

Summary
Tex is a highly conserved bacterial protein that likely functions in a variety of transcriptional processes. Here we describe two crystal structures of the 86 kDa Tex protein from Pseudomonas aeruginosa at 2.3 Å and 2.5 Å resolution, respectively. These structures reveal a relatively flat and elongated protein, with several potential nucleic-acid binding motifs clustered at one end, including an S1 domain near the C-terminus that displays considerable structural flexibility. Tex binds nucleic acids, with a preference for ssRNA, and the Tex S1 domain is required for this binding activity. Point mutants further demonstrate that the primary nucleic acid binding site corresponds to a surface of the S1 domain. Sequence alignment and modeling indicate that the eukaryotic Spt6 transcription factor adopts a similar core structure. Structural analysis further suggests that the RNA polymerase and nucleosome interacting regions of Spt6 flank opposite sides of the Tex-like scaffold. Therefore, the Tex structure may represent a conserved scaffold that binds ssRNA to regulate transcription in both eukaryotic and prokaryotic organisms.
Keywords: transcription, S1 domain, RNA, x-ray crystallography, Spt6
Introduction
The Tex (toxin expression) protein was originally described in Bordetella pertussis as an essential protein involved in expression of critical toxin genes.1 Tex is a relatively large protein with a domain architecture consisting of several nucleic acid binding domains predicted from primary sequence. The presence of these domains supports the proposal that Tex is a transcription factor that functions in toxin expression and/or pathogen fitness.1-3 Tex displays a remarkably high degree of identity and similarity across a host of significant pathogens. For example, Tex from Pseudomonas aeruginosa shares 65% identity and 78% similarity (at the amino acid level) with Tex from Vibrio cholerae (the causative agent of cholera). Similar degrees of identity are seen with Tex proteins from Shigella flexneri (the causative agent of dysentery) and Yersinia pestis (the causative agent of plague).
Despite being ubiquitous and extremely well-conserved, the molecular function(s) of Tex remains enigmatic. Insight into Tex function is derived from several bacterial studies. Aside from its role in expression of toxin gene products in B. pertussis the tex gene from Pseudomonas aeruginosa (PA5201) appears to play an important role in pathogenesis, being required for lung infection in a chronic disease model.4 In Streptococcus pneumoniae, Tex does not effect expression of the major pneumococcus toxin pneumolysin, but does appear to be a transcription factor involved in pathogen fitness.3 These studies indicate that Tex may play a role in gene expression or transcript maintenance of either specific toxin or general housekeeping genes.
Tex domain architecture and sequence conservation may extend beyond prokaryotes to the essential eukaryotic transcription elongation factor Spt6.5-7 Tex is approximately half the size of Spt6 (e.g. 86 kDa for P. aeruginosa Tex vs. 168 kDa for S. cerevisiae Spt6), with sequence homology spanning the central region of Spt6. The flanking non-homologous regions of Spt6 include a highly charged N-terminal region and a C-terminal SH2-like domain. Within the region of homology, Tex and Spt6 share ∼ 25% pairwise sequence identity and have a similar predicted domain architecture; primary sequence analysis identified YqgF, HhH, and S1 RNA binding domains in both proteins.7 This level of sequence similarity falls in Doolittle’s “twilight zone,”8 indicating that Tex and Spt6 may have similar structures, although direct evidence is lacking.
The sequence similarity may also indicate that Tex and Spt6 have related cellular functions. Although current evidence suggests that Spt6 is a nucleosome chaperone,9-11 a function unique to eukaryotes, recent studies have shown that Spt6 also interacts directly with both RNA polymerase12 and mRNA processing factors, including the exosomal RNA degradation machinery.13 Thus, beyond its role in nucleosome maintenance, Spt6 appears to provide a physical link between transcription and pre-mRNA surveillance, although the relationship between these critical processes is lacking in structural detail. Interestingly, we have recently observed similar interactions with Tex. P. aeruginosa Tex co-purifies with RNA polymerase (RNAP), RNase E, and PNPase (I.V.-G. and S.L.D., unpublished data); RNase E and PNPase are components of the prokaryotic RNA degradosome, a 3′-5′ RNA degradation complex analogous to the eukaryotic exosome.14
In an effort to better understand the molecular function of Tex, and possibly to gain insight into Spt6, we have determined high-resolution crystal structures of the P. aeurginosa Tex protein in two crystal forms. These reveal four putative nucleic acid binding/modifying domains including a helix-turn-helix (HtH) domain that was not predicted from primary sequence. In addition, we have quantitatively examined the ability of Tex to bind various nucleic acid substrates and have found that Tex has a strong preference for single-stranded RNA. Binding appears to be sequence non-specific and mutagenesis studies indicate that this interaction is mediated by the flexible S1 domain. In contrast to an earlier proposal,1,2 we do not observe significant nuclease function associated with the Tex YqgF domain. Our findings provide a structural foundation for understanding Tex function, and can guide future studies on the structure and function of Spt6.
Results & Discussion
Structure Determination and Overall Description
The full-length Pseudomonas aeruginosa Tex protein was expressed recombinantly in E. coli and purified by Ni-chelate, Heparin affinity, and gel filtration chromatography. The C-terminal hexahistidine tag was retained for the structural and biochemical studies. The structure was determined by SAD phasing and density modification using data collected to 2.7 Å resolution from a selenomethionine-substituted crystal. This structure was refined against data collected from an isomorphous native crystal (crystal form I, 2.5 Å resolution) and from a second native crystal that belonged to the same space group but had substantially different cell dimensions (form II, 2.3 Å). The native structures were refined to R/Rfree values of 24.0/27.4 % (form I) and 23.1/27.0 % (form II), with good geometry (Table I). In both crystal forms, residues 1-730 are clearly observed in the electron density, with the exception of a short loop region (residues 246-251) in crystal form II. The C-terminal 55 residues of Tex and the hexahistidine tag are disordered and are not included in the final model.
Table1.
