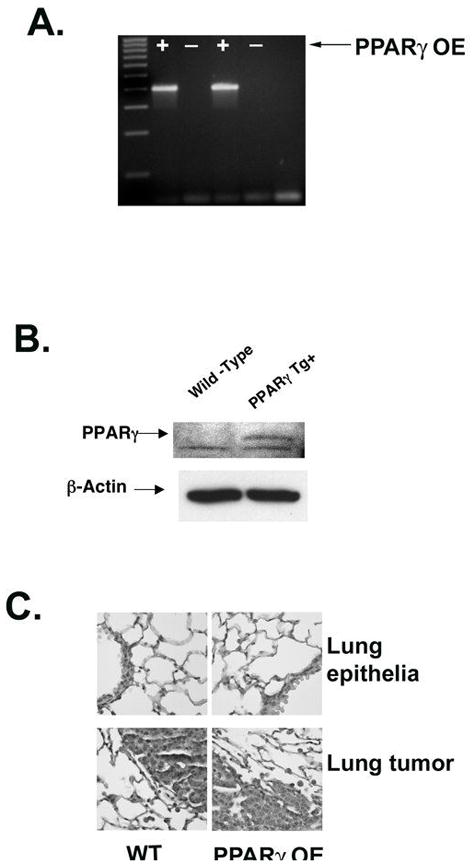
A. Agarose gel electrophoresis of the PCR product for the PPARγ transgene (400bp band) indicating transgene positive mice. B. Western blot of isolated Type II cells from PPARγ overexpressing transgenic mice (PPARγ OE) and wild-type littermates (WT) confirms increased expression of PPARγ (50 kDa) in the overexpressing mice. C. Representative hematoxylin stained lung tissue samples from wild-type (WT) and PPARγ transgenic positive (PPARγ OE) mice. Specimens show that PPARγ OE mouse lungs display no anatomic differences and the tumors that develop are similar to those in wild-type mice (magnification, ×40). Tumors from the animals exposed to urethane also show no differences.
PPARγ OE mice and wild-type littermates were injected with 1 mg/g ethyl carbamate, and tumors were counted 20 weeks after treatment. PPARγ OE mice developed 2.6 ±0.53 lung tumors, a 73% reduction (p<0.05) in tumor multiplicity as compared to wildtype mice (9.6 ±1.4) (Figure 4), similar to what we have previously reported (29). To determine if iloprost supplementation could further enhance the chemopreventive effects of targeted PPARγ overexpression, transgenics were given 3% iloprost-impregnated chow one week prior to the ethyl carbamate injection and throughout the remainder of the study, while the control animals for these experiments received anti-oxidant free control chow. While iloprost decreased tumor number in wild-type mice (6.5 ± 0.6 vs. 9.6 ± 1.4 tumors/mouse, p<0.05), PPARγ OE mice fed iloprost chow showed no further reduction in tumor number when compared to control chow (2.6 ± 0.73 vs. 2.6 ± 0.53 tumors/mouse). The lack of additive effects of iloprost and PPARγ overexpression suggest that iloprost and PPARγ may inhibit tumorigenesis through a common pathway, and that the presence of the PPARγ ligand iloprost does not augment the chemoprevention. Tumor burden was similar in all groups, but there was a trend towards smaller tumors in PPARγ overexpressors (data not shown).
Figure 4. Mice fed iloprost and PPARγ overexpressing transgenic mice develop fewer lung tumors than wild-type controls.
