

Abstract
Protease activated receptor-1 (PAR1) mediates barrier protective signaling of activated protein C (APC) in human endothelial cells in vitro and may contribute to APC’s beneficial effects in patients with severe sepsis. Mouse models are of key importance for translational research but species differences may limit conclusions for the human system. We analyzed whether mouse APC can cleave, activate and induce signaling through murine PAR1 and tested in newly established mouse models if long term infusion of APC prevents from vascular leakage. Cell surface immunoassays demonstrated efficient cleavage of endogenous murine endothelial PAR1 by either murine or human APC. Pharmacological concentrations of APC of either species had powerful barrier protective effects on cultured murine endothelial cells that required PAR1 cleavage. Vascular endothelial growth factor-mediated hyperpermeability in the skin was reduced by either endogenously generated as well as directly infused recombinant mouse APC in wild type mice. However APC did not significantly alter the vascular barrier function in PAR1-deficient mice. In endotoxin challenged mice, infused APC significantly prevented from pulmonary fluid accumulation in the wild type but not in mice lacking PAR1. Our results directly show that murine APC cleaves and signals through PAR1 in mouse endothelial cells. APC reduces vascular permeability in mouse models and PAR1 plays a major role in mediating these effects. Our in vitro and in vivo data support the paradigm that PAR1 contributes to protective effects of APC on vascular barrier integrity in sepsis.
Keywords: activated protein C; endothelial cells; protease-activated receptor-1; sepsis, vascular barrier
Infusion of recombinant human activated protein C (APC) can reduce mortality in patients with severe sepsis (1). APC downregulates the generation of thrombin, the key procoagulant enzyme of the blood coagulation system (2, 3) but these anticoagulant effects of APC are unlikely to explain its benefit in septic patients because other anticoagulants improved the sepsis related coagulopathy but failed to improve survival of septic patients (4, 5). Consistent with this conclusion, recent results show that a non-anticoagulant variant of APC can protect mice in an endotoxemia model similarly well as wildtype APC (6).
APC can mediate a number of potentially protective effects in human endothelial cells that include stabilization of barrier integrity, anti-apoptotic effects, and downregulation of adhesion receptors (7). These cytoprotective activities of APC require binding to the endothelial protein C receptor (EPCR) and are at least in part mediated by signaling through the thrombin receptor protease activated receptor-1 (PAR1), a G protein-coupled receptor that is activated by proteolytic cleavage of its N-terminal exodomain (8, 9). PAR1 has been implicated in protective in vivo effects of APC in mouse models, including increased survival of APC treated mice in endotoxemia (6). Both human and mouse APC has been used in mouse models but it is unknown whether significant species differences exist in APC signaling. PAR1 is known to play distinct roles in different species, e.g. PAR1 mediates platelet activation in humans but not in mice (8). So far, it has never been tested directly whether human or mouse APC can cleave and activate PAR1 on mouse endothelial cells. Indirect evidence that in the mouse APC cleaves and activates PAR1 was drawn from in vivo studies (10) using PAR1 blocking antibodies. However, recent reports show that APC binding to EPCR can mediate protective PAR1-dependent signaling even if this receptor gets cleaved by another protease such as thrombin (11). Since mouse models are widely used in translational research to understand how APC therapy improves survival of septic patients more direct insights in whether the APC-PAR1 pathway is conserved between mouse and humans is important.
Dysfunction of the vascular barrier is a key event in the pathogenesis of sepsis and plays an important role in the development of organ dysfunction, such as the lung injury-triggered development of acute respiratory distress syndrome. Previous studies have shown that APC can attenuate acute lung injury (12, 13). Enhancement of endothelial barrier integrity is a highly sensitive downstream effect of APC-PAR1 signaling in cultured human endothelial cells that requires crossactivation of sphingosine 1-phosphate (S1P) receptors (14, 15). Given that S1P can reduce the vascular leak in animal models of acute lung injury (16, 17), it is tempting to speculate that vascular barrier protection may contribute to beneficial effects of APC treatment in sepsis.
