

Abstract
CDDO-Im is a synthetic triterpenoid recently shown to induce cytoprotective genes through the Nrf2-Keap1 pathway, an important mechanism for the induction of cytoprotective genes in response to oxidative stress. Upon oxidative or electrophilic insult, the transcription factor Nrf2 translocates to the nucleus, heterodimerizes with small Maf proteins, and binds to antioxidant response elements (AREs) in the upstream promoter regions of various cytoprotective genes. To further elucidate the hepatoprotective effects of CDDO-Im, wild-type and Nrf2-null mice were pretreated with CDDO-Im (1 mg/kg, i.p.) or vehicle (DMSO), and then administered acetaminophen (500 mg/kg, i.p.). Pretreatment of wild-type mice with CDDO-Im reduced liver injury caused by acetaminophen. In contrast, hepatoprotection by CDDO-Im was not observed in Nrf2-null mice. CDDO-Im increased Nrf2 protein expression and Nrf2-ARE binding in wild-type, but not Nrf2–null mice. Furthermore, CDDO-Im increased the mRNA expression of the Nrf2 target genes NAD(P)H: quinone oxidoreductase-1 (Nqo1); glutamate-cysteine ligase, catalytic subunit (Gclc); and heme-oxygenase-1 (Ho-1), in both a dose- and time-dependent manner. Conversely, CDDO-Im did not induce Nqo1, Gclc, and Ho-1 mRNA expression in Nrf2-null mice. Collectively, the present study shows that CDDO-Im pretreatment induces Nrf2-dependent cytoprotective genes and protects the liver from acetaminophen-induced hepatic injury.
Keywords: Nrf2, CDDO-Im, oxidative stress, acetaminophen, liver
INTRODUCTION
Oxidative stress is an injurious phenomenon that results from an imbalance in the ratio of pro-oxidant species to anti-oxidative defense mechanisms within a cell that ultimately damages three major critical macromolecules in cells: DNA, lipids, and proteins. Oxidative damage can lead to a variety of pathologies, including cancer, Parkinson’s disease, atherosclerosis, diabetes, Alzheimer’s disease, and even the aging process (Valko et al., 2007). One, or a combination of the following three factors, manifests oxidative stress. The first factor is an increase in the amount of oxidants, which include any number of highly reactive species possessing a single unpaired electron, capable of damaging DNA, lipids, and proteins. In addition to the amount of oxidants, there can be a decrease in the antioxidant status. Antioxidants include free radical scavengers, such as glutathione (GSH), ascorbic acid, and α-tocopherol. There are also detoxifying enzymes that contribute to antioxidant status, such as glutathione-S-transferases (Gsts); UDP-glucuronyl transferases (Ugts); NAD(P)H: quinone oxidoreductase 1 (Nqo1); catalytic and modifier subunits of γ-glutamyl cysteine ligase (Gclc, Gclm), which synthesize GSH; and heme oxygenase-1 (Ho-1), which catabolizes heme into iron, carbon monoxide, and the free radical scavenger bilirubin (Cho et al., 2006). Finally, an inability to repair oxidized DNA, lipids, and proteins also may contribute to oxidative stress. Examples of macromolecule repair include DNA repair by base or nucleotide exclusion, and protein repair by thioredoxin and glutaredoxin.
Over the past decade, the Nrf2-Keap1 pathway has been characterized as an important endogenous mechanism for combating oxidative stress. Nuclear factor-erythroid 2-related factor 2 (Nrf2) is a transcription factor that induces the expression of various cytoprotective enzymes possessing an antioxidant response element (ARE) in the promoter region. Nrf2 activation and subsequent cytoprotective gene induction promotes the restoration of the balance between oxidants and antioxidants after oxidative insult. Under conditions where oxidative stress is low, kelch-like ECH-associated protein-1 (Keap1) sequesters Nrf2 in the cytosol and acts as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2 (Tong et al., 2006). Increased oxidative stress promotes Nrf2 avoidance of Keap1-mediated proteasomal degradation by Cul3 and subsequent translocation of Nrf2 into the nucleus. Once in the nucleus, Nrf2 heterodimerizes with a small musculo-aponeurotic fibrosarcoma (Maf) protein and binds to the ARE, promoting transcription of various cytoprotective genes (Itoh et al., 1997). Nrf2-target genes include, but are not limited to Gsts, Nqo1, Ugts, Gclc, Gclm, and Ho-1, collectively being referred to as the “Nrf2 regulon” (Kobayashi and Yamamoto, 2006). Furthermore, Nrf2-null mice are susceptible to injury from acetaminophen (Enomoto et al., 2001), benzo[a]pyrene (Ramos-Gomez et al., 2001), diesel exhaust (Aoki et al., 2001), hyperoxia (Cho et al., 2002), hydrogen peroxide (Kraft et al., 2004), and other types of oxidative stress-induced injury.
