

Abstract
Embryonic stem cells are envisioned as a viable source of pluripotent cells for use in regenerative medicine applications when donor tissue is not available. However, most current harvest techniques for embryonic stem cells require the destruction of embryos, which has led to significant political and ethical limitations on their usage. Parthenogenesis, the process by which an egg can develop into an embryo in the absence of sperm, may be a potential source of embryonic stem cells that may avoid some of the political and ethical concerns surrounding embryonic stem cells. Here we provide the technical aspects of embryonic stem cell isolation and expansion from the parthenogenetic activation of oocytes. These cells were characterized for their stem-cell properties. In addition, these cells were induced to differentiate to the myogenic, osteogenic, adipogenic, and endothelial lineages, and were able to form muscle-like and bony-like tissue in vivo. Furthermore, parthenogenetic stem cells were able to integrate into injured muscle tissue. Together, these results demonstrate that parthenogenetic stem cells can be successfully isolated and utilized for various tissue engineering applications.
Keywords: Parthenogenesis, Stem Cells, Tissue Engineering, Regenerative Medicine
1. Introduction
For the replacement of damaged or diseased tissue, most current strategies for tissue engineering depend upon a sample of autologous cells from the diseased organ of the host (1-3). However, in the setting of end-stage organ failure, a tissue biopsy may not yield enough normal cells for expansion and transplantation. In other instances, primary autologous human cells can not be adequately expanded from a particular organ for replacement purposes. In these situations, embryonic stem cells are envisioned as a viable source of pluripotent cells from which the desired tissue can be derived. Combining regenerative medicine techniques with this potentially endless source of versatile cells could lead to novel sources of replacement organs that would alleviate the chronic shortage of available donor organs.
Embryonic stem cells exhibit two remarkable properties: the ability to proliferate in a undifferentiated, but pluripotent state (self-renew), and the ability to differentiate into many specialized cell types (4). However, the destruction of embryos required for embryonic stem cell research has led to limitations in the United States, where federally-funded embryonic stem cell research has been restricted to the use of a limited number of cell lines under current federal regulations (5, 6). Recent studies describing induced pluripotent stem (iPS) cells present the possibility of embryonic stem cell research without the need for destruction of embryos (7-9). However, iPS cell research is still in its infancy, and it is still unclear whether these cells will be safe for use in clinical applications such as tissue engineering (10).
Parthenogenesis, the process by which an egg can develop into a blastocyst in the absence of sperm, has previously been identified as a potential source for embryonic stem cells (11). The ability to have embryonic stem cells, without sacrificing conception human embryos, would be valuable for patients who require the replacement of damaged or diseased tissue. In addition, this alternative source of embryonic stem cells may avoid some of the political and ethical concerns that surround current human embryonic stem cell techniques. Previous studies have described the isolation of embryonic stem cells from parthenogenesis (11-18); however, the adaptation of parthenogenetic stem cells for tissue engineering applications have not been previously described.
The aim of our study was to investigate whether multipotent stem cells could be isolated from parthenogenesis-derived blastocysts for tissue engineering applications. We have described a protocol for the isolation and expansion of embryonic stem cells derived from the parthenogenetic activation of rabbit oocytes, and we characterized these cells for their stem-cell properties. In addition, we induced these cells to differentiate into multiple lineages, and have demonstrated their survival in various in vivo environments. We anticipate that these methods will lead to eventual scale-up of these techniques to produce the large quantity of cells and tissue required for regenerative medicine applications.
2. Materials and Methods
2.1 Retrieval and Parthenogenetic Activation of Oocytes
Oocytes were collected from the oviducts of superovulated New Zealand rabbits approximately 16 hours after intravenous injection of 500 units of human chorionic gonadotropin (19). All procedures were approved by the institutional animal care committee.
Meiosis II oocytes were activated with two sets of electrical stimulation applied 30 minutes apart with an ECM 2001 stimulator (BTX, Holliston, Massachusetts). For each set, a total of three direct current (DC) pulses of 1.2 kV/cm for 60 μsec each were applied while the oocytes were in a solution containing mannitol 0.3 M in water with 0.1 mM CaCl2, 0.1 mM MgCl2, 10 mM HEPES, and 0.1% poly-vinyl alcohol (PVA). Electrically stimulated oocytes were then incubated for 2 hours at 37 °C in Potassium Simply Optimized Medium (KSOM - Specialty Media, Phillipsburg, New Jersey) containing 5 μg/ml cyclohexamide (CHX) and 1 mM 6-D-methyl aminopyridine (DMAP), and then cultured in a 500 μl drop of KSOM medium under mineral oil in a 37°C, 5% CO2 environment. In general, CHX is used to maintain interphase in the cell cycle, and DMAP is a catalyst for acylation reactions.
2.2. Isolation and Culture of Parthenogenesis-derived Stem Cells
Once the activated oocytes reached the blastocyst stage, microsurgery was used to mechanically dissect the inner cell mass from the blastocyst. The inner cell mass was placed initially on mitotically inactive mouse feeder layers for expansion in Dulbecco's minimal essential medium (Invitrogen) and 15% fetal calf serum (Invitrogen) for passage P0 (19). The expanded cells were then microsurgically dissected and transferred to Petri dishes for further expansion (P1 and beyond).
For immunoisolation, selection for c-kitpos cells was performed with magnetic cell sorting (Mini-MACS -Miltenyi Biotec Inc., Auburn, CA) as previously described (20). Magnetic sorting with the Mini-MACS system was utilized to minimize the contamination risk to the cells.
