

Abstract
In an effort to find novel agents which selectively target the kappa opioid receptor (KOPR), we modified the furan ring of the highly potent and selective KOPR agonist salvinorin A. Introduction of small substituents at C-16 was well tolerated. 12-epi-Salvinorin A, synthesized in four steps from salvinorin A, was a selective partial agonist at the KOPR. No clear SAR patterns were observed for C-13 aryl ketones. Introducing a hydroxymethylene group between C-12 and the furan ring was tolerated. Small C-13 esters and ethers gave weak KOPR agonists, while all C-13 amides were inactive. Finally, substitution of oxadiazoles for the furan ring abolished affinity for the KOPR. None of the compounds displayed any KOPR antagonism or any affinity for mu or delta opioid receptors.
Keywords: Kappa opioid receptor agonists, Salvinorin, Binding, [35S]GTPγS
1. Introduction
Selective kappa opioid receptor (KOPR) ligands with activity in the central nervous system may be useful in the treatment of various disorders, including drug abuse,1 mood abnormalities,2, 3 and anxiety disorders.4 Although there are many selective KOPR agonists available for preclinical studies, few selective KOPR antagonists have been identified. The prototypical selective KOPR antagonist norBNI5 has long been used in preclinical studies. However, recent reports suggest that, in vivo, it may display some mu opioid receptor (MOPR) antagonism in addition to its effects at KOPR.6, 7 GNTI8 and ANTI,9 two structurally simplified analogues of norBNI, reportedly possess increased selectivity7 but have not been extensively studied, perhaps due to the complexity of their synthesis and purification. The more recently developed JDTic10 is a highly potent and selective KOPR antagonist. However, its slow onset (48 h) and long duration (several weeks) of action in vivo can be a limitation for some studies.11 The development of additional selective KOPR agents, in particular antagonists, will provide alternative tools that enable a better understanding of KOPR mediated effects. Recently, the natural product salvinorin A (1),12 a high affinity, highly selective agonist at the KOPR,13 has been used as a lead compound for the design of selective KOPR agonists (full or partial) and antagonists.14
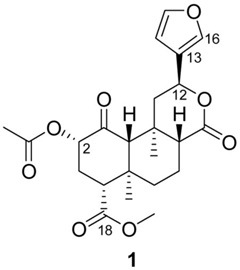
In vivo, salvinorin A has a fast onset of action and (at least in some tests) a relatively brief duration of effects,15–17 probably due to rapid metabolism of its labile acetate. In previous studies,18, 19 we modified the C-2 substituent and obtained potent and selective KOPR agonists with improved metabolic stability, longer lasting in vivo effects, and oral efficacy.17
Most early SAR studies focused on the modification of the acetate and methyl ester.14 Briefly, some small C-2 substituents confer potent KOPR agonism, while bulkier substituents tend to reduce KOPR affinity and in some cases increase MOPR affinity. At C-18, most modifications tested to date substantially reduce binding affinity, suggesting that this part of the molecule binds tightly to a complementary pocket of the KOPR. Only a few modifications of the furan ring have been studied, but preliminary reports indicate that (a) bromination at C-1620 or saturation20, 21 are well tolerated, (b) replacement of the furan ring by a methyl substituted oxadiazole or a γ-hydroxybutenolide produce weak nonselective antagonists,20 and (c) other modifications lead to loss of affinity.20, 22, 23 In our search for structurally different KOPR antagonists, we were intrigued by reports that some alterations of the furan ring of salvinorin A appeared to confer KOPR antagonist properties. To further investigate the SAR at this position, we first studied the effect of C-16 substitution. In addition, to assess whether C-12 configuration had any effect on binding, we synthesized 12-epi-salvinorin A. We subsequently introduced various linkers between C-12 and the furan ring and prepared related C-13 esters, amides, and ketones. Simultaneously, considering the KOPR antagonist properties reported for methyloxadiazole 28a23 (Scheme 6), we selected it as a lead compound and prepared a set of derivatives containing various 3-substituted oxadiazoles.
Scheme 6.

Syntheses of C-13 oxadiazoles. Reagents and conditions: (a) RC(=NOH)NH2, EDCI, HOBt, CH2Cl2, r.t.; (b) toluene, 110°C, 10–45%, over two steps.
2. Chemistry
We used a modification of the published procedure to prepare 16-bromosalvinorin A (Scheme 1, 2).22 The reported conditions (NBS in CH3CN) formed a complex mixture containing only traces of 2. We found that using CHCl3 as solvent was more effective, although the yields were highly variable (10–62%). The highest yield was obtained when the reaction was carried out at room temperature overnight using an old bottle of NBS. Addition of AIBN did not affect the progression of the reaction, and absence of light led to the formation of an unknown side-product. Stille coupling24 of 2 and tributylvinyltin in the presence of Pd(PPh3)4 provided 3 in 58% yield.
Scheme 1.

Syntheses of 16-substituted salvinorin derivatives. Reagents and conditions: (a) NBS, CHCl3, r.t., 10–62%; (b) tributylvinyltin, Pd(PPh3)4, toluene, 80°C, 58%.
Bikbulatov and coworkers recently reported that refluxing 1 in aqueous KOH gave diacid 4 in 69% yield.25 The structure of 4, featuring inverted configuration at C-12, was firmly established by X-ray crystallography.25 Interestingly, related diacids have been prepared under the same conditions from diterpenoids lacking the C-1 ketone.26 The inverted C-12 configuration of 4 raised the possibility of preparing 12-epi-salvinorin A. We prepared 4 in 95% yield using a slight modification of the published procedure (Scheme 2).25 Standard acetylation then afforded monoacetate 5 in moderate yield. The hemiacetal cleavage conditions reported by Bikbulatov et al (catalytic AcOH or PTSA, CH2Cl2, r.t.)25 were without effect on our substrate. However, refluxing AcOH promoted cleavage and lactonization. Subsequent methylation with TMSCHN2 provided 12-epi-salvinorin A (6). The structure of 6 was established by NMR experiments. The crucial H-10 singlet overlapped with other multiplets in CDCl3 or (CD3)2CO, but a 1:1 mixture of these solvents gave full resolution. The H-8 signal included a diaxial coupling constant (11.7 Hz), establishing an axial orientation. Irradiation of H-8 gave strong nOe enhancements of both H-10 and H-12, establishing a shared β-configuration. Consistent with this, irradiation of H-12 gave no enhancement of H-20. The proposed structure was firmly established by X-ray crystallography (Figure 1). The absolute stereochemistry shown for 6 is taken from that of 1.27 The lactone ring adopts a boat conformation with the furan equatorial. The crystallographic data have been deposited with the Cambridge Crystallographic Data Centre (CCDC 697740).
Scheme 2.

Synthesis of 12-epi-salvinorin A. Reagents and conditions: (a) 5% aqueous KOH, 80°C, 95%; (b) Ac2O, pyridine, r.t., 48%; (c) AcOH, 118°C; (d) TMSCHN2, CH3CN, r.t., 19%, over two steps.
Figure 1.

(stereoview). Crystal structure of 12-epi-salvinorin A (6) showing 50% probability displacement ellipsoids. Hydrogen atoms are shown as spheres of arbitrary radius.
Replacement of the furan with other substituents at C-12 utilized the acid precursor 7,22 prepared by the published procedure (Scheme 3). We found that substituting CH2Cl2 for CCl4, a potent carcinogen, did not affect the yield. Reduction of 7 using BH3·THF21 afforded alcohol 8 in 46% yield. Methyl and ethyl ethers (9 and 10) were prepared from 8 in the presence of silver oxide, and iodomethane or iodoethane, respectively. Alkylation with 2-iodopropane did not proceed under these conditions. Finally, 8 was acetylated to give 11 in 79% yield.
Scheme 3.

Syntheses of C-13 alcohol and ethers. Reagents and conditions: (a) NaIO4, RuCl3.3H2O, CH2Cl2/CH3CN/H2O, 63%; (b) BH3.THF, THF, 55°C, 46%; (c) RI, Ag2O, CH3CN, 60°C, 12–15%; (d) Ac2O, Et3N, CH2Cl2, r.t., 79%.
Acid 7 was converted into the unstable acyl chloride using oxalyl chloride in CH2Cl2 (Scheme 4; thionyl chloride was ineffective under the same conditions). The crude product was directly cross-coupled with various aryltributyl tin reagents under Stille conditions24 to afford C-13 aryl or heteroaryl ketones 12–16 in low to moderate yields. The corresponding ketothiazole derivative decomposed during purification. Subsequently, furan 12 was reduced (NaBH4) to form 17 as a mixture of C-13 epimers in 19% yield. Salvinorins are known to epimerize at C-8 under basic conditions.28 The possibility that epimerization of 17 had occurred at C-8, rather than the expected C-13, was excluded on the basis of NMR data. The H-12 multiplets were coincident; epimerization at C-8 typically causes a large upfield shift of the H-12 multiplet. The H-20 singlets were nearly coincident (1.35 and 1.37 ppm), and closer to the value for 1 (1.45 ppm) than 8-epi-1 (1.62 ppm).21 Irradiation of H-12 caused nOe enhancements of H-20 and H-11α, as expected, rather than H-8. Two distinct H-8 multiplets were apparent, both axial as indicated by diaxial coupling constants (11.0 and 11.4 Hz respectively); the orientation being equatorial in 8-epi-1. Irradiation of H-8 gave nOe enhancements of H-10 and H-11β. Providing further evidence for epimerization at C-13, the broad H-13 resonances were widely separated (4.89 and 4.60 ppm), and on D2O exchange gave doublets with different coupling constants (2.5 and 4.1 Hz). Yields from reduction of other ketones (13–16) under the same conditions were too low to permit pharmacological evaluation. Inspection of alternative synthetic routes to these secondary alcohols is underway.
Scheme 4.

Syntheses of C-13 arylketones and alcohol. Reagents and conditions: (a) (COCl)2, CH2Cl2, r.t.; (b) RSn(nBu)3, Pd(PPh3)4toluene, 80–100°C, 7–57%, over two steps; (c) NaBH4, CH3OH, 0°C, 19%.
Analogues with ester and amide groups connected to the C-13 position were readily obtained from 7 as depicted in Scheme 5. Methyl ester 18 was prepared by treatment of 7 with TMSCHN2. All other esters 19–22 were synthesized in 26–54% yield using the appropriate alcohol, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI), and DMAP. Commercial amines were coupled to 7 in the presence of EDCI and 1-hydroxybenzotriazole (HOBt) to yield amides 23–27.
Scheme 5.

