

Abstract
Epidermal growth factor receptor variant III (EGFRvIII) is a constitutively active mutant form of EGFR that is expressed in 40% to 50% of gliomas and several other malignancies. Here, we describe the therapeutic effects of silencing EGFRvIII on glioma cell lines in vitro and in vivo. A small interfering RNA molecule against EGFRvIII was introduced into EGFRvIII-expressing glioma cells (U87Δ) by electroporation resulting in complete inhibition of expression of EGFRvIII as early as 48 h post-treatment. During EGFRvIII silencing, a decrease in the proliferation and invasiveness of U87Δ cells was accompanied by an increase in apoptosis (P < 0.05). Notably, EGFRvIII silencing inhibited the signal transduction machinery downstream of EGFRvIII as evidenced by decreases in the activated levels of Ras and extracellular signal-regulated kinase. A lentivirus capable of expressing anti-EGFRvIII short hairpin RNA was also able to achieve progressive silencing of EGFRvIII in U87Δ cells in addition to inhibiting cell proliferation, invasiveness, and colony formation in a significant manner (P < 0.05). Silencing EGFRvIII in U87Δ cultures with this virus reduced the expression of factors involved in epithelial-mesenchymal transition including N-cadherin, β-catenin, Snail, Slug, and paxillin but not E-cadherin. The anti-EGFRvIII lentivirus also affected the cell cycle progression of U87Δ cells with a decrease in G1 and increase in S and G2 fractions. In an in vivo model, tumor growth was completely inhibited in severe combined immunodeficient mice (n = 10) injected s.c. with U87Δ cells treated with the anti-EGFRvIII lentivirus (P = 0.005). We conclude that gene specific silencing of EGFRvIII is a promising strategy for treating cancers that contain this mutated receptor.
Introduction
The tyrosine kinase family of growth factor receptors plays a major role in the etiology of many human malignancies. The epidermal growth factor receptor (EGFR) is composed of an extracellular binding domain, a transmembrane lipophilic segment, and an intracellular domain with protein kinase activity (1-3). On binding to ligands such as EGF or transforming growth factor-α, EGFR undergoes dimerization, which triggers the kinase activity of this receptor and initiates a cascade of downstream signaling events.
EGFR can exert oncogenic effects by different mechanisms, such as autocrine growth factor loops, amplification of the EGFR gene, and deletions/mutations that render the receptor constitutively active (4-9). The most common mutation of the EGFR gene is an in-frame deletion of exons 2 to 7, generating a mRNA that is 801 nucleotides shorter than its wild-type counterpart (10, 11). The product of this mutation is known as EGFR variant III (EGFRvIII; de2-7 EGFR or EGFRΔ) and lacks amino acids 6 to 273 of the extracellular domain and is constantly active. This mutant receptor has a molecular mass of 145 kDa, 25 kDa less than wild-type EGFR. EGFRvIII has been reported to be expressed in many types of cancer, including 40% to 50% of gliomas, 86% of medulloblastomas, 78% of breast carcinomas, and 73% of ovarian carcinomas (12-14). High-grade gliomas show a predominant membrane and cytoplasmic staining pattern for EGFRvIII as detected by immunohistochemical staining using specific monoclonal antibodies (9, 15, 16). EGFRvIII is also present in squamous cell, adenosquamous cell, and undifferentiated non-small cell lung cancer (4, 12, 17-20).
EGFR has a well-characterized role in regulating the Ras signaling pathway. Ras is a guanine nucleotide-binding protein that is active when bound to GTP and located at the plasma membrane (21, 22). Retroviral introduction of EGFRvIII into the U87 human malignant glioma cell line or murine NR6 cell line (23) results in expression of a truncated receptor with a ligand-independent kinase and enhanced tumorigenicity in nude mice (9). The molecular mechanism by which the EGFRvIII transfectants acquire transforming activity is not yet clear. EGFRvIII has been found constitutively associated with signaling adapter proteins Shc and Grb2, which are involved in the recruitment of Ras to activated receptors, even if dimerization does not occur (24, 25). Studies by Prigent et al. (25) and Feldkamp et al. (26) showed that constitutively active EGFRvIII enhanced the growth of glioblastoma cells through increased activity of Ras-GTP. Fan et al. reported that silencing EGFRvIII reduced the levels of phospho-Akt, increased apoptosis, and partially arrested the cell cycle at the G2-M phase (27). Because EGFRvIII is only expressed in cancer tissues and not normal tissues, it has been long considered as a target for a range of modalities including monoclonal antibody therapy (15, 28, 29).
Considering the studies mentioned above, silencing of EGFRvIII expression appears to be a rational strategy for targeting a range of tumors. In this study, we have used small interfering RNA (siRNA) to target and silence EGFRvIII expression. Mechanistically, dicer-processed siRNAs (21 nucleotides) and synthetic siRNAs undergo an ATP-dependent unwinding step before being incorporated into a high-molecular-weight protein complex termed RNA-induced silencing complex that contains single-stranded siRNAs (23, 30). Thereafter, the RNA-induced silencing complex is remodeled into its active form, which contains the proteins necessary for cleaving the target mRNA at the site where the guide antisense siRNA binds. To target EGFRvIII, we have designed a 21-nucleotide siRNA with sequence similarity to the breakpoint generated by deletion of exons 2 to 7. Considering the fact that such a breakpoint is unique to the cells containing EGFRvIII, siRNA molecules designed against it would specifically target cells expressing EGFRvIII, providing significant cell specificity for this strategy. On treatment of glioma cells that expressed EGFRvIII with this siRNA or with a lentivirus expressing anti-EGFRvIII short hairpin RNA (shRNA), EGFRvIII was efficiently silenced. Although silencing existed, treated glioma cells showed decreased proliferation rate, increased apoptosis, decreased invasiveness and colony formation capabilities and reduction of a series of epithelial-mesenchymal transition (EMT) markers such as Snail, Slug, and β-catenin. Expression of E-cadherin, an acting suppressor of invasion and growth of many epithelial cancers (31-34), was not changed on silencing EGFRvIII, whereas expression of N-cadherin, a promoter of tumor invasiveness, was reduced. We have also evaluated the effects of anti-EGFRvIII lentivirus on cell cycling progression of glioma cells and found a decrease in the fraction of G1 on silencing EGFRvIII. In vivo tumorigenesis of U87Δ cells was also diminished in a significant manner (P = 0.001) on treatment with anti-EGFRvIII virus as studied in severe combined immunodeficient (SCID) mouse model.
Materials and Methods
Cell Culture and siRNA Production
The sequence for EGFRvIII mRNA was analyzed with siRNA designing software available on the worldwide Web. The oligonucleotide was synthesized in the double-stranded form in conjugation with a FITC molecule on the 3′-OH of the sense strand. Commercially available siRNA was used as a negative control (Qiagen). Human glioma cell lines U87MG (or U87) and its derivative U87MGΔ (or U87Δ expressing EGFRvIII) were generous gifts of Dr. Antonio Chiocca (Ohio State University). U87 cells were grown in DMEM supplemented with 10% fetal bovine serum and additional antibiotics. U87Δ were grown in the same medium conditions as U87 with the addition of G418 at a concentration of 0.4 mg/mL to continuously select for EGFRvIII-expressing cells.