2.3. Flow Cytometry
Cells were trypsinized, washed with PBS, then incubated with antibodies against oct-4, bmp-4, cmet, ckit, tra-1-60, and tra-1-80 (Santa Cruz Biotechnology, Santa Cruz, California), as well as ssea-3 and ssea-4 (Developmental Studies Hybridoma Bank).
For cell cycle analysis, cells were fixed in 90% ethanol, then incubated in a 50ug/ml propidium iodide solution to stain the DNA.
All analyses were performed with a FACScalibur flow cytometer (Becton Dickinson).
2.4. Karyotype
Cells were cultured in T-25 culture flasks (flask method) and/or on glass coverslips (in situ method) for 3 - 5 days. Standard cytogenetic harvesting, fixation, and GTG banding techniques were used.
2.5. Induction toward Multiple Cell Lineages
The cells were induced to osteogenic (21), myogenic (22, 23), and adipogenic (24) lineages according to previously described protocols. For endothelial differentiation, the cells were maintained in EGM-2 endothelial cell medium (Cambrex Corporation, East Rutherford, NJ) supplemented with 10% fetal bovine serum (FBS), and basic fibroblast growth factor (bFGF).
2.6. Immunocytochemistry and Histochemistry
Single and double immunolabelling was performed using indirect immunofluorescence. Cell cultures were fixed in methanol for 10 minutes, incubated in PBS with 10% of appropriate serum for 45 minutes to block non-specific antibody binding, incubated overnight at 4°C with primary antibodies, washed three times with PBS, and incubated for 30 minutes at room temperature with secondary antibodies. Primary antibodies used were desmin, myo D, actinin, sarcomeric tropomyosin, osteocalcin, and osteopontin (Santa Cruz Biotechnology, Santa Cruz, California), KDR (Imclone Systems, New York, NY), P1H12 (Chemicon, Temecula, CA), and Von Willebrand Factor (VWF) (Dako, Denmark). Secondary antibodies were biotinylated anti-mouse and biotinylated anti-goat immunoglobulins (Vector Laboratories, Burlingame, CA). The secondary biotinylated antibodies were incubated with Fluorescein - Avidin D (Vector).
Von Kossa staining was used to detect the presence of mineralization in cell culture. The cell culture plates were fixed with 10% formaldehyde for 1 hour, incubated with 2% silver nitrate solution for 10 minutes in the dark, washed thoroughly with deionized water, and then exposed to ultraviolet (UV) light for 15 minutes.
For Oil Red O staining, cells were fixed with 2% paraformaldehyde for 10 minutes at room temperature and rinsed with PBS. Samples were incubated in Oil Red O solution (Sigma) for 15 minutes and rinsed with distilled water. Hematoxylin was used for counterstaining.
2.7. Reverse-transcriptase polymerase chain reaction (RT-PCR)
RNA was isolated from cultured cells and cell pellets with RNA-Bee RNA Isolation Reagent (Tel-Test Inc., Friendswood, TX) according to the manufacturer's protocol. RNA (2μg) was processed for c-DNA synthesis with Superscript II reverse transcriptase with random hexamers (Invitrogen). c-DNA was used for each PCR reaction, in a final volume of 50 μl with 10mM dNTP, 15pM of each primer, 0.4U Taq-DNA-polymerase, reaction buffer, and MgCl2 (Invitrogen), in a Mastercycler (Eppendorf). The cycling conditions consisted of annealing at various temperatures for 30 seconds, elongation at 72°C for 1 minute, and denaturation at 92°C for 45 seconds. Cycle numbers varied between 28 and 35 cycles and were chosen in the exponential phase of the RT-PCR reaction. Primer sequences and fragment sizes are listed in Table 1. All primers were obtained from Invitrogen. PCR products were resolved on a 2% agarose gel and stained with ethidium bromide. The amplified DNA was visualized under UV light.
Table 1.
List of primers used for PCR
| Gene | Primer (sense; anti-sense) | Segment length (bp) | Cycles | Annealing Temp (°C) |
|---|---|---|---|---|
| G3PD | 5′GTCTTCACCACCATGGAG3′ 5′CCACCCTGTTGCTGTAGC3′ | 673 | 35 | 58 |
| cbfa1 | 5′GGCCTTCCACTCTCAGTAAGA3′ 5′GATTCATCCATTCTGCCACTA3′ | 474 | 35 | 63 |
| lipoprotein lipase | 5′CTGGTCGAAGCATTGGAAT3′ 5′TGTAGGGCATCTGAGAACGAG3′ | 366 | 35 | 64 |
| mrf4 | 5′CGACAGCAGCGGAGAGG3′ 5′GGAATGATCGGAAACACTTGG3′ | 421 | 35 | 58 |
| pparγ2 | 5′AGGGCGATCTTGACAGGAAAG3′ 5′CTCGATGGGCTTCACATTCAG3′ | 504 | 35 | 58 |
| VCAM | 5′AATGGGCAGGTGACAAGTGAA3′ 5′CAGAACGGCCTTCCTCCAC3′ | 504 | 35 | 60 |
2.8. In vivo placement of labelled-cell-seeded scaffolds
The outer membrane of undifferentiated cells were labelled using PKH26 Red (Sigma) according to the manufacturer's protocol with the exception of using 40 ul dye with each ml of diluent. For the myogenic cells, polyglycolic acid (PGA) scaffolds measuring 0.5cm × 0.5cm × 0.5cm were seeded with undifferentiated cells, and for the osteogenic cells, hydroxyapatite-collagen scaffolds measuring 0.5cm × 0.5cm × 0.5 cm were seeded with undifferentiated cells, both at a density of 60 million cells per cm3. The cell-seeded scaffolds were incubated in their respective differentiation media for 14 days, then implanted into the dorsal subcutaneous area of athymic mice. After in vivo formation, tissue was surgically retrieved and frozen in Tissue-Tek OCT compound (Miles Scientific, Naperville, IL) at -80 degrees Celsius. Frozen sections were cut using a cryotome, fixed onto glass slides, then processed for hematoxylin and eosin (H&E) staining using standard techniques.