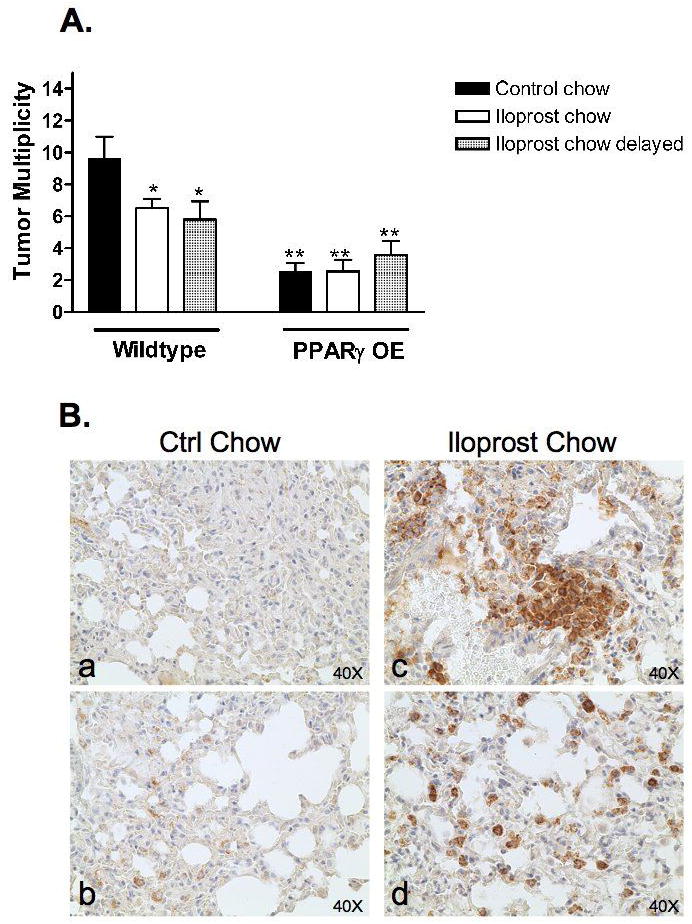
A. Wild-type or PPARγ OE mice were injected with 1mg/g mouse weight ethyl carbamate. Twenty weeks later, mice were sacrificed and lung tumors counted. Black bars represent mice fed control chow; white bars represent mice fed chow containing 3% iloprost from 1wk prior to injection through the end of the study; and hatched bars represent mice fed control chow until 5 wks after the injection and then changed to chow containing 3% iloprost for the remainder of the study (*P<0.05 vs. wild-type mice fed control chow; **P<0.001 vs. wild-type mice fed control chow). The difference between wild-type mice fed iloprost chow and PPARγ mice fed control chow or iloprost chow was also significant (P<0.05).
B: Increased accumulation of macrophages in lungs of iloprost fed mice.
Wild type mice were injected with 1mg/g weight ethyl carbamate. Mice were fed control chow or chow containing 3% iloprost one week prior to injection through the end of the study. Shown are representative lung tissues from control-fed mice (a&b) compared to iloprost-fed mice (c&d) immunohistochemically stained for the macrophage marker, F4/80 (brown reaction color).
To determine whether there is a critical timepoint for iloprost tumor inhibition, separate groups of both wild-type and PPARγ OE mice were maintained on control chow for the first 5 weeks after ethyl carbamate administration, and were then switched to iloprost-containing chow for the remaining 15 weeks of the experiment. In wild-type mice, iloprost administration 5 weeks after ethyl carbamate decreased tumor size to the same extent as seen in animals exposed to iloprost throughout the entire 20 week regimen (Figure 4A). In PPARγ OE mice, iloprost given at 5 weeks after ethyl carbamate had no effect on tumor number, which was still markedly decreased compared to wild-type controls.
To determine if iloprost treatment altered pulmonary inflammatory cell content, lungs from iloprost and control chow fed animals were examined for macrophage presence. Immunohistochemistry with the pan macrophage cell-surface marker F4/80 revealed a marked increase in pulmonary macrophages in iloprost exposed animals (Figure 4B).
Discussion
Pulmonary-specific PGIS overexpression (and resulting elevations in pulmonary PGI2 levels) significantly decreases both tumor incidence and multiplicity in multiple lung tumor models, and we have shown that pharmacologic supplementation with the oral prostacyclin analogue iloprost also decreases tumor multiplicity. The observed chemoprevention is independent of the IP membrane receptor, as IP KO animals that lack PGIS overexpression do not differ from wild-type mice in lung tumor development. Importantly, the combination of overexpression of PGIS and deletion of the IP receptor is still protective, indicating that PGI2 must act through an alternative pathway or receptor. These findings are in contrast to studies showing that the protective effects of prostacyclin against the development of pulmonary hypertension are in fact mediated through the IP receptor (30). Thus the beneficial effects of prostacyclin are mediated through distinct pathways in different disease states.