Syntheses of C-13 esters and amides. Reagents and conditions: (a) TMSCHN2, CH3OH, toluene, r.t., 48%; (b) ROH, EDCI, DMAP, CH2Cl2, r.t., 26–54%; (c) RNH2, EDCI, HOBt, CH2Cl2, r.t., 45–72%.
Finally, oxadiazoles 28–34 were obtained in two steps from acid 7 using commercially available amidoximes (Scheme 6). As in previous reports,23 these conditions led to epimerization at C-8: each epimer was purified by HPLC before in vitro evaluation.
3. In vitro binding and functional assays
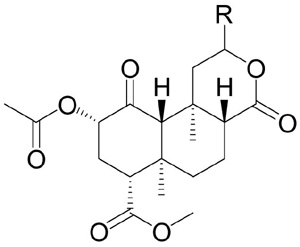
The affinities of compounds 1–3 and 6–34 for opioid receptors were determined by competitive inhibition of [3H]diprenorphine binding to KOPR, MOPR, and delta opioid receptors (DOPR) in membranes prepared from Chinese hamster ovary cells (CHO) stably transfected with the human KOPR (hKOPR), rat MOPR (rMOPR), and mouse DOPR (mDOPR).29 The rMOPR and the mDOPR have very high sequence homology to the respective human orthologs and share similar binding and functional properties. The potencies and efficacies of compounds 1–3 and 6–34 on hKOPR were determined by their abilities to regulate [35S]GTPγS binding to membranes of CHO-hKOPR cells.30 The selective KOPR full agonist, U50,488H, was used as a reference compound, with its efficacy designated as 100%. Receptor binding and functional assay data were analyzed using Prism (GraphPad Software Inc., San Diego, CA). Ki, EC50 (potency) and Emax (efficacy) values were determined using the same software. The in vitro pharmacological data for those derivatives with detectable KOPR binding affinity (Ki < 1,000 nM) (1–3, 6–7, 9–11, 13, 16–20) are listed in Table 1. The dose-response curves of compound 6 and U50,488 in the [35S]GTPγS functional assay are shown in Figure 2.
Table 1.
Affinities (Ki), potencies (EC50), and efficacies (Emax) of C12-substituted salvinorins at the KOPR.
 | ||||
|---|---|---|---|---|
| Compound | R | Ki, nMa,b | EC50, nMb,c | Emaxd |
| 1, SalvA |

|
2.5 ± 0.6 | 2.1 ± 0.6 | 105 ± 4 |
| 2e |

|
2.9 ± 0.3 | 2.4 ± 0.2 | 108 ± 5 |
| 3e |

|
7.1 ± 0.1 | 4.6 ± 0.1 | 120 ± 6 |
| 6 |
|
41 ±5 | 84 ± 8 | 67 ± 5 |
| 7 |
|
55 ± 23 | 167 ± 35 | 99 ± 1 |
| 9 |

|
498 ± 71 | 330 ± 30 | 98 ± 2 |
| 10 |

|
497 ± 13 | > 1,000 | – |
| 11 |

|
555 ± 97 | 299 ± 13 | 113 ± 3 |
| 13 |

|
38 ± 10 | 101 ± 6 | 103 ± 1 |
| 16 |

|
83 ± 28 | 195± 6 | 103 ± 3 |
| 17 |

|
20 ± 2 | 36 ± 5 | 111 ± 4 |
| 18 |

|
154 ± 27 | 361 ± 25 | 99 ± 2 |
| 19 |

|
196 ± 23 | 508 ± 8 | 94 ± 2 |
| 20 |

|
109 ± 12 | 337 ± 54 | 94 ± 2 |
|
| ||||
| U50,488H | 2.2 ± 0.2 | 2.9 ± 0.2 | 100 | |
Ki values in inhibiting [3H]diprenorphine binding to hKOPR.
Each value represents the mean of at least three independent experiments performed in duplicate.
EC50 values in activating the hKOPR to enhance [35S]GTPγS binding.
Efficacy determined as the % of maximal response produced by U50,488 run in parallel experiments.
U50,488H values for these assays: Ki = 1.6 ± 0.6 nM; EC50 = 3.2 ± 0.7 nM.
Figure 2.

12-epi-Salvinorin A (6) exhibits partial agonist properties in the [35S]GTPγS assay. At 10 μM, compound 6 significantly reduced [35S]GTPγS binding induced by U50,488 ranging from 3 × 10−8 to 10−5 M (Student’s t test, *, P < 0.01).
4. Results and discussion
None of the compounds evaluated in this study showed any affinity for the MOPR or DOPR (Ki > 1,000 nM). Compound 6 showed partial agonist effects in the [35S]GTPγS assay. All other compounds exhibiting detectable agonism in the KOPR functional assay were full agonists. As reported by Simpson et al.,20 monobrominated analogue 2 was a highly potent KOPR agonist. Similarly, introduction of a 16-vinyl group (3) did not compromise binding affinity, potency or selectivity (Ki = 7.1 nM, EC50 = 4.6 nM).
Two groups have recently achieved total syntheses of salvinorin A.31, 32 Total synthesis allows the preparation of derivatives that may not be achievable by semi-synthetic modification of salvinorin A. In particular, Nozawa et al.32 suggested that a slight modification of their multi-step synthetic pathway would produce 12-epi-salvinorin A (6). We used a semi-synthetic approach to prepare 6: cleavage of hemiacetal 4 (Scheme 2) and concomitant lactonization being the key transformations. The binding affinity and potency of 6 at the KOPR (Ki = 41 nM, EC50 = 84 nM) were moderately reduced relative to the natural epimer. Surprisingly, 6 was a partial agonist at KOPR in the [35S]GTPγS assay with 67 ± 5% efficacy relative to U50,488 (Figure 2), a difference of statistical significance (p < 0.001). In addition, compound 6 partially blocked the effect of U50,488 in the same assay. The next step will be to evaluate if partial agonism and selectivity are maintained in vivo. Inversion at C-12 has surprisingly little effect on the position of the furan ring: superimposition with the crystal structure of 1 (Figure 3)12 shows that C-13 and the furan oxygen atom are almost coincident in the two structures. That this subtle change in position and orientation should convert a full agonist into a partial one merits further investigation.
Figure 3.