2.9. Implantation of parthenogenesis-derived stem cells into a muscle injury model
Parthenogenetic cells were labeled using a retroviral vector encoding a bacterial lac Z reporter gene. Labeled parthenogenetic stem cells were used for the engineered muscle transplantation studies (1,500-2,000 MOI) (Harvard Gene Therapy Initiative). Mice were anesthetized by isoflurane inhalation. The tibialis muscle was injected with 50 μl of 1mM cardiotoxin (Calbiochem) diluted in PBS. After 24 hours, 1×106 LacZ-parthenogenesis-derived stem cells were injected into the injured tibialis muscle of nude mice. Muscle was harvested at 1 and 2 weeks after injection.
3. Results
3.1. Isolation and Characterization of Parthenogenesis-derived Stem Cells
Parthenogenetically-activated oocytes were able to be grown to the blastocyst stage after electrical stimulation. Although a feeder layer was used for passage 0, populations of activated cells were then grown on plastic without feeder layers for all subsequent passages. After adequate expansion of the cells to allow for the use of Mini-MACS cell sorting, candidate cells were then immunoisolated from the rest of the cell population using stem cell markers, and were noted to constitute approximately 10% of the total cell population. We noted that these cells were homogenously diploid after cell cycle and karyotype analysis. Cell cycle analysis with propidium iodide revealed that these cells were of a homogenous ploidy, as only one peak associated with the G1 phase was noted. Karyotyping confirmed the diploid nature of these stem cells.
These cells were able to be expanded with a doubling time of approximately 20 hours, a high self-renewal rate that would allow for an adequate number of cells to be available for reconstructive applications.
Several important early embryonic stem cell markers were noted to be present in these cells after FACS analysis of early passage cells, including oct-4, a transcription factor unique to pluripotent stem cells that is essential for the establishment and maintenance of early pluripotent stem cells; bone morphogenetic protein–4 (bmp-4), a growth and differentiation factor that is expressed during early mesoderm formation and differentiation; c-kit, a cell surface receptor found on hematopoietic and mesenchymal stem cells; and stage-specific embryonic antigen-4 (ssea-4), which is a glycoprotein specifically expressed in early embryonic development and by undifferentiated pluripotent stem cells (Figure 1). Other stem cell markers, such as tra-1-60, tra-1-81, and stage-specific embryonic antigen-1 (ssea-1), were not identified in these cells. Other stem cell markers were identified in these cells by immunohistochemistry, including stage-specific embryonic antigen-3 (ssea-3), another glycoprotein specifically expressed in early embryonic development and by undifferentiated pluripotent stem cells; alpha fetoprotein (AFP), a protein expressed during primitive endoderm development and which reflects endodermal differentiation; noggin, a neuron-specific gene that is expressed during the development of neurons; and vimentin, which is found in ectoderm, neural and progenitor cells and which is characteristic of primitive neuroectoderm formation (Figure 2).
Figure 1.
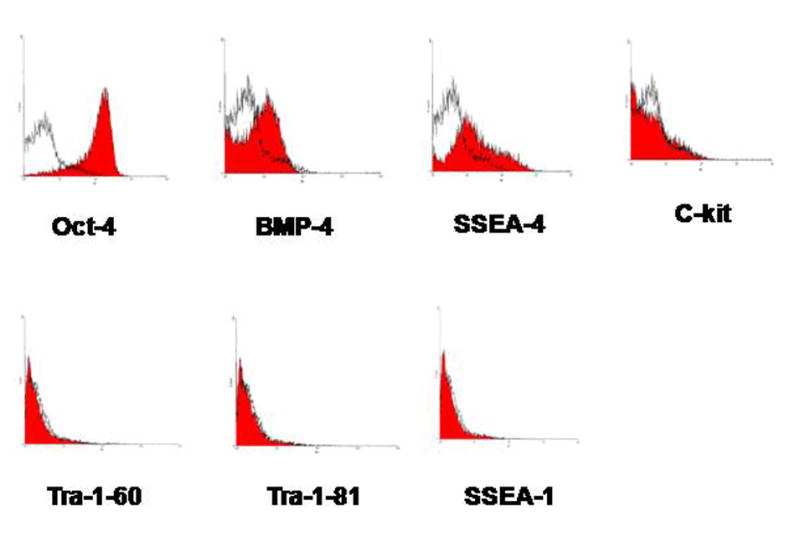
FACS analysis for stem cells markers. By FACS analysis, the following stem cell markers were found in these cells: oct-4, bone morphogenetic protein–4 (bmp-4), stage-specific embryonic antigen-4 (ssea-4), and c-kit. Other stem cell markers such as tra-1-60, tra-1-81, and stage-specific embryonic antigen-1 (ssea-1) were not identified in these cells.
Figure 2.