In the studies described above, we provide evidence that the observed chemoprotection is mediated by the nuclear transcription activator PPARγ. Several studies have indicated that PGI2 analogs such as iloprost can activate PPARδ (27). We did not observe this activation in non-transformed lung epithelial cells using a PPARδ-specific response element. This could be a result of low baseline expression of PPARδ in non-malignant lung epithelial cells, or the absence of critical co-activators. The role of PPARδ in lung cancer chemoprevention remains to be determined. In contrast to PPARδ, prostacyclin analogues like iloprost selectively increases PPARγ activity both in non-transformed epithelial cells and in NSCLC. In human NSCLC cell lines, activation of PPARγ by pharmacological agents, or by molecular overexpression, strongly inhibits transformed growth, as assessed by colony formation in soft agar (31, 32). Additionally, NSCLC cells over-expressing PPARγ exhibit significantly less invasiveness and metastasis compared to control cells both in vitro and in a rat orthotopic lung xenograft model (31). Similar to PGIS overexpression and oral iloprost, selective pulmonary overexpression of PPARγ decreases tumor multiplicity, and this reduction is not further enhanced by the addition of the PPARγ ligand iloprost (Figure 4), suggesting that PPARγ is a major effector for the chemopreventive effects of iloprost. In fact, the observed decrease in tumor number was greater in PPARγ OE mice than in mice treated with iloprost. This could be a result of lower PPARγ expression in wild-type mice, or alternatively, the level of PPARγ activation achieved with systemic iloprost is lower than activation achieved through lung-specific overexpression.
Our data also indicate that iloprost will have systemic effects which may be critical for tumor chemoprevention. Iloprost increased the number of macrophages in the lung of tumor bearing animals. Macrophages play a complex role in cancer progression (33). While initially mediating direct cytotoxic effects on tumors, the presence of tumor associated-macrophages (TAM) has been implicated in promoting tumor growth and metastasis. In lung cancer some studies have reported an association of increased tumor associated macrophages and tumor regression (34). The location of macrophages appears to be important. Increased numbers of macrophages within the tumor itself was associated with a favorable prognosis, whereas increased numbers of macrophages in the stroma surrounding the tumor was associated with an unfavorable prognosis (35). Further studies elucidating the role of macrophages in chemoprevention are required.
Additional support that PPARγ may play an important role in human lung tumorigenesis is found in recent epidemiologic studies of diabetics treated with the thiazolidinediones (TZDs) rosiglitazone and pioglitazone. Lung cancer incidence in diabetics on these PPARγ agonists was decreased 33% when compared to diabetics on non-PPARγ modulating treatments (36). TZDs have been shown to induce 15-hydroxyprostaglandin dehydrogenase (15-PGDH), the enzyme that inactivates the anti-apoptotic and immunosuppressive PGE2 by conversion to 15-keto prostaglandins (37). Therefore, unlike COX inhibition which would act to decrease PGE2 and PGI2, TZDs would be predicted to enhance 15-PGDH and selectively lower PGE2 levels, while preserving PGI2. TZDs have also been shown to inhibit angiogenesis by blocking ELR + CLC chemokine production in NSCLC (29, 38) and to inhibit primary tumor growth and spontaneous lung metastases in animal models (39). The finding by Govindarajan and colleagues of decreased lung cancer in diabetics on TZDs must be tempered against a recent meta-analysis suggesting that diabetics on PPARγ agonists have significantly increased risks of myocardial infarction (40). A more recent interim analysis of clinical events in the RECORD (Rosiglitazone Evaluated for Cardiac Outcomes and Regulation of Glycaemia in Diabetes) study was inconclusive regarding the effects of rosiglitazone on cardiovascular mortality and rosiglitazone was associated with an increase in heart failure but not death from cardiovascular causes (41). One critical issue in assessing PPARγ activators will be defining whether the suggested adverse cardiovascular events are mediated through PPARγ or through off-target drug effects. A recent report has shown that an IP mutation resulting in defective adenylyl cyclase activation, thereby promoting platelet aggregation and atherothrombosis (10).