Oleanolic acid is a naturally-occurring triterpenoid used in China for the treatment of hepatitis and is protective against chemical-induced liver injury in mice (Liu et al., 1993; 1995a; 1995b). Although, the limited efficacy of many naturally-occurring substances often precludes clinical use, but they may serve as a critical foundation for drug development. In an effort to create a more efficacious and potent triterpenoid, 2-cyano-3,12 dioxooleana-1,9 diene-28-imidazolide (CDDO-Im) was synthesized (Honda et al., 1998). CDDO-Im has been characterized as being effective in diminishing iNOS production in an in vivo model of inflammation (Place et al., 2003). CDDO-Im also activates the Nrf2-Keap1 pathway in both in vitro and in vivo models (Liby et al., 2005). Furthermore, pretreatment with CDDO-Im is protective against the LPS-inflammatory response and aflatoxin-induced liver tumorigenesis (Thimmulappa et al., 2006; Yates et al., 2006). The ability of CDDO-Im to act as both an anti-inflammatory and antioxidant chemical renders this compound a prime candidate for chemoprevention.
The purpose of the present study was to evaluate the protective effects of CDDO-Im in an acute chemical model of acetaminophen-induced hepatotoxicity. Because oxidative stress is a main contributor to the hepatotoxic effects of acetaminophen (Knight et al., 2001; Jaeschke et al., 2003), and CDDO-Im has been previously shown to activate Nrf2, the hepatoprotective effects of CDDO-Im against acetaminophen hepatotoxicity were evaluated in both wild-type and Nrf2-null mice.
METHODS
Materials
Acetaminophen and DMSO were purchased from Sigma-Aldrich (St. Louis, MO). CDDO-Im was a generous gift from Dr. Michael Sporn (Dartmouth College, Hanover, New Hampshire). β-actin antibody was purchased from Abcam (Ab8227, Cambridge, MA). Nrf2 antibody was purchased from Santa Cruz Biotechnology (sc-30915, Santa Cruz, CA).
Animals and Husbandry
Eight-week-old male C57BL/6 mice were purchased from Charles River Laboratories, Inc. (Wilmington, MA). Nrf2-null mice on a mixed C57BL/6 and AKR background were obtained from Dr. Jefferson Chan (University of California-Irvine, Irvine, CA). Nrf2-null mice were then backcrossed seven generations into C57BL/6 mice and >99% congenicity was determined by Jackson Laboratories (Bar Harbor, ME). Animals were housed in a temperature-, light-, and humidity-controlled environment and had access to Teklad Rodent Diet #8604 (Harlan Laboratories, Madison, WI) and water ad libitum. The housing facility is an American Animal Associations Laboratory Animal Care-accredited facility at the University of Kansas Medical Center, and all procedures were preapproved in accordance with Institutional Animal Care and Use Committee guidelines.
Hepatotoxicity Study
Mice were treated with CDDO-Im (1 mg/kg, i.p.) or vehicle (DMSO, 5 mL/kg, i.p.) once daily for three days. On the fourth day, mice were administered acetaminophen (500 mg/kg, i.p.) or vehicle (saline, pH 8, 20 mL/kg, i.p.). Six hours after acetaminophen administration, blood samples were taken for quantification of serum ALTs and liver samples were taken for histopathology. A sample of liver was also frozen in liquid nitrogen and stored at -80°C.
Serum alanine aminotransferase (ALT) concentrations
Serum ALT concentrations were determined as a biochemical indicator of hepatocellular necrosis using Pointe Scientific Liquid ALT Reagent Set (Canton, MI) according to the manufacturer’s protocol.
Histopathology
Samples were taken consistently as cross-sections of the largest lobe of the liver. Liver sections, approximately 5 μm thick, were fixed in 10% neutral-buffered zinc formalin, processed, stained with hematoxylin and eosin, and analyzed by light microscopy for liver injury. Samples were blinded before analysis. Grade of liver injury was analyzed semiquantitatively with six scores of severity: 0, none; 1, minimal (>2 foci of single cell necrosis per section); 2, mild (at least 5 areas of focal necrosis per section); 3, moderate (at least five foci of zonal necrosis per section); 4, severe (lobular damage, with many viable lobules per section); and 5, global (severe lobular damage, few areas of viability per section). A section was defined as a single viewpoint at low power (x100) magnification.
Nrf2 Protein Expression
Nuclear extracts were prepared with the NE-PER nuclear extraction kit according to the manufacturer’s directions (Pierce Biotechnology, Rockford, IL). Protein concentrations were determined with the BCA Assay Kit from Pierce Biotechnology (Rockford, IL). Nuclear proteins (60 μg protein/lane) were electrophoretically resolved using polyacrylamide gels (4% stacking and 7.5% resolving). Gels were transblotted overnight at 4°C onto a polyvinylidene fluoride membrane. Membranes were then washed with PBS–buffered saline containing 0.05% Tween-20 (PBS-T). Membranes were blocked for 1 h at room temperature with 5% non-fat milk in PBS–T. Blots were then incubated with primary antibody (1:1000 dilution, in 2% non-fat milk in PBS-T) for 3 h at room temperature. Blots were then washed in PBS-T and incubated with secondary antibody conjugated with horseradish peroxidase (1:2000 dilution, in 2% non-fat milk in PBS-T) for 1 hr at room temperature. Blots were then washed with PBS-T. Protein-antibody complexes were detected using an enhanced chemiluminescent kit (Pierce Biotechnology, Rockford, IL) and exposed to X-ray film (Denville Scientific, Metuchen, NJ). Intensity of protein bands was quantified using the Discovery Series Quantity One 1-D Analysis software (Bio-Rad Laboratories, Hercules, CA). Intensity values were expressed as protein expression relative to control. β-actin was utilized as a loading control.