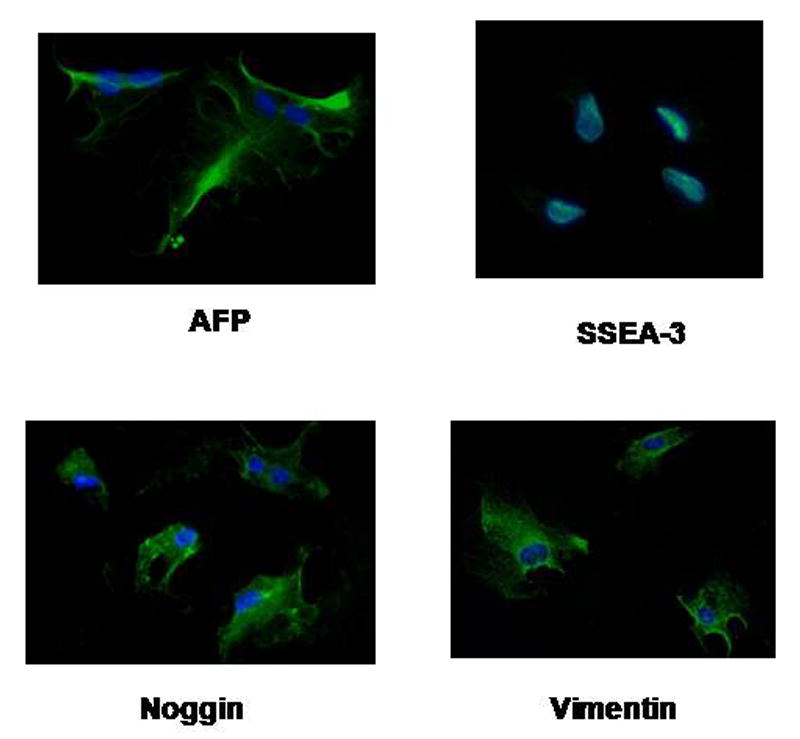
Immunohistochemistry for stem cell markers. Immunohistochemistry identified the presence of other stem cell markers: stage-specific embryonic antigen-3 (ssea-3), alpha fetoprotein (AFP), noggin, and vimentin.
3.2. Differentiation of Parthenogenesis-derived Stem Cells into Multiple Lineages
The stem cells were inducible to different cell lineages under specific growth conditions. Differentiation was confirmed by phenotypic changes, immunocytochemistry, gene expression, and functional analyses. When the cells were directed toward the myogenic lineage (Figure 3), the presence of several early muscle markers, such as desmin, myoD, actinin, and sarcomeric tropomyosin, was detected in the induced cells by immunohistochemistry. RT-PCR revealed the presence of mrf4, a muscle-specific transcription factor that is important in the regulation of muscle cell development.
Figure 3.
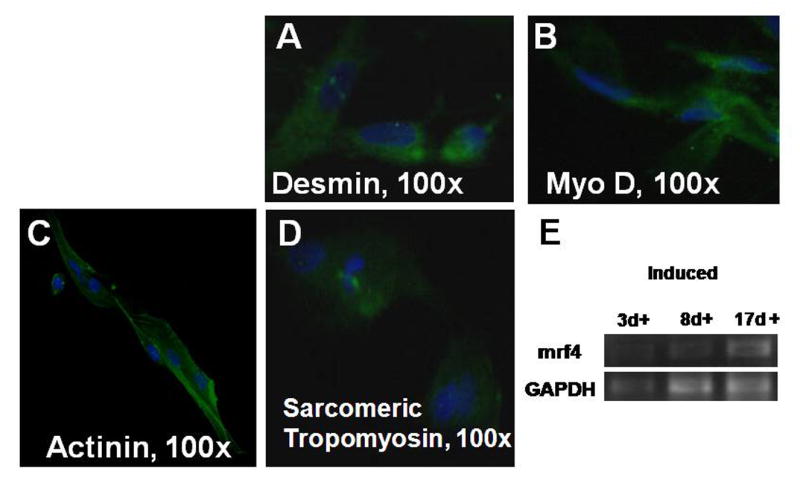
Myogenic differentiation. Induced cells stained positively for desmin (A), myo D (B), actinin (C), and sarcomeric tropomyosin (D). RT-PCR noted the presence of the muscle gene mrf4 at days 3, 7, and 14 after induction.
When directed toward the osteogenic lineage, the induced cells stained positively for osteocalcin, a mineral-binding protein uniquely synthesized by the osteoblast, and osteopontin, which is also known as bone sialoprotein-1 (Figure 4). Von Kossa staining demonstrated the presence of calcium mineralization, and RT-PCR indicated the presence of cbfa-1, which is also known as osteoblast specific transcription factor.
Figure 4.
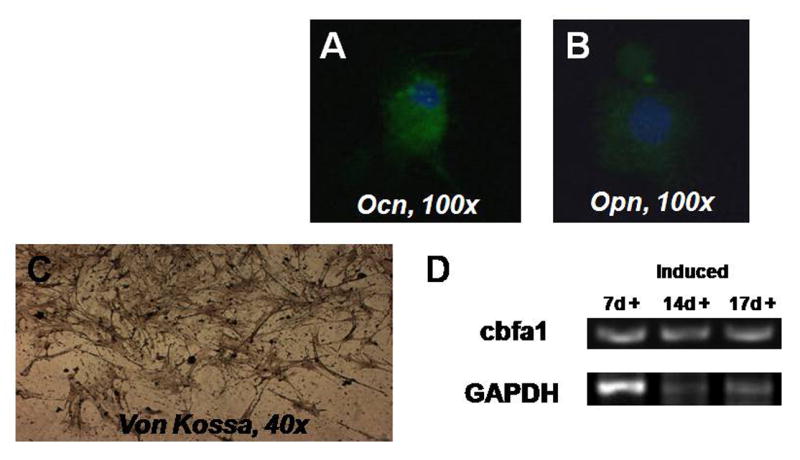
Osteogenic differentiation. Induced cells stained positively for osteocalcin (A) and osteopontin (B). Von Kossa staining revealed the presence of calcium mineralization. RT-PCR noted the presence of the osteogenic gene cbfa-1, which is also known as osteoblast specific transcription factor.