(stereoview). Superimposition of the crystal structures of 1 (dark grey) and 6 (light grey).
Acid 7 is known22 but pharmacological data have not previously been reported. Interestingly, we found that 7 retained some affinity for the KOPR (Ki = 55 nM) and was a weak agonist in the [35S]GTPγS functional assay (EC50 = 167 nM). Reduction of acid 7, giving alcohol 8, resulted in complete loss of KOPR binding affinity. Some affinity was recovered upon O-alkylation: methyl and ethyl ethers 9 and 10 have comparable in vitro pharmacological profiles. Similarly, acetylation of 8 improved KOPR binding affinity: ester 11 is a weak KOPR agonist (Ki = 555 nM, EC50 = 299 nM).
We then studied the effect of inserting a linker between C-12 and the furan (or other aromatic) rings. No clear SAR patterns were observed for the ketoaryl derivatives 12–16. The fact that the ketothiophene (13) and ketopyrazine (16) analogues display moderate KOPR binding affinity (13: Ki = 38 nM, 16: Ki = 83 nM), while the other ketones display none is puzzling. The lack of SAR patterns may be due to instability of these arylketones under the assay conditions. Significantly, secondary alcohols 17 (Ki = 20 nM) were considerably more potent than the corresponding ketone 12 (Ki > 1,000 nM). Since 13 and 16 showed enhanced potencies when compared to 12, reduction of their C-13 ketone might lead to even greater potency. An alternative synthetic route to these alcohols is currently being examined.
Alkyl esters (18–20) demonstrated low KOPR binding affinity (Ki = 109–196 nM). Increasing the size of the ester substituent (21, 22) led to complete loss of KOPR affinity. All secondary amides prepared for this study (23–27) were devoid of KOPR affinity (Ki > 1,000 nM).
Since a previous report20 identified methyloxadiazole 28a as a weak KOPR antagonist, we evaluated it along with a few related oxadiazoles (29–34). Under our binding assay conditions, none of the oxadiazoles displayed any appreciable KOPR binding affinity (Ki > 1,000 nM). By contrast, Simpson et al.20 reported high affinity for 28a (Ki = 56 nM). This may be due to differences in the binding assay protocols, such as the choice of radioligand and buffer composition. Simpson et al. used [125I]IOXY as the radioligand while we used the structurally related [3H]diprenorphine, also a non-selective opioid antagonist (see supplementary information for the structures of diprenorphine and IOXY). Since [125I]IOXY was not commercially available, we ran a second binding assay with 28a and 28b using [3H]U69,593 as the radioligand (see supplementary information). In another control experiment, we substituted our buffer (50 mM Tris-HCl buffer (pH 7.4) containing 1 mM EGTA) with the one reported by Simpson et al. [50 mM Tris-HCl buffer (pH 7.4) containing a protease inhibitor cocktail (bacitracin (100 μg/mL), bestatin (10 μg/mL), leupeptin (4 μg/mL), and chymostatin (2 μg/mL))].20 In each case, 28a and 28b were found to have negligible KOPR binding affinity (see supplementary information). Despite their lack of affinity in our KOPR binding assays, 28a and 28b were also tested in the [35S]GTPγS functional assay. As expected, reference compound U50,488 induced maximum elevation of [35S]GTPγS levels at low concentration. By comparison, 28a and 28b showed KOPR agonist properties only at high concentrations, the natural 8-configuration being preferred (see supplementary information). Finally, 28a failed to inhibit the U50,488H-induced increases in [35S]GTPγS specific binding, demonstrating that this compound did not act as a KOPR antagonist in our functional assay.
5. Conclusions
Our results suggest that it is possible to alter the C-12 substituent of salvinorin A and retain selective KOPR agonism. We developed a four-step synthesis of 12-epi-salvinorin A (6) and found that it induced KOPR partial agonist effects in vitro. Under our conditions, the methyloxadiazole 28a was devoid of antagonist properties as were related oxadiazoles (29–34). Further modifications will be necessary to better understand the SAR at this position.
6. Experimental section
6.1. General methods
Commercial reagents and solvents were used without further purification. Reactions were monitored by thin-layer chromatography (TLC) using either an ethanolic solution of vanillin and H2SO4 or an aqueous solution of ammonium molybdate, cerium sulfate, and H2SO4, and heat as developing agents. Products were purified using automated flash chromatography (50 μm silica gel), manual flash chromatography (230–400 mesh silica gel), or a Waters HPLC system (ELSD detector, Novapak column [6 μm silica, 7.8 × 300 mm]). 1H NMR and 13C NMR chemical shifts are referenced to residual solvent peaks as internal standards: CDCl3 (7.26 and 77 ppm) or CD3OD (3.30 and 49 ppm).
6.2. Method A
A solution of 7 in oxalyl chloride (2.0 M in CH2Cl2) was stirred at room temperature (3 h). The reaction solvent was evaporated and the crude was used immediately without purification for the Stille coupling reaction. The appropriate tributyl tin reagent (1.1 equiv) was added to a mixture of crude acyl chloride and Pd(PPh3)4 (catalytic amount) in anhydrous toluene and the reaction was stirred at 80–100 °C (2–18 h). A saturated aq KF solution and Et2O were added to the reaction mixture. The organic layer was dried (MgSO4) and the residue purified by column chromatography to yield the desired product.
6.3. Method B
To a solution of 7, EDCI (1.2 equiv), and DMAP (catalytic amount) in CH2Cl2 was added the appropriate alcohol (2.0 equiv). The reaction was stirred at room temperature (3 h). The reaction was washed with an aq 1 M HCl solution, brine, dried (MgSO4), and concentrated in vacuo. The residue was purified by column chromatography to obtain the desired product.
6.4. Method C
To a CH2Cl2 solution of 7, EDCI (1.2 equiv), and HOBt (1.2 equiv) was added the appropriate amine (1.5 equiv) and the solution was stirred at room temperature (5–20 min). The reaction was washed with H2O, dried (MgSO4), and concentrated in vacuo. The residue was purified by column chromatography (silica gel; CH2Cl2/CH3OH, hexanes/EtOAc, or CH2Cl2/EtOAc) to obtain the desired product.
6.5. Method D
Using a modification of the reported procedure,23 a mixture of 7, EDCI (1.2 equiv), and HOBt (1.3 equiv) in CH2Cl2 was stirred at room temperature. After 5 minutes, the appropriate oxime was added and the reaction stirred at room temperature. Upon completion, the reaction was washed with a saturated aq NaHCO3 solution and brine. The organic layer was dried (MgSO4) and concentrated to give the crude ester. Toluene was added and the solution was refluxed (17–43 h), concentrated, and the crude residue was purified by normal phase HPLC (hexanes/EtOAc).
6.6. Hemiacetal 4
A solution of 1 (750 mg, 1.7 mmol) in aq KOH (5%, 50 mL) was refluxed for 2 h. The solution was cooled to room temperature and acidified to pH ~ 2 with aq HCl (5 M). The cloudy mixture was then extracted into EtOAc. Drying (MgSO4) and concentration in vacuo gave 425 (650 mg, 95%) as a yellow foam. Rf 0.21 (9:1, CH2Cl2/CH3OH); 1H NMR (300 MHz, CD3OD): δ 7.47 (dt, J = 1.6, 0.8 Hz, 1H), 7.40 (t, J = 1.7 Hz, 1H), 6.44 (dd, J = 1.8, 0.7 Hz, 1H), 5.09 (dd, J = 11.8, 1.6 Hz, 1H), 3.53 (dd, J = 11.9, 5.0 Hz, 1H), 2.23 (dd, J = 13.2, 2.7 Hz, 1H), 2.15 (dd, J = 12.9, 3.0 Hz, 1H), 2.02 – 1.89 (m, 3H), 1.82 (dt, J = 13.3, 3.1 Hz, 1H), 1.74 – 1.55 (m, 3H), 1.53 (s, 3H), 1.43 – 1.30 (m, 1H), 1.34 (s, 1H), 1.23 (s, 3H); 13C NMR (75 MHz, CD3OD): δ 177.3, 176.6, 144.0, 140.6, 128.9, 110.1, 98.3, 75.2, 62.6, 56.8, 55.3, 55.3, 51.2, 41.4, 37.8, 37.1, 30.4, 22.4, 18.9, 16.1.