Electroporation and Fluorescence-Activated Cell Sorting
Anti-EGFRvIII siRNA (5′-CUGGAGGAAAAGAAAGGUAAU-3′) and negative control siRNA (Qiagen) were introduced to cells by electroporation. This method is considered transient because the concentration of siRNA eventually drops to nonefficient levels. Cells (2 × 107) were resuspended in 270 μL Opti-MEM and mixed with 30 mL of 20 μmol/L siRNA stock solution generating a final concentration of 2 μmol/L siRNA. This cocktail was then transferred to an electroporation cuvette while on ice and electroporated at 300 mV for a single 10 s pulse with a mammalian cell electroporation system (BTX). The cuvettte containing cells were then incubated at 37°C for 2 h. Cells were then transferred to a flow cytometry facility for sorting when applicable. The sorting of cells containing siRNA was done based on Alexa Fluor 488 tag using FACSVantage with FACSDiVa option. Cell cycle analysis has been done after cells were fixed in 80% ethanol on ice for 20 min and washed with PBS and treated with 5 g/mL propidium iodide (Sigma).
SDS-PAGE and Western Blot Analysis
Different cells were lysed with a single detergent lysis buffer [50 mmol/L Tris (pH 8.0), 150 mmol/L NaCl, 0.02% sodium azide, 100 μg/mL phenylmethylsulfonyl fluoride, 1 μg/mL aprotinin, and 1% Triton X-100], normalized for the amount of total protein, and subjected to SDS-PAGE using mini-cell protein-II system (Bio-Rad; using precast 10% discontinuous gels) followed by electroblotting onto nitrocellulose paper. The membrane was washed and incubated with the different primary antibody against EGFRvIII (Zymed), EGFR, and β-actin (Cell Signaling) followed by the horseradish peroxidase-conjugated secondary antibody. The blot was exposed to Lumigel detection solution and subjected to autoradiography. Antibodies used for probing EMT markers (Snail, Slug, β-catenin, E-cadherin, and N-cadherin), focal adhesion (paxillin), cell survival pathway (Bad and Bcl-2), and cyclin A were used per manufacturer’s instructions (Cell Signaling).
Affinity Pull-Down Assays for Ras and Nonradioactive Extracellular Signal-Regulated Kinase Activation Assay
In accordance with the manufacturer’s instructions (Ras activation assay kit; Upstate), cells were grown in 10 cm tissue culture dishes and lysed at 75% to 80% confluency with 300 μL of 1× magnesium-containing lysis buffer. Following determination of protein concentration, 7.5 μg Ras assay reagent (Raf-1 RBD, agarose) was added to 200 μg total cell protein in 200 μL magnesium-containing lysis buffer. The reaction mixture was gently rocked at 4°C for 3 h. The activated (GTP) forms of Ras bound to the agarose beads were collected by centrifugation, loaded to a 10% SDS-PAGE gel (Bio-Rad), and blotted with a pan-Ras antibody.
To do extracellular signal-regulated kinase (ERK) activation assay, 15 μL immobilized phospho-ERK antibody (p42/44 mitogen-activated protein kinase nonradioactive assay; Cell Signaling) was added to 200 μg total cell protein in 200 μL cell lysis buffer. The mixture was incubated overnight at 4°C. The beads with bound active ERK were collected by centrifugation for 1 min and washed on ice with 300 μL of 1× lysis buffer. The beads were then washed twice with 1× kinase buffer and resuspended in 50 μL of 1× kinase buffer supplemented with 200 μmol/L ATP and either 2 μg ELK-1 fusion protein as the substrate. The kinase reaction was incubated at 30°C for 1 h. The beads were collected by centrifugation and resuspended in 20 μL of 2× Laemmli reducing sample buffer (Bio-Rad) and boiled for 5 min, loaded to a 10% SDS-PAGE gel (Bio-Rad), and then blotted with phospho-ELK-1 antibody (1:1,000).
Cell Proliferation Assay
The cell proliferation assay was done using a kit (WST-1 cell proliferation assay; Chemicon) according to the manufacturer’s instructions. The assay is based on the cleavage of the tetrazolium salt WST-1 to formazan by cellular mitochondrial dehydrogenases. Expansion in the number of viable cells results in an increase in the overall activity of the mitochondrial dehydrogenases resulting in an increase in the amount of formazan dye formed. Briefly, 104 cells per well were seeded in a 96-well microplate in volume of 100 μL/well. At different times, 10 μL WST-1/ ECS solution was added to each well. The plates were incubated for 4 h in standard culture conditions. The plates were then shaken thoroughly for 1 min and absorbance was measured at 480 nm.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labeling Assay for Detection of Apoptosis in Late Phase
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling assay was done according to the recommendations of the manufacturer (TdT-Mediated dUTP Nick End Labeling Apoptosis Detection Kit; Chemicon). Briefly, cells were fixed in paraformaldehyde and alcohol and then incubated at −20°C overnight. Cells were rehydrated in TBS and then permeabilized by using proteinase K and equilibrated by using TdT buffer. Cells were labeled by using the TdT labeling reaction mix and TdT enzyme at 37°C for 1.5 h in dark. The reaction was then stopped by washing with 1× TBS, and cells were analyzed by a flow cytometer using argon 488 nm argon ion laser source.
Cell Invasion Assay
To evaluate cell invasiveness, a commercial kit was used (BD BioCoat Matrigel invasion assay; BD Biosciences). Briefly, cells (50,000 control or test cells) were introduced into Matrigel-coated inserts fitting 24-well plates. As cells invaded through the layer of Matrigel, the fraction of invaded cells were detected by staining them with crystal violet and quantifying them by spectroscopy. Invaded cells were fixed with 5% paraformaldehyde and stained with a 5% solution of crystal violet and then photographed to obtain a visual representation of their density. The cells were then solubilized in a 3% detergent (NP-40) solution, and the absorbance was measured by spectrophotometry at 590 nm.
Soft-Agar Colony Formation Assay
A soft-agar colony formation assay was done using 6-well plates. Each well contained 2 mL of 0.8% agarose base layer in complete medium (DMEM with 10% fetal bovine serum and 1% antibiotics) as the bottom layer and 1 mL of 0.4% agarose in complete medium and 3,000 cells (control and anti-EGFRvIII lentivirus-infected cells) as the top layer. Cultures were maintained under standard conditions for 21 days. The colonies were stained with cell stain solution (Chemicon) overnight at 37°C and counted the following morning. The number of colonies was determined with a microscope at ×100 magnification; a group of >20 cells were counted as a colony. For colony quantification 1.4 mL cell quantification solution (Chemicon) was added to each well and incubated for 4 h at 37°C. Absorbance was measured at 490 nm.
Lentivirus Production
The anti-EGFRvIII shRNA was designed based on the siRNA oligonucleotide using Invitrogen software. The shRNA was then cloned into pENTR/H1/TO (BLOCK-iT lentiviral RNAi expression system; Invitrogen) for production of the entry clone. A recombination was done using the entry clone and pLenti4/BLOCK-iT-DEST vector to generate an expression vector. The ampicillin-positive and chloramphenicol-negative expression clone was then selected and confirmed further by sequencing. Lentiviruses were collected 48 h after transfection of the 293FT producer cells with expression vector containing EGFRvIII shRNA using ViraPower Packaging Mix.
S.c. Model for In vivo Tumorigenicity in SCID Mouse
Transduced U87Δ cells were trypsinized and counted using a hemocytometer. Cells were diluted in 75% Matrigel and 25% U87Δ to a final concentration of 1.67 × 105/mL Matrigel/media. Diluted cells (0.3 mL) were injected s.c. into the right and left flanks of 5- to 6-week-old male Fox Chase SCID outbred male mice (Charles River Labs). Animals were checked for tumor development every other day and tumor size was measured using a digital caliper. Tumor volume was determined with the following formula: tumor volume (mm3) = [length (mm)] × [width (mm)] 3 × ϕ.