Nrf2-ARE binding
Nrf2-ARE binding was determined using the ELISA-based TransAM™ Nrf2 Kit from Active Motif, according to manufacturer specifications (Carlsbad, CA). Briefly, the kit contains a 96-well plate to which oligonucleotides containing the consensus ARE have been immobilized. Nrf2 from nuclear extracts then binds to the oligonucleotide and is detected through use of an antibody against Nrf2. A secondary antibody conjugated to horseradish peroxidase is then added and allowed to bind to the primary antibody. The signal is detected at 450 nm using a spectrophotomer. The data is displayed as mean optical density (OD) at 450 nm.
Dose-Response Study
Wild-type mice were dosed (i.p.) with CDDO-Im at 0.1, 1.0, 3.0, or 10.0 mg/kg or vehicle (DMSO, 5 mL/kg). Nrf2-null mice were dosed (i.p.) with CDDO-Im at 1.0 mg/kg or vehicle (DMSO, 5 mL/kg). Livers were removed 6 hrs after dosing, frozen in liquid nitrogen, and stored at -80°C until RNA isolation. GSH.
Time-Course Study
Wild-type mice were dosed with CDDO-Im (1 mg/kg, i.p.) or vehicle (DMSO, 5 mL/kg, i.p.). Livers were removed 6, 12, 18, 24, 48, and 72 hrs after dosing, frozen in liquid nitrogen, and stored at -80°C until RNA isolation.
Total RNA Isolation
Total RNA was isolated using RNA-Bee reagent (Tel-Test, Inc., Friendswood, TX) according to the manufacturer’s protocol. Total RNA concentrations were determined spectrophotometrically at 260 nm. Two hundred ng/μl RNA solutions were prepared by dilution with diethyl pyrocarbonate-treated deionized water.
Branched DNA Signal Amplification (bDNA) Analysis
Specific mRNAs were quantified using the bDNA assay (1.0 Quantigene bDNA signal amplification kit; Panomics, Inc., Fremont, CA). Gene sequences of interest were accessed from GenBank. Target sequences were analyzed using ProbeDesigner software v1.0 (Bayer Corp., Emeryville, CA) to design oligonucleotide probe sets (capture, label, and blocker probes). All probes were designed with a melting temperature of 63°C, enabling hybridization conditions to be held constant (i.e., 53°C) during each hybridization step. Probe sets for Ho-1 and Nqo1 were developed as described previously (Aleksunes et al., 2005). Probe sets for Gclc are listed in Table 1. Total RNA was added to each well of a 96-well plate containing 50 μL of each diluted probe set. RNA was allowed to hybridize at 53°C with the probe sets overnight. Subsequent hybridization steps were carried out according to the manufacturer’s protocol, and luminescence was quantified with a Quantiplex 320 bDNA luminometer interfaced with Quantiplex Data Management software v5.02. Data are presented as relative light units (RLU) or as normalized to control.
Table 1.
Oligonucleotide probes used for analysis of mouse gene expression by bDNA signal amplification assay.
| Gene | GenBank AC# | Target a | Function b | Probe Sequence |
|---|---|---|---|---|
| Gclc | NM_012815 | 1548-1566 | CE | atggctcggagctggtctgTTTTTctcttggaaagaaagt |
| 1697-1724 | CE | cttaattagcttcaggtagttcagaataTTTTTctcttggaaagaaagt | ||
| 1725-1749 | CE | tcattagttctccagatgctctcttTTTTTctcttggaaagaaagt | ||
| 1848-1870 | CE | tcattagttctccagatgctctcttTTTTTctcttggaaagaaagt | ||
| 1896-1917 | CE | acttcgcttttctaaagcctgaTTTTTctcttggaaagaaagt | ||
| 1528-1547 | LE | ggccttgctacacccatccaTTTTTaggcataggacccgtgtct | ||
| 1567-1588 | LE | atgagcgtgtactcctctgcagTTTTTaggcataggacccgtgtct | ||
| 1589-1612 | LE | ccattgatgatggtgtctatgctcTTTTTaggcataggacccgtgtct | ||
| 1656-1678 | LE | tcgacttccatgttttcaaggtaTTTTTaggcataggacccgtgtct | ||
| 1679-1696 | LE | ctgcatcgggtgtccacgTTTTTaggcataggacccgtgtct | ||
| 1750-1770 | LE | ctctcatccacctggcaacagTTTTTaggcataggacccgtgtct | ||
| 1771-1794 | LE | agtcaggatggtttgcaataaactTTTTTaggcataggacccgtgtct | ||
| 1822-1847 | LE | tttcaaaatgaggctatagttgatctTTTTTaggcataggacccgtgtct | ||
| 1918-1939 | LE | gggtcgcttttacctccactgtTTTTTaggcataggacccgtgtct | ||
| 1613-1632 | BL | caggaaacacgccttccttc | ||
| 1633-1655 | BL | ggagttcagaatggggatgagtc | ||
| 1795-1821 | BL | catcagttattacactgtcttgcttgt | ||
| 1871-1895 | BL | tccaagtaactctggacattcacac |
Target refers to the sequence of the mRNA transcript as enumerated in the GenBank file.