6.7. Monoacetate 5
A solution of 4 (309 mg, 784 μmol) and Ac2O (90 μL, 942 μmol, 1.2 equiv) in pyridine (6 mL) was stirred at room temperature (42 h). The reaction was concentrated in vacuo and the residue purified by column chromatography (silica gel; 0→10% MeOH/CH2Cl2) to yield 5 (170 mg, 48%) as a white powder: Rf 0.32 (9:1, CH2Cl2/CH3OH); 1H NMR (300 MHz, CD3OD): δ 7.38 (t, J = 1.7 Hz, 1H), 7.32 (dd, J = 0.8, 1.5 Hz, 1H), 6.35 (dd, J = 0.7, 1.8 Hz, 1H), 5.06 (d, J = 10.8 Hz, 1H), 4.80 (dd, J = 4.8, 12.1 Hz, 1H), 2.31 (dd, J = 2.7, 13.2 Hz, 1H), 2.20–1.99 (m, 6H), 1.94 (dd, J = 2.0, 13.3 Hz, 1H), 1.90–1.82 (m, 1H), 1.73 (ddd, J = 2.7, 4.6, 12.8 Hz, 1H), 1.67–1.57 (m, 2H), 1.55 (s, 3H), 1.47–1.30 (m, 2H), 1.25 (s, 3H); 13C NMR (75 MHz, CD3OD): δ 177.2, 176.2, 172.2, 144.0, 140.1, 129.1, 109.8, 97.7, 77.4, 63.0, 56.7, 55.6, 55.2, 51.4, 41.4, 37.9, 37.1, 28.3, 22.4, 21.1, 18.8, 16.2.
6.8. 12-epi-Salvinorin A (6)
A solution of 5 (102 mg, 234 μmol) in AcOH (2.3 mL) was refluxed (18 h). The reaction was concentrated in vacuo and the residue was diluted with i-PrOH and concentrated to remove excess AcOH. The residue was then dissolved in MeCN (2 mL) and TMSCHN2 (2.0 M in hexane, 6 equiv, 700 μL, 1.4 mmol) added. The solution was stirred at room temperature (25 min), concentrated in vacuo, and the residue purified by column chromatography (silica gel; 0→50% EtOAc/hexanes). Slow evaporation from EtOAc/hexanes yielded 6 (11 mg, 11%) as colorless needles, mp. 212–217 °C (dec); Rf 0.27 (1:1, hexanes/EtOAc); 1H NMR (300 MHz, CDCl3): δ 7.45–7.41 (m, 1H), 7.39 (t, J = 1.7 Hz, 1H), 6.41 (dd, J = 0.7, 1.8 Hz, 1H), 5.30 (dd, J = 6.1, 11.6 Hz, 1H), 5.22–5.11 (m, 1H), 3.73 (s, 3H), 2.78 (dd, J = 7.6, 9.2 Hz, 1H), 2.49–2.36 (m, 2H), 2.36–2.30 (m, 2H), 2.28 (dd, J = 4.7, 7.0 Hz, 1H), 2.17 (s, 3H), 2.07–1.72 (m, 4H), 1.64 (dd, J = 4.9, 14.0 Hz, 1H), 1.38 (s, 3H), 1.07 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.8, 173.1, 171.5, 170.0, 143.7, 139.8, 123.8, 108.7, 75.0, 70.3, 65.8, 53.4, 52.0, 47.3, 44.7, 42.3, 37.7, 35.2, 30.6, 21.2, 20.6, 18.2, 16.1; HRMS–ESI (m/z): [M + H]+ calcd for C23H28O8, 433.1862; found, 433.1847.
6.9. Alcohol 8
To a THF (3 mL) solution of 7 (157 mg, 382 μmol) was added BH3·THF (1.0 M in THF, 0.5 mL, 0.5 mmol) dropwise, and the reaction was stirred at 55 °C. After 1 h, the reaction was cooled to room temperature, water (2 mL) was added dropwise and the solution was evaporated. The residue was taken up in a saturated aq NaHCO3 solution and extracted with CH2Cl2. The organic layer was dried (MgSO4) and concentrated in vacuo. The residue was purified by column chromatography (silica gel; 19:1, CH2Cl2/MeOH) to obtain 8 (70 mg, 46%) as a white powder: Rf 0.27 (19:1, CH2Cl2/MeOH); 1H NMR (300 MHz, CDCl3): δ 5.19–5.05 (m, 1H), 4.67–4.49 (m, 1H), 3.84 (dd, J = 2.7, 12.4 Hz, 1H), 3.72 (s, 3H), 3.52 (dd, J = 4.2, 12.4 Hz, 1H), 2.80–2.66 (m, 1H), 2.35–2.23 (m, 2H), 2.21–2.08 (m, 6H), 2.00 (dd, J = 2.7, 11.5 Hz, 1H), 1.81–1.73 (m, 1H), 1.68–1.46 (m, 3H), 1.37 (s, 3H), 1.08 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 202.1, 171.6, 171.6, 170.0, 77.2, 75.1, 64.7, 64.0, 53.5, 52.0, 50.9, 42.1, 38.1, 37.4, 34.8, 30.7, 20.6, 18.1, 16.3, 15.2; HRMS–ESI (m/z): [M + H]+ calcd for C20H28O8, 397.1862; found, 397.1859.
6.10. Methyl ether 9
To a CH3CN solution of 8 (20 mg, 52 μmol) was added Ag2O (150 mg, 647 μmol) and iodomethane (70 μL, 1.1 mmol). The reaction was stirred at 60 °C (5 d). The reaction was concentrated and the residue purified by column chromatography (silica gel; 0→5% MeOH/CH2Cl2) followed by a second column (silica gel; 0→25% EtOAc/CH2Cl2, then 5% MeOH/CH2Cl2) to yield 9 (2.5 mg, 15%) as an orange resin: Rf 0.50 (4:1, CH2Cl2/EtOAc); 1H NMR (300 MHz, CDCl3): δ 5.13 (t, J = 9.9 Hz, 1H), 4.68–4.53 (m, 1H), 3.79–3.53 (m, 4H), 3.49–3.29 (m, 4H), 2.81–2.67 (m, 1H), 2.39–2.24 (m, 2H), 2.24–2.06 (m, 6H), 2.05–1.96 (m, 1H), 1.84–1.70 (m, 1H), 1.69–1.47 (m, 3H), 1.35 (s, 3H), 1.08 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 202.1, 171.6, 171.5, 170.0, 76.0, 75.1, 74.0, 64.2, 59.3, 53.5, 52.0, 50.6, 42.1, 38.1, 37.8, 34.9, 30.7, 20.6, 18.1, 16.2, 15.1; HRMS–ESI (m/z): [M + H]+ calcd for C21H30O8, 411.2019, found, 411.2036.
6.11. Ethyl ether 10
To a CH3CN solution of 8 (16 mg, 40 μmol) was added Ag2O (122 mg, 526 μmol) and iodoethane (94 μL, 1.2 mmol). The reaction was stirred at 60 °C (4 d). Additional Ag2O (136 mg, 586 μmol) and iodoethane (94 μL, 1.2 mmol) were added. The reaction was stirred at room temperature (8 d) and then concentrated. The residue purified by column chromatography (silica gel; 0→5% MeOH/CH2Cl2) followed by a second column (silica gel; 0→20% EtOAc/CH2Cl2) to yield 10 (2.0 mg, 12%) as a clear resin: Rf 0.50 (19:1, CH2Cl2/MeOH); 1H NMR (300 MHz, CDCl3): δ 5.18–5.07 (m, 1H), 4.60 (d, J = 5.8 Hz, 1H), 3.71 (d, J = 5.1 Hz, 3H), 3.64–3.37 (m, 4H), 2.82–2.67 (m, 1H), 2.31 (d, J = 10.2 Hz, 2H), 2.23–2.07 (m, 6H), 2.01 (d, J = 8.4 Hz, 1H), 1.76 (dd, J = 3.2, 9.8 Hz, 1H), 1.55 (d, J = 14.6 Hz, 3H), 1.42–1.03 (m, 9H); 13C NMR (75 MHz, CDCl3): δ 202.1, 171.6, 171.6, 170.0, 76.2, 75.1, 72.0, 67.1, 64.2, 53.5, 52.0, 50.7, 42.1, 38.1, 38.0, 34.9, 30.7, 20.6, 18.2, 16.2, 15.1, the OCH2CH3 signal was not detected; HRMS–ESI (m/z): [M + H]+ calcd for C22H32O8, 425.2175, found, 425.2168.
6.12. Acetate 11
To a CH2Cl2 solution of 8 (19 mg, 48 μmol) was added Ac2O (5.4 μL, 57 μmol) and Et3N (8 μL, 57 μmol). The reaction was stirred at room temperature (25 h). DMAP (catalytic amount) was added and the reaction stirred for an additional 4 h. The reaction was concentrated and the residue purified by column chromatography (silica gel; 0→4% MeOH/CH2Cl2) to obtain 11 (17 mg, 79%) as a white foam: Rf 0.35 (19:1, CH2Cl2/MeOH); 1H NMR (300 MHz, CDCl3): δ 5.22–5.08 (m, 1H), 4.80–4.65 (m, 1H), 4.22 (dd, J = 3.2, 12.1 Hz, 1H), 4.03 (dd, J = 5.2, 12.1 Hz, 1H), 3.72 (s, 3H), 2.83–2.66 (m, 1H), 2.36–2.20 (m, 3H), 2.21–2.12 (m, 5H), 2.09 (s, 3H), 1.97 (dd, J = 2.7, 11.7 Hz, 1H), 1.79 (dd, J = 2.8, 9.8 Hz, 1H), 1.68–1.53 (m, 3H), 1.37 (s, 3H), 1.09 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.9, 171.5, 170.8, 170.6, 170.0, 75.0, 74.5, 65.9, 64.1, 53.5, 52.0, 51.1, 42.0, 38.2, 38.0, 34.9, 30.7, 20.8, 20.6, 18.1, 16.3, 15.0; HRMS–ESI (m/z): [M + H]+ calcd for C22H30O9, 439.1968; found, 439.1956.
6.13. Furanyl ketone 12
Compound 12 (7.3 mg, 33%) was prepared as a white powder from 7 (20 mg, 49 μmol), oxalyl chloride (1 mL, 2 mmol), Pd(PPh3)4 (catalytic amount), and 2-(tributylstannyl)furan (17 μL, 54 μmol) utilizing method A, stirring the reaction at 80 °C (18 h), and using column chromatography (silica gel; 0→50% EtOAc/hexanes): Rf 0.31 (1:1, hexanes/EtOAc); 1H NMR (300 MHz, CDCl3): δ 7.74–7.61 (m, 1H), 7.37 (dd, J = 0.6, 3.6 Hz, 1H), 6.59 (dd, J = 1.7, 3.6 Hz, 1H), 5.62 (t, J = 8.3 Hz, 1H), 5.12 (t, J = 10.0 Hz, 1H), 3.71 (s, 3H), 2.78–2.69 (m, 1H), 2.63 (dd, J = 8.0, 13.6 Hz, 1H), 2.38–2.22 (m, 2H), 2.22–2.05 (m, 6H), 1.82–1.50 (m, 4H), 1.44 (s, 3H), 1.08 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.9, 184.0, 171.5, 170.7, 169.8, 149.9, 147.8, 120.3, 112.9, 76.0, 74.9, 64.6, 53.2, 51.9, 49.6, 42.0, 38.0, 37.7, 35.5, 30.7, 20.6, 18.2, 16.4, 16.0; HRMS–ESI (m/z): [M + NH4]+ calcd for C24H28O9, 478.2077; found, 478.2084.