Here we show that in cultured mouse endothelial cell lines APC directly cleaves and activates endogenous PAR1 which leads to reduced permeability of an endothelial cell monolayer. Infused or in vivo generated APC significantly enhanced vascular barrier integrity in wildtype mice but not in PAR1-deficient mice. The findings support the concept that PAR1-dependent protection of vascular barrier integrity contributes to beneficial effects of APC in sepsis.
METHODS
Reagents and Antibodies
Human plasma-derived APC and PAR1 agonist peptide were as described previously (9, 14, 18-20). Recombinant mouse APC was made as described (21). Human WE-thrombin was kindly provided by Dr. Di Cera (Washington University). All in vitro experiments involving stimulation with APC included hirudin (Calbiochem, La Jolla, CA). Mouse thrombin was from Haematologic Technologies (Essex Junction, VT). The S-19 polyclonal goat anti-mouse PAR1 (Lot# L1205) and its commercial blocking peptide (RSFFLRNPSENTFELVPLGDE) were from Santa Cruz (Santa Cruz, CA). Additional peptides corresponding to the N-terminus of mature mouse PAR1 were custom synthesized (CHI Scientific Inc., MA USA) and used for mapping the PAR1 epitope recognized by the S-19 anti-PAR1 antibody. Mouse plasma IL-6 was quantified by DuoSet ELISA (R&D Systems, Minneapolis, MN) and thrombin antithrombin complexes by the ELISA for TAT complexes (Enzyme Research Laboratories, South Bend, IN) following the protocols of the manufactures. Mouse APC plasma levels were determined as described (22). In brief, mouse blood was collected into vials containing 0.1 M citrate and 10 mM benzamidine (final concentrations). After centrifugation, the plasma samples were loaded onto wells precoated with monoclonal anti-mouse PC AMGDPC1587 (kindly provided by Dr. Esmon; Oklahoma Health Sciences Center), incubated for 2 h, and extensively washed with Tris buffer containing 0.05% Tween 20. Serial dilutions of recombinant mouse APC were used to prepare a standard curve. Amidolytic activity of pulled down proteins was quantified by Spectrozyme PCa (#336, American Diagnostica, Stamford, CT).
Cell Culture
Transduced mouse endothelial cell lines MS1 and b.End3 were from the American Type Culture Collection (Manassas, VA) and were grown in Dulbecco’s Modified Eagle Media (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT). Results obtained with both mouse cell lines were similar throughout all in vitro assays except for permeability assays where only the b.End3 sufficiently attached to the microporous membrane. Plasmid transfection was performed in human EA.hy926 cells (23). Cells were plated in tissue culture medium together with plasmids and Lipofectamine™2000 (Invitrogen, Carlsbad, CA) which was replaced with growth medium 6 h after transfection and the cells were used for experiments two days later. The plasmids contained the full length cDNA coding for mouse PAR1 in the pcDNA3.1/Zeo(+) vector (Invitrogen, Carlsbad, CA). Sequence analysis confirmed the consistency with gene bank entry #W20934. Full length human PAR4 in the same vector was used as a control.
Permeability Assay and Surface Immunoassay
The macromolecular monolayer permeability was assessed as described (14). Briefly b.End3 endothelial cells were grown to subconfluence for two days on polycarbonate membrane transwells (Costar, 3 μM pore size) serum starved for 30 min in absence or presence of the PAR1 blocking antibody S19. After a 3 h incubation with agonists all media were removed and DMEM containing 0.67 mg/mL of Evans Blue and 4% bovine serum albumin was added to the transwell compartment above the microporous membrane. Changes in endothelial permeability were quantified by measuring the increase in absorbance at 650 nm of the medium (DMEM containing 0.4% bovine serum albumin) placed into the bottom well. Cell surface PAR1 was quantified by an immunoassay as described (20) except that S-19 goat anti-mouse PAR1 at 2 μg/ml and a peroxidase-coupled rabbit anti-goat IgG (Calbiochem) were used. Modifications of this assay were used in order to map (the) binding site(s) of S-19 on mouse PAR1. On one hand, it was tested whether an excess (100 nM) of peptide can compete with endogenously expressed mouse PAR1 on MS1 cells for S-19 binding. On the other hand, it was tested whether the immobilized peptides are able to bind to S-19.