Statistical Analysis
Results are reported as mean ± SD. Student’s t test was used to analyze statistical differences between groups. The α level was set at 0.05.
Results
Anti-EGFRvIII siRNA Molecule
To knockdown EGFRvIII by siRNA, we designed a 21-nucleotide siRNA oligomer with sequence similarity to the breakpoint generated by deletion of exons 2 to 7 at the 88th base pair after the ATG starting codon of EGFRvIII mRNA (Fig. 1A). A highly specific siRNA sequence was identified within EGFRvIII cDNA starting at the 73rd base pair after the ATG starting codon (Fig. 1B). According to the projected analysis, this sequence would not bind efficiently to any mRNA sequence from the normal human genome. Negative control siRNA had no sequence similarity to human genome.
Figure 1. cDNA sequence of human EGFR and EGFRvIII and the design of anti-EGFRvIII siRNA.

A, cDNA sequence of wild-type EGFR: the sequence corresponding to the deletion resulting in generation of the EGFRvIII is highlighted in yellow. B, cDNA sequence of EGFRvIII: the junction point generated by the deletion highlighted in A and the sequence used for the design of anti-EGFRvIII siRNA is boxed in red.
Anti-EGFRvIII siRNA Silences EGFRvIII Expression in U87Δ Cells
Anti-EGFRvIII-siRNA was introduced into U87Δ cells expressing EGFRvIII by using electroporation at the final concentration of 2 μmol/L. The expression of EGFRvIII was suppressed in U87Δ cells as early as 48 h post-electroporation (Fig. 2A, top) as evaluated by Western blotting. Efficient EGFRvIII suppression continued in U87Δ cells until 72 h post-electroporation and a return to full expression was observed by 144 h post-electroporation. Treatment of U87Δ cells with the negative control siRNA had no effect on the expression of EGFRvIII. EGFRvIII expression was not detected by Western blotting in U87 cells at any time point, thus proving the specificity of the antibody for EGFRvIII. Anti-EGFRvIII siRNA did not silence the expression of wild-type EGFR or β-actin in U87Δ cells (results shown at 48 h), thus confirming the specificity of this siRNA molecule (Fig. 2A, bottom). β-Actin also served as protein concentration and gel loading control for Western blots.
Figure 2. Silencing EGFRvIII in glioma cells inhibits Ras and ERK activation.
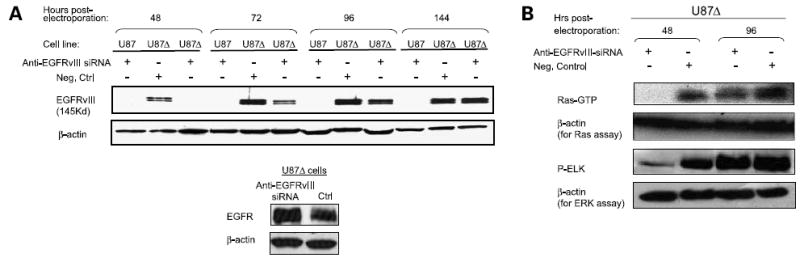
A, effects of anti-EGFRvIII and negative control siRNAs on the expression of EGFRvIII in U87Δ cells at different times post-electroporation. siRNA molecules were introduced to the cells by electroporation (2 μmol/L final concentration). U87 cells do not express EGFRvIII and were used as a negative control for the specificity of anti-EGFRvIII antibody. Expression of EGFRvIII was effectively silenced as early as 48 h post-electroporation. Expression of EGFR in U87Δ cells at 48 h post-electroporation with anti-EGFRvIII siRNA was detected (bottom). Representative of three independent experiments. B, activation of Ras was evaluated in U87Δ cells following EGFRvIII silencing by performing an affinity pull-down assay for Ras-GTP (active Ras). Top bands represent Ras-GTP; bottom bands represent the expression of β-actin as a control. The activity of the ERK-pathway was evaluated in U87Δ cells following EGFRvIII silencing by performing an in vitro kinase reaction in the presence of ELK as a substrate for active-ERK. Top bands show the levels of phospho-ELK as an indicator of ERK activation; bottom bands represent the expression of β-actin as a control.
Silencing EGFRvIII Decreases Activated Fraction of Ras and ERK
To determine if the amount of Ras-GTP (activated form of Ras) was altered by the silencing of EGFRvIII, we performed an affinity pull-down assay for Ras-GTP. The level of Ras-GTP was decreased on EGFRvIII knockdown in U87Δ cells at 48 h post-electroporation (Fig. 2B). Activation of ERK in U87Δ cells was also evaluated by performing an in vitro kinase assay detecting phospho-ELK. A decrease in phospho-ELK was observed at 48 h post-electroporation (Fig. 2B). Both Ras-GTP and phospho-ELK returned to control levels after the expression of EGFRvIII returned close to baseline at 96 h. β-Actin levels remained unchanged for all of these experiments.
Anti-EGFRvIII siRNA Inhibits Proliferation of U87Δ Cells but Not U87 Cells
EGFRvIII is thought to contribute to an increased rate of cell proliferation that provides these cells with a growth advantage (35). To determine whether silencing EGFRvIII influences cell proliferation, U87Δ cells electroporated with anti-EGFRvIII siRNA were compared with U87Δ cells treated with a negative control siRNA and U87 cells treated with anti-EGFRvIII siRNA. To quantitatively assess the proliferation rate of these cells, we performed an assay based on the cleavage of the tetrazolium salt WST-1 to formazan by cellular mitochondrial dehydrogenases. When U87Δ cells were treated with anti-EGFRvIII siRNA, their growth was significantly reduced by 15% to 20% between days 2 and 4 compared with control-treated U87Δ cells (Fig. 3A; P < 0.005). By day 5, however, there was no difference in the proliferation rate between treated and untreated cells, which correlates with the time point in which the silencing effects of siRNA are diminished. Figure 3A shows the result of cell proliferation assay in a mixed population of cells (cells that may or may not contain the anti-EGFRvIII siRNA). Interestingly, when the subpopulation of cells carrying the anti-EGFRvIII siRNA were sorted by flow cytometry, a much wider gap was observed between the growth curve of anti-EGFRvIII siRNA-treated and control U87Δ cells (Fig. 3B). For sorted cells, the retardation in growth started to rebound toward values for the control cells at day 4 post-electroporation. In contrast, the growth rate of U87 cells, which do not express EGFRvIII, was not significantly affected by treatment with anti-EGFRvIII siRNA (Fig. 3C).
Figure 3. Anti-EGFRvIII siRNA reduces proliferation and increases apoptosis in glioma cells.