6.14. Thienyl ketone 13
Compound 13 (8.8 mg, 36%) was prepared as a white powder from 7 (21 mg, 51 μmol), oxalyl chloride (1 mL, 2 mmol), Pd(PPh3)4 (catalytic amount), and 2-(tributylstannyl)thiophene (18 μL, 57 μmol) utilizing method A, stirring the reaction at 100 °C (2 h), and using column chromatography (silica gel; 0→33% acetone/hexanes) followed by a second column (silica gel; 0→10% EtOAc/CH2Cl2): Rf 0.21 (2:1, hexanes/acetone); 1H NMR (300 MHz, CDCl3): δ 7.82 (dd, J = 1.1, 3.9 Hz, 1H), 7.75 (dd, J = 1.1, 5.0 Hz, 1H), 7.18 (dd, J = 3.9, 5.0 Hz, 1H), 5.62 (t, J = 8.2 Hz, 1H), 5.21–5.04 (m, 1H), 3.71 (s, 3H), 2.78–2.68 (m, 1H), 2.62 (dd, J = 8.1, 13.7 Hz, 1H), 2.34–2.23 (m, 2H), 2.23–2.06 (m, 6H), 1.81–1.53 (m, 4H), 1.44 (s, 3H), 1.08 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 202.1, 188.4, 171.7, 170.8, 170.0, 140.1, 136.0, 134.2, 128.9, 76.9, 75.0, 64.7, 53.4, 52.1, 49.7, 42.1, 38.6, 37.8, 35.6, 30.8, 20.7, 18.3, 16.8, 16.2; HRMS–ESI (m/z): [M + NH4]+ calcd for C24H28O8S, 494.1849; found, 494.1831.
6.15. Oxazolyl ketone 14
Compound 14 (3.1 mg, 6.5%) was prepared as a clear resin from 7 (21 mg, 52 μmol), oxalyl chloride (1 mL, 2 mmol), Pd(PPh3)4 (catalytic amount), and 2-(tributylstannyl)oxazole (12 μL, 57 μmol) utilizing method A, stirring the reaction at 100 °C (2 h), and using column chromatography (silica gel; 0→40% acetone/hexanes) followed by a second column (silica gel; 0→50% EtOAc/hexanes): Rf 0.21 (2:1, hexanes/acetone): Rf 0.09 (1:1, hexanes/EtOAc); 1H NMR (300 MHz, CDCl3): δ 7.89 (d, J = 0.6 Hz, 1H), 7.39 (d, J = 0.6 Hz, 1H), 5.93 (dd, J = 7.7, 10.0 Hz, 1H), 5.20–4.99 (m, 1H), 3.72 (s, 3H), 2.82–2.67 (m, 2H), 2.34–2.19 (m, 3H), 2.18–2.12 (m, 5H), 1.83–1.51 (m, 4H), 1.47 (s, 3H), 1.09 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.7, 183.2, 171.7, 170.4, 170.1, 142.8, 129.9, 77.4, 76.8, 75.0, 64.6, 53.5, 52.2, 50.0, 42.2, 38.3, 38.0, 35.8, 30.8, 20.8, 18.3, 16.2, 16.0; HRMS–ESI (m/z): [M + NH4]+ calcd for C23H27NO9, 479.203; found, 479.2019.
6.16. Phenyl ketone 15
Compound 15 (20 2 mg, 59% yield) was prepared as a white powder from 7 (30 mg, 73 μmol), oxalyl chloride (1 mL, 2 mmol), Pd(PPh3)4 (catalytic amount), and 2-(tributylstannyl)phenyl (26 μL, 80 μmol) utilizing method A, stirring the reaction at 100 °C (2 h), and using column chromatography (silica gel; 0→33% acetone/hexanes): Rf 0.29 (2:1, hexanes/acetone); 1H NMR (300 MHz, CDCl3): δ 7.90 (dd, J = 1.3, 8.4 Hz, 2H), 7.68–7.57 (m, 1H), 7.55–7.43 (m, 2H), 5.87 (t, J = 8.3 Hz, 1H), 5.15–5.03 (m, 1H), 3.70 (s, 3H), 2.76–2.59 (m, 2H), 2.34–2.22 (m, 2H), 2.21–2.06 (m, 6H), 1.82–1.52 (m, 4H), 1.44 (s, 3H), 1.06 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 202.0, 195.2, 171.6, 171.0, 169.8, 134.3, 133.3, 129.1, 128.9, 75.2, 74.8, 64.6, 53.1, 51.9, 49.2, 41.9, 38.2, 37.6, 35.5, 30.6, 20.6, 18.2, 16.7, 16.0; HRMS–ESI (m/z): [M + NH4]+ calcd for C26H30O8, 488.2284; found, 488.2297.
6.17. Pyrazi nyl ketone 16
Compound 16 (19.9 mg, 57% yield) was prepared as an off-white powder from 7 (30 mg, 73 μmol), oxalyl chloride (1 mL, 2 mmol), Pd(PPh3)4 (catalytic amount), and 2-(tributylstannyl)pyrazine (26 μL, 81 μmol) utilizing method A, stirring the reaction at 100 °C (2 h), and using column chromatography (silica gel; 0→40% acetone/hexanes): Rf 0.08 (2:1, hexanes/acetone); 1H NMR (300 MHz, CDCl3): δ 9.24 (d, J = 1.4 Hz, 1H), 8.80 (d, J = 2.4 Hz, 1H), 8.72–8.59 (m, 1H), 6.28 (dd, J = 8.3, 9.2 Hz, 1H), 5.16–4.97 (m, 1H), 3.70 (s, 3H), 2.81 (dd, J = 8.1, 13.3 Hz, 1H), 2.75–2.66 (m, 1H), 2.32–2.21 (m, 2H), 2.21–2.09 (m, 6H), 1.81–1.53 (m, 3H), 1.49–1.35 (m, 4H), 1.07 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 202.0, 196.1, 171.8, 171.1, 170.1, 149.0, 145.3, 144.9, 144.1, 75.9, 75.0, 64.8, 53.5, 52.3, 49.9, 42.2, 38.5, 38.0, 35.9, 30.9, 20.9, 18.5, 16.3, 16.2; HRMS–ESI (m/z): [M + NH4]+ calcd for C24H28N2O8, 490.2189; found, 490.2209.
6.18. Furanyl alcohols 17
To a MeOH solution of 12 (82 mg, 178 μmol) was added NaBH4 (5.8 mg, 153 μmol) and the reaction was stirred at 0 °C (1.5 h), concentrated and the residue purified by column chromatography (silica gel; 0→4% MeOH/CH2Cl2) followed by a second column (silica gel, 0→40% EtOAc/CH2Cl2) and a third column (silica gel, 0→50% EtOAc/hexanes) to yield 17 (14 mg, 19% BOSM) as a clear resin and as a ~1:1 mixture of 13-epimers: Rf 0.16 (4:1, CH2Cl2/EtOAc); 1H NMR (300 MHz, CDCl3): δ 7.43–7.35 (m, 2H), 6.44–6.27 (m, 4H), 5.13 (dd, J = 8.6, 11.3 Hz, 2H), 4.94–4.76 (m, 3H), 4.61 (br s, 1H), 3.71 (d, J = 2.6 Hz, 6H), 2.80–2.64 (m, 2H), 2.38–2.22 (m, 4H), 2.21–2.04 (m, 12H), 1.99 (dd, J = 3.1, 10.7 Hz, 1H), 1.89 (dd, J = 3.0, 11.2 Hz, 1H), 1.81–1.69 (m, 2H), 1.66–1.47 (m, 6H), 1.43–1.23 (m, 8H), 1.07 (d, J = 3.9 Hz, 6H); 13C NMR (75 MHz, CDCl3): δ 201.9, 201.9, 171.6, 171.5, 171.4, 171.0, 169.9, 169.8, 151.8, 151.4, 142.6, 142.6, 110.5, 110.5, 108.3, 108.2, 78.7, 78.2, 77.2, 74.9, 74.9, 70.2, 69.7, 64.1, 53.5, 53.4, 52.0, 51.9, 50.9, 50.6, 42.0, 42.0, 38.3, 38.3, 38.0, 38.0, 34.9, 34.7, 30.8, 30.8, 20.6, 20.6, 18.1, 16.2, 16.2, 15.2, 15.1, one signal was not detected; HRMS–ESI (m/z): [M + H]+ calcd for C24H30O9, 463.1968; found, 463.1971.
6.19. Methyl ester 18
To a toluene (3 mL)/MeOH (2 mL) solution of 7 (21 mg, 51 μmol) was added TMSCHN2 (36 μL, 72 μmol) dropwise. The solution was stirred at room temperature (45 min), concentrated, and the residue purified by column chromatography (silica gel; 1:1, EtOAc/hexanes) to obtain 18 (10 mg, 48%) as a white powder: Rf 0.31 (1:1, hexanes/EtOAc); 1H NMR (300 MHz, CDCl3): δ 5.19–5.08 (m, 1H), 4.98 (dd, J = 7.3, 9.7 Hz, 1H), 3.77 (s, 3H), 3.72 (s, 3H), 2.78–2.69 (m, 1H), 2.61 (dd, J = 7.3, 13.6 Hz, 1H), 2.35–2.24 (m, 2H), 2.17 (s, 3H), 2.16–2.06 (m, 3H), 1.81–1.73 (m, 1H), 1.71–1.48 (m, 3H), 1.36 (s, 3H), 1.07 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.7, 171.5, 170.5, 170.0, 169.9, 74.9, 73.6, 64.3, 53.4, 52.9, 52.0, 50.0, 42.0, 38.9, 37.8, 35.2, 30.6, 20.6, 18.1, 16.1, 15.8; HRMS–ESI (m/z): [M + NH4]+ calcd for C21H28O9, 442.2077; found, 442.2088.
6.20. Ethyl ester 19
Compound 19 (5.1 mg, 49%) was prepared as a white powder from 7 (26 mg, 63 μmol), EDCI (14 mg, 73 μmol), DMAP (catalytic amount), and EtOH (7.4 μL, 127 μmol) utilizing method B and using column chromatography (silica gel; 1:1, EtOAc/hexanes): Rf 0.63 (1:1, hexanes/EtOAc); 1H NMR (300 MHz, CDCl3): δ 5.21–5.07 (m, 1H), 4.95 (dd, J = 7.2, 9.7 Hz, 1H), 4.33–4.14 (m, 2H), 3.72 (s, 3H), 2.79–2.68 (m, 1H), 2.60 (dd, J = 7.2, 13.5 Hz, 1H), 2.38–2.23 (m, 2H), 2.18 (s, 3H), 2.16–2.06 (m, 3H), 1.79–1.74 (m, 1H), 1.72–1.47 (m, 3H), 1.36 (s, 3H), 1.29 (t, J = 7.1 Hz, 3H), 1.08 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.7, 171.5, 170.1, 170.1, 169.9, 74.9, 73.8, 64.4, 62.1, 53.4, 52.0, 50.0, 42.0, 38.9, 37.8, 35.2, 30.7, 20.6, 18.1, 16.1, 15.8, 14.1; HRMS–ESI (m/z): [M + NH4]+ calcd for C22H30O9, 456.2234; found, 456.2246.
6.21. Isopropyl ester 20