Additionally, we demonstrate that iloprost given five weeks after tumor initiation continues to protect against lung tumorigenesis. Five weeks after urethane administration microadenomas have formed (42), and the ability of iloprost to block the progression of these microadenomas to visible adenomas suggests that this agent may have chemotherapeutic activity and may be a potential chemopreventive agent in former smokers who have accumulated airway damage. Further studies are in progress to test the ability of iloprost to inhibit NSCLC growth, and to define whether this is mediated through PPARγ activation.
Our studies indicate that the protective effect of overexpression of PGIS can be reproduced using a bioavailable prostacyclin analogue, providing rationale for clinical lung cancer chemoprevention studies in high risk humans. Potential prevention agents must have excellent safety profiles, and chronic COX-2 administration has been associated with increased cardiovascular mortality(8). Prostacyclin has known atheroprotective properties that may make it a more attractive prevention agent (9). To better investigate this, we have an ongoing clinical chemoprevention trial where patients at high risk for lung cancer are given oral iloprost and assessed for the histologic progression of endobronchial dysplasia. Our data continue to confirm a pivotal role for PGI2 in preventing lung tumorigenesis (the overwhelming majority of which is tobacco smoking associated), and indicate that PPARγ activation is one mechanism and PPARγ agonists (as single agents or in combination with other agents) may be ready for chemoprevention and chemotherapeutic trials.
Acknowledgments
This work was supported by grants from the NIH (CA103618, CA108610, and CA58187). RLK was supported by VA Advanced Career Development and Merit Review Awards from the Department of Veterans Affairs.
References
- 1.Michaud CM, Murray CJ, Bloom BR. Burden of disease--implications for future research. Jama. 2001;285:535–9. doi: 10.1001/jama.285.5.535. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–30. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 3.Schreinemachers DM, Everson RB. Aspirin use and lung, colon, and breast cancer incidence in a prospective study. Epidemiology. 1994;5:138–46. doi: 10.1097/00001648-199403000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Moody TW, Leyton J, Zakowicz H, et al. Indomethacin reduces lung adenoma number in A/J mice. Anticancer Res. 2001;21:1749–55. [PubMed] [Google Scholar]
- 5.Rioux N, Castonguay A. Prevention of NNK-induced lung tumorigenesis in A/J mice by acetylsalicylic acid and NS-398. Cancer Res. 1998;58:5354–60. [PubMed] [Google Scholar]
- 6.Kisley LR, Barrett BS, Dwyer-Nield LD, Bauer AK, Thompson DC, Malkinson AM. Celecoxib reduces pulmonary inflammation but not lung tumorigenesis in mice. Carcinogenesis. 2002;23:1653–60. doi: 10.1093/carcin/23.10.1653. [DOI] [PubMed] [Google Scholar]
- 7.Nussmeier NA, Whelton AA, Brown MT, et al. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N Engl J Med. 2005;352:1081–91. doi: 10.1056/NEJMoa050330. [DOI] [PubMed] [Google Scholar]
- 8.Solomon SD, McMurray JJ, Pfeffer MA, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352:1071–80. doi: 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- 9.Egan KM, Lawson JA, Fries S, et al. COX-2-derived prostacyclin confers atheroprotection on female mice. Science. 2004;306:1954–7. doi: 10.1126/science.1103333. [DOI] [PubMed] [Google Scholar]