Function refers to the use of the oligonucleotide probe in the assay (CE, capture extender; LE, label extender; BL, blocker probe).
Statistical Analysis
All data were analyzed using one-way analysis of variance (ANOVA) followed by Duncan’s multiple range test (p < 0.05). ALT values were log transformed before statistical analysis. Histopathological data were rank ordered prior to ANOVA analysis, which was followed by Newman-Keuls multiple range test (p ≤ 0.05).
RESULTS
CDDO-Im pretreatment lowers serum ALT concentrations and necrosis in livers of wild-type but not Nrf2-null mice after acetaminophen administration
CDDO-Im or vehicle (DMSO) did not increase serum ALT concentrations in wild-type or Nrf2-null mice (data not shown). Wild-type mice administered acetaminophen had elevated serum ALT concentrations (481 IU/L). Acetaminophen administered to CDDO-Im pretreated wild-type mice resulted in less of an increase in serum ALT concentrations (110 IU/L) (Fig 1). After acetaminophen administration, Nrf2-null mice had much higher serum ALT concentrations (2103 IU/L) than wild-type mice, as previously reported (Enomoto et al., 2001). Acetaminophen administered to CDDO-Im pretreated Nrf2-null mice resulted in serum ALT concentrations similar to Nrf2-null mice not pretreated with CDDO-Im.
Fig 1.
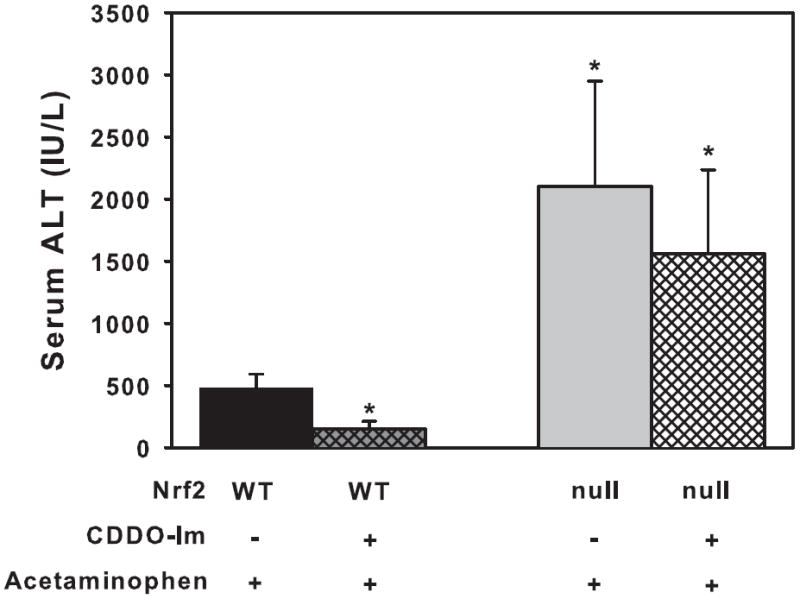
Serum alanine transaminase (ALT) levels in wild-type and Nrf2-null mice (n=5) pretreated with vehicle (DMSO) or CDDO-Im and then administered acetaminophen. ALT concentrations are expressed as International Units/Liter. Values are expressed as mean ± S.E.M. ALT values were log transformed before statistical analysis. Asterisks (*) indicate a statistically significant difference from DMSO pretreated wild-type mice (p ≤ 0.05).
CDDO-Im did not produce histologically observable abnormalities in either wild-type or Nrf2-null mice (data not shown). Grading of liver necrosis after acetaminophen is shown in Table 2. Acetaminophen produced moderate hepatic necrosis in wild-type mice, however, CDDO-Im pretreatment reduced the acetaminophen-induced hepatic necrosis. Acetaminophen produced much more severe hepatotoxicity in the Nrf2-null mice, and CDDO-Im pretreatment did not protect Nrf2-null mice from acetaminophen-induced necrosis.
Table 2.
Histological analysis of livers from vehicle- and CDDO-Im-pretreated wild-type and Nrf2-null mice after AA challenge.
| Histological Grade | |||||||
|---|---|---|---|---|---|---|---|
| Treatment Group | 0 | 1 | 2 | 3 | 4 | 5 | p≤5 |
| Wild-type VC-AA | 0 | 0 | 1 | 4 | 0 | 0 | |
| Wild-type CDDO-Im-AA | 0 | 2 | 3 | 0 | 0 | 0 | * |
| Nrf2-null VC-AA | 0 | 0 | 0 | 0 | 3 | 2 | * |
| Nrf2-null CDDO-Im-AA | 0 | 0 | 0 | 2 | 1 | 2 | |
Grade of liver injury was analyzed at low power (x100) with six scores of severity: 0, none; 1, minimal (>2 foci of single cell necrosis per section); 2, mild (at least 5 areas of focal necrosis per section); 3, moderate (at least five foci of zonal necrosis per section); 4, severe (lobular damage, with many viable lobules per section); and 5, global (severe lobular damage, few areas of viability per section).