Compound 20 (19 mg, 38%) was prepared as a clear resin from 7 (25 mg, 60 μmol), EDCI (15 mg, 78 μmol), DMAP (catalytic amount), and i-PrOH (9 μL, 118 μmol) utilizing method B and using column chromatography (silica gel; 1:2, EtOAc/hexanes): Rf 0.45 (1:1, hexanes/EtOAc); 1H NMR (300 MHz, CDCl3): δ 5.20–5.11 (m, 1H), 5.11–4.99 (m, 1H), 4.91 (dd, J = 7.2, 9.8 Hz, 1H), 3.72 (s, 3H), 2.78–2.69 (m, 1H), 2.57 (dd, J = 7.1, 13.5 Hz, 1H), 2.34–2.29 (m, 2H), 2.17 (s, 3H), 2.17–2.06 (m, 3H), 1.80–1.71 (m, 1H), 1.71–1.46 (m, 3H), 1.36 (s, 3H), 1.27 (d, J = 6.7 Hz, 3H), 1.26 (d, J = 6.7 Hz, 3H), 1.08 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.8, 171.5, 170.2, 169.9, 169.6, 74.8, 74.0, 70.0, 64.3, 53.3, 52.0, 50.1, 42.0, 38.9, 37.8, 35.2, 30.7, 21.6, 21.6, 20.6, 18.1, 16.1, 15.8; HRMS–ESI (m/z): [M + NH4]+ calcd for C23H32O9, 470.2390; found, 470.2404.
6.22. Benzyl ester 21
Compound 21 (14 mg, 54%) was prepared as a clear resin from 7 (21 mg, 50 μmol), EDCI (12 mg, 63 μmol), DMAP (catalytic amount) and benzyl alcohol (11 μL, 101 μmol) utilizing method B and using column chromatography (silica gel; 1:2, EtOAc/hexanes): Rf 0.29 (1:1, hexanes/EtOAc); 1H NMR (300 MHz, CDCl3): δ 7.44–7.33 (m, 5H), 5.20 (dd, J = 12.0, 27.9 Hz, 2H), 5.14–5.05 (m, 1H), 4.99 (dd, J = 7.6, 8.7 Hz, 1H), 3.72 (s, 3H), 2.71–2.63 (m, 1H), 2.59 (dd, J = 7.6, 13.7 Hz, 1H), 2.32–2.22 (m, 2H), 2.17 (s, 3H), 2.11–1.97 (m, 3H), 1.78–1.68 (m, 1H), 1.66–1.43 (m, 3H), 1.34 (s, 3H), 1.05 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.7, 171.5, 170.2, 170.0, 169.9, 134.8, 128.8, 128.8, 128.7, 74.8, 73.6, 67.6, 64.5, 53.3, 52.0, 49.7, 41.9, 38.9, 37.7, 35.2, 30.6, 20.6, 18.1, 16.3, 16.1; HRMS–ESI (m/z): [M + NH4]+ calcd for C27H32O9, 518.2390; found, 518.2407.
6.23. Furfuryl ester 22
Compound 22 (7.8 mg, 26%) was prepared as a white powder from 7 (25 mg, 61 μmol), EDCI (14 mg, 75 μmol), DMAP (catalytic amount), and furfuryl alcohol (11 μL, 121 μmol) utilizing method B and using column chromatography (silica gel; 1:2, EtOAc/hexanes): Rf 0.37 (1:1, hexanes/EtOAc); 1H NMR (300 MHz, CDCl3): δ 7.44 (dd, J = 0.8, 1.9 Hz, 1H), 6.45 (dd, J = 0.6, 3.3 Hz, 1H), 6.38 (dd, J = 1.9, 3.3 Hz, 1H), 5.24–5.06 (m, 3H), 4.98 (dd, J = 7.4, 9.2 Hz, 1H), 3.72 (s, 3H), 2.76–2.67 (m, 1H), 2.59 (dd, J = 7.4, 13.7 Hz, 1H), 2.34–2.23 (m, 2H), 2.17 (s, 3H), 2.14–2.04 (m, 3H), 1.79–1.71 (m, 1H), 1.70–1.48 (m, 3H), 1.34 (s, 3H), 1.06 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.7, 171.5, 170.1, 169.9, 169.8, 148.3, 143.7, 111.6, 110.7, 74.8, 73.6, 64.4, 59.2, 53.4, 52.0, 49.9, 41.9, 38.9, 37.8, 35.2, 30.7, 20.6, 18.1, 16.1, 16.1; HRMS–ESI (m/z): [M + NH4]+ calcd for C25H30O10, 508.2183; found, 508.2161.
6.24. Methyl amide 23
Compound 23 (15 mg, 70%) was prepared as a clear resin from 7 (21 mg, 50 μmol), EDCI (12 mg, 61 μmol), HOBt (7.9 mg, 59 μmol), and CH3NH2 (2.0 M in CH2Cl2, 36 μL, 72 μmol) utilizing method C, stirring at room temperature (5 min), and using column chromatography (silica gel; 19:1, CH2Cl2/MeOH): Rf 0.08 (19:1, CH2Cl2/CH3OH); 1H NMR (300 MHz, CDCl3): δ 6.43 (br d, J = 4.6 Hz, 1H), 5.15 (dd, J = 8.4, 11.5 Hz, 1H), 4.89 (dd, J = 6.3, 10.6 Hz, 1H), 3.71 (s, 3H), 2.83 (d, J = 4.9 Hz, 3H), 2.77–2.65 (m, 2H), 2.35–2.21 (m, 2H), 2.16 (s, 3H), 2.04–2.16 (m, 2H), 2.00 (dd, J = 2.8, 11.3 Hz, 1H), 1.65 (ddd, J = 10.7, 24.2, 24.9 Hz, 4H), 1.37 (s, 3H), 1.08 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.4, 171.5, 170.3, 169.8, 169.6, 75.8, 74.7, 64.0, 53.4, 52.0, 51.0, 41.9, 39.1, 37.8, 35.3, 30.8, 26.0, 20.6, 18.1, 16.3, 15.5; HRMS–ESI (m/z): [M + NH4]+ calcd for C21H29NO8, 441.2237; found, 441.2245.
6.25. Ethyl amide 24
Compound 24 (11 mg, 51%) was prepared as a black resin from 7 (21 mg, 51. μmol), EDCI (12 mg, 61 μmol), HOBt (8.1 mg, 60 μmol), and EtNH2 (2.0 M in THF, 64 μL, 0.13 mmol) utilizing method C, stirring at room temperature (20 min), and using column chromatography (silica gel; 19:1, CH2Cl2/MeOH): Rf 0.26 (19:1, CH2Cl2/CH3OH); 1H NMR (300 MHz, CDCl3): δ 6.38 (br s, 1H), 5.15 (dd, J = 8.6, 11.7 Hz, 1H), 4.88 (dd, J = 6.2, 10.8 Hz, 1H), 3.71 (s, 3H), 3.40–3.20 (m, 2H), 2.78–2.65 (m, 2H), 2.36–2.21 (m, 2H), 2.19–2.05 (m, 5H), 2.01 (dd, J = 2.9, 11.5 Hz, 1H), 1.83–1.73 (m, 1H), 1.72–1.49 (m, 3H), 1.38 (s, 3H), 1.14 (t, J = 7.3 Hz, 3H), 1.09 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.4, 171.5, 170.4, 169.6, 169.0, 75.8, 74.7, 64.0, 53.4, 52.0, 51.0, 41.9, 39.1, 37.8, 35.3, 34.2, 30.8, 20.6, 18.1, 16.3, 15.5, 14.7; HRMS–ESI (m/z): [M + NH4]+ calcd for C22H31NO8, 455.2393; found, 455.2380.
6.26. Isopropyl amide 25
Compound 25 (16 mg, 69%) was prepared as a white powder from 7 (21 mg, 51 μmol), EDCI (12 mg, 63 μmol), HOBt (8.0 mg, 59 μmol), and i-PrNH2 (6.4 μL, 75 μmol) utilizing method C, stirring at room temperature (20 min), and using column chromatography (silica gel; 1:1, EtOAc/hexanes): Rf 0.11 (1:1, hexanes/EtOAc); 1H NMR (300 MHz, CDCl3): δ 6.20 (br d, J = 8.0 Hz, 1H), 5.15 (dd, J = 8.6, 11.6 Hz, 1H), 4.85 (dd, J = 6.0, 10.9 Hz, 1H), 4.17–3.99 (m, 1H), 3.71 (s, 3H), 2.80–2.61 (m, 2H), 2.37–2.20 (m, 2H), 2.19–2.06 (m, 5H), 2.02 (dd, J = 2.3, 11.4 Hz, 1H), 1.83–1.46 (m, 4H), 1.37 (s, 3H), 1.20–1.11 (m, 6H), 1.09 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.4, 171.5, 170.4, 169.6, 168.2, 75.9, 74.7, 63.9, 53.4, 52.0, 51.0, 41.9, 41.4, 39.2, 37.9, 35.3, 30.8, 22.6, 22.5, 20.6, 18.1, 16.3, 15.4; HRMS–ESI (m/z): [M + NH4]+ calcd for C23H33NO8, 469.2544; found, 469.2646.
6.27. Benzyl amide 26
Compound 26 (17 mg, 72%) was prepared as a white powder from 7 (20 mg, 49 μmol), EDCI (12 mg, 62 μmol), HOBt (8.0 mg, 59 μmol), and benzylamine (8.0 μL, 73 μmol) utilizing method C, stirring at room temperature (5 min), and using column chromatography (silica gel; 1:1, EtOAc/hexanes): Rf 0.22 (1:1, hexanes/EtOAc); 1H NMR (300 MHz, CDCl3): δ 7.42–7.19 (m, 5H), 6.76 (br t, J = 5.6 Hz, 1H), 5.16 (dd, J = 8.4, 11.6 Hz, 1H), 4.91 (dd, J = 6.1, 10.8 Hz, 1H), 4.43 (d, J = 5.9 Hz, 2H), 3.71 (s, 3H), 2.72 (dd, J = 5.5, 12.8 Hz, 2H), 2.34–2.21 (m, 2H), 2.18–2.02 (m, 5H), 1.99 (dd, J = 2.9, 11.2 Hz, 1H), 1.80–1.70 (m, 1H), 1.68–1.49 (m, 3H), 1.36 (s, 3H), 1.08 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.4, 171.5, 170.3, 169.6, 169.1, 137.4, 128.8, 127.9, 127.8, 75.9, 74.7, 63.9, 53.4, 51.9, 51.0, 43.3, 41.8, 39.2, 37.8, 35.3, 30.8, 20.6, 18.0, 16.3, 15.5; HRMS–ESI (m/z): [M + NH4]+ calcd for C27H33NO8, 517.2550; found, 517.2567.
6.28. Furfuryl amide 27
Compound 27 (11. mg, 45%) was prepared as a white powder from 7 (21 mg, 51 μmol), EDCI (12 mg, 62 μmol), HOBt (7.9 mg, 59 μmol), and furfurylamine (7.0 μL, 76 μmol) utilizing method C, stirring at room temperature (20 min), and using column chromatography (silica gel; 19:1, CH2Cl2/MeOH) followed by a second column (silica gel; 2:1, CH2Cl2/EtOAc): Rf 0.26 (2:1, CH2Cl2/EtOAc); 1H NMR (300 MHz, CDCl3): δ 7.35 (dd, J = 0.8, 1.8 Hz, 1H), 6.71 (br t, J = 5.6 Hz, 1H), 6.32 (dd, J = 1.9, 3.2 Hz, 1H), 6.23 (d, J = 3.2 Hz, 1H), 5.15 (dd, J = 8.5, 11.6 Hz, 1H), 4.91 (dd, J = 6.2, 10.8 Hz, 1H), 4.51–4.36 (m, 2H), 3.71 (s, 3H), 2.78–2.65 (m, 2H), 2.35–2.21 (m, 2H), 2.20–1.96 (m, 6H), 1.82–1.72 (m, 1H), 1.70–1.50 (m, 3H), 1.37 (s, 3H), 1.08 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.4, 171.5, 170.2, 169.6, 169.0, 150.2, 142.5, 110.5, 108.0, 75.8, 74.7, 63.9, 53.4, 52.0, 51.0, 41.8, 39.1, 37.8, 36.2, 35.3, 30.8, 20.6, 18.0, 16.3, 15.5; HRMS–ESI (m/z): [M + NH4]+ calcd for C25H31NO9, 507.2343; found, 507.2350.