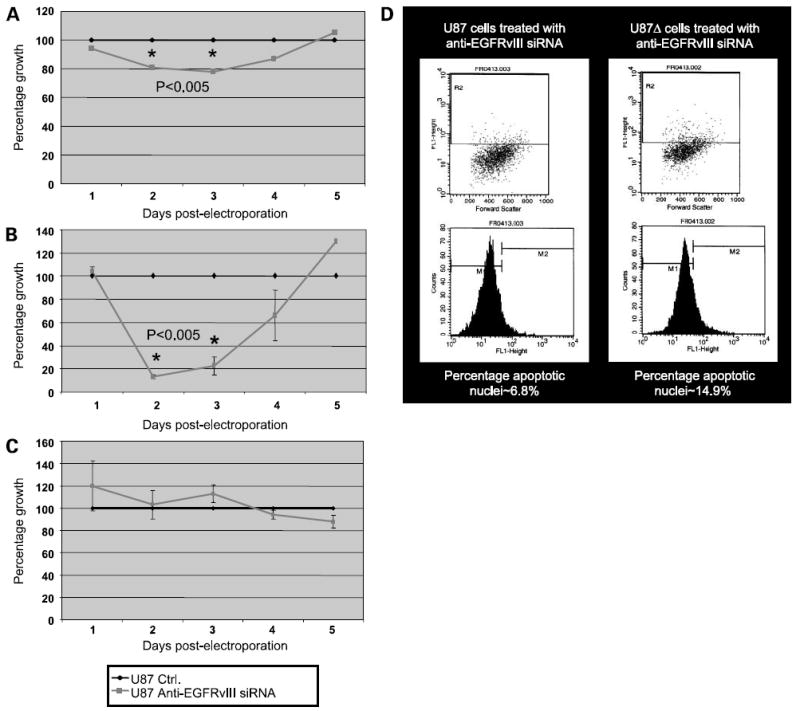
A, cell proliferation assay for U87Δ cells electroporated with and without anti-EGFRvIII siRNA. Representative of a mixed population of cells that may or may not contain siRNA. The growth rate of control electroporated cells was plotted as the maximum capability of growth for these cells at each time point and represented by a value of 100% (blue line); the growth rate of anti-EGFRvIII siRNA electroporated cells was then calculated in comparison with the growth rate of control cells and plotted for each time point (pink line; P < 0.005). Representative of three independent experiments. B, cell proliferation assay for U87Δ cells electroporated with and without anti-EGFRvIII siRNA and sorted for Alexa Fluor 488 as an indicator for existence of siRNA in these cells (P < 0.005). C, cell proliferation assay for U87 cells electroporated with and without anti-EGFRvIII siRNA and sorted for Alexa Fluor 488 as an indicator for existence of siRNA in these cells. D, induction of apoptosis in U87 (left) and U87Δ (right) cells at 24 h post-electroporation with anti-EGFRvIII siRNA as measured by TdT-mediated dUTP nick end labeling assay. The basal level of apoptosis in control cells was ~6%.
Anti-EGFRvIII siRNA Increases Apoptosis in U87Δ Cells but Not in U87 Cells
To evaluate whether silencing EGFRvIII induced apoptosis, U87 and U87Δ cells were electroporated with the anti-EGFRvIII siRNA and then plated in 6-cm culture dishes for 24 h before being processed by TdT-mediated dUTP nick end labeling assay. Over twice as many U87Δ cells were apoptotic compared with U87 cells (Fig. 3D). Therefore, the decreased proliferation rate observed in U87Δ cells is at least partly due to induction of apoptosis on silencing EGFRvIII. Apoptosis in U87 cells remained close to the levels observed for cells that were electroporated as control (~%6, data not shown).
Anti-EGFRvIII siRNA Reduces the Invasiveness of U87Δ Cells
Glioma is a highly invasive malignancy with the capability of establishing a high number of metastatic foci in a short period. The effect of silencing EGFRvIII on U87Δ cell invasiveness was assessed using a Matrigel-based cell invasiveness assay. As Fig. 4A and B show, cell migration through the Matrigel layer was reduced in U87Δ cells treated with anti-EGFRvIII siRNA compared with control-treated cells (light microscopy). To quantify the difference in invasiveness between test and control treated cells, the population of invaded U87Δ cells were dissolved using a detergent (NP-40) solution, and the absorbance of this solution was measured with a spectrophotometer in comparison with a blank. A significant reduction (P = 0.001) was observed in the invasiveness of U87Δ cells treated with anti-EGFRvIII siRNA versus control treated cells (Fig. 4C; P = 0.001, results shown in percentages).
Figure 4. Anti-EGFRvIII siRNA reduces the invasiveness of glioma cells.
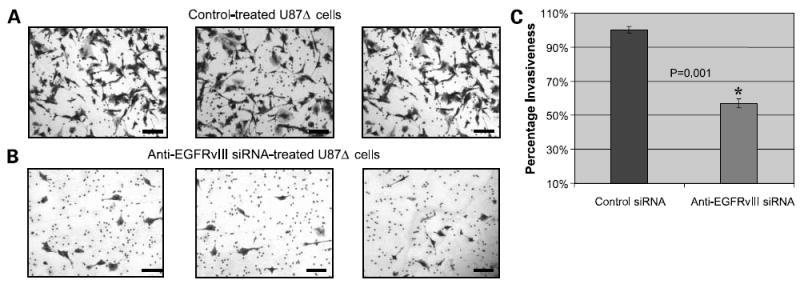
A, light microscopy (×40) images of the density of invaded U87Δ cells on the third day following control electroporation. Invasiveness was evaluated by a Matrigel-based cell invasion assay. Representative of three independent experiments done at 72 h post-electroporation. Cells were fixed and stained with crystal violet. Bar, 300 μm. B, light microscopy (×40) images of the density of invaded U87Δ cells on the third day following electroporation with anti-EGFRvIII siRNA. Invasiveness was evaluated by a Matrigel-based cell invasion assay. Bar, 300 μm. C, quantification of invaded U87Δ cells following electroporation with and without anti-EGFRvIII siRNA. This was achieved by dissolving U87Δ cells in NP-40 solution and performing spectrophotometry.
Anti-EGFRvIII Lentivirus, but Not the Control Virus, Inhibits EGFRvIII Expression and Proliferation of U87Δ Cells
To establish a translational tool for treating glioma in preclinical experiments, we developed a lentivirus capable of silencing EGFRvIII through the stable expression of anti-EGFRvIII shRNA. The structure of this anti-EGFRvIII lentivirus is illustrated in Fig. 5A.
Figure 5. Effects of anti-EGFRvIII lentivirus on proliferation and invasiveness of U87Δ cells.