After induction toward the adipogenic lineage, Oil-Red-O staining demonstrated the presence of lipid droplets in these cells (Figure 5). RT-PCR noted the presence of the adipogenic genes, lipoprotein lipase and pparγ-2.
Figure 5.
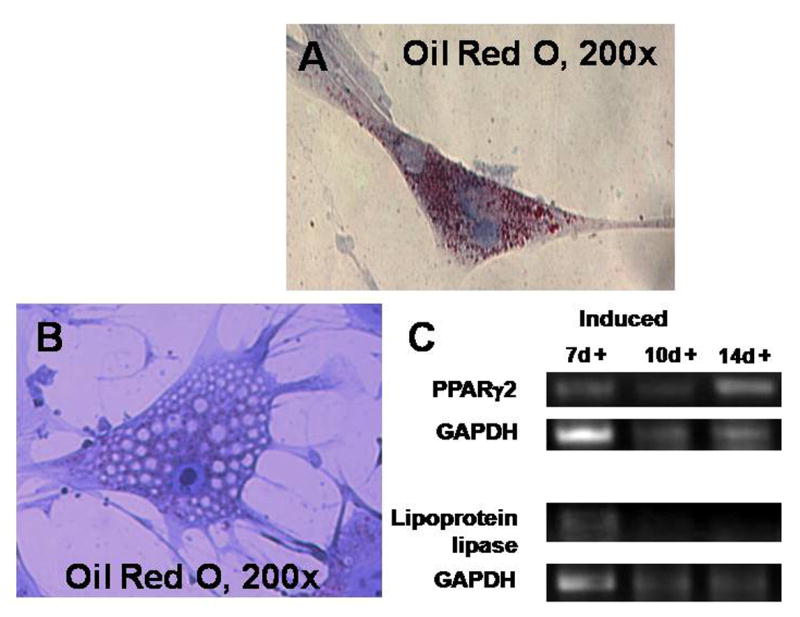
Adipogenic differentiation. After induction toward the adipogenic lineage, the presence of fat was noted by Oil-Red-O staining. RT-PCR noted the presence of the adipogenic genes, lipoprotein lipase and pparγ-2.
When the cells were induced toward the endothelial lineage, the cells were noted to have a cobblestone appearance on phase microscopy (Figure 6). By immunohistochemistry, the following endothelial markers were noted to be present: KDR, VWF, and P1H12. In addition, the presence of the gene for vascular cell adhesion molecule (VCAM) was noted by RT-PCR.
Figure 6.
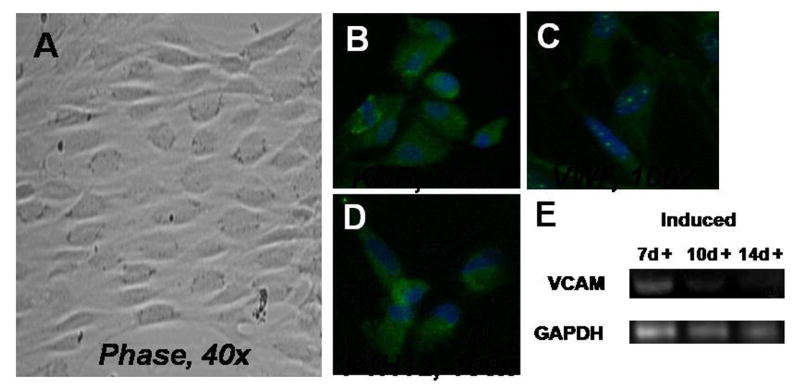
Endothelial differentiation. Induced cells had a cobblestone appearance on phase microscopy (A). The cells stained positively for KDR (B), VWF (C), and P1H12 (D). In addition, RT-PCR noted the presence of the gene for vascular cell adhesion molecule (VCAM).
3.3. In Vivo Studies
Labelled undifferentiated cells were seeded onto PGA scaffolds, placed in myogenic induction media for 2 weeks, then implanted into the dorsal subcutaneous space in athymic mice. After 2 weeks in vivo, labelled muscle-like tissue was retrieved (Figure 7A and 7B).
Figure 7.
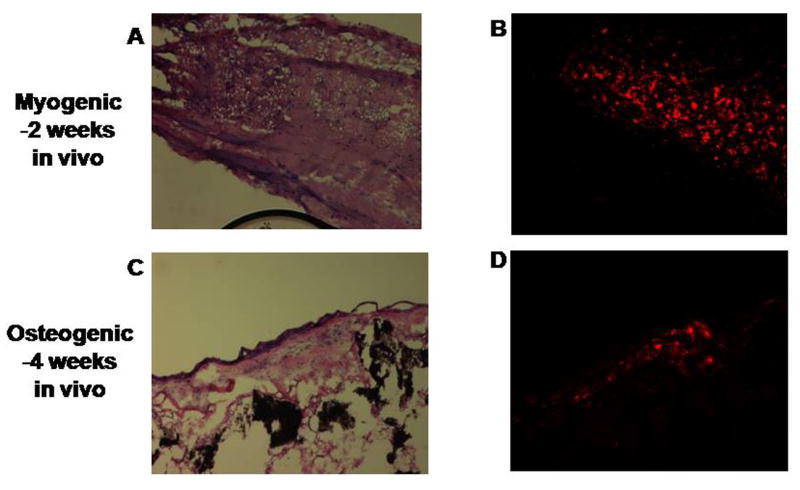
In vivo formation of tissue from labeled induced cells. Muscle-like tissue (A) was retrieved after 2 weeks in vivo with maintenance of the PKH26 red label (B). Bone-like tissue (C) was retrieved after 4 weeks in vivo with maintenance of the PKH26 red label (D).