Asterisks indicate a statistically significant difference from DMSO pretreated wild-type mice (p ≤ 0.05).
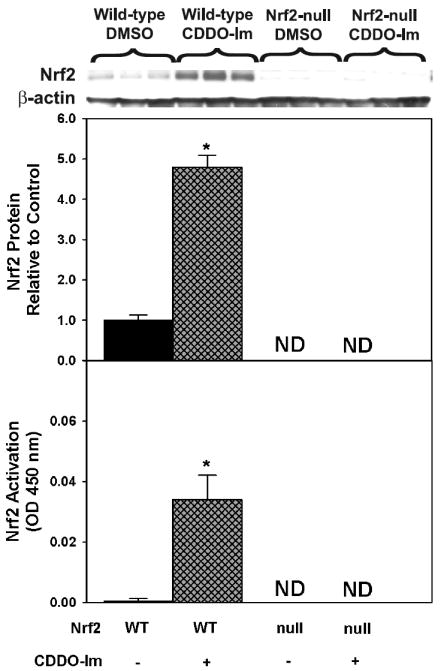
Effect of CDDO-Im on translocation of Nrf2 to the nucleus and binding to the ARE
CDDO-Im increased Nrf2 protein expression 380% in hepatic nuclear fractions from wild-type mice (Fig 2, upper). No Nrf2 protein was detected in vehicle or CDDO-Im-administered Nrf2-null mice. Activated Nrf2 (or Nrf2 capable of binding to the ARE) in hepatic nuclear fractions was determined via ELISA (Fig 2, lower). CDDO-Im activated Nrf2 in wild-type mice, whereas no activated Nrf2 was detected in vehicle or CDDO-Im treated Nrf2-null mice.
Fig 2.
GSH, GSSG, and total GSH concentrations in livers from wild-type and Nrf2-null mice pretreated with vehicle or CDDO-Im (1 mg/kg, i.p.) and then administered acetaminophen (500 mg/kg, i.p.). Livers were removed 6 h after acetaminophen administration. Data is expressed as relative to control mean ± S.E.M. Asterisks (*) indicate statistically significant differences between CDDO-Im treated and control groups (p ≤ 0.05).
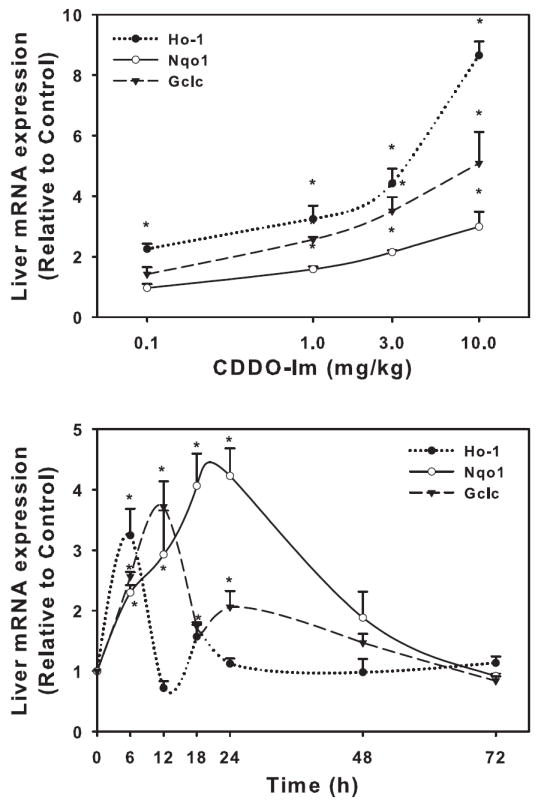
Dose- and time- dependent effects of CDDO-Im on Ho-1, Nqo1, and Gclc induction in wild-type mice
Wild-type mice were administered various doses of CDDO-Im from 0.1 to 10 mg/kg, i.p., and hepatic Ho-1, Nqo1, and Gclc mRNA expression were quantified (Fig 3, upper). CDDO-Im induced Ho-1 mRNA expression at all dosages used, with a 125% increase after 0.1 mg/kg and 765% increase at 10 mg/kg. CDDO-Im did not induce Nqo1 or Gclc at 0.1 mg/kg. However, CDDO-Im did increase mRNA expression of both Nqo1 and Gclc at 1.0 mg/kg (58 and 156%, respectively) with much more induction observed after the highest dose of 10 mg/kg (199 and 408%, respectively).
Fig 3.
Upper. Protein expression of Nrf2 determined by western blot in livers of wild-type and Nrf2-null mice after treatment with CDDO-Im (1 mg/kg, i.p.) once daily for three days. Also shown is the quantification of specific band intensity, expressed relative to control as mean ± S.E.M. Lower. Activated Nrf2 (or Nrf2 capable of binding to the ARE) in hepatic nuclear fractions was determined via ELISA. Values are expressed as mean optical density (OD) at 450 nm ± S.E.M.