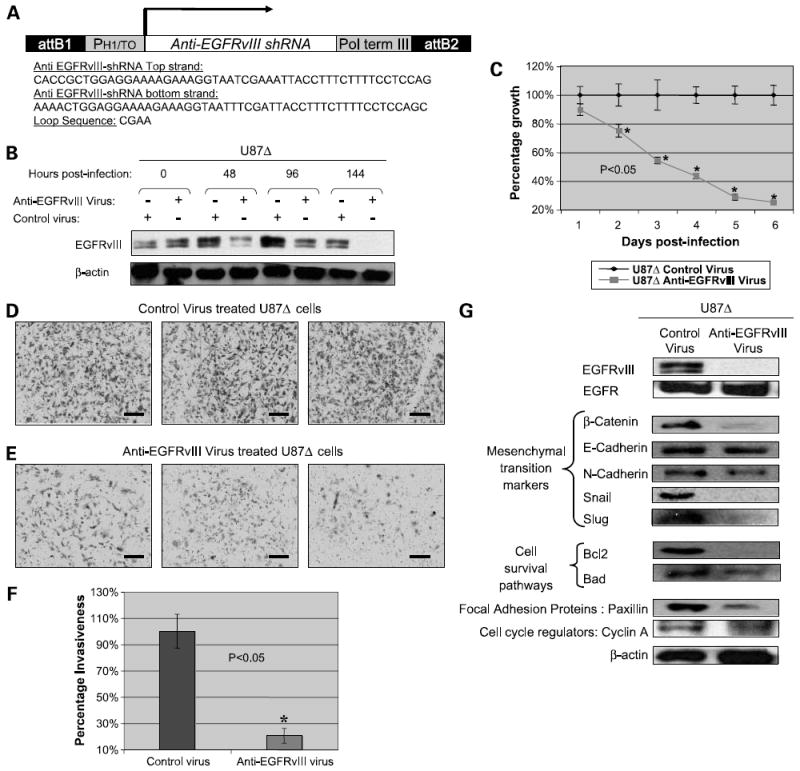
A, structure of anti-EGFRvIII lentivirus expressing anti-EGFRvIII shRNA and the sequence for this shRNA along with loop sequence. B, silencing of EGFRvIII on exposure of U87Δ cells to anti-EGFRvIII virus. Cells were exposed to control and anti-EGFRvIII virus and a complete inhibition of expression was observed at 144 h post-infection. Representative of three independent experiments. C, cell proliferation assay done on U87Δ cells exposed to anti-EGFRvIII and control virus up to 6 days post-infection. D and E, light microscopy (×20) images of the density of invaded U87Δ cells on the third day following electroporation with anti-EGFRvIII siRNA. Invasiveness was evaluated by a Matrigel-based cell invasion assay. Bar, 600 μm. F, quantification of invaded U87Δ cells following exposure to the anti-EGFRvIII siRNA and control viruses. This was achieved by dissolving U87Δ cells in NP-40 solution and performing spectrophotometry. G, Western blotting results for a series of EMT markers (β-catenin, E-cadherin, N-cadherin, Snail, and Slug), focal adhesion kinase protein paxillin, and cyclin A confirm reduction of many of these factors on exposure of cells to the anti-EGFRvIII virus silencing EGFRvIII. An exception was found in case of E-cadherin, EGFR, and β-actin where no significant changes were observed.
On exposure of U87Δ cells to this virus, we examined the expression of EGFRvIII by Western blotting. As shown in Fig. 5B, a progressive silencing of EGFRvIII was observed as early as 48 h post-infection leading to a complete blockade of expression of EGFRvIII at 144 h post-infection. Proliferation rate of U87Δ cells exposed to the anti-EGFRvIII virus was progressively reduced (P < 0.05) and did not rebound as was observed before for cells electroporated with anti-EGFRvIII siRNA (Fig. 5C).
Anti-EGFRvIII Lentivirus, but Not Control Lentivirus, Reduces Invasiveness of U87Δ Cells
Once U87Δ cells were exposed to the anti-EGFRvIII and control lentivirus, a significant (P < 0.05) reduction was observed in invasiveness of these cells only in case of the exposure to anti-EGFRvIII lentivirus. Figure 5D and E shows the density of invaded cells after 3 days of exposure to the anti-EGFRvIII and control viruses for three independent experiments studied by light microscopy. Figure 5F shows quantification of invaded cells as explained before. The fraction of invaded cells treated with control virus was plotted as 100% and the fraction of invaded cells treated with the anti-EGFRvIII virus was plotted in comparison with the control.
Expression of EMT Markers, Cell Survival Pathway Factors, and Paxillin Are Altered on EGFRvIII Silencing
Considering the importance of EMT markers in the loss of cell adhesion and increased cell mobility, we were interested to evaluate the expression levels of such markers under the silencing conditions for EGFRvIII. As shown in Fig. 5G, on silencing EGFRvIII (but not EGFR) at day 5, the levels of β-catenin, Snail, and Slug (transcription factors involved in EMT) were all reduced. Whereas E-cadherin remained relatively unchanged, the level of N-cadherin was also reduced. Paxillin, an important protein in the focal adhesion complex, was also reduced on EGFRvIII silencing. Additionally, we probed for expression of an antiapoptotic protein, Bcl-2, and a proapoptotic molecule, Bad. Although the expression of Bcl-2 was completely diminished on silencing EGFRvIII by the lentivirus, the expression level of Bad was moderately decreased in such conditions.
Silencing EGFRvIII Reduces Anchorage-Independent Colony Formation of U87Δ Cells and Increases Necrosis
An important characteristic of malignancy is the capability of cells to form colonies in soft agar in an anchorage-independent manner. To investigate whether silencing EGFRvIII affects colony formation, U87Δ cells were treated with anti-EGFRvIII or control lentivirus for 24 h and then introduced to colony formation assay. The number of U87Δ colonies that had developed after 21 days of treatment with this virus (at multiplicity of infection of ~1) was reduced by ~80% (P < 0.001) of U87Δ cells treated with the control virus (Fig. 6A). The rate of necrosis was also increased significantly in cells treated with the anti-EGFRvIII virus (Fig. 6B; P < 0.05).
Figure 6. Effects of the anti-EGFRvIII lentivirus on colony formation, cell death, cell cycle progression, and in vivo tumorigenesis of U87Δ cells.
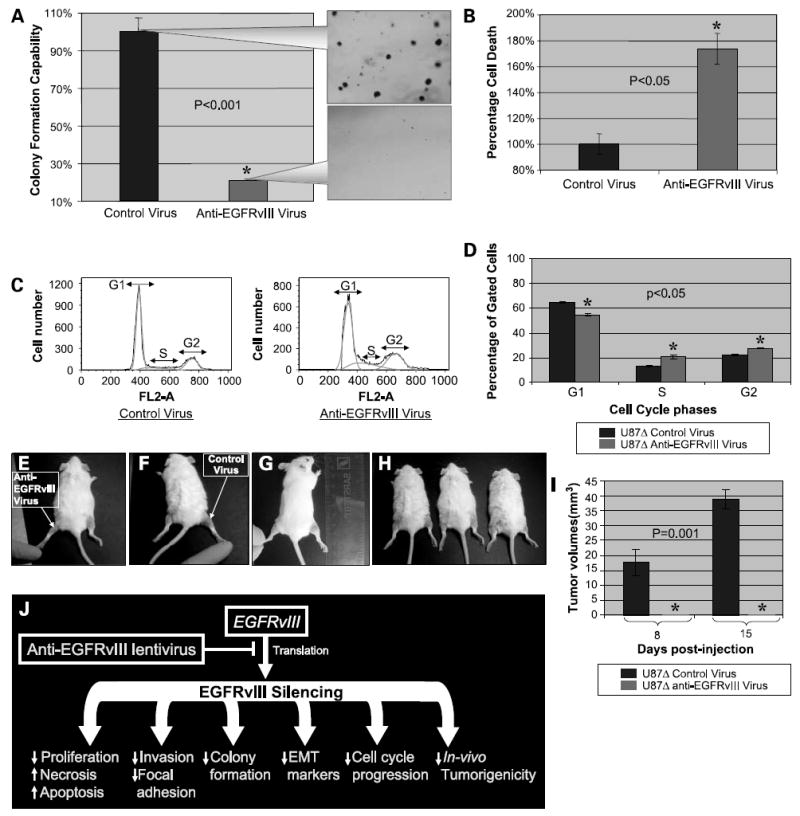
A, anchorage-independent colony formation was evaluated after 3 weeks of exposure of U87Δ cells to anti-EGFRvIII and control viruses. The callout panel illustrates the density of colonies. The mean ± SD of six independent experiments for cells exposed to control virus was considered as 100%. Values for anti-EGFRvIII-treated cells were graphed comparatively. B, rate of necrosis was increased on treatment of U87Δ cells with the anti-EGFRvIII virus in a significant manner (P < 0.05). C, cell cycle progression was evaluated on exposure of U87Δ cells to anti-EGFRvIII virus. A significant decrease was observed in the G1 fraction, but an increase was observed in the S and G2 fractions. D, quantification of cells in each step of cell cycle shows a decrease in G1 and increase in S and G2. Mean ± SD of three independent experiments in percentages (P < 0.05). E to H, in vivo tumorigenicity of U87Δ cells was evaluated on injection of anti-EGFRvIII and control treated U87Δ cells to the flank of SCID mice. E, representative image of the left flank injected with U87Δ cells treated with the anti-EGFRvIII virus. F, representative image of the right flank injected with U87Δ cells treated with control virus. G and H, other test subjects with growth of tumor on the right flank. I, tumors growth was recorded at days 8 and 15 post-injection. At both time points, no growth is observed for the U87Δ cells treated with anti-EGFRvIII virus, whereas the average size of tumors reached an average of 17.5 and 38 mm3 on days 8 and 15, respectively. J, schematic of the biological effect of EGFRvIII silencing on glioma cells. Silencing EGFRvIII reduces proliferation, invasiveness, cell cycle progression, and colony formation capabilities of glioma cells. The EMT markers seem to be repressed significantly on silencing this mutant receptor, reversing the “E-cadherin to N-cadherin switch.” In vivo tumorigenicity of glioma cells is also completely blocked by such treatment.