Data Collection and Refinement Statistics
| Tex Se-met | Tex native 1 | Tex native 2 | |
|---|---|---|---|
| crystal form I | crystal form I | crystal form II | |
| Data Collection | |||
| Beamline | NSLS X29 | NSLS X29 | home source |
| Wavelength (Å) | 0.978 | 1.10000 | 1.54178 |
| Resolution (Å) | 40-2.7 | 50-2.5 | 20-2.3 |
| Outer shell (Å) | 2.8-2.7 | 2.59-2.5 | 2.38-2.3 |
| No. reflections | |||
| Unique | 23,640 | 35,199 | 38347 |
| Total | 273,923 | 448,770 | 343965 |
| Mean I/σI* | 21.1 (4.8) | 30.0 (4.2) | 19.0 (3.3) |
| Completeness (%)* | 91.2 (61.7) | 90.4 (64.8) | 99.4 (99.3) |
| Rsym*a | 9.0 (32.0) | 8.6 (40.3) | 8.4 (50.7) |
| Space group | P212121 | P212121 | P212121 |
| Unit cell dimensions (Å) | a = 57.0 | a = 57.2 | a = 56.2 |
| b = 135.1 | b = 131.8 | b = 106.7 | |
| c = 144.5 | c = 144.0 | c = 139.7 | |
| Phasing (40-3.4 Å) | |||
| FOM, before DM (Solve) | 0.320 | ||
| FOM, after DM (Resolve) | 0.720 | ||
| Refinement | |||
| Rcrystb /Rfreec (%) | 24.0/27.4 | 22.1/26.6 | |
| Non-hydrogen atoms | |||
| Total | 5692 | 5884 | |
| Solvent | 73 | 256 | |
| RMSD from ideal geometry | |||
| Bond lengths (Å) | 0.004 | 0.004 | |
| Bond angles (°) | 0.6 | 0.6 | |
| Average Isotropic B Value (Å2) | 66.7 | 27.4 | |
| Protein geometryd | |||
| Ramachandran outliers (%) | 0.0 | 0.0 | |
| Ramachandran favored (%) | 96.2 | 98.5 | |
| Rotamer outliers (%) | 0.2 | 0.2 |
Values in parentheses correspond to those in the outer resolution shell
Rsym= (Σ|(I-<I>)|)/(ΣI), where <I> is the average intensity of multiple measurements.
Rcryst = (Σ|Fobs-Fcalc|)/(Σ|Fobs|).
Rfree is the R-factor based on 5% of the data excluded from refinement.
Geometry statistics were determined by Molprobity.51
Tex is ∼ 53% α-helical and 10 % β-sheet. The overall structure is notably flat and elongated, with approximate dimensions of 27 × 72 × 107 Å (Figure 1). The most striking structural feature is a long, central helix (H15) spanning ∼72 Å and comprising amino acid residues 274-322. The rest of the protein wraps around the central helix at both the N-terminal and C-terminal ends. The resulting structure has a distinctive question-mark-like appearance (Figure 1C). The structure closely resembles a lower resolution Tex structure that was recently submitted to the protein data bank by the New York Structural GenomiX Research Consortium (NYSGXRC) (PDB 2OCE).
Figure 1.

Crystal structure of Tex. (A) Structure of Tex. The structure is colored from N-terminus (blue) to C-terminus (red). (B) Tex amino acid sequence. The observed secondary structure is indicated above the sequence. Identified domains are indicated below the sequence. (C) Tex surface representation in two orthogonal views. Left hand view is the same as panel A.
Although the Tex structure is fairly compact, it can be largely described as a series of distinct domains. The first 84 residues adopt a helix-turn-helix (HtH) structure (Figure 2), although primary sequence analysis has not previously identified this motif in Tex. The next 189 residues (85-273) wrap around the bottom of the central helix (H15; residues 274-322), which extends nearly the full length of the structure, with a 30° kink occurring at lysine 288. Extending from the C-terminal end of this helix, the rest of the structure forms three domains that had been previously predicted from analysis of the amino acid sequence:7 YqgF (residues 329-455), HhH (501-557), and S1 (654-730) domains. The S1 domain is tethered to the rest of the structure by a stretch of 25 residues (629-653) that traverses the top of the molecule and contains little defined secondary structure but is clearly ordered in the electron density.
Figure 2.

Tex domain arrangement. (A-B) Orthogonal views of ribbon diagram of the Tex structure. Structural motifs identified from primary sequence and structural analyses are colored. Blue, helix-turn-helix motif. Red, YqgF homologous domain. Yellow, tandem helix-hairpin-helix motif. Green, S1 domain. (C) Tex domain structure, colored as in (A-B). The segment boxed with dotted lines, comprising residues 1-730, indicates the region of Tex sequence observed in the crystal structure.
Helix-turn-helix (HtH)
Despite very low (< 15%) sequence identity, the N-terminal HtH domain overlaps with other HtH structures (e.g. DNA helicase hel308, PDB 2P6U) with a root mean square deviation (RMSD) of 2.2 Å over 64 Cα atoms. HtH motifs typically bind dsDNA by inserting a helix into the major groove of the DNA duplex.15 The observed conformation of the HtH region in Tex does not appear to be competent for canonical dsDNA binding because the DNA would significantly clash with the surrounding Tex structure. However Tex may be capable of binding a single-stranded nucleic acid substrate through this domain. The strongest ‘hit’ from the DALI search (z-score = 5.6, RMSD = 2.2 Å) is to the ratchet domain of an archeal DNA helicase, Hel308.16 Hel308 unwinds dsDNA and uses the third helix of the HtH motif to bind a single strand of the DNA. Superposition of the DNA-bound Hel308 complex (PDB 2P6R) onto the Tex structure places the ssDNA through a narrow, elongated cavity (∼ 6 × 20 Å) that passes through the center of the Tex structure (Figure 3). Although we currently have no direct evidence that Tex binds nucleic acid substrates in this region, there is nothing obvious from the Tex structure that would preclude single-stranded nucleic acid binding at this site in some contexts.
Figure 3.
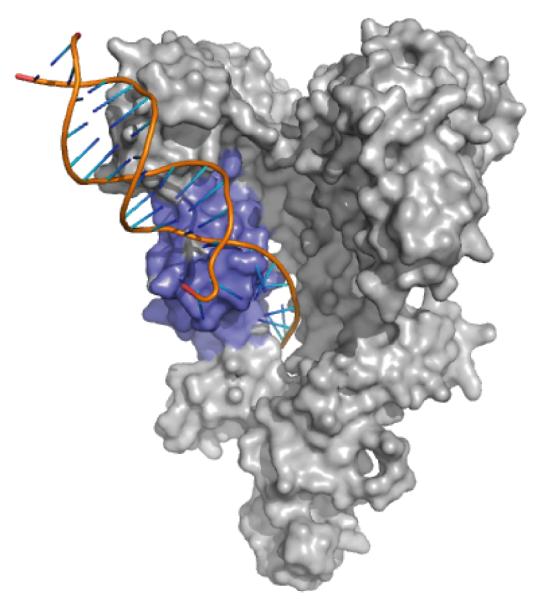
Model for single-stranded nucleic acid binding to Tex HtH motif. The archeal Hel308 structure (PDB 2P6R; only the DNA is shown here) was superimposed on the Tex structure (gray) by aligning HtH regions (blue). The path of the superimposed Hel308-bound ssDNA projects through a hole in the core of the Tex structure. The structure is oriented as in Figure 1A.