To determine the time-response effects of CDDO-Im, wild-type mice were administered CDDO-Im (1 mg/kg, i.p.), and livers removed at various times thereafter (6-72 hrs) (Fig 3, lower). CDDO-Im induced Ho-1 mRNA expression, which was induced only at 6 hrs (224%). Nqo1 peaked between 18-24 hrs (323%) and returned back to control levels by 48 hrs, whereas Gclc mRNA induction peaked at 12 hrs (272%) and returned to control levels by 48 hrs.
Effect of CDDO-Im and acetaminophen on Ho-1, Nqo1, and Gclc-1 mRNA induction in wild-type and Nrf2-null mice
CDDO-Im or vehicle was administered to wild-type and Nrf2-null mice. CDDO-Im induced Ho-1 (411%), Nqo1 (153%), and Gclc (248%) mRNA expression in wild-type mice (Fig 4). Wild-type mice pretreated with CDDO-Im or vehicle and administered acetaminophen had increased Gclc and Nqo1 mRNA expression compared to wild-type mice receiving CDDO-Im or vehicle only. Interestingly, wild-type mice pretreated with CDDO-Im and then administered acetaminophen had lower Ho-1 mRNA expression than wild-type mice pretreated with vehicle and administered acetaminophen. Furthermore, CDDO-Im pretreatment had no effect on Nqo1 or Gclc mRNA expression when acetaminophen was also administered. Neither CDDO-Im, acetaminophen, nor the combination induced Ho-1, Nqo1, or Gclc mRNA expression in Nrf2-null mice.
Fig 4.
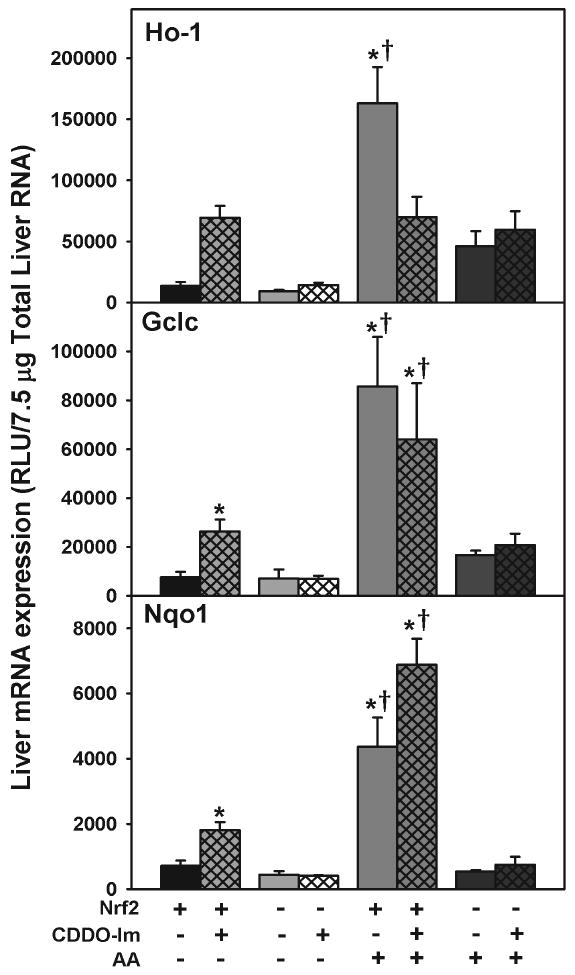
Upper. Hepatic mRNA expression of Nqo1, Gclc, and Ho-1 in wild-type mice after various doses of CDDO-Im. Livers were removed 6 h after dosing and mRNA quantified by the bDNA assay. Lower. Hepatic mRNA expression of Nqo1, Gclc, and Ho-1 in wild-type mice at different time points after CDDO-Im (1 mg/kg, i.p.) administration. mRNA was quantified by the bDNA assay. Data are expressed as relative to control mean ± S.E.M. Asterisks (*) indicate statistically significant differences between CDDO-Im treated and control groups (p ≤ 0.05).
DISCUSSION
CDDO-Im has emerged in recent years as a possible protective compound against oxidative stress. CDDO-Im decreases the inflammatory response in a variety of in vitro and in vivo models. For example, CDDO-Im decreases iNOS formation in vivo in primary mouse macrophages after interferon-γ administration (Place et al., 2003). Also, related triterpenoid compounds have been shown to decrease the inflammatory response through direct inactivation of IκBα-kinase, resulting in a decrease in NFκB activation (Shishodia et al., 2006).
CDDO-Im activates the Nrf2-Keap1 pathway (Liby et al., 2005). Activation of the Nrf2-Keap1 pathway leads to a cytoprotective response through the up-regulation of genes containing an antioxidant response element in their promoter regions (Itoh et al., 1997). Cytoprotective genes exert their effects by decreasing the amount of oxidative stress and restoring the balance between oxidants and antioxidants in the cell. Pretreatment with CDDO-Im protects the liver from aflatoxin-induced tumorigenesis (Yates et al., 2006). Furthermore, it has also been shown that CDDO-Im protects from the LPS-inflammatory response via a Nrf2-dependent mechanism (Thimmulappa et al., 2006).