Immunoprecipitation, and Western Blotting
Cell surface proteins were biotinylated as described (20). PAR1 was immunoprecipitated with S-19 (0.5 μg/ml) and protein G-agarose (Calbiochem). Following extensive washing, eluted proteins were separated by SDS-PAGE, transferred to Immobilon-P-membranes (Millipore, Bedford, MA) and biotinylated proteins were visualized with Streptavidin-HRP (Zymed, San Francisco, CA) and the Femto detection system (Pierce). Western blotting for mitogen-activated protein kinase phosphorylation was as reported (18). Stripped membranes were re-probed with anti-β-actin (Sigma) and analyzed as loading controls.
Mouse Models for Vascular Barrier Integrity
All studies were approved by The Scripps Research Institute Animal Care and Use Committee and comply with National Institutes of Health guidelines. Wildtype C57BL/6 and PAR1-deficient mice, backcrossed for >10 generations into the C57BL/6 background, were as described (24) and bred in-house. Jugular vein catheters were surgically implanted in age-matched 8 to 10 week old male mice 4-7 days before experiments. Implanted mice were free to move and without any signs of distress and systemic markers for inflammation (IL-6) and coagulation (TAT complexes). A microprocessor controlled syringe pump infused 300 μL per hour of vehicle (physiological saline) either alone or containing WE (200 μg/kg/h) or recombinant mouse APC (120 μg/kg/h). Up to 16 mice were infused simultaneously. LPS (E. coli serotype O111:B4; Sigma, St. Louis, MO) injected mice were sacrificed at the end of the infusion. To quantify vascular leakage in the skin, intravascular albumin was labeled with Evans blue (1 mg in 100 μl intravenous). Recombinant murine vascular endothelial growth factor (VEGF165; Peprotech, Rocky Hill, NJ) was intradermally injected (25 μl; 1 μg/ml) into the preshaved abdominal skin and the injection sites were removed by dermal punch biopsies (diameter 6 mm) 30 min later after the animal was sacrificed. The skin samples were incubated in 200 μl of formamide at 56°C for 24 h and the extracted Evans blue content was quantified by measuring the OD at 620 nm. Results from six samples of each animal were averaged and used as a single data point for further data analysis. In endotoxin (LPS) challenged mice (20 mg/kg; E.coli serotype O111:B4: Sigma, St. Louis, MO) pulmonary edema was quantified by measuring the wet to dry weight ratio of the left lung (weight obtained immediately after extirpation divided by the weight assessed after 4 days of drying at 60°C). For histological analysis the cardiac lung lobe was formaldehyde fixed, paraffin embedded and 10 μm microsections stained with Hematoxylin Eosin were analyzed by light microscopy (Olympus BX60, Olympus America INC, San Diego, CA).
Statistical Analysis
Data analysis was performed using the NCSS Statistical & Power Analysis Software. A two-sample two-tailed homoscedastic t-test was used to compare groups of two, parametric ANOVA was used to compare equally powered groups of more than two.
RESULTS
APC Induces Barrier Function in Mouse Endothelial Cells
In a first series of experiments we analyzed effects of APC on endothelial barrier integrity in a dual chamber system. Mouse APC enhanced the barrier function in a dose dependent manner in a mouse brain endothelial cell line (Fig. 1). Mouse APC was slightly more efficient than human APC in this assay. The anti human PAR1 antibody H-111 (Santa Cruz) has been previously reported to block cleavage and was used to establish that APC-PAR1 signaling is required for anti apoptotic effects of APC in mouse neuronal cells (10). Unfortunately, recent lots of this polyclonal antibody did not bind to PAR1 in cultured mouse endothelial cell lines (data not shown), likely due to a change in the properties of this polyclonal antibody raised against the N-terminus of human PAR1. Thus, novel tools had to be established in order to test whether APC can directly cleave PAR1 and thereby enhance the endothelial barrier function in mouse cells.