- 10.Arehart E, Stitham J, Asselbergs FW, et al. Acceleration of cardiovascular disease by a dysfunctional prostacyclin receptor mutation: potential implications for cyclooxygenase-2 inhibition. Circ Res. 2008;102:986–93. doi: 10.1161/CIRCRESAHA.107.165936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dannenberg AJ, Altorki NK, Boyle JO, et al. Cyclo-oxygenase 2: a pharmacological target for the prevention of cancer. [Review] [90 refs] Lancet Oncology. 2001;2:544–51. doi: 10.1016/S1470-2045(01)00488-0. [DOI] [PubMed] [Google Scholar]
- 12.Vane JR. Prostacyclin: a hormone with a therapeutic potential. The Sir Henry Dale Lecture for 1981. J Endocrinol. 1982;95:3P–43P. [PubMed] [Google Scholar]
- 13.Honn KV, Cicone B, Skoff A. Prostacyclin: a potent antimetastatic agent. Science. 1981;212:1270–2. doi: 10.1126/science.7015512. [DOI] [PubMed] [Google Scholar]
- 14.Schirner M, Schneider MR. Inhibition of metastasis by cicaprost in rats with established SMT2A mammary carcinoma growth. Cancer Detect Prev. 1997;21:44–50. [PubMed] [Google Scholar]
- 15.Keith RL, Miller YE, Hoshikawa Y, et al. Manipulation of Pulmonary Prostacyclin Synthase Expression Prevents Murine Lung Cancer. Cancer Res. 2002;62:734–40. [PubMed] [Google Scholar]
- 16.Keith RL, Miller YE, Hudish TM, et al. Pulmonary prostacyclin synthase overexpression chemoprevents tobacco smoke lung carcinogenesis in mice. Cancer Res. 2004;64:5897–904. doi: 10.1158/0008-5472.CAN-04-1070. [DOI] [PubMed] [Google Scholar]
- 17.Namba T, Oida H, Sugimoto Y, et al. cDNA cloning of a mouse prostacyclin receptor. Multiple signaling pathways and expression in thymic medulla. J Biol Chem. 1994;269:9986–92. [PubMed] [Google Scholar]
- 18.Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S. Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br J Pharmacol. 1997;122:217–24. doi: 10.1038/sj.bjp.0701367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 20.Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci U S A. 1997;94:4312–7. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geraci MW, Gao B, Shepherd DC, et al. Pulmonary prostacyclin synthase overexpression in transgenic mice protects against development of hypoxic pulmonary hypertension. J Clin Invest. 1999;103:1509–15. doi: 10.1172/JCI5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korfhagen TR, Glasser SW, Wert SE, et al. Cis-acting sequences from a human surfactant protein gene confer pulmonary-specific gene expression in transgenic mice. Proc Natl Acad Sci U S A. 1990;87:6122–6. doi: 10.1073/pnas.87.16.6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murata T, Ushikubi F, Matsuoka T, et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388:678–82. doi: 10.1038/41780. [DOI] [PubMed] [Google Scholar]
- 24.Rice WR, Conkright JJ, Na C-L, Ikegami M, Shannon JM, Weaver TE. Maintenance of the mouse type II cell phenotype in vitro. Am J Physiol Lung Cell Mol Physiol. 2002;283:L256–64. doi: 10.1152/ajplung.00302.2001. [DOI] [PubMed] [Google Scholar]
- 25.Roberts PE, Phillips DM, Mather JP. A novel epithelial cell form neonatal rat lung: isolation and differentiated phenotype. Am J Physiol. 1990;259:L415–L25. doi: 10.1152/ajplung.1990.259.6.L415. [DOI] [PubMed] [Google Scholar]
- 26.He TC, Chan TA, Vogelstein B, Kinzler KW. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99:335–45. doi: 10.1016/s0092-8674(00)81664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gupta RA, Tan J, Krause WF, et al. Prostacyclin-mediated activation of peroxisome proliferator-activated receptor delta in colorectal cancer. Proc Natl Acad Sci U S A. 2000;97:13275–80. doi: 10.1073/pnas.97.24.13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim H, Dey SK. A novel pathway of prostacyclin signaling-hanging out with nuclear receptors. Endocrinology. 2002;143:3207–10. doi: 10.1210/en.2002-220159. [DOI] [PubMed] [Google Scholar]