YqgF homologous domain
The central YqgF domain of Tex (residues 329-455) belongs to the YqgFc domain family (SMART SM0073217), a domain described as RNase H-like and typified by the E.coli protein YqgF.2,7 Little functional or biochemical data are available for YqgF domain-containing proteins, even though these proteins are highly conserved and occur across a wide-variety of bacterial genomes.18 YqgF domain family proteins are predicted to be ribonucleases or resolvases based on homology to RuvC Holliday Junction Resolvases.2,19,20 Like RuvC, which is structurally and biochemically well characterized, YqgF nuclease domains preserve the overall topology and the majority of structural and sequence elements characteristic of the RNase H fold.2,21-23 The Tex YqgF domain maintains these core structural elements and aligns especially possess nuclease activity (see below).
Helix-hairpin-helix (HhH)
The Tex structure contains two adjacent HhH motifs, comprising residues 502-531 and 537-557, both of which were predicted from earlier sequence analysis (Figure 4).5,6 The two HhH motifs are related by an approximately 90° rotation with respect to each other and pack together through extensive conserved hydrophobic interactions to generate a single, compact unit called a (HhH)2 domain.24 In contrast to a canonical (HhH)2 structure in which the two HhH motifs are connected by an extra helix, the Tex (HhH)2 domain makes the connection with a short 5-residue loop. The typical function of (HhH)2 domains is to bind dsDNA, mediated through nonspecific interactions with nitrogen atoms in the protein backbone and oxygen atoms in the DNA phosphate groups.24 In Tex, the binding face of this domain lies on the surface of the structure and would be accessible to a potential nucleic acid substrate.
Figure 4.

The Tex (HhH)2 domain stereoview. Tandem helix-hairpin-helix (HhH) motifs (yellow and orange) are linked by a short loop (gray) and pack together to form a single (HhH)2 domain. Conserved hydrophobic residues that comprise the core of the HhH packing surface are indicated. The view is looking down on the surface that binds dsDNA in other structures.
S1 domain
The Tex S1 domain (Figure 5) adopts the canonical topology characteristic of the S1 RNA-binding domain family.25 First identified as a motif of the ribosome essential for translational initiation, S1 domains are ubiquitous and found primarily in proteins that bind RNA and/or have nuclease activity.26,27 The Tex S1 domain adopts the overall five-stranded antiparallel β-barrel topology representative of the ubiquitous oligonucleotide/oligosaccharide binding (OB) fold. OB-fold proteins, including S1 domains, present a common binding cleft for interaction with a variety of different ligands, the most common being nucleic acids.3,27 This cleft runs perpendicular to the axis of the β-barrel where nucleic acids almost always bind with common polarity. The Tex S1 domain contains a short 310 helix (H35) adjacent to the binding cleft that, along with a strong preference for ssRNA, distinguishes S1 domains from other OB-fold proteins.28,29
Figure 5.

The Tex S1 domain. (A) Two orthogonal cartoon representations with conserved residues R718, H683, F668, and F671 shown in green. Right, a close-up view of an alignment of conserved residues from structurally related S1domains: green=Tex, orange= PNPaseS1 (PDB 1SRO), yellow= archealRPB4/7 (PDB 1GO3), blue=archealIFα (PDB 1ZY6). (B) A hypothetical model for RNA binding to the S1 domain binding cleft. The crystal structures of S1 domains with bound RNA from RNaseE (PDB 2COB, red) and RNaseII (PDB 2IX1, blue) were aligned with Tex S1 using DALI.33 As illustrated, ssRNA binds the same face of the different S1 domains but considerable differences in detail are apparent. (C) Surface representation showing primary sequence conservation as assigned by the CONSURF server.37 Conservation is indicated as a gradient from magenta (high) to white (low). In contrast to the view shown here, minimal conservation is observed on the opposite face of the S1 domain.
Evidence for Flexibility
Comparison of the two crystal forms, which differ by 25 Å in b-axis length and each contain one molecule in the asymmetric unit, indicates that Tex displays flexibility in the disposition of its C-terminal S1 domain. The two Tex structures superimpose closely throughout (RMSD = 1.596 over 510 Cα atoms), with the exception of a small rotation in the YqgF domain and a 14 Å displacement of the S1 domain (Figure 6). The S1 domain rearrangement is accomplished by rotation in the loop that tethers the domain to the rest of the Tex structure. C-terminal to the S1 domain, there is no discernable electron density for residues 731-785 in either crystal form, indicating a high degree of flexibility. In addition, very few contacts are observed in either structure between the S1 domain and the rest of the Tex protein. These observations indicate that the S1 domain is relatively unrestrained and able to adopt a range of orientations in solution.
Figure 6.

S1 domain mobility. Significant rearrangement of the S1 domain is observed in different crystal forms of the Tex structure. The 2.5 Å (crystal form I, gray) and 2.3 Å (crystal form II, green) crystal structures are superimposed. A side view of the superimposed crystal structures (inset) highlights a 14 Å shift between the two S1 domains.
Tex binds oligonucleic acids
Although the precise function of Tex is not known, the structural motifs observed in the Tex structure suggest binding to nucleic acids. It has recently been demonstrated by southwestern and northwestern analysis that recombinant S. pneumoniae Tex can interact with RNA and DNA,3 although Tex-nucleic acid interactions have not been explored in detail. We therefore quantified the ability of Tex to bind various nucleic acid substrates using electrophoretic mobility shift assays. Random 25-mer single stranded (ss) and double stranded (ds) DNA and RNA sequences were tested for binding (Figure 7). Tex bound all four types of nucleic acids, with a strong preference for ssRNA (Kd 210 ± 50 nM, n=9), and binding affinities were not altered by the presence of Mg2+. Binding to dsDNA (Kd 3720 ± 150 nM, n = 3), dsRNA (Kd 4200 ± 147 nM, n = 4), and ssDNA (Kd 5100 ± 150 nM, n = 4) was more than 10-fold weaker (Table II). Binding to an RNA/DNA hybrid was also confirmed though not quantified. Because Tex binds RNA and DNA chosen at random, including a poly-U ssRNA sequence (data not shown), binding appears to be sequence-independent, although the possibility of some strong sequence preferences can not be excluded at this time.
Figure 7.