6.29. Methyl oxadiazoles 28a and 28b
Compound 28a (3.6 mg, 8%) was prepared as a clear resin from 7 (43 mg, 105 μmol), EDCI (25 mg, 130 μmol), HOBt (19 mg, 142 μmol), and acetamide oxime (12 mg, 157 μmol) utilizing method D, stirring at room temperature (23 h), and using HPLC purification (silica gel, 20→55% EtOAc/hexanes): Rf 0.32 (1:1, hexanes/EtOAc); HRMS–ESI (m/z): [M + H]+ calcd for C22H28N2O8, 449.1924; found, 449.1907. Compound 28b (5.9 mg, 13%) was also isolated as a clear resin: Rf 0.23 (1:1, hexanes/EtOAc); 1H NMR(300 MHz, CDCl3): δ 5.49 (dd, J = 2.1, 12.1 Hz, 1H), 5.16–5.05 (m, 1H), 3.71 (s, 3H), 2.75 (dd, J = 6.5, 10.3 Hz, 1H), 2.53 (dd, J = 2.4, 15.1 Hz, 1H), 2.47–2.43 (m, 1H), 2.41 (s, 3H), 2.33–2.18 (m, 4H), 2.15 (s, 3H), 1.97 (dd, J = 2.7, 13.3 Hz, 1H), 1.93–1.84 (m, 1H), 1.78 (dd, J = 12.2, 15.1 Hz, 1H), 1.66 (s, 3H), 1.62–1.51 (m, 1H), 1.09 (s, 3H); HRMS–ESI (m/z): [M + H]+ calcd for C22H28N2O8, 449.1924; found, 449.1920.
6.30. Ethyl oxadiazole 29a
Compound 29a (4.9 mg, 10%) was prepared as a white resin from 7 (43 mg, 104 μmol), EDCI (25 mg, 131 μmol), HOBt (20 mg, 144 μmol), and N-hydroxypropionamidine (12 μL, 146 μmol) in CH2Cl2 (2.4 mL) utilizing method D, stirring at room temperature (42 h), and using HPLC purification (silica gel, 20→50% EtOAc/hexanes): Rf 0.38 (1:1, hexanes/EtOAc); 1H NMR(300 MHz, CDCl3): δ 5.73 (dd, J = 6.3, 11.0 Hz, 1H), 5.21–5.06 (m, 1H), 3.73 (s, 3H), 2.82–2.71 (m, 3H), 2.64 (dd, J = 6.3, 13.5 Hz, 1H), 2.36–2.20 (m, 4H), 2.20–2.13 (m, 4H), 1.91 (dd, J = 11.3, 13.4 Hz, 1H), 1.85–1.77 (m, 1H), 1.72–1.52 (m, 2H), 1.46 (s, 3H), 1.32 (t, J = 7.6 Hz, 3H), 1.11 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.7, 175.6, 171.8, 171.4, 169.9, 169.3, 74.9, 69.6, 63.9, 53.4, 52.0, 50.8, 42.0, 39.9, 37.9, 35.4, 30.6, 20.6, 19.7, 18.0, 16.3, 15.4, 11.3; HRMS–ESI (m/z): [M + H]+ calcd for C23H30N2O8, 463.2080; found, 463.2089.
6.31. Isopropyl oxadiazoles 30a and 30b
Compound 30a (20 mg, 41%) was prepared as a clear resin from 7 (42 mg, 102 μmol), EDCI (25 mg, 129 μmol), HOBt (20 mg, 146 μmol), and N-hydroxy 2-methylpropionamide (15 mg, 149 μmol) utilizing method D, stirring at room temperature (19 h), and using HPLC purification (silica gel, 20→50% EtOAc/hexanes): Rf 0.48 (1:1, hexanes/EtOAc); 1H NMR(300 MHz, CDCl3): δ 5.73 (dd, J = 6.1, 11.1 Hz, 1H), 5.18–5.09 (m, 1H), 3.72 (s, 3H), 3.17–3.02 (m, 1H), 2.80–2.69 (m, 1H), 2.62 (dd, J = 6.1, 13.5 Hz, 1H), 2.36–2.22 (m, 4H), 2.21–2.12 (m, 4H), 1.91 (dd, J = 11.4, 13.3 Hz, 1H), 1.85–1.76 (m, 1H), 1.75–1.52 (m, 2H), 1.46 (s, 3H), 1.33 (dd, J = 0.6, 7.0 Hz, 6H), 1.11 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.7, 175.4, 175.3, 171.4, 169.9, 169.0, 74.9, 69.7, 63.8, 53.4, 52.0, 50.9, 41.9, 39.9, 37.9, 35.4, 30.6, 26.7, 20.5, 20.4, 20.4, 18.0, 16.3, 15.3; HRMS–ESI (m/z): [M + H]+ calcd for C24H32N2O8, 477.2237; found, 477.2214.
Compound 30b (2.0 mg, 4%) was also isolated as a clear resin: Rf 0.52 (1:1, hexanes/EtOAc); 1H NMR(300 MHz, CDCl3): δ 5.50 (dd, J = 2.4, 12.2 Hz, 1H), 5.19–5.05 (m, 1H), 3.71 (s, 3H), 3.20–3.02 (m, 1H), 2.75 (dd, J = 5.7, 11.1 Hz, 1H), 2.53 (dd, J = 2.1, 15.2 Hz, 1H), 2.47–2.41 (m, 1H), 2.34–2.17 (m, 4H), 2.15 (s, 3H), 1.97 (dd, J = 3.1, 12.9 Hz, 1H), 1.93–1.84 (m, 1H), 1.78 (dd, J = 12.6, 15.0 Hz, 1H), 1.66 (s, 3H), 1.61–1.52 (m, 1H), 1.33 (d, J = 7.0 Hz, 6H), 1.09 (s, 3H); (HRMS–ESI (m/z): [M + H]+ calcd for C24H32N2O8, 477.2237; found, 477.2249.
6.32. Propyl oxadiazole 31a
Compound 31a (9.6 mg, 21%) was prepared as a white powder from 7 (40 mg, 97 μmol), EDCI (24 mg, 125 μmol), HOBt (19 mg, 141 μmol), and N-hydroxy-butyramidine (17 μL, 146 μmol) utilizing method D, stirring at room temperature (18 h), and using HPLC purification (silica gel, 20→50% EtOAc/hexanes): Rf (.42, 1:1, hexanes/EtOAc); 1H NMR(300 MHz, CDCl3): δ 5.74 (dd, J = 6.2, 10.9 Hz, 1H), 5.22–5.06 (m, 1H), 3.73 (s, 3H), 2.83–2.57 (m, 4H), 2.37–2.21 (m, 4H), 2.20–2.12 (m, 4H), 1.92 (dd, J = 11.3, 13.1 Hz, 1H), 1.85–1.73 (m, 3H), 1.72–1.54 (m, 2H), 1.47 (s, 3H), 1.12 (s, 3H), 0.99 (t, J = 7.4 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ 201.7, 175.5, 171.4, 170.7, 169.9, 169.3, 74.9, 69.6, 63.9, 53.4, 52.0, 50.8, 42.0, 39.9, 37.9, 35.4, 30.7, 27.8, 20.5, 20.3, 18.1, 16.3, 15.4, 13.6; HRMS–ESI (m/z): [M + H]+ calcd for C24H32N2O8, 477.2237; found, 477.2218.
6.33. Phenyl oxadiazoles 32a and 32b
Compound 32a (9.2 mg, 17 %) was prepared as a clear resin from 7 (42 mg, 103 μmol), EDCI (27 mg, 140 μmol), HOBt (22 mg, 163 μmol), and benzamidoxime (25 mg, 181 μmol) utilizing method D, stirring at room temperature (22 h), and using HPLC purification (silica gel, 20→25% EtOAc/hexanes): Rf 0.55 (1:1, Hexanes/EtOAc); 1H NMR(300 MHz, CDCl3): δ 8.12–8.04 (m, 2H), 7.58–7.44 (m, 3H), 5.83 (dd, J = 6.3, 10.8 Hz, 1H), 5.22–5.09 (m, 1H), 3.73 (s, 3H), 2.80–2.67 (m, 2H), 2.37–2.24 (m, 4H), 2.24–2.12 (m, 4H), 2.00 (dd, J = 10.5, 12.8 Hz, 1H), 1.86–1.56 (m, 3H), 1.49 (s, 3H), 1.12 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.7, 176.0, 171.4, 169.9, 169.3, 168.6, 131.5, 128.9, 127.5, 126.1, 74.9, 69.7, 63.9, 53.4, 52.0, 50.8, 42.0, 39.9, 37.8, 35.5, 30.7, 20.6, 18.1, 16.3, 15.5; HRMS–ESI (m/z): [M + H]+ calcd for C27H30N2O8, 511.2080; found, 511.2069.
Compound 32b (7.9 mg, 15%) was also isolated as a clear resin: Rf 0.62 (1:1, Hexanes/EtOAc); 1H NMR(300 MHz, CDCl3): δ 8.14–8.01 (m, 2H), 7.59–7.41 (m, 3H), 5.59 (dd, J = 2.0, 12.4 Hz, 1H), 5.23–5.07 (m, 1H), 3.71 (s, 3H), 2.78 (dd, J = 6.5, 10.4 Hz, 1H), 2.62 (dd, J = 2.3, 15.1 Hz, 1H), 2.52–2.47 (m, 1H), 2.36–2.19 (m, 4H), 2.15 (s, 3H), 1.99 (dd, J = 3.0, 13.2 Hz, 1H), 1.95–1.80 (m, 2H), 1.69 (s, 3H), 1.63–1.54 (m, 1H), 1.11 (s, 3H); HRMS–ESI (m/z): [M + H]+ calcd for C27H30N2O8, 511.2080; found, 511.2059.
6.34. 4-Fluorophenyl oxadiazoles 33a and 33b
Compound 33a (3.9 mg, 7%) was prepared as a white powder from 7 (43 mg, 104 μmol), EDCI (27 mg, 140 μmol), HOBt (19 mg, 143 μmol), and 4-fluorobenzamidoxime (23 mg, 150 μmol) utilizing method D, stirring at room temperature (2 h), and using HPLC purification (silica gel, 20→25% EtOAc/hexanes): Rf 0.58 (1:1, Hexanes/EtOAc); 1H NMR(300 MHz, CDCl3): δ 8.1–8.03 (m, 2H), 7.21–7.13 (m, 2H), 5.82 (dd, J = 6.3, 10.9, 1H), 5.21–5.10 (m, 1H), 3.73 (s, 3H), 2.84–2.64 (m, 2H), 2.35–2.25 (m, 4H), 2.22–2.13 (m, 4H), 1.99 (dd, J = 11.3, 13.4, 1H), 1.85–1.79 (m, 1H), 1.72–1.56 (m, 2H), 1.50 (s, 3H), 1.13 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.7, 176.1, 171.4, 169.9, 169.2, 167.8, 129.8, 129.7, 116.3, 116.0, 74.9, 69.7, 63.9, 53.5, 52.0, 50.9, 42.0, 40.0, 37.9, 35.5, 30.7, 20.6, 18.1, 16.3, 15.4; HRMS–ESI (m/z): [M + H]+ calcd for C27H29FN2O8, 529.1986; found, 529.1999.
Compound 33b (9.6 mg, 17%) was also isolated as a white resin: Rf 0.69 (1:1, Hexanes/EtOAc); 1H NMR(300 MHz, CDCl3): δ 8.07 (dd, J = 5.4, 8.6 Hz, 2H), 7.16 (t, J = 8.7 Hz, 2H), 5.57 (dd, J = 1.0, 11.9 Hz, 1H), 5.21–5.09 (m, 1H), 3.71 (s, 3H), 2.85–2.69 (m, 1H), 2.60 (dd, J = 1.6, 15.1 Hz, 1H), 2.53–2.44 (m, 1H), 2.36–2.19 (m, 4H), 2.15 (s, 3H), 2.03–1.94 (m, 1H), 1.94–1.78 (m, 2H), 1.68 (s, 3H), 1.64–1.52 (m, 1H), 1.10 (s, 3H); HRMS–ESI (m/z): [M + H]+ calcd for C27H29FN2O8, 529.1986; found, 529.1996.