Silencing EGFRvIII Alters Cell Cycle Progression of U87Δ Cells
To evaluate the effects of the anti-EGFRvIII virus on cell cycle progression, U87Δ cells were treated with the anti-EGFRvIII or control lentivirus and evaluated with propidium iodide staining. As shown in Fig. 6C and D, on exposure of cells to anti-EGFRvIII virus, a significant decrease was observed in G1 population of cells, whereas the S- and G2-phase fraction of cells showed an increase (P < 0.05). Figure 6C represents a sample of flow cytometry analysis of such cells. Figure 6D shows the average number of cells from each fraction in cells treated with the therapeutic (anti-EGFRvIII) and control viruses. One potential mechanism underlying such alterations may be explained by a moderate decrease observed in the expression of cyclin A on silencing EGFRvIII (Fig. 5G).
In vivo Tumorigenicity of U87Δ Cells Is Reduced on Exposure toAnti-EGFRvIII Virus
To evaluate the effects of silencing EGFRvIII on in vivo tumorigenicity of U87Δ cells, we have exposed U87Δ cells to anti-EGFRvIII or control lentiviruses (multiplicity of infection not to exceed 1.0 pfu/cell). SCID mice (n = 10) were injected in left flank with 10,000 U87Δ cells treated with the anti-EGFRvIII virus and in the right flank by same number of U87Δ cells treated with the control virus. Figure 6E to H (each panel is a different mouse) shows the formation, location, and relative size of these tumors. Figure 6I shows quantification of tumor volumes (mm3). No tumors were formed in the left flank (anti-EGFRvIII virus-treated cells) of animals, whereas the average size of tumors in the right flank (control virus-treated cells) reached 17.5 ± 5 mm3 by day 10 and 38 ± 3 mm3 by day 15 post-injection.
Discussion
The expression of EGFRvIII in several human cancers has been implicated in the stimulation of EGFR signaling pathways that drive the progression of neoplastic events. To evaluate the effects of EGFRvIII suppression, we employed siRNA technology to silence gene expression of EGFRvIII in vitro in U87Δ glioma cells as a model. In this study, we have shown the effect of siRNA-mediated suppression of EGFRvIII on biological characteristics such as proliferation, invasiveness, apoptosis, colony formation, and cell signaling pathways such as Ras/ERK and expression of EMT and focal adhesion markers. We also developed an anti-EGFRvIII lentivirus and studied its effects on the biology of glioma cells as a potential translational tool for targeting this malignancy and other related caners expressing this truncated receptor. When EGFRvIII was silenced in a temporary manner (electro-poration), we showed that proliferation of U87Δ cells was reduced significantly during the window in which EGFR-vIII was silenced. Additionally, the invasiveness of glioma cells and activation of Ras and ERK was reduced in these cells.
Based on the successful silencing of EGFRvIII expression in this and other studies, we concluded that the development of a translational strategy for the treatment of tumors that express EGFRvIII is warranted. siRNA molecules can be efficiently introduced into cells in a permanent manner through expression systems such as lentiviruses that are capable of integration into the cellular genome. The later approach for permanent delivery of siRNA has the potential of being translated into important clinical applications. Lentiviruses are considered as powerful integrative vector systems (36) and are highly efficient for in vivo application (37). The latest generation of lentiviral vectors is one of the safest and most efficient tools for stable gene transfer, even eliminating the risk of vector mobilization, due to infection with a wild-type HIV-1 (36-39). The anti-EGFRvIII lentivirus, but not the control virus, was efficiently capable of silencing EGFRvIII, which resulted in a complete suppression at 144 h post-infection. Such silencing of EGFRvIII was accompanied by loss of viability, invasiveness, and colony formation for U87Δ cells.
There is growing evidence suggesting that the change in tumor tissue architecture allowing the cancer cells to acquire invasive properties takes place through a peculiar phenotype modulation known as EMT (40, 41). The important characteristics of EMT are the deregulation of intercellular contacts and the increased of cell mobility resulting in the release of cells from the parent epithelial tissue. Considering the observed loss of invasiveness on EGFRvIII silencing, we were interested in investigating the outcome of EGFRvIII silencing on the expression of EMT-related markers and cadherins. On EGFRvIII silencing, we observed down-regulation of a series of transcription factors involved in EMT such as β-catenin, Snail, and Slug. The loss of E-cadherin expression in epithelial carcinomas has been studied as a primary factor in disruption of tight epithelial cell contacts and release of invasive tumor cells from the primary tumor site. Indeed, E-cadherin serves as a widely acting suppressor of invasion and growth of epithelial cancers, and its functional elimination represents a key step in the acquisition of the invasive phenotype for many tumors. Recent evidence indicates, however, that, in addition to the loss of the invasion-suppressor E-cadherin, another adhesion molecule, N-cadherin, is up-regulated in invasive tumor cell lines (32, 42). Such a “E-cadherin to N-cadherin switch” seems to be disrupted in glioma cells on silencing EGFRvIII as the expression of E-cadherin remains unchanged, but the expression of N-cadherin was found to be decreased.
Although the molecular bases of EMT are still being studied, several signal transduction pathways and several signaling molecules have been indentified to be involved. These include growth factors, receptor tyrosine kinases, Ras and other small GTPases, Src, β-catenin, and integrins. On such basis, we anticipated that the elimination of EGFRvIII would have potent effects on important signaling molecules such as Ras and ERK and would thereby influence EMT. This expectation has been shown by our studies to be correct.