In a similar fashion, labelled undifferentiated cells were seeded onto hydroxyapatite/collagen scaffolds, placed in osteogenic induction media for 2 weeks, then implanted into athymic mice. After 4 weeks in vivo, labelled bony-like tissue was obtained (Figure 7C and 7D).
LacZ-parthenogenetic stem cells were injected into a cardiotoxin-injured tibialis muscle in mice. After 2 weeks in vivo, histolopathologic evaluation revealed the survival and integration of the LacZ-parthenogenetic stem cells into the injured muscle (Figure 8).
Figure 8.
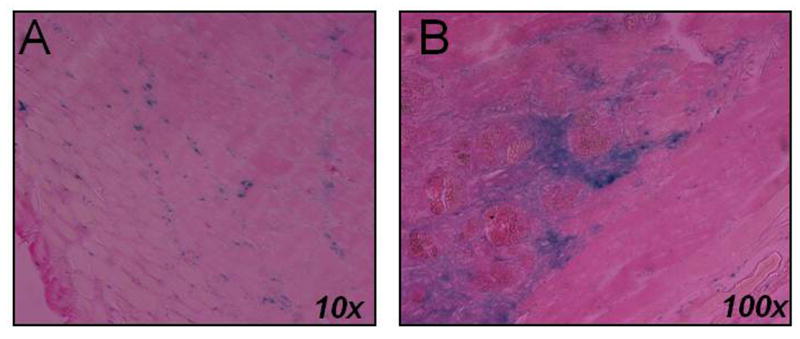
In vivo integration of labeled cells into muscle. Cardiotoxin-injured tibialis muscle was retrieved after 2 weeks in vivo with evidence of LacZ parthenogenetic-derived stem cell survival and integration.
4. Discussion
The fields of regenerative medicine and tissue engineering seek to relieve the suffering of patients from the consequences of disease and injury by restoring the form and function of their damaged tissue and organs. Many disorders, such as congenital anomalies, cancer, trauma, infection, inflammation, iatrogenic injuries, and other conditions, can lead to organ damage or loss and to the eventual need for reconstruction. The majority of current reconstructive techniques rely on donor tissue for replacement; however, a shortage of donor tissue may limit these types of reconstruction, and usually significant morbidity is associated with the harvest procedure.
Particularly in cases when donor tissue is unavailable, embryonic stem cells has been envisioned as a potentially endless source of versatile cells for use in regenerative medicine applications because of their ability to proliferate in a undifferentiated, but pluripotent state (self-renew) and to differentiate into specialized cells from all three embryonic germ layers. Skin and neurons have been formed, indicating ectodermal differentiation (25-28). Blood, cardiac cells, cartilage, endothelial cells, and muscle have been formed, indicating mesodermal differentiation (29-31). And pancreatic cells have been formed, indicating endodermal differentiation (32). In addition, as further evidence of their pluripotency, embryonic stem cells have formed embryoid bodies, which are cell aggregations that contain all three embryonic germ layers, while in culture, and have formed teratomas in vivo (33).
However, ethical and political concerns have placed limitations upon embryonic stem cell research in the United States, since most current techniques require the destruction of human embryos in order to harvest the embryonic stem cells (34). Current federal policy limits the use of federal research funding to a limited number of approved cell lines; however, only a handful of these cell lines are currently available for use (6). Additional cell lines are available from non-federally-funded laboratories, but the usage of these cell lines is limited to the small percentage of laboratories that have separate non-federally-funded facilities and personnel (5). The recent studies describing iPS cells do present a possibility of embryonic stem cell research without the need for destruction of embryos (7-9). However, iPS cell research is still in its infancy, and it is still unclear whether these cells will be safe for use in humans, or whether the cells can be adequately expanded in large quantities necessary for tissue engineering applications (10).
Parthenogenesis (Greek : “virgin birth”) is the reproductive process in which a female gamete can develop into a new individual without fertilization by a male gamete, and occurs naturally in certain plants, arthopods, and insects. Artificial parthenogenesis with frog eggs was first described by Loeb in 1900, and was first described in mammalian eggs by Pincus in 1936. As further evidence of the multipotency potential of these cells, Kuno et al (35) noted the presence of fully differentiated tissue of all three germ layers in a mature ovarian cystic teratoma of a virginal woman, thereby suggesting that the Information for the organization of the body plan is already present in the oocyte. More recently, the isolation and characterization of parthenogenetic stem cells from several oocyte sources, including human and non-human primates, have been described (11-18).
Our study describes the adaptation of parthenogenesis-derived stem cells for tissue engineering applications, as they are capable of differentiation into multiple lineages and can form tissue in vivo. Oocytes, the sources of these stem cells, can be harvested with the use of minimally invasive techniques with minimal donor morbidity. In addition, these stem cells have a rapid proliferative rate that would allow adequate expansion of these cells for tissue replacement therapy, and do not require the use of feeder layers for expansion, thereby avoiding the exposure of these cells to xenogenic viruses and proteins.