Because liver is the major site of biotransformation for exogenous chemicals, a model of hepatotoxicity that involves electrophilic stress was utilized. Acetaminophen is primarily metabolized in the liver by sulfotransferases (Sults) and Ugts. High doses of acetaminophen saturate sulfation and glucuronidation detoxification pathways and result in a larger fraction of acetaminophen being biotransformed to the reactive intermediate, N-acetyl-p-benzoquinone imine (NAPQI), by cytochrome p450 2e1 (CYP2e1). NAPQI is detoxified by GSH conjugation, however, upon GSH depletion, NAPQI covalently binds to critical macromolecules and produces hepatotoxicity (Parkinson et. al, 2008). NQO1 is not only capable of detoxifying NAPQI (Moffit et al., 2007), but is induced in livers of patients who died from acetaminophen overdose (Aleksunes et al., 2006), suggesting that NQO1 induction may be an adaptive response to acetaminophen hepatotoxicity. Because Nrf2 target genes, such as Nqo1, are involved in detoxification of highly reactive intermediates, a chemical that activates Nrf2 might be a good cytoprotective compound, protecting from the hepatotoxicity of acetaminophen and other chemicals.
The present study was aimed at characterizing the hepatoprotective effects of CDDO-Im against a well-known hepatotoxicant, acetaminophen. CDDO-Im pretreatment protected wild-type mice from acetaminophen-induced hepatic injury, as determined by serum ALTs and histopathology. In contrast, CDDO-Im pretreatment did not protect from acetaminophen hepatotoxicity in Nrf2-null mice.
CDDO-Im facilitated Nrf2 translocation to the nucleus, which correlated with increased Nrf2 binding to consensus AREs (Fig 2)
Furthermore, the Nrf2-dependent genes Ho-1, Nqo1, and Gclc were induced in both a dose- and time-dependent manner by CDDO-Im. Ho-1 mRNA expression was up-requlated the most by CDDO-Im, followed by Gclc, and then Nqo1. The induction of Ho-1, Nqo1, and Gclc in livers of wild-type mice by CDDO-Im was not observed in Nrf2-null mice, strongly suggesting the induction of cytoprotective genes and subsequent hepatoprotective effects of CDDO-Im are Nrf2-dependent.
Acetaminophen induced Ho-1, Nqo1, and Gclc in wild-type but not Nrf2 – null mice, similar to a previous report (Aleksunes et al., 2008)
Pretreatment of wild-type mice with CDDO-Im did not further induce Nrf2 target genes in response to acetaminophen, suggesting that CDDO-Im-mediated hepatoprotection is not related to the adaptive response to acetaminophen injury. Rather, CDDO-Im pretreatment enhanced Nrf2 cytoprotective gene expression prior to acetaminophen exposure, which likely contributed to detoxification of the reactive electrophilic intermediate NAPQI, as well as resulting oxidative stress, thereby protecting against hepatic damage. In conclusion, CDDO-Im pretreatment induces Nrf2-dependent genes, in both a dose- and time-dependent manner, which protects from acetaminophen-induced hepatotoxicity.
Acknowledgments
The authors would like to thank Xiaohong Lei and the rest of the Klaassen laboratory for technical and manuscript revision assistance. Nrf2-null breeding pairs were graciously provided by Dr. Jefferson Chan (University of California-Irvine, Irvine, CA). CDDO-Im was provided by Dr. Michael Sporn (Dartmouth College, Hanover, New Hampshire). This research was financially supported by NIH Grants ES07079, ES09649, ES09716, ES013714, and RR021940.
Abbreviations
- ALT
Alanine transaminase
- CDDO-Im
2-cyano-3,12 dioxooleana-1,9 diene-28-imidazolide
- Gclc
Glutamate cysteine ligase catalytic subunit
- GSH
Glutathione
- Ho-1
Heme oxygenase-1
- Keap1
Kelch-like ECH associated protein 1
- Maf
Musculo-aponeurotic fibrosarcoma
- Nqo1, NAD(P)H
quinone oxidoreductase 1
- Nrf2
Nuclear factor-erythroid 2-related factor 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aleksunes LM, Goedken M, Manautou JE. Up-regulation of NAD(P)H quinone oxidoreductase 1 during human liver injury. World J Gastroenterol. 2006;12:1937–1940. doi: 10.3748/wjg.v12.i12.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksunes LM, Slitt AM, Cherrington NJ, Thibodeau MS, Klaassen CD, Manautou JE. Differential expression of mouse hepatic transporter genes in response to acetaminophen and carbon tetrachloride. Toxicol Sci. 2005;83:44–52. doi: 10.1093/toxsci/kfi013. [DOI] [PubMed] [Google Scholar]
- Aleksunes LM, Slitt AL, Maher JM, Augustine LM, Goedken MJ, Chan JY, Cherrington NJ, Klaassen CD, Manautou JE. Induction of Mrp3 and Mrp4 transporters during acetaminophen hepatotoxicity is dependent on Nrf2. Toxicol Appl Pharmacol. 2008;226:74–83. doi: 10.1016/j.taap.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, Sato H, Nishimura N, Takahashi S, Itoh K, Yamamoto M. Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol Appl Pharmacol. 2001;173:154–160. doi: 10.1006/taap.2001.9176. [DOI] [PubMed] [Google Scholar]
- Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 2002;26:175–182. doi: 10.1165/ajrcmb.26.2.4501. [DOI] [PubMed] [Google Scholar]
- Cho HY, Reddy SP, Kleeberger SR. Nrf2 defends the lung from oxidative stress. Antioxid Redox Signal. 2006;8:76–87. doi: 10.1089/ars.2006.8.76. [DOI] [PubMed] [Google Scholar]
- Enomoto A, Itoh K, Nagayoshi E, Haruta J, Kimura T, O’Connor T, Harada T, Yamamoto M. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci. 2001;59:169–177. doi: 10.1093/toxsci/59.1.169. [DOI] [PubMed] [Google Scholar]
- Honda T, Rounds BV, Gribble GW, Suh N, Wang Y, Sporn MB. Design and synthesis of 2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid, a novel and highly active inhibitor of nitric oxide production in mouse macrophages. Bioorg Med Chem Lett. 1998;8:2711–2714. doi: 10.1016/s0960-894x(98)00479-x. [DOI] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- Jaeschke H, Knight TR, Bajt ML. The role of oxidant stress and reactive nitrogen species in acetaminophen hepatotoxicity. Toxicol Lett. 2003;144:279–288. doi: 10.1016/s0378-4274(03)00239-x. [DOI] [PubMed] [Google Scholar]
- Knight TR, Kurtz A, Bajt ML, Hinson JA, Jaeschke H. Vascular and hepatocellular peroxynitrite formation during acetaminophen toxicity: role of mitochondrial oxidant stress. Toxicol Sci. 2001;62:212–220. doi: 10.1093/toxsci/62.2.212. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46:113–140. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci. 2004;24:1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liby K, Hock T, Yore MM, Suh N, Place AE, Risingsong R, Williams CR, Royce DB, Honda T, Honda Y, Gribble GW, Hill-Kapturczak N, Agarwal A, Sporn MB. The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res. 2005;65:4789–4798. doi: 10.1158/0008-5472.CAN-04-4539. [DOI] [PubMed] [Google Scholar]
- Liu J, Liu Y, Klaassen CD. Protective effect of oleanolic acid against chemical-induced acute necrotic liver injury in mice. Zhongguo Yao Li Xue Bao. 1995a;16:97–102. [PubMed] [Google Scholar]
- Liu J, Liu Y, Madhu C, Klaassen CD. Protective effects of oleanolic acid on acetaminophen-induced hepatotoxicity in mice. J Pharmacol Exp Ther. 1993;266:1607–1613. [PubMed] [Google Scholar]
- Liu J, Liu Y, Parkinson A, Klaassen CD. Effect of oleanolic acid on hepatic toxicant-activating and detoxifying systems in mice. J Pharmacol Exp Ther. 1995b;275:768–774. [PubMed] [Google Scholar]
- Moffit JS, Aleksunes LM, Kardas MJ, Slitt AL, Klaassen CD, Manautou JE. Role of NAD(P)H:quinone oxidoreductase 1 in clofibrate-mediated hepatoprotection from acetaminophen. Toxicology. 2007;230:197–206. doi: 10.1016/j.tox.2006.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson A, Ogilvie B. Biotransformation. In: Klaassen CD, editor. Casarett and Doull’s Toxicology: The Basic Science of Poisons. McGraw-Hill; New York: 2008. pp. 161–304. [Google Scholar]
- Place AE, Suh N, Williams CR, Risingsong R, Honda T, Honda Y, Gribble GW, Leesnitzer LM, Stimmel JB, Willson TM, Rosen E, Sporn MB. The novel synthetic triterpenoid, CDDO-imidazolide, inhibits inflammatory response and tumor growth in vivo. Clin Cancer Res. 2003;9:2798–2806. [PubMed] [Google Scholar]
- Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, Kensler TW. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001;98:3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishodia S, Sethi G, Konopleva M, Andreeff M, Aggarwal BB. A synthetic triterpenoid, CDDO-Me, inhibits IkappaBalpha kinase and enhances apoptosis induced by TNF and chemotherapeutic agents through down-regulation of expression of nuclear factor kappaB-regulated gene products in human leukemic cells. Clin Cancer Res. 2006;12:1828–1838. doi: 10.1158/1078-0432.CCR-05-2044. [DOI] [PubMed] [Google Scholar]
- Thimmulappa RK, Scollick C, Traore K, Yates M, Trush MA, Liby KT, Sporn MB, Yamamoto M, Kensler TW, Biswal S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem Biophys Res Commun. 2006;351:883–889. doi: 10.1016/j.bbrc.2006.10.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong KI, Kobayashi A, Katsuoka F, Yamamoto M. Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol Chem. 2006;387:1311–1320. doi: 10.1515/BC.2006.164. [DOI] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Yates MS, Kwak MK, Egner PA, Groopman JD, Bodreddigari S, Sutter TR, Baumgartner KJ, Roebuck BD, Liby KT, Yore MM, Honda T, Gribble GW, Sporn MB, Kensler TW. Potent protection against aflatoxin-induced tumorigenesis through induction of Nrf2-regulated pathways by the triterpenoid 1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole. Cancer Res. 2006;66:2488–2494. doi: 10.1158/0008-5472.CAN-05-3823. [DOI] [PubMed] [Google Scholar]