6.35. Benzyl oxadiazoles 34a and 34b
Compound 34a (8.5 mg, 17%) was prepared as a white powder from 7 (39 mg, 95 μmol), EDCI (25 mg, 130 μmol), HOBt (19 mg, 138 μmol), N′-hydroxy-2-phenylethanimidamide (22 mg, 148 μmol) utilizing method D, stirring at room temperature (18 h), and using HPLC purification (silica gel, 20→25% EtOAc/hexanes): Rf 0.50 (1:1, Hexanes/EtOAc); 1H NMR(300 MHz, CDCl3): δ 7.37–7.24 (m, 5H), 5.72 (dd, J = 6.2, 10.8 Hz, 1H), 5.16–5.07 (m, 1H), 4.08 (s, 2H), 3.73 (s, 3H), 2.77–2.68 (m, 1H), 2.61 (dd, J = 6.2, 13.6 Hz, 1H), 2.35–2.24 (m, 2H), 2.23–2.08 (m, 6H), 1.89 (dd, J = 10.8, 13.5 Hz, 1H), 1.82–1.74 (m, 1H), 1.70–1.49 (m, 2H), 1.44 (s, 3H), 1.10 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 201.7, 176.0, 171.4, 169.9, 169.7, 169.3, 134.8, 129.0, 128.8, 127.3, 74.9, 69.6, 63.8, 53.4, 52.0, 50.8, 41.9, 39.8, 37.8, 35.4, 32.2, 30.6, 20.6, 18.0, 16.3, 15.4; HRMS–ESI (m/z): [M + H]+ calcd for C28H32N2O8, 525.2237; found, 525.2248.
Compound 34b (12 mg, 24%) was also isolated as a clear resin: Rf 0.56 (1:1, Hexanes/EtOAc); 1H NMR(300 MHz, CDCl3): δ 7.42–7.25 (m, 5H), 5.48 (dd, J = 2.3, 12.2 Hz, 1H), 5.17–5.05 (m, 1H), 4.08 (s, 2H), 3.70 (s, 3H), 2.74 (dd, J = 6.4, 10.4 Hz, 1H), 2.50 (dd, J = 2.4, 15.1 Hz, 1H), 2.45–2.39 (m, 1H), 2.38–2.11 (m, 7H), 1.95 (dd, J = 4.1, 14.1 Hz, 1H), 1.91–1.72 (m, 2H), 1.63 (s, 3H), 1.61–1.52 (m, 1H), 1.08 (s, 3H); (HRMS–ESI (m/z): [M + H]+ calcd for C28H32N2O8, 525.2237; found, 525.2217.
Supplementary Material
Crystal structure and structure factors of 6 in CIF format. 1H NMR spectra for compounds 6, 17, 28a, and 28b. In vitro pharmacological profiles of 28a and 28b. Structures of diprenorphine and IOXY. Author contributions.
Acknowledgments
We thank Eric P. Brown for isolating salvinorin A. This work was supported by the Stanley Medical Research Institute, NARSAD and NIH grants DA 17302 and P30 DA 13429 (to LYLC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shippenberg TS, Zapata A, Chefer VI. Pharmacol Ther. 2007;116:306. doi: 10.1016/j.pharmthera.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jr, Jones RM, Portoghese PS, Carlezon WA., Jr J Pharmacol Exp Ther. 2003;305:323. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- 3.Nestler EJ, Carlezon WA., Jr Biol Psychiatry. 2006;59:1151. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 4.Knoll AT, Meloni EG, Thomas JB, Carroll FI, Carlezon WA., Jr J Pharmacol Exp Ther. 2007;323:838. doi: 10.1124/jpet.107.127415. [DOI] [PubMed] [Google Scholar]
- 5.Portoghese PS, Lipkowski AW, Takemori AE. Life Sci. 1987;40:1287. doi: 10.1016/0024-3205(87)90585-6. [DOI] [PubMed] [Google Scholar]
- 6.Broadbear JH, Negus SS, Butelman ER, de Costa BR, Woods JH. Psychopharmacology (Berlin, Ger) 1994;115:311. doi: 10.1007/BF02245071. [DOI] [PubMed] [Google Scholar]
- 7.Negus SS, Mello NK, Linsenmayer DC, Jones RM, Portoghese PS. Psychopharmacology (Berlin, Ger) 2002;163:412. doi: 10.1007/s00213-002-1038-x. [DOI] [PubMed] [Google Scholar]
- 8.Jones RM, Portoghese PS. Eur J Pharmacol. 2000;396:49. doi: 10.1016/s0014-2999(00)00208-9. [DOI] [PubMed] [Google Scholar]
- 9.Stevens WC, Jr, Jones RM, Subramanian G, Metzger TG, Ferguson DM, Portoghese PS. J Med Chem. 2000;43:2759. doi: 10.1021/jm0000665. [DOI] [PubMed] [Google Scholar]
- 10.Thomas JB, Atkinson RN, Rothman RB, Fix SE, Mascarella SW, Vinson NA, Xu H, Dersch CM, Lu Y, Cantrell BE, Zimmerman DM, Carroll FI. J Med Chem. 2001;44:2687. doi: 10.1021/jm015521r. [DOI] [PubMed] [Google Scholar]
- 11.Carroll I, Thomas JB, Dykstra LA, Granger AL, Allen RM, Howard JL, Pollard GT, Aceto MD, Harris LS. Eur J Pharmacol. 2004;501:111. doi: 10.1016/j.ejphar.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 12.Ortega A, Blount JF, Manchand PS. J Chem Soc, Perkin Trans 1. 1982:2505. [Google Scholar]
- 13.Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Proc Natl Acad Sci USA. 2002;99:11934. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prisinzano TE, Rothman RB. Chem Rev (Washington, DC, US) 2008;108:1732. doi: 10.1021/cr0782269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butelman ER, Harris TJ, Kreek MJ. Psychopharmacology (Berlin, Ger) 2004;172:220. doi: 10.1007/s00213-003-1638-0. [DOI] [PubMed] [Google Scholar]
- 16.McCurdy CR, Sufka KJ, Smith GH, Warnick JE, Nieto MJ. Pharmacol Biochem Behav. 2006;83:109. doi: 10.1016/j.pbb.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 17.Beguin C, Potter DN, Dinieri JA, Munro TA, Richards MR, Paine TA, Berry L, Zhao Z, Roth BL, Xu W, Liu-Chen LY, Carlezon WA, Jr, Cohen BM. J Pharmacol Exp Ther. 2008;324:188. doi: 10.1124/jpet.107.129023. [DOI] [PubMed] [Google Scholar]
- 18.Beguin C, Richards MR, Wang Y, Chen Y, Liu-Chen LY, Ma Z, Lee DY, Carlezon WA, Jr, Cohen BM. Bioorg Med Chem Lett. 2005;15:2761. doi: 10.1016/j.bmcl.2005.03.113. [DOI] [PubMed] [Google Scholar]
- 19.Beguin C, Richards MR, Li JG, Wang Y, Xu W, Liu-Chen LY, Carlezon WA, Jr, Cohen BM. Bioorg Med Chem Lett. 2006;16:4679. doi: 10.1016/j.bmcl.2006.05.093. [DOI] [PubMed] [Google Scholar]
- 20.Simpson DS, Katavic PL, Lozama A, Harding WW, Parrish D, Deschamps JR, Dersch CM, Partilla JS, Rothman RB, Navarro H, Prisinzano TE. J Med Chem. 2007;50:3596. doi: 10.1021/jm070393d. [DOI] [PubMed] [Google Scholar]
- 21.Munro TA, Rizzacasa MA, Roth BL, Toth BA, Yan F. J Med Chem. 2005;48:345. doi: 10.1021/jm049438q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harding WW, Schmidt M, Tidgewell K, Kannan P, Holden KG, Gilmour B, Navarro H, Rothman RB, Prisinzano TE. J Nat Prod. 2006;69:107. doi: 10.1021/np050398i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harding WW, Schmidt M, Tidgewell K, Kannan P, Holden KG, Dersch CM, Rothman RB, Prisinzano TE. Bioorg Med Chem Lett. 2006;16:3170. doi: 10.1016/j.bmcl.2006.03.062. [DOI] [PubMed] [Google Scholar]
- 24.John KS. Angew Chem, Int Ed. 1986;25:508. [Google Scholar]
- 25.Bikbulatov RV, Stewart J, Jin W, Yan F, Roth BL, Ferreira D, Zjawiony JK. Tetrahedron Lett. 2008;49:937. doi: 10.1016/j.tetlet.2007.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Overton KH, Weir NG, Wylie A. J Chem Soc C. 1966:1482. [Google Scholar]
- 27.Koreeda M, Brown L, Valdes LJ., III Chem Lett. 1990;19:2015. [Google Scholar]
- 28.Munro TA, Duncan KK, Staples RJ, Xu W, Liu-Chen LY, Beguin C, Carlezon WA, Jr, Cohen BM. Beilstein J Org Chem. 2007;3:1. doi: 10.1186/1860-5397-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Tang K, Inan S, Siebert D, Holzgrabe U, Lee DY, Huang P, Li JG, Cowan A, Liu-Chen LY. J Pharmacol Exp Ther. 2005;312:220. doi: 10.1124/jpet.104.073668. [DOI] [PubMed] [Google Scholar]
- 30.Zhu J, Luo LY, Li JG, Chen C, Liu-Chen LY. J Pharmacol Exp Ther. 1997;282:676. [PubMed] [Google Scholar]
- 31.Scheerer JR, Lawrence JF, Wang GC, Evans DA. J Am Chem Soc. 2007;129:8968. doi: 10.1021/ja073590a. [DOI] [PubMed] [Google Scholar]
- 32.Nozawa M, Suka Y, Hoshi T, Suzuki T, Hagiwara H. Org Lett. 2008;10:1365. doi: 10.1021/ol800101v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure and structure factors of 6 in CIF format. 1H NMR spectra for compounds 6, 17, 28a, and 28b. In vitro pharmacological profiles of 28a and 28b. Structures of diprenorphine and IOXY. Author contributions.