Electrophoretic mobility shift data for Tex binding different substrates. (A) Full-length Tex protein was added in increasing concentrations to 5′-fluorescein labeled ssRNA (top left), dsRNA (top right), ssDNA (bottom left), or dsDNA (bottom right). Nucleic acid bound Tex complexes were resolved from free substrate by native gel electrophoresis. (B) Representative binding isotherms for the gel shifts represented in (A). Shifts and respective isotherms were repeated at least three times for each substrate. Resulting Kd values with standard error (in μM) were 0.21 ± 0.05 for 25bp ssRNA, 4.2 ± 1.47 for 25bp dsRNA, 5.1 ± 1.5 for 25bp ssDNA, and 3.7 ± 0.15 for 25bp ssDNA. (C) Fluorescence Polarization (FP) binding isotherm for Tex Binding to fluorescein labeled 25bp ssRNA. Data points with error bars (standard error) represent average with Polarization values (P) were measured for increasing concentrations of Tex. Kd and standard error for Tex binding ssRNA based on FP experiments is 0.057 ± 0.006 μM.
Table 2.
Tex binding affinities from gel mobility shift assays.
| Protein | Kda (uM) | Percent Binding Relative to WT |
|---|---|---|
| 25 nt ssRNA | ||
| WT Tex | 0.21 ± 0.05 | 100% |
| ΔS1 Tex | ndb | < 0.1% |
| R718E | 28.4 ± 11.4 | 0.74 ± 0.30 % |
| F668D/F671D | 13.7 ± 0.7 | 1.53 ± 0.08 % |
| F668D | 15.5 ± 5.5 | 1.35 ± 0.48 % |
| F671D | 9.8 ± 1.7 | 2.14 ± 0.37 % |
| H683E | 10.7 ± 3.5 | 1.96 ± 0.64 % |
| 25 bp dsRNA | ||
| WT Tex | 4.2 ± 1.47 | 100% |
| ΔS1 Tex | nd | < 0.1% |
| R718E | 46.9 ± 6.3 | 8.96 ± 1.20 % |
| F668D/F671D | 21.2 ± 11.5 | 19.8 ± 10.6 % |
| 25 bp dsDNA | ||
| WT Tex | 3.7 ± 0.15 | 100% |
| ΔS1 Tex | nd | < 0.1% |
| R718E | 48.3 ± 5.2 | 7.66 ± 0.82 % |
| F668D/F671D | 20.4 ± 3.8 | 18.1 ± 3.38 % |
| 25 bp dsDNA | ||
| WT Tex | 5.1 ± 1.5 | 100% |
| ΔS1 Tex | nd | < 0.1% |
| R718E | 118.7 ± 10.7 | 4.30 ± 0.39 % |
| F668D/F671D | 55.3 ± 6.0 | 9.22 ± 1.00 % |
Values represent the average Kd from multiple experiments (2 ≤ n ≤ 9) ± standard error
No binding detected
To supplement our electrophoretic mobility shift data and provide a solution-based estimate of binding, we used fluorescence polarization (FP). The Kd for 25 nt ssRNA binding to Tex was 56.6 ± 6.2 nM (n = 5). The ∼4-fold decrease in Kd compared to the value determined by electrophoretic mobility shift likely results from differences in equilibrium considerations in solution studies versus gel-based electrophoretic methods.30
The length dependence of ssRNA binding was examined using 10-mer, 13-mer, 16-mer, 20-mer, and 25-mer sequences. Negligible differences in binding were observed between the 20-mer and 25-mer sequences, and a modest reduction in binding was observed for the 16-mer sequence. In contrast, the 13 nucleotide ssRNA bound with markedly reduced affinity, and binding was not detected with the 10-mer sequence.
Our electrophoretic mobility shift assays reveal two distinct, shifted bands for single-stranded RNA and DNA (Figure 7). Although this may be an artifact of the native gel electrophoresis, it may also suggest non-stoichiometric binding or protein multimerization during the binding event. Studies of other proteins, such as RNase E and PNPase, have shown that S1 domain-mediated multimerization may be critical for substrate binding and enzymatic activity.28,31 We therefore investigated Tex RNA binding stoichiometry using Fluorescence Polarization (FP) and gel filtration. Apo Tex elutes from a gel filtration column as a single peak with a retention time expected for a monomer, even at a very high concentration. Similarly, a Tex:ssRNA complex, prepared by premixing Tex and an excess of 20 nt ssRNA, elutes from the sizing column as a single peak with the retention time expected for a 1:1 stoichiometry. FP was also used to estimate stoichiometry by titrating Tex protein into a solution containing a saturating concentration (> 20-fold above Kd) of 25 nt ssRNA. Polarization values (P) were read at each Tex concentration and the values were plotted as P versus the molar ratio of Tex to ssRNA. The inflection in this plot represents the point at which Tex has saturated all the binding sites on the RNA substrate. The inflection point for this experiment occurred at a molar ratio of 1:1 indicating that a single molecule of Tex binds one 25nt ssRNA molecule (data not shown).
Oligonucleic acids bind the Tex S1 domain
Tex’s preference for binding ssRNA made the S1 domain an obvious candidate for mediating binding of nucleic acid. In support of this possibility, we found that protein lacking the S1 domain (TexΔS1) was unable to bind ssRNA, dsRNA, ssDNA, or dsDNA in our electrophoretic mobility shift assays (Table II). It is unlikely that the loss of nucleic acid binding is due to mis-folding because the TexΔS1 protein behaves similarly to the full-length protein throughout the purification process, including gel filtration. Furthermore, significant repositioning of the S1 domain (with respect to the rest of the protein) in different Tex crystal forms has little effect on the rest of the Tex structure. These data indicate that although Tex binds a variety of nucleic acids and is composed of a number of putative nucleic acid binding motifs, binding does not occur in the absence of the S1 domain.
In the numerous S1/OB-fold co-structures determined to date, nucleic acid substrate is coordinated in the binding cleft via surface-exposed aromatic side chains such as phenylalanine and more polar groups such as lysine or arginine.27,32 The structure of Tex reveals the characteristic S1/OB binding cleft and candidate contact residues: F668, F671, H683, and R718 (Figure 5). When using DALI33 to align the Tex S1 domain with other S1 structures, Tex residues F668, F671, H683, and R718 superimpose closely with comparable residues in the S1 domain RNA-binding clefts of PNPase (PDB 1SRO, 73 Cα, RMSD 2.0 Å , 79 Cα, RMSD 1.8 Å), and the archeal homolog of the RNAP II subunit RPB7 (PDB 1GO3 72 Cα, RMSD 1.8 Å) (Figure 5). These proteins are all reported to bind RNA transcripts in a sequence non-specific manner with the interaction being important for RNA decay,31 general translation initiation,34 and transcription initiation.35,36 Given the close alignment of these conserved residues, it is likely that cellular substrate for Tex is also sequence non-specific RNA transcripts.