Figure 1. Effect of APC on endothelial barrier permeability in mouse endothelial cells in tissue culture.
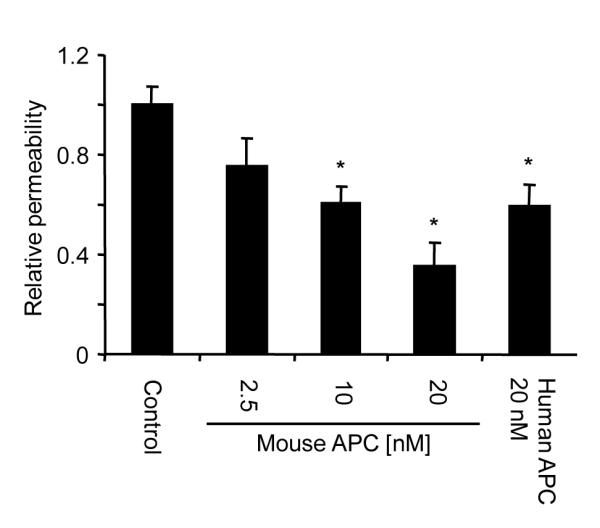
Subconfluent murine endothelial cells (b.End3) were incubated for 3 h with the indicated agonists in a dual chamber system followed by analysis of permeability. Values are means±SEM, n=6 to 10, *P<0.05.
APC Cleaves Mouse PAR1 and the APC-cleaved Receptor is Retained on the Cell Surface
To test whether APC directly cleaves mouse PAR1 we established an immunoassay to quantify binding of anti-PAR1 to the endothelial cell surface. Transfection studies overexpressing mouse PAR1 in the human endothelial cell line EA.hy926 established specificity of the S-19 polyclonal goat anti-mouse PAR1 (Fig. 2A). Mouse thrombin efficiently removed anti-PAR1 epitopes from cultured mouse endothelial cells whereas mouse APC had no effect (Fig. 2B). These results either suggest that APC cannot cleave mouse PAR1 or that APC-cleaved receptors are not efficiently removed from the cell surface, as described recently for human PAR1 (20). To clarify if cleavage or internalization of mouse PAR1 explains the reduced binding of S-19 in thrombin treated cells we mapped the S-19 epitope(s) (Fig. 2C). The commercially available S-19 blocking peptide (blocked S-19 — used throughout this study), was sequenced and found to contain 20 amino acids beginning with R41 at the scissile bond that leads to receptor activation. This peptide as well as the shorter, custom synthesized peptide NE-15 (amino acids 47 to 61) competes with S-19 binding to endothelial expressed PAR1 and support S-19 binding to the immobilized peptide. In contrast neither the peptide corresponding to the full length of the mouse PAR1 upstream of the scissile bond (amino acid 22 to 41) nor any other of the custom made peptides bound to S-19 in both assays. These data show that S-19 goat anti-mouse PAR1 antibody is not expected to be cleavage sensitive and that it specifically binds to an epitope containing V56 and P57. Biotinylated cell surface exposed receptors were analyzed by SDS-PAGE to assess agonist dependent cleavage. Densitometric analysis of immunoreactive bands confirmed that the total amount of mouse PAR1 on the cell surface is neither affected by mouse nor by human APC but significantly reduced upon incubation with mouse thrombin (Fig. 2D), consistent with the findings from the surface immunoassay. Importantly, both thrombin and APC led to the appearance of a smaller cleavage product with an average apparent size of approximately 60 kDa. In order to estimate cleavage efficiency we determined the proportion of immunoreactive products above and below PAR1’s native average size of 82 kDa. This proportion was significantly reduced not only upon incubation with thrombin but also after APC incubation, indicating that APC does cleave mouse PAR1. As found previously for human PAR1 (20), APC-cleaved mouse PAR1 was surface retained even after a short incubation with high dose thrombin (Fig. 2E) and when cells were coincubated with thrombin and APC or zymogen protein C (not shown). Taken together, these data show for the first time directly, that mouse PAR1 can in fact be cleaved by APC and APC-cleaved receptors are retained on the cell surface similar as described for human PAR1.