- 29.Bren-Mattison Y, Meyer AM, Van Putten V, et al. Antitumorigenic Effects of Peroxisome Proliferator-Activated Receptor-{gamma} in Non-Small-Cell Lung Cancer Cells Are Mediated by Suppression of Cyclooxygenase-2 via Inhibition of Nuclear Factor-{kappa}B. Mol Pharmacol. 2008;73:709–17. doi: 10.1124/mol.107.042002. [DOI] [PubMed] [Google Scholar]
- 30.Hoshikawa Y, Voelkel NF, Gesell TL, et al. Prostacyclin receptor-dependent modulation of pulmonary vascular remodeling. Am J Respir Crit Care Med. 2001;164:314–8. doi: 10.1164/ajrccm.164.2.2010150. [DOI] [PubMed] [Google Scholar]
- 31.Bren-Mattison Y, Van Putten V, Chan D, Winn R, Geraci MW, Nemenoff RA. Peroxisome proliferator-activated receptor-gamma (PPAR(gamma)) inhibits tumorigenesis by reversing the undifferentiated phenotype of metastatic non-small-cell lung cancer cells (NSCLC) Oncogene. 2005;24:1412–22. doi: 10.1038/sj.onc.1208333. [DOI] [PubMed] [Google Scholar]
- 32.Wick M, Hurteau G, Dessev C, et al. Peroxisome Proliferator-Activated Receptor-gamma Is a Target of Nonsteroidal Anti-Inflammatory Drugs Mediating Cyclooxygenase-Independent Inhibition of Lung Cancer Cell Growth. Mol Pharmacol. 2002;62:1207–14. doi: 10.1124/mol.62.5.1207. [DOI] [PubMed] [Google Scholar]
- 33.Lewis CE, Pollard JW. Distinct Role of Macrophages in Different Tumor Microenvironments. Cancer Res. 2006;66:605–12. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 34.Kerr KM, Johnson SK, King G, Kennedy MM, Weir J, Jeffrey R. Partial regression in primary carcinoma of the lung: does it occur? Histopathology. 1998;33:55–63. [PubMed] [Google Scholar]
- 35.Welsh TJ, Green RH, Richardson D, Waller DA, O’Byrne KJ, Bradding P. Macrophage and Mast-Cell Invasion of Tumor Cell Islets Confers a Marked Survival Advantage in Non-Small-Cell Lung Cancer. J Clin Oncol. 2005;23:8959–67. doi: 10.1200/JCO.2005.01.4910. [DOI] [PubMed] [Google Scholar]
- 36.Govindarajan R, Ratnasinghe L, Simmons DL, et al. Thiazolidinediones and the Risk of Lung, Prostate, and Colon Cancer in Patients With Diabetes. J Clin Oncol. 2007;25:1476–81. doi: 10.1200/JCO.2006.07.2777. [DOI] [PubMed] [Google Scholar]
- 37.Hazra S, Batra RK, Tai HH, Sharma S, Cui X, Dubinett SM. Pioglitazone and Rosiglitazone Decrease Prostaglandin E2 in Non-Small-Cell Lung Cancer Cells by Up-Regulating 15-Hydroxyprostaglandin Dehydrogenase. Mol Pharmacol. 2007;71:1715–20. doi: 10.1124/mol.106.033357. [DOI] [PubMed] [Google Scholar]
- 38.Keshamouni VG, Arenberg DA, Reddy RC, Newstead MJ, Anthwal S, Standiford TJ. PPAR-gamma activation inhibits angiogenesis by blocking ELR+CXC chemokine production in non-small cell lung cancer. Neoplasia. 2005;7:294–301. doi: 10.1593/neo.04601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keshamouni VG, Reddy RC, Arenberg DA, et al. Peroxisome proliferator-activated receptor-gamma activation inhibits tumor progression in non-small-cell lung cancer. Oncogene. 2004;23:100–8. doi: 10.1038/sj.onc.1206885. [DOI] [PubMed] [Google Scholar]
- 40.Nissen SE, Wolski K. Effect of Rosiglitazone on the Risk of Myocardial Infarction and Death from Cardiovascular Causes. N Engl J Med. 2007;356:245–71. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 41.Home PD, Pocock SJ, Beck-Nielsen H, et al. Rosiglitazone evaluated for cardiovascular outcomes--an interim analysis. N Engl J Med. 2007;357:28–38. doi: 10.1056/NEJMoa073394. [DOI] [PubMed] [Google Scholar]
- 42.Malkinson AM. Primary lung tumors in mice as an aid for understanding, preventing, and treating human adenocarcinoma of the lung. Lung Cancer. 2001;32:265–79. doi: 10.1016/s0169-5002(00)00232-4. [DOI] [PubMed] [Google Scholar]