Although putative RNA-binding residues align well amongst some S1 proteins, precise alignment of critical residues does not appear to be required for binding similar RNA substrates. The crystal structures of the ribonucleases RNase E (PDB 2C0B) and RNase II (PDB 2IX1) represent co-structures of RNA-bound S1 domains that interact with RNA independent of sequence. S1 residues making significant contacts in these structures do not align precisely with one another even though the general composition of side-chains is maintained and the interaction occurs across the same S1/OB binding cleft. When aligning Tex S1 with these structures, a similar theme is observed where F668, F671, H683, and R718 are clustered in the same binding cleft as equivalent RNase E and RNase II residues. Based on these observations, we propose a model for Tex binding to single-stranded RNA via the S1/OB binding cleft (Figure 5B).
In order to map the nucleic acid binding surface further and to test the binding model, a variety of Tex S1 domain point mutants were assayed for RNA binding. Mutation of the conserved S1 binding cleft residues F668, F671, H683, and R718 to aspartate or glutamate disrupts ssRNA binding by at least 46-fold (< 2.2% binding relative to WT Tex) (Table II), with a 135 ± 50 fold reduction (0.74 ± 0.30% of WT Tex) in ssRNA binding when the mutant R718E was assayed by gel shift. The double mutation F668D/F671D results in a 65 ± 3 fold reduction (1.53 ± 0.08% of WT Tex) in ssRNA binding and implies that hydrophobic base-stacking and/or packing interactions on the S1 surface are additionally important. Consistent with the model that all nucleic acid binding in our assay conditions is to the S1 domain and that ssRNA is the preferred ligand, affinities for all substrates were reduced in the S1 point mutants, with the greatest effect seen on binding of ssRNA.
Overall, the structural and binding data indicate that the S1 domain is a highly dynamic module that is required for nucleic acid binding and displays a strong preference for ssRNA. Binding is likely to be sequence independent, as has been found for the majority of other described S1 domain-containing proteins, including those that align well with Tex. Although S1 is the primary nucleic acid binding domain, we cannot rule out minor contributions to binding from other regions, and it is possible that specific in vivo contexts, such as binding to another partner, might open additional surfaces for binding to nucleic acid substrates.
Consideration of minimal oligonucleotide length required for Tex binding suggests that S1 is not the only region that binds the various nucleic acid substrates. 10 nucleotide oligonucleotides do not bind Tex in our assay and a significant decrease in binding affinity observed for ssRNA lengths less than 20 nucleotides. A ssRNA molecule of 10 nucleotides or longer would extend beyond the available binding surface of the S1 domain implying that additional contacts outside the S1 domain occur in our assay. This is a familiar theme for S1-containing proteins. For example, the RNase E S1 domain is a dynamic module that serves as a molecular clamp for correctly orienting ssRNA substrate.32 Given the modularity of the Tex S1 domain, the presence of multiple other nucleic-acid binding domains, and a minimal substrate length that spans a surface larger than that offered by the S1 domain, one attractive possibility is that Tex may utilitze an S1 molecular clamp binding model similar to RNase E.32
Putative nuclease activity
Based on the presence of the RNase H fold YqgF domain and the observation that Tex negatively regulates transcription when overexpressed, Tex is predicted to have ribonuclease activity.1,2 In conflict with this prediction, however, we have not detected nuclease activity. There is no indication of nucleic acid degradation in our gel shift experiments. We performed a qualitative nuclease assay in which the positive controls (RNase T1 and Micrococcal Nuclease) were active, but activity was at background levels for Tex (data not shown). In addition, we observe no differences in nucleic acid binding or the very low background nuclease activity between wild-type Tex and a double mutant in which two putative catalytic residues in the YqgF domain (Asp335 and Glu421) were changed to alanine (data not shown). The lack of nuclease activity could be explained by the fact that the Tex YqgF domain lacks a critical and highly conserved carboxylate residue that is required for metal coordination in known RNase H-fold ribonucleases (for example, D141 of E.coli RuvC) (Figure 8). Therefore, in contrast to the earlier prediction,1,2 these data suggest that Tex is not a ribonuclease.
Figure 8.

The Tex Yqgf (red) superimposed on the E. coli RuvC (sand) (PDB 1HJR, 120 Cα, 3.0 RMSD) and an exploded view of the catalytic center. The RuvC catalytic residues D7, E66, D141, and D138 are shown in stick representation aligned with Tex residues D335, D421, and D441 that share the same basic geometric orientation in the catalytic center. Although three of the four conserved catalytic residues are present in Tex, there is no equivalent acidic residue present at the location corresponding to RuvC D141.
Implications for Spt6
Many S1 domain-containing proteins are factors involved in general processes such as transcription, translation initiation, and mRNA decay.25,28,34,35 Consistent with this idea, comparative genomic and evolutionary studies suggested that Tex represents a bacterial ortholog of the eukaryotic transcription elongation factor Spt6.5,7 Although Spt6 is twice the size of Tex, it is predicted to possess YqgF, HhH, and S1 domains in the same order, and to possess 15% sequence identity (27% similarity) over these regions (Figure 9A). The predicted secondary structural elements of the Spt6 sequence also show good agreement with the Tex structure (data not shown).
Figure 9.

Model for Spt6 structure. (A) Comparison of Tex and Spt6 domain structures. (B) The Tex structure is used to model the central portion of the Spt6 structure (surface representation). A proposed nucleosome binding domain (magenta, inset panel) is modeled based on structural alignment with the C-terminal portion of the IswI nucleosome interacting domain (PDB 1OFC). An SH2-like domain (orange, PDB 1PIC) is modeled at the C-terminal end of the S1 domain.
The nature of the structural similarity between Tex and Spt6 is further clarified by aligning the Spt6 sequence with the Tex structure. In particular, all of the differences in sequence length between Tex and Spt6 (e.g. Spt6 insertion sequence) occur on the surface of the Tex structure, which appears able to accommodate additional sequences without disrupting the core structural scaffold (data not shown). Additionally, evolutionary conservation scores based on Tex- and Spt6-related sequences were assigned for each Tex amino acid residue, and mapped onto the Tex structure using the Consurf server.37 Most of the conserved residues identified by this method appear to be involved in packing interactions in the Tex structure. These observations suggest that Spt6 retains the core Tex structure, with variations on the periphery, along with additional domain features at the N- and C-terminal ends (described below).
Unlike Tex, Spt6 possesses an SH2-like domain C-terminal to the S1 domain.38 Given the proximity of the SH2-like sequence to the S1 domain, it is likely that this domain lies along one face of the Spt6 structure, as indicated in Figure 9. The Spt6 SH2-like domain is reported to mediate interactions with the C-terminal domain (CTD) of RNA polymerase II (RNAPII).12 Based on the mobility of the S1 domain observed in the Tex structures, it is likely that Spt6 binds RNAPII via a flexible tether.