Figure 2. Cleavage of cell surface-exposed mouse PAR1 by APC.

A) The human EA.hy926 endothelial cells were transfected with cDNA encoding mouse PAR1 (mPAR1) or control cDNA followed two days later by a 30 minutes agonist incubation. Cell surface binding of S-19 anti-mouse PAR1 and immunizing peptide-blocked S-19 is shown. B) Expression of PAR1 on the surface of a monolayer of murine MS1 endothelial cells was analyzed after a 3 h incubation with the indicated agonists by cell surface ELISA using anti-mouse PAR1 S-19. APC was used at 20 nM. C) The indicated peptides corresponding to the N-terminal amino acid sequence of mouse PAR1 were analyzed for binding to S-19. In a competition assay S-19 binding to cell surface expressed PAR1 was tested in the presence of excess peptide. Direct binding of S-19 to immobilized peptides was analyzed in a peptide binding assay. D) Following a 3 h incubation with vehicle, 20nM of APC or 1nM of mouse thrombin the cell surface proteins were biotinylated and PAR1 was immunoprecipitated using S-19. Biotinylated proteins were detected on blots with peroxidase-coupled streptavidin. Bands are shown from a representative experiment in the upper panel and density between 55 and 120 kDa was quantified in three independent repeat experiments (middle panel). The ratio of density above and below PAR1’s native average size of 82 kDa is shown in the lower panel. E) MS1 cells were incubated for 3 h with control or APC followed by an additional 30 min incubation with vehicle alone or mouse thrombin (0.5 nM). Binding of S-19 anti-PAR1 was analyzed by cell surface ELISA. Means±SEM are shown in all panels, n=3 (A), 4 (B), 3 (C,D) and 16 (E), P values are indicated.
APC Signaling and Barrier Protection in Mouse Endothelial Cells Require PAR1 Cleavage
To test the hypothesis that PAR1 cleavage is indeed necessary for APC’s effects on the barrier function of cultured mouse endothelial cells, we screened several antibodies raised against PAR1 for their ability to block PAR1 cleavage-mediated signaling. Only S-19 significantly reduced thrombin- but not PAR1 agonist peptide-induced phosphorylation of Erk1/2 in mouse endothelial cells (Fig. 3A). As expected, preincubation of S-19 with its immunizing peptide rendered the antibody inefficient and ruled out non specific effects of S-19. Mouse APC but not human APC induced detectable Erk1/2 phosphorylation in our assay conditions (Fig. 3B), consistent with less efficient signaling of human APC in mouse cells. Preincubation with S-19 blocked Erk1/2 phosphorylation in response to mouse APC. Most importantly, the barrier enhancing effect of mouse APC in cultured mouse endothelial cells was found to be blocked in the presence of anti PAR1 S-19 (Fig. 3C), indicating that PAR1 cleavage is required for the induction of endothelial barrier function by APC in cultured mouse endothelial cells.
Figure 3. Anti-PAR1 S-19 blocks PAR1 signaling and APC-mediated barrier protection in mouse endothelial cells.

A) ERK1/2 phosphorylation was analyzed after 7 min incubation with mouse thrombin (1 nM) or the PAR1 agonist peptide TFLLRNPNDK (PAR1 AP; 20 μM). Where indicated anti PAR1 S-19 (25 μg/ml) alone or blocked by preincubation with excess of immunizing peptide was added 15 min prior to the agonists. A representative blot is shown on the left side, quantitative analyzes of S-19 immunoreactive bands are given in the right part of the figure (means±SEM) and ANOVA reviled that S-19 only significantly affects the thrombin response (*P<0.05). B) ERK1/2 phosphorylation in response to the indicated agonists (7 min) was analyzed in the absence or presence of S-19. APC was used at 20 nM. Representative blot shown in the upper part, quantification (means±SEM) of 4 independent experiments in the lower part, P value indicated. C) Subconfluent murine endothelial cells (b.End3) were incubated for 3 h with mouse APC (20 nM) in the absence or presence of S-19 in a dual chamber system followed by analysis of permeability. Means±SEM, n=9; P value indicated.