Brain tumors as with many other malignancies are characterized by invasiveness into surrounding tissue. We observed that EGFRvIII knockdown markedly diminished the invasiveness of U87Δ glioma cells through a membrane in a Boyden chamber assay. As the metastatic capability of glioma cells is a major cause of mortality, any reduction in the invasiveness of glioma cells is of important consequence in evaluating the potential for clinical application. Further, we observed that silencing EGFRvIII significantly reduced the number of glioma cell colonies, suggesting that siRNA technology is an efficient strategy for mitigating the malignant phenotype of brain cancer cells. Another important factor in the survival of tumor cells is the impaired balance between prosurvival and antisurvival factors. Bcl-2 exerts a survival function in response to a wide range of apoptotic stimuli through inhibition of mitochondrial cytochrome c release (43). It has been implicated in modulating mitochondrial calcium homeostasis and proton flux (43, 44). On the other hand, Bad is a known proapoptotic member of the Bcl-2 family, which acts by promoting cell death by displacing Bax from binding to Bcl-2 and Bcl-xL (45, 46). We have found the balance between these two survival mediators altered toward a prodeath status on exposure of U87Δ cells to the anti-EGFRvIII virus. A significant suppression in the expression of Bcl-2 (below detection levels for Western blotting) was found, whereas Bad expression was only moderately reduced.
Alterations in the course of the cell cycle on silencing EGFRvIII resulted in a decrease in G1 and an increase in S and G2 fractions. This shift also reflects the changes observed in overall biology of glioma cells under EGFRvIII silencing conditions. In this regard, the down-regulation of cyclin A observed on EGFRvIII silencing can partly explain the increased S and G2 fraction on treatment of cells with anti-EGFRvIII virus.
In this study, we have reported the biological outcomes of transient and permanent silencing of EGFRvIII in U87Δ cells in vitro, which has been summarized in the model proposed in Fig. 6J. Future studies should be conducted in vivo to evaluate the efficiency and specificity of the siRNA strategy for treatment of brain tumors and other malignancies. One step toward this was taken by us in investigating the reducing effects of anti-EGFRvIII virus on the size of U87Δ s.c. tumor implants. A suitable vehicle for the in vivo delivery of siRNA is a lentivirus. By integrating into the animal or human genome, lentiviruses could be used to stably express the anti-EGFRvIII shRNA and induce stronger antineoplastic effects. One step toward this was taken in our study by investigating the reducing effects of anti-EGFRvIII virus on the size of s.c. tumors in SCID mice. Future preclinical experiments could be designed using intracranial injection of glioma cells lines or primary glioma cells expressing EGFRvIII in which tumors could then be treated with an anti-EGFRvIII-expressing lentivirus. Such studies will provide valuable data for designing clinical strategies that employ EGFRvIII silencing as the primary method for treating glioma or other related malignancies. Another perspective from this study can be described as providing new evidence and mechanistic insights for the concept of “oncogenic signaling addiction” (47, 48). Oncogene addiction describes an unexplained dependency of cancer cells to rely on a particular cellular pathway for survival or proliferation. This study shows that, even in the presence of EGFR, silencing of EGFRvIII is not tolerable to the glioma cells and causes a significant decrease in their viability. This concept has been studied for lung cancer (48) and now is supported by our data to be involved in glioma.
Acknowledgments
This work is dedicated to the loving memory of Eleanor M. Rawan. We thank Michael J. Franklin for editorial assistance with this article.
Grant support: American Brain Tumor Association and Institute for Science and Health.
Footnotes
Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed.
References
- 1.Zwick E, Hackel PO, Prenzel N, Ullrich A. The EGF receptor as central transducer of heterologous signalling systems. Trends Pharmacol Sci. 1999;20:408–12. doi: 10.1016/s0165-6147(99)01373-5. [DOI] [PubMed] [Google Scholar]
- 2.Hackel PO, Zwick E, Prenzel N, Ullrich A. Epidermal growth factor receptors: critical mediators of multiple receptor pathways. Curr Opin Cell Biol. 1999;11:184–9. doi: 10.1016/s0955-0674(99)80024-6. [DOI] [PubMed] [Google Scholar]
- 3.Woodburn JR. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol Ther. 1999;82:241–50. doi: 10.1016/s0163-7258(98)00045-x. [DOI] [PubMed] [Google Scholar]
- 4.Fontanini G, Vignati S, Bigini D, et al. Epidermal growth factor receptor (EGFr) expression in non-small cell lung carcinomas correlates with metastatic involvement of hilar and mediastinal lymph nodes in the squamous subtype. Eur J Cancer. 1995;31A:178–83. doi: 10.1016/0959-8049(93)00421-m. [DOI] [PubMed] [Google Scholar]
- 5.Chaffanet M, Chauvin C, Laine M, et al. EGF receptor amplification and expression in human brain tumours. Eur J Cancer. 1992;28:11–7. doi: 10.1016/0959-8049(92)90374-b. [DOI] [PubMed] [Google Scholar]
- 6.Turner T, Chen P, Goodly LJ, Wells A. EGF receptor signaling enhances in vivo invasiveness of DU-145 human prostate carcinoma cells. Clin Exp Metastasis. 1996;14:409–18. doi: 10.1007/BF00123400. [DOI] [PubMed] [Google Scholar]
- 7.Verbeek BS, Adriaansen-Slot SS, Vroom TM, Beckers T, Rijksen G. Overexpression of EGFR and c-erbB2 causes enhanced cell migration in human breast cancer cells and NIH3T3 fibroblasts. FEBS Lett. 1998;425:145–50. doi: 10.1016/s0014-5793(98)00224-5. [DOI] [PubMed] [Google Scholar]
- 8.Todd R, Wong DT. Epidermal growth factor receptor (EGFR) biology and human oral cancer. Histol Histopathol. 1999;14:491–500. doi: 10.14670/HH-14.491. [DOI] [PubMed] [Google Scholar]
- 9.Nishikawa R, Ji XD, Harmon RC, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci U S A. 1994;91:7727–31. doi: 10.1073/pnas.91.16.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamazaki H, Ohba Y, Tamaoki N, Shibuya M. A deletion mutation within the ligand binding domain is responsible for activation of epidermal growth factor receptor gene in human brain tumors. Jpn J Cancer Res. 1990;81:773–9. doi: 10.1111/j.1349-7006.1990.tb02644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moscatello DK, Holgado-Madruga M, Godwin AK, et al. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 1995;55:5536–9. [PubMed] [Google Scholar]
- 12.Garcia de Palazzo IE, Adams GP, Sundareshan P, et al. Expression of mutated epidermal growth factor receptor by non-small cell lung carcinomas. Cancer Res. 1993;53:3217–20. [PubMed] [Google Scholar]
- 13.Chakravarti A, Dicker A, Mehta M. The contribution of epidermal growth factor receptor (EGFR) signaling pathway to radioresistance in human gliomas: a review of preclinical and correlative clinical data. Int J Radiat Oncol Biol Phys. 2004;58:927–31. doi: 10.1016/j.ijrobp.2003.09.092. [DOI] [PubMed] [Google Scholar]
- 14.Pedersen MW, Tkach V, Pedersen N, Berezin V, Poulsen HS. Expression of a naturally occurring constitutively active variant of the epidermal growth factor receptor in mouse fibroblasts increases motility. Int J Cancer. 2004;108:643–53. doi: 10.1002/ijc.11566. [DOI] [PubMed] [Google Scholar]