Structural comparisons suggest that a region toward the Spt6 N-terminus may possess histone chaperone activity. A DALI search of the Tex HtH domain identified significant structural homology (z—score = 5.2, RMSD = 2.4 Å) with the nucleosome-binding SLIDE domain of ISWI (PDB 1OFC).39 While there is insufficient sequence for Tex to form a complete SLIDE-like nucleosome binding domain, Spt6 contains additional sequence N-terminal to the region of Tex homology that could potentially fulfill this role. In this model, histone chaperone activity would be located on the face of Spt6 opposite from the S1 and SH2-like domains (Figure 9).
Spt6 has demonstrated eukaryotic exosome recruiting faculties12 and we have observed that Tex co-purifies with RNase E and PNPase, which in E. coli are components of the RNA degradosome (I.V.-G. and S.L.D., unpublished data). This may at least partially explain why Tex appears to negatively effect transcription when overexpressed1,2 but does not itself appear to possess ribonuclease activity in our assays; it may be coordinating the recruitment, or influencing the activities, of degradosome-associated ribonucleases. Further, Spt6 interacts with an elongating RNA polymerase at the CTD based on elongation specific phosphorylation.12 The observation that Tex from P. aeruginosa co-purifies with components of RNA polymerase (I.V.-G. and S.L.D., unpublished data) suggests it may be associated, either directly or indirectly, with the transcription machinery; functional parallels may therefore exist between Spt6 and Tex.
In summary, the Tex crystal structures reveal an elongated, helical protein comprising several putative nucleic acid binding domains. Biochemical characterization revealed that Tex binds single-stranded and double-stranded DNA and RNA substrates with a preference for ssRNA, with a primary interface being mediated by interactions along the canonical OB-fold binding cleft of the Tex S1 domain. The Tex structure provides a model for the core of the eukaryotic transcription factor Spt6, and raises the possibility that the N-terminal portion of Spt6 constitutes a nucleosome-binding domain that evolved from an HtH domain.
Materials and methods
Tex protein expression and purification
Full-length Tex from Pseudomonas aeruginosa strain PAO1 was cloned into a pET24 kan expression vector containing a C-terminal 6-histidine tag. The plasmid was transformed into cells of E. coli BL21-codonplus-(DE3)-RP (Stratagene). Cells were grown in LB media and induced with 1 mg/ml IPTG at an OD600 of 0.6 or alternatively grown using an autoinduction method as described in.40 In both cases, cells were grown at 37 °C for 5 hours then transferred to 20 °C and grown to saturation. Harvested cells were stored at -80 °C.
Cells were thawed and resuspended in lysis buffer (50 mM Tris, pH 7.5; 500 mM NaCl; 10 % glycerol; 10 mM imidazole; 2 mM BME) in the presence of lysozyme and protease inhibitors. Following sonication and centrifugation (25,000 × g), the soluble fraction was applied to Ni-NTA agarose resin (Qiagen) and eluted with 300mM imidazole. Protein was dialyzed (50 mM Tris, pH 7.0; 10% glycerol; 2 mM BME; 35 mM NaCl), applied to a heparin column (5 ml HiTrap Heparin HP, GE Healthcare), and eluted over a NaCl gradient. ΔS1 Tex was purified in the same way but a Q ion-exchange column (5 ml HiTrap Q HP, GE Healthcare) was used instead of the Heparin column as the ΔS1 construct did not bind Heparin. Peak fractions from either Heparin or Q columns were pooled, dialyzed (50 mM Tris, pH 7.5; 5 % glycerol; 100 mM NaCl; 2 mM BME), and run over a size exclusion column (Superdex 200 26/70; GE Healthcare). Selenomethionine-substituted Tex protein was expressed41 and purified using the same protocol as native protein.
Tex crystallization and structure determination
Crystals (Se-met and crystal form I) were grown by sitting drop vapor diffusion in 19 % (w/v) PEG 3350, 0.1M Bis-Tris pH 5.5, 0.17 M ammonium sulfate, and 10 % (v/v) glycerol. A second crystal form (crystal form II) was grown in 18 % w/v PEG 4000, and 100 mM Sodium Acetate pH 4.6. Data were collected at the National Synchrotron Light Source (NSLS) at Brookhaven National Lab (BNL) for crystal form I and on a home source (Rigaku Raxis IV) for crystal form II (Table I). X-ray diffraction data were processed using HKL2000.42
Phases were determined for the form I crystals by the single anomalous dispersion (SAD) method using seleonomethionine-substituted Tex. The programs Solve43 and Resolve44 were used to identify selenium positions (12 out of 13 potential sites were identified) to calculate initial maps to 3.4 Å resolution followed by phase extension to 2.7 Å. Data from native crystals grown in the same conditions (crystal form I) were subsequently collected to extend the resolution of the Tex structure to 2.5 Å. A 2.3 Å structure (crystal form II) was determined using crystal form I as a model for molecular replacement in PHASER.45 Refinement was performed using CNS46, Phenix47, and Refmac48. O49, Coot50 and Molprobitys51 were used for model building and validation. PyMol52 was used to prepare figures, superimpose the Tex structures, and perform electrostatic calculations (APBS tools). The Consurf server37 was used to calculate evolutionary based conservation scores.
Nucleic acid binding assays
Oligonucleotides were designed using random sequence and purchased from the University of Utah DNA/Peptide Core facility. Oligonucleotide sequences used in this study are (5′ to 3′): UCUUUUCCUGUGUUUUUCCGCAAUC (25 nt ssRNA and 25 bp dsRNA, sense), GATTGCGGAAAAACACAGGAAAAGA (25 nt ssDNA), CGCAGGCCCGGCGCGAGGCCGAGGG (25 bp dsDNA, sense), UCCUGUGUUUUUCCGCAAUC (20 nt ssRNA), UUGUUUUUCCGCAAUC (16 nt ssRNA), UUUUUCCGCAAUC (13 nt ssRNA), UUCCGCAAUC (10 nt ssRNA). Prior to binding studies, oligonucleotides were gel purified on a 20% acrylamide/7M Urea denaturing gel. Double-stranded substrates were mixed in equimolar amounts in 10 mM Tris-HCl pH 7.5 and 40 mM KCl with one strand being end-labeled with fluorescein. Samples were annealed by boiling (5 min) and slow (2 hour) cooling to room temperature, and gel purified using a non-denaturing 20% acrylamide gel.