Vascular Barrier Protection by APC in the Skin Requires PAR1
APC-PAR1 has powerful endothelial barrier protective effects in vitro but relatively high concentrations of APC (>2.5 nM) were required (Fig. 1). Thus, we first attempted to test whether APC similarly affects the vascular barrier in vivo. Based upon our in vitro studies we expected that a prolonged exposition for several hours to APC may be necessary and that endogenously generated APC may be more efficient compared to exogenous APC (19). Efficient endothelial generation of APC was reported from baboons infused with mutated, anticoagulant thrombin (WE) and WE has a much longer half life in the circulation than APC (25, 26). Human WE efficiently activated mouse protein C on cultured endothelial cells (Fig. 4A) and it was about 500 times less efficient in cleaving mouse PAR1 if compared to non mutated mouse thrombin (Fig. 4B). Bolus injections of WE in mice resulted in efficient and sustained in vivo generation of APC (Fig. 4C) which was comparable to reports from baboons (26). A continuous infusion with 200 μg/kg/h of WE (without bolus) yielded plasma levels of around 20 ng/ml, comparable to levels achieved in septic patients treated with recombinant APC (Fig. 4D).
Figure 4. Human anticoagulant mutant thrombin (WE) efficiently activates mouse protein C in vitro and in vivo but poorly activates PAR1.

A) Mouse endothelial cells (MS1) were incubated for 3 h with indicated concentrations of WE in the presence and absence of 80 nM of mouse protein C (means±SEM, n=6). B) Expression of PAR1 on the surface of a monolayer of murine MS1 endothelial cells was analyzed after a 3 h incubation with the indicated agonists by cell surface ELISA using anti-mouse PAR1 S-19 (means±SEM, n=3). C) Plasma APC levels after a bolus injection of the indicated amounts of WE (means±SEM, n=3) D) In two mice plasma APC levels were assessed immediately before, during and after a 4.5 h infusion with WE.
To directly test whether exogenous and endogenously generated APC can protect vascular barrier integrity, we analyzed vascular endothelial growth factor (VEGF)-mediated hyperpermeability in a skin model derived from the classic Miles assay (Fig. 5A). The dermal vascular bed is known to highly express EPCR as well as PAR1 and is therefore expected to support local APC generation and APC-PAR1 signaling. Evans blue was used to label intravascular albumin and control experiments demonstrated that the results indeed reflect time-dependent extravasation in response to increased vascular permeability (not shown). Infusion with WE at a dose that leads to plasma APC levels comparable to APC-treated patients had a highly significant protective effect against VEGF-induced hyperpermeability (Fig. 5B). Direct infusion of mouse APC at a dose (120 μg/kg/h) comparable to the one licensed for treating septic patients also had a significant vascular barrier protective effect. To test if effects of APC depend on PAR1 a set of wildtype and PAR1 deficient mice were parallel infused with either saline alone or APC. Only in mice expressing PAR1 APC had significant effects (Fig. 5C). These results demonstrate that APC can enhance vascular barrier integrity in vivo and that PAR1 plays a major role mediating this effect.
Figure 5. PAR1 dependent vascular barrier protection by APC in a modified Miles assay.

A) Male mice were infused with saline, murine APC or WE for 4.5 h and Evans blue was intravenously injected 30 min prior to the end of the infusion. At the end of the infusion 25 ng of murine VEGF was intradermally injected and 30 min later the injection sites were removed by punch biopsies. B) C57BL/6 wildtype mice were infused in parallel. Evans blue content in VEGF-injected skin is shown. C) Evans blue content of an additional set of parallel infused C57BL/6 wildtype and PAR1 deficient (PAR1 KO) animals. Means±SEM are shown in all panels, n=10 per group (B, C), P values are indicated.