The use of parthenogenesis-derived stem cells may avoid some of the political and ethical controversy surrounding human embryonic stem cells. Several attempts to produce viable individuals from parthenogenesis in the past have been unsuccessful (36-38). While Kono et al has recently shown that some parthenogenetic mice can survive into adulthood (39), they noted the predominant mortality of these embryos in that 99.4% of the embryos perished prior to adulthood, with the majority of the deaths occurring during embryonic development. This may help to relieve some of the potential ethical and political concerns with the use of parthenogenetic stem cells in that the parthenogenetic embryos rarely survive beyond the embryo stage, but do survive long enough where the embryonic stem cells can be harvested for tissue engineering purposes.
Acknowledgments
Evelyn Flynn for histological assistance, Alan Flint for the FACS analysis, and Mark J. Pettenati, MD and Stewart Wilson for the karyotype analysis
Note: This study was supported by grant 1-T32-DK60442-01 (NIH / NIDDK)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Oberpenning F, Meng J, Yoo JJ, Atala A. De novo reconstitution of a functional mammalian urinary bladder by tissue engineering. Nature Biotechnology. 1999;17(2):149–55. doi: 10.1038/6146. see comment. [DOI] [PubMed] [Google Scholar]
- 2.De Filippo RE, Yoo JJ, Atala A. Engineering of vaginal tissue in vivo. Tissue Engineering. 2003;9(2):301–6. doi: 10.1089/107632703764664765. [DOI] [PubMed] [Google Scholar]
- 3.De Filippo RE, Yoo JJ, Atala A. Urethral replacement using cell seeded tubularized collagen matrices. Journal of Urology. 2002;168(4 Pt 2):1789–92. doi: 10.1097/01.ju.0000027662.69103.72. discussion 92-3. [DOI] [PubMed] [Google Scholar]
- 4.Brivanlou AH, Gage FH, Jaenisch R, Jessell T, Melton D, Rossant J. Stem cells. Setting standards for human embryonic stem cells. Science. 2003;300(5621):913–6. doi: 10.1126/science.1082940. comment. [DOI] [PubMed] [Google Scholar]
- 5.Cowan CA, Klimanskaya I, McMahon J, et al. Derivation of embryonic stem-cell lines from human blastocysts. New England Journal of Medicine. 2004;350(13):1353–6. doi: 10.1056/NEJMsr040330. see comment. [DOI] [PubMed] [Google Scholar]
- 6.Kennedy D. Stem cells: still here, still waiting. Science. 2003;300(5621):865. doi: 10.1126/science.300.5621.865. comment. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72. doi: 10.1016/j.cell.2007.11.019. see comment. [DOI] [PubMed] [Google Scholar]
- 8.Park IH, Zhao R, West JA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451(7175):141–6. doi: 10.1038/nature06534. see comment. [DOI] [PubMed] [Google Scholar]
- 9.Meissner A, Wernig M, Jaenisch R. Direct reprogramming of genetically unmodified fibroblasts into pluripotent stem cells. Nature Biotechnology. 2007;25(10):1177–81. doi: 10.1038/nbt1335. [DOI] [PubMed] [Google Scholar]
- 10.Holden C, Vogel G. CELL BIOLOGY: A Seismic Shift for Stem Cell Research. Science. 2008;319(5863):560–3. doi: 10.1126/science.319.5863.560. [DOI] [PubMed] [Google Scholar]
- 11.Cibelli JB, Grant KA, Chapman KB, et al. Parthenogenetic stem cells in nonhuman primates. Science. 2002;295(5556):819. doi: 10.1126/science.1065637. [DOI] [PubMed] [Google Scholar]
- 12.Cheng L. More new lines of human parthenogenetic embryonic stem cells. Cell Research. 2008;18(2):215–7. doi: 10.1038/cr.2008.19. [DOI] [PubMed] [Google Scholar]
- 13.Mai Q, Yu Y, Li T, et al. Derivation of human embryonic stem cell lines from parthenogenetic blastocysts. Cell Research. 2007;17(12):1008–19. doi: 10.1038/cr.2007.102. [DOI] [PubMed] [Google Scholar]
- 14.Lin G, OuYang Q, Zhou X, et al. A highly homozygous and parthenogenetic human embryonic stem cell line derived from a one-pronuclear oocyte following in vitro fertilization procedure. Cell Research. 2007;17(12):999–1007. doi: 10.1038/cr.2007.97. [DOI] [PubMed] [Google Scholar]
- 15.Brevini TAL, Gandolfi F. Parthenotes as a source of embryonic stem cells. Cell Proliferation. 2008;41(s1):20–30. doi: 10.1111/j.1365-2184.2008.00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dighe V, Clepper L, Pedersen D, et al. Heterozygous Embryonic Stem Cell Lines Derived from Nonhuman Primate Parthenotes. Stem Cells. 2008;26(3):756–66. doi: 10.1634/stemcells.2007-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sritanaudomchai H, Pavasuthipaisit K, Kitiyanant Y, Kupradinun P, Mitalipov S, Kusamran T. Characterization and multilineage differentiation of embryonic stem cells derived from a buffalo parthenogenetic embryo. Molecular Reproduction & Development. 2007;74(10):1295–302. doi: 10.1002/mrd.20592. [DOI] [PubMed] [Google Scholar]