Gel mobility shift assays were performed by mixing varying concentrations of protein with nucleic acid substrate in binding buffer (15 mM Tris-HCl pH 7.5, 100 mM NaCl, 10% Glycerol, and 1U/μl RNasin (Promega) or 1U/μl RNaseOUT (Invitrogen). The final concentration of nucleic acid used in each experiment was at least 20-fold below the Kd for the respective substrate/protein. Reactions were incubated at room temperature for 30 minutes then assayed by electrophoresis using 4-20% TBE native gels (Bio-rad Laboratories) at room temperature. Gels were imaged and quantified using a TYPHOON imaging system with Imagequant software (GE Healthsciences). The fraction bound was calculated by quantifying the RNA/DNAtotal (total fluorescence in entire lane) and RNA/DNAfree. All RNA/DNAs of slower mobility than the RNA/DNAfree were considered bound. The fraction bound = 1 — ([RNA/DNA]free/[RNA/DNA]total). Dissociation constants (Kds) were calculated by plotting data points and curve fitting using the Hill Formalism where Fraction Bound = 1/(1 + (Kdn/[P ]n)). Average Kd values were determined by fitting data points from individual experiments then averaging the calculated dissociation constants. All plots and curve fits were performed using the program Kaleidagraph (Synergy Software).
Fluorescence Polarization
Fluorescence Polarization (FP) was performed in 96 well format using a Tecan fluorimeter with the same fluorescein-labeled substrates and buffer conditions as the shift analysis. Tex concentrations were varied in individual wells and mixed with RNA at a final concentration that was at least 10-fold below the Kd. Samples were incubated at room temperature for at least 30 minutes prior to measuring polarization. Polarization values (P) were measured and plotted as a function of Tex concentration. Data points were fit using P = ((Pbound - Pfree)[Tex]/Kd + [Tex]) + Pfree. The free and total protein concentrations are assumed to be equal because the RNA concentration is at least ten-fold lower than the Kd.
When using FP to evaluate binding stoichiometry, polarization measurements were performed following the procedure described in53. RNA was mixed in binding buffer solution at a concentration 20-fold above the Kd. Protein was titrated into the RNA solution until polarization values leveled off, and polarization values were plotted as a function of the concentration ratio of Tex protein vs. RNA substrate. The Tex /RNA ratio where an inflection in the data occurs represents the binding stoichiometry, as this is the point where Tex has been saturated by RNA and polarization values change modestly as protein concentration is increased.
Protein Data Bank accession numbers
Coordinates and structure factors for Tex crystal form I and crystal form II have been deposited in the Protein Data Bank with accession numbers 3BZC and 3BZK, respectively.
Supplementary Material
Acknowledgements
We thank Brenda Bass and members of the Bass Lab for use of materials and reagents as well as help with binding studies. This work was supported by NIH grants GM074368 (S.J.J.), T32-GM008537 (D.C.), AI057754 (I.V.-D. and S.L.D.), and GM076242 (C.P.H.). Financial support for use of the NSLS comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy, and from the National Center for Research Resources of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fuchs TM, Deppisch H, Scarlato V, Gross R. A new gene locus of Bordetella pertussis defines a novel family of prokaryotic transcriptional accessory proteins. J Bacteriol. 1996;178:4445–52. doi: 10.1128/jb.178.15.4445-4452.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aravind L, Makarova KS, Koonin EV. SURVEY AND SUMMARY: holliday junction resolvases and related nucleases: identification of new families, phyletic distribution and evolutionary trajectories. Nucleic Acids Res. 2000;28:3417–32. doi: 10.1093/nar/28.18.3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He X, Thornton J, Carmicle-Davis S, McDaniel LS. Tex, a putative transcriptional accessory factor, is involved in pathogen fitness in Streptococcus pneumoniae. Microb Pathog. 2006;41:199–206. doi: 10.1016/j.micpath.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Potvin E, Lehoux DE, Kukavica-Ibrulj I, Richard KL, Sanschagrin F, Lau GW, Levesque RC. In vivo functional genomics of Pseudomonas aeruginosa for high-throughput screening of new virulence factors and antibacterial targets. Environ Microbiol. 2003;5:1294–308. doi: 10.1046/j.1462-2920.2003.00542.x. [DOI] [PubMed] [Google Scholar]
- 5.Anantharaman V, Koonin EV, Aravind L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002;30:1427–64. doi: 10.1093/nar/30.7.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaplan CD, Morris JR, Wu C, Winston F. Spt5 and spt6 are associated with active transcription and have characteristics of general elongation factors in D. melanogaster. Genes Dev. 2000;14:2623–34. doi: 10.1101/gad.831900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ponting CP. Novel domains and orthologues of eukaryotic transcription elongation factors. Nucleic Acids Res. 2002;30:3643–52. doi: 10.1093/nar/gkf498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doolittle RF. Urfs and Orfs: A Primer on How to Analyze Derived Amino Acid Sequences. University Science Books; Mill Valley: 1986. [Google Scholar]
- 9.Adkins MW, Tyler JK. Transcriptional Activators Are Dispensable for Transcription in the Absence of Spt6-Mediated Chromatin Reassembly of Promoter Regions. Molecular Cell. 2006;21:405–416. doi: 10.1016/j.molcel.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 10.Bortvin A, Winston F. Evidence that Spt6p controls chromatin structure by a direct interaction with histones. Science. 1996;272:1473–6. doi: 10.1126/science.272.5267.1473. [DOI] [PubMed] [Google Scholar]
- 11.Kaplan CD, Laprade L, Winston F. Transcription elongation factors repress transcription initiation from cryptic sites. Science. 2003;301:1096–9. doi: 10.1126/science.1087374. [DOI] [PubMed] [Google Scholar]
- 12.Yoh SM, Cho H, Pickle L, Evans RM, Jones KA. The Spt6 SH2 domain binds Ser2-P RNAPII to direct Iws1-dependent mRNA splicing and export. Genes Dev. 2007;21:160–74. doi: 10.1101/gad.1503107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andrulis ED, Werner J, Nazarian A, Erdjument-Bromage H, Tempst P, Lis JT. The RNA processing exosome is linked to elongating RNA polymerase II in Drosophila. Nature. 2002;420:837–41. doi: 10.1038/nature01181. [DOI] [PubMed] [Google Scholar]
- 14.Carpousis AJ. The Escherichia coli RNA degradosome: structure, function and relationship in other ribonucleolytic multienzyme complexes. Biochem Soc Trans. 2002;30:150–5. [PubMed] [Google Scholar]
- 15.Aravind L, Anantharaman V, Balaji S, Babu MM, Iyer LM. The many faces of the helix-turn-helix domain: transcription regulation and beyond. FEMS Microbiol Rev. 2005;29:231–62. doi: 10.1016/j.femsre.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Buttner K, Nehring S, Hopfner KP. Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat Struct Mol Biol. 2007;14:647–52. doi: 10.1038/nsmb1246. [DOI] [PubMed] [Google Scholar]