- 15.Ohman L, Gedda L, Hesselager G, et al. A new antibody recognizing the vIII mutation of human epidermal growth factor receptor. Tumour Biol. 2002;23:61–9. doi: 10.1159/000059704. [DOI] [PubMed] [Google Scholar]
- 16.Jungbluth AA, Stockert E, Huang HJ, et al. A monoclonal antibody recognizing human cancers with amplification/overexpression of the human epidermal growth factor receptor. Proc Natl Acad Sci U S A. 2003;100:639–44. doi: 10.1073/pnas.232686499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Damstrup L, Rygaard K, Spang-Thomsen M, Poulsen HS. Expression of the epidermal growth factor receptor in human small cell lung cancer cell lines. Cancer Res. 1992;52:3089–93. [PubMed] [Google Scholar]
- 18.Ohtsuka K, Ohnishi H, Fujiwara M, et al. Abnormalities of epidermal growth factor receptor in lung squamous-cell carcinomas, adenosquamous carcinomas, and large-cell carcinomas: tyrosine kinase domain mutations are not rare in tumors with an adenocarcinoma component. Cancer. 2007;109:741–50. doi: 10.1002/cncr.22476. [DOI] [PubMed] [Google Scholar]
- 19.Sasaki H, Kawano O, Endo K, Yukiue H, Yano M, Fujii Y. EGFRvIII mutation in lung cancer correlates with increased EGFR copy number. Oncol Rep. 2007;17:319–23. [PubMed] [Google Scholar]
- 20.Ji H, Zhao X, Yuza Y, et al. Epidermal growth factor receptor variant III mutations in lung tumorigenesis and sensitivity to tyrosine kinase inhibitors. Proc Natl Acad Sci U S A. 2006;103:7817–22. doi: 10.1073/pnas.0510284103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vojtek AB, Der CJ. Increasing complexity of the Ras signaling pathway. J Biol Chem. 1998;273:19925–8. doi: 10.1074/jbc.273.32.19925. [DOI] [PubMed] [Google Scholar]
- 22.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 23.Zhou D, He QS, Wang C, Zhang J, Wong-Staal F. RNA interference and potential applications. Curr Top Med Chem. 2006;6:901–11. doi: 10.2174/156802606777303630. [DOI] [PubMed] [Google Scholar]
- 24.Moscatello DK, Montgomery RB, Sundareshan P, McDanel H, Wong MY, Wong AJ. Transformational and altered signal transduction by a naturally occurring mutant EGF receptor. Oncogene. 1996;13:85–96. [PubMed] [Google Scholar]
- 25.Prigent SA, Nagane M, Lin H, et al. Enhanced tumorigenic behavior of glioblastoma cells expressing a truncated epidermal growth factor receptor is mediated through the Ras-Shc-Grb2 pathway. J Biol Chem. 1996;271:25639–45. doi: 10.1074/jbc.271.41.25639. [DOI] [PubMed] [Google Scholar]
- 26.Feldkamp MM, Lala P, Lau N, Roncari L, Guha A. Expression of activated epidermal growth factor receptors, Ras-guanosine triphosphate, and mitogen-activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery. 1999;45:1442–53. doi: 10.1097/00006123-199912000-00034. [DOI] [PubMed] [Google Scholar]
- 27.Fan QW, Weiss WA. RNA interference against a glioma-derived allele of EGFR induces blockade at G2M. Oncogene. 2005;24:829–37. doi: 10.1038/sj.onc.1208227. [DOI] [PubMed] [Google Scholar]
- 28.Mischel PS, Cloughesy TF. Targeted molecular therapy of GBM. Brain Pathol. 2003;13:52–61. doi: 10.1111/j.1750-3639.2003.tb00006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Modjtahedi H, Moscatello DK, Box G, et al. Targeting of cells expressing wild-type EGFR and type-III mutant EGFR (EGFRvIII) by anti-EGFR MAb ICR62: a two-pronged attack for tumour therapy. Int J Cancer. 2003;105:273–80. doi: 10.1002/ijc.11055. [DOI] [PubMed] [Google Scholar]
- 30.Gaur RK. RNA interference: a potential therapeutic tool for silencing splice isoforms linked to human diseases. Biotechniques. 2006;(Suppl):15–22. doi: 10.2144/000112165. [DOI] [PubMed] [Google Scholar]
- 31.Christofori G. Changing neighbours, changing behaviour: cell adhesion molecule-mediated signalling during tumour progression. EMBO J. 2003;22:2318–23. doi: 10.1093/emboj/cdg228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hazan RB, Qiao R, Keren R, Badano I, Suyama K. Cadherin switch in tumor progression. Ann N Y Acad Sci. 2004;1014:155–63. doi: 10.1196/annals.1294.016. [DOI] [PubMed] [Google Scholar]
- 33.Wheelock MJ, Johnson KR. Cadherins as modulators of cellular phenotype. Annu Rev Cell Dev Biol. 2003;19:207–35. doi: 10.1146/annurev.cellbio.19.011102.111135. [DOI] [PubMed] [Google Scholar]
- 34.Wheelock MJ, Johnson KR. Cadherin-mediated cellular signaling. Curr Opin Cell Biol. 2003;15:509–14. doi: 10.1016/s0955-0674(03)00101-7. [DOI] [PubMed] [Google Scholar]
- 35.Pedersen MW, Meltorn M, Damstrup L, Poulsen HS. The type III epidermal growth factor receptor mutation. Biological significance and potential target for anti-cancer therapy. Ann Oncol. 2001;12:745–60. doi: 10.1023/a:1011177318162. [DOI] [PubMed] [Google Scholar]
- 36.Zufferey R, Dull T, Mandel RJ, et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998;72:9873–80. doi: 10.1128/jvi.72.12.9873-9880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naldini L. In vivo gene delivery by lentiviral vectors. Thromb Haemost. 1999;82:552–4. [PubMed] [Google Scholar]
- 38.Evans CH, Ghivizzani SC, Robbins PD. Gene therapy for arthritis: what next? Arthritis Rheum. 2006;54:1714–29. doi: 10.1002/art.21886. [DOI] [PubMed] [Google Scholar]
- 39.Montini E, Cesana D, Schmidt M, et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol. 2006;24:687–96. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- 40.Tse JC, Kalluri R. Mechanisms of metastasis: epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J Cell Biochem. 2007;101:816–29. doi: 10.1002/jcb.21215. [DOI] [PubMed] [Google Scholar]
- 41.Hugo H, Ackland ML, Blick T, et al. Epithelial-mesenchymal and mesenchymal-epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374–83. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- 42.Agiostratidou G, Hulit J, Phillips GR, Hazan RB. Differential cadherin expression: potential markers for epithelial to mesenchymal transformation during tumor progression. J Mammary Gland Biol Neoplasia. 2007;12:127–33. doi: 10.1007/s10911-007-9044-6. [DOI] [PubMed] [Google Scholar]
- 43.Murphy KM, Ranganathan V, Farnsworth ML, Kavallaris M, Lock RB. Bcl-2 inhibits Bax translocation from cytosol to mitochondria during drug-induced apoptosis of human tumor cells. Cell Death Differ. 2000;7:102–11. doi: 10.1038/sj.cdd.4400597. [DOI] [PubMed] [Google Scholar]
- 44.Murphy KM, Streips UN, Lock RB. Bcl-2 inhibits a Fas-induced conformational change in the Bax N terminus and Bax mitochondrial translocation. J Biol Chem. 2000;275:17225–8. doi: 10.1074/jbc.C900590199. [DOI] [PubMed] [Google Scholar]
- 45.Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–91. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 46.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–28. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 47.Sharma SV, Fischbach MA, Haber DA, Settleman J. “Oncogenic shock”: explaining oncogene addiction through differential signal attenuation. Clin Cancer Res. 2006;12:4392–5s. doi: 10.1158/1078-0432.CCR-06-0096. [DOI] [PubMed] [Google Scholar]
- 48.Sharma SV, Gajowniczek P, Way IP, et al. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell. 2006;10:425–35. doi: 10.1016/j.ccr.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]