- 18.Fang ZF, Gai H, Huang YZ, et al. Rabbit embryonic stem cell lines derived from fertilized, parthenogenetic or somatic cell nuclear transfer embryos. Experimental Cell Research. 2006;312(18):3669–82. doi: 10.1016/j.yexcr.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 19.Vrana KE, Hipp JD, Goss AM, et al. Nonhuman primate parthenogenetic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(Suppl 1):11911–6. doi: 10.1073/pnas.2034195100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Coppi P, Bartsch G, Jr, Siddiqui MM, et al. Isolation of amniotic stem cell lines with potential for therapy. Nature Biotechnology. 2007;25(1):100–6. doi: 10.1038/nbt1274. [DOI] [PubMed] [Google Scholar]
- 21.Jaiswal N, Haynesworth SE, Caplan AI, Bruder SP. Osteogenic differentiation of purified, culture-expanded human mesenchymal stem cells in vitro. Journal of Cellular Biochemistry. 1997;64(2):295–312. [PubMed] [Google Scholar]
- 22.Ferrari G, Cusella-De Angelis G, Coletta M, et al. Muscle regeneration by bone marrow-derived myogenic progenitors. Science. 1998;279(5356):1528–30. doi: 10.1126/science.279.5356.1528. see comment erratum appears in. [DOI] [PubMed] [Google Scholar]; Science. Aug 14;281(5379):923. [Google Scholar]
- 23.Rosenblatt JD, Lunt AI, Parry DJ, Partridge TA. Culturing satellite cells from living single muscle fiber explants. In Vitro Cellular & Developmental Biology Animal. 1995;31(10):773–9. doi: 10.1007/BF02634119. [DOI] [PubMed] [Google Scholar]
- 24.Poliard A, Nifuji A, Lamblin D, Plee E, Forest C, Kellermann O. Controlled conversion of an immortalized mesodermal progenitor cell towards osteogenic, chondrogenic, or adipogenic pathways. Journal of Cell Biology. 1995;130(6):1461–72. doi: 10.1083/jcb.130.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reubinoff BE, Itsykson P, Turetsky T, et al. Neural progenitors from human embryonic stem cells. Nature Biotechnology. 2001;19(12):1134–40. doi: 10.1038/nbt1201-1134. comment. [DOI] [PubMed] [Google Scholar]
- 26.Schuldiner M, Eiges R, Eden A, et al. Induced neuronal differentiation of human embryonic stem cells. Brain Research. 2001;913(2):201–5. doi: 10.1016/s0006-8993(01)02776-7. [DOI] [PubMed] [Google Scholar]
- 27.Schuldiner M, Yanuka O, Itskovitz-Eldor J, Melton DA, Benvenisty N. Effects of eight growth factors on the differentiation of cells derived from human embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(21):11307–12. doi: 10.1073/pnas.97.21.11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang SC, Wernig M, Duncan ID, Brustle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nature Biotechnology. 2001;19(12):1129–33. doi: 10.1038/nbt1201-1129. comment. [DOI] [PubMed] [Google Scholar]
- 29.Kaufman DS, Hanson ET, Lewis RL, Auerbach R, Thomson JA. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(19):10716–21. doi: 10.1073/pnas.191362598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kehat I, Kenyagin-Karsenti D, Snir M, et al. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. Journal of Clinical Investigation. 2001;108(3):407–14. doi: 10.1172/JCI12131. comment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levenberg S, Golub JS, Amit M, Itskovitz-Eldor J, Langer R. Endothelial cells derived from human embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(7):4391–6. doi: 10.1073/pnas.032074999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Assady S, Maor G, Amit M, Itskovitz-Eldor J, Skorecki KL, Tzukerman M. Insulin production by human embryonic stem cells. Diabetes. 2001;50(8):1691–7. doi: 10.2337/diabetes.50.8.1691. [DOI] [PubMed] [Google Scholar]
- 33.Itskovitz-Eldor J, Schuldiner M, Karsenti D, et al. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Molecular Medicine. 2000;6(2):88–95. [PMC free article] [PubMed] [Google Scholar]
- 34.Vogel G. Biomedical policy. Bush squeezes between the lines on stem cells. Science. 2001;293(5533):1242–5. doi: 10.1126/science.293.5533.1242. [DOI] [PubMed] [Google Scholar]
- 35.Kuno N, Kadomatsu K, Nakamura M, Miwa-Fukuchi T, Hirabayashi N, Ishizuka T. Mature ovarian cystic teratoma with a highly differentiated homunculus: a case report. Birth Defects Res Part A Clin Mol Teratol. 2004;70(1):40–6. doi: 10.1002/bdra.10133. [DOI] [PubMed] [Google Scholar]
- 36.Fukui Y, Sawai K, Furudate M, Sato N, Iwazumi Y, Ohsaki K. Parthenogenetic development of bovine oocytes treated with ethanol and cytochalasin B after in vitro maturation. Molecular Reproduction & Development. 1992;33(3):357–62. doi: 10.1002/mrd.1080330318. [DOI] [PubMed] [Google Scholar]
- 37.Kaufman MH, Barton SC, Surani MA. Normal postimplantation development of mouse parthenogenetic embryos to the forelimb bud stage. Nature. 1977;265(5589):53–5. doi: 10.1038/265053a0. [DOI] [PubMed] [Google Scholar]
- 38.McGrath J, Solter D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell. 1984;37(1):179–83. doi: 10.1016/0092-8674(84)90313-1. [DOI] [PubMed] [Google Scholar]
- 39.Kono T, Obata Y, Wu Q, et al. Birth of parthenogenetic mice that can develop to adulthood. Nature. 2004;428(6985):860–4. doi: 10.1038/nature02402. see comment. [DOI] [PubMed] [Google Scholar]