- 17.Letunic I, Copley RR, Pils B, Pinkert S, Schultz J, Bork P. SMART 5: domains in the context of genomes and networks. Nucleic Acids Res. 2006;34:D257–60. doi: 10.1093/nar/gkj079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rocha EP, Cornet E, Michel B. Comparative and evolutionary analysis of the bacterial homologous recombination systems. PLoS Genet. 2005;1:e15. doi: 10.1371/journal.pgen.0010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ichiyanagi K, Iwasaki H, Hishida T, Shinagawa H. Mutational analysis on structure-function relationship of a holliday junction specific endonuclease RuvC. Genes Cells. 1998;3:575–86. doi: 10.1046/j.1365-2443.1998.00213.x. [DOI] [PubMed] [Google Scholar]
- 20.Yoshikawa M, Iwasaki H, Shinagawa H. Evidence that phenylalanine 69 in Escherichia coli RuvC resolvase forms a stacking interaction during binding and destabilization of a Holliday junction DNA substrate. J Biol Chem. 2001;276:10432–6. doi: 10.1074/jbc.M010138200. [DOI] [PubMed] [Google Scholar]
- 21.Ariyoshi M, Vassylyev DG, Iwasaki H, Nakamura H, Shinagawa H, Morikawa K. Atomic structure of the RuvC resolvase: a holliday junction-specific endonuclease from E. coli. Cell. 1994;78:1063–72. doi: 10.1016/0092-8674(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 22.Saito A, Iwasaki H, Ariyoshi M, Morikawa K, Shinagawa H. Identification of four acidic amino acids that constitute the catalytic center of the RuvC Holliday junction resolvase. Proc Natl Acad Sci U S A. 1995;92:7470–4. doi: 10.1073/pnas.92.16.7470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshikawa M, Iwasaki H, Kinoshita K, Shinagawa H. Two basic residues, Lys-107 and Lys-118, of RuvC resolvase are involved in critical contacts with the Holliday junction for its resolution. Genes Cells. 2000;5:803–13. doi: 10.1046/j.1365-2443.2000.00371.x. [DOI] [PubMed] [Google Scholar]
- 24.Shao X, Grishin NV. Common fold in helix-hairpin-helix proteins. Nucleic Acids Res. 2000;28:2643–50. doi: 10.1093/nar/28.14.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bycroft M, Hubbard TJ, Proctor M, Freund SM, Murzin AG. The solution structure of the S1 RNA binding domain: a member of an ancient nucleic acid-binding fold. Cell. 1997;88:235–42. doi: 10.1016/s0092-8674(00)81844-9. [DOI] [PubMed] [Google Scholar]
- 26.Subramanian AR. Structure and functions of ribosomal protein S1. Prog Nucleic Acid Res Mol Biol. 1983;28:101–42. doi: 10.1016/s0079-6603(08)60085-9. [DOI] [PubMed] [Google Scholar]
- 27.Theobald DL, Mitton-Fry RM, Wuttke DS. Nucleic acid recognition by OB-fold proteins. Annu Rev Biophys Biomol Struct. 2003;32:115–33. doi: 10.1146/annurev.biophys.32.110601.142506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amblar M, Barbas A, Gomez-Puertas P, Arraiano CM. The role of the S1 domain in exoribonucleolytic activity: substrate specificity and multimerization. Rna. 2007;13:317–27. doi: 10.1261/rna.220407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schubert M, Edge RE, Lario P, Cook MA, Strynadka NC, Mackie GA, McIntosh LP. Structural characterization of the RNase E S1 domain and identification of its oligonucleotide-binding and dimerization interfaces. J Mol Biol. 2004;341:37–54. doi: 10.1016/j.jmb.2004.05.061. [DOI] [PubMed] [Google Scholar]
- 30.Hill JJ, Royer CA. Fluorescence approaches to study of protein-nucleic acid complexation. Methods Enzymol. 1997;278:390–416. doi: 10.1016/s0076-6879(97)78021-2. [DOI] [PubMed] [Google Scholar]
- 31.Stickney LM, Hankins JS, Miao X, Mackie GA. Function of the conserved S1 and KH domains in polynucleotide phosphorylase. J Bacteriol. 2005;187:7214–21. doi: 10.1128/JB.187.21.7214-7221.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Callaghan AJ, Marcaida MJ, Stead JA, McDowall KJ, Scott WG, Luisi BF. Structure of Escherichia coli RNase E catalytic domain and implications for RNA turnover. Nature. 2005;437:1187–91. doi: 10.1038/nature04084. [DOI] [PubMed] [Google Scholar]
- 33.Holm L, Sander C. Touring protein fold space with Dali/FSSP. Nucleic Acids Res. 1998;26:316–9. doi: 10.1093/nar/26.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yatime L, Schmitt E, Blanquet S, Mechulam Y. Structure-function relationships of the intact aIF2alpha subunit from the archaeon Pyrococcus abyssi. Biochemistry. 2005;44:8749–56. doi: 10.1021/bi050373i. [DOI] [PubMed] [Google Scholar]
- 35.Todone F, Brick P, Werner F, Weinzierl RO, Onesti S. Structure of an archaeal homolog of the eukaryotic RNA polymerase II RPB4/RPB7 complex. Mol Cell. 2001;8:1137–43. doi: 10.1016/s1097-2765(01)00379-3. [DOI] [PubMed] [Google Scholar]
- 36.Meka H, Werner F, Cordell SC, Onesti S, Brick P. Crystal structure and RNA binding of the Rpb4/Rpb7 subunits of human RNA polymerase II. Nucleic Acids Res. 2005;33:6435–44. doi: 10.1093/nar/gki945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-Tal N. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maclennan AJ, Shaw G. A yeast SH2 domain. Trends Biochem Sci. 1993;18:464–5. doi: 10.1016/0968-0004(93)90006-9. [DOI] [PubMed] [Google Scholar]
- 39.Grune T, Brzeski J, Eberharter A, Clapier CR, Corona DF, Becker PB, Muller CW. Crystal structure and functional analysis of a nucleosome recognition module of the remodeling factor ISWI. Mol Cell. 2003;12:449–60. doi: 10.1016/s1097-2765(03)00273-9. [DOI] [PubMed] [Google Scholar]
- 40.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–34. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 41.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–24. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 42.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter JCW, Sweet RM, editors. Methods in Enzymology. Vol. 276: Macromolecular Crystallography, part A. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 43.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55:849–61. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56:965–72. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–64. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 46.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 47.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 48.Collaborative Computational Project The CCP4 suite: programs for protein crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 49.Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 50.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 51.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–83. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; Palo Alto, CA, USA: 2002. [Google Scholar]
- 53.Hoffmann KM, Williams D, Shafer WM, Brennan RG. Characterization of the multiple transferable resistance repressor, MtrR, from Neisseria gonorrhoeae. J Bacteriol. 2005;187:5008–12. doi: 10.1128/JB.187.14.5008-5012.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.