PAR1-dependent Protection from Pulmonary Edema by APC in an Endotoxemia Model
To test if APC has similar vascular barrier protective effects in systemic inflammation we assessed pulmonary edema formation at an early time point in an endotoxemia model (Fig. 6A). Intraperitoneal injection of a lethal dose of lipopolysaccharide (LPS) led to a strong increase of pulmonary fluid content after 5 h and APC infusion significantly reduced fluid accumulation in wildtype animals whereas it lacked a significant protective effect in PAR1 deficient mice (Fig. 6B). Histological analysis of the lungs performed on a subset of the animals revealed interstitial and focal alveolar edema in saline infused wild type mice as well as in both APC treated and untreated PAR1 deficient mice whereas APC infused wild type mice showed minimal to no alterations (Fig. 6C), consistent with the measurements of the wet to dry weight ratio. Interleukin-6 (IL-6) plasma levels were similarly high in all groups (Fig. 6D). These results indicate that APC leads to reduced fluid extravasation without affecting a marker of systemic inflammation showing that PAR1 plays a central role in APC mediated enhancement of endothelial barrier integrity in a sepsis model.
Figure 6. Infusion of APC reduces pulmonary fluid accumulation in a mouse endotoxemia model dependent on PAR1.

A) Male C57BL/6 wildtype or PAR1-deficient (PAR1 KO) mice were infused with either saline alone or with murine APC (120 μg/kg/h) for 5 h via a central venous catheter (10-12 mice per group). LPS (20 mg/kg) was injected intraperitoneally one hour after the start of the infusion. Lungs were dissected for wet/dry weight ratio determination at the end of the infusion period. B) Lung wet weight/dry weight ratios are shown (means±SEM, P values indicated). Results from non LPS injected animals are shown as a reference. C) Hematoxylin-Eosin stained representative pulmonary tissue sections show normal alveolar tissue (arrow) alveolar edema (gray arrow) and alveolar flooding (arrowhead). D) IL-6 plasma levels at the end of the infusion are shown (means±SEM).
DISCUSSION
The mouse has become a unique and important tool for studying human diseases and for screening and testing putative drugs. However, species differences limit projecting conclusions drawn in one species onto the other. For example, thrombin signaling is mediated by PAR1 in human platelets and by PAR3 and PAR4 in mouse platelets (8). Here we demonstrate for the first time directly that mouse APC cleaves mouse PAR1 in endothelial cells. Using an antibody that blocks PAR1 cleavage-mediated signaling we provide evidence that APC can directly enhance the barrier of cultured endothelial cells by signaling through PAR1. We further show that APC-cleaved mouse PAR1 in contrast to thrombin cleaved mouse PAR1 is cell surface retained as described in human cell lines (20). Thus, PAR1 activation is linked to the protein C pathway similarly in humans and mice.
Previous studies in mouse models of cerebral ischemia demonstrated protective signaling of APC through PAR1 (10, 27). Furthermore, APC treatment improved survival in an endotoxemia model in wildtype mice but not in PAR1-deficient mice (6). In wildtype mice APC treatment altered endotoxin-induced vascular hyperpermeability in the kidney, resulting in decreased permeability in small vessels but increased or unchanged permeability in medium sized or bigger vessels (6). However, this study did not analyze whether APC’s effects on vascular permeability require PAR1. Here we extend these observations by demonstrating that in mouse models APC infusion protects from endotoxin-mediated fluid accumulation in a whole organ, i.e. the lung or the skin. In addition, we show for the first time that PAR1 plays a key role mediating the vascular barrier protective properties of infused APC in vivo. Taken together with our finding that PAR1-dependent APC signaling reduces monolayer permeability in cultured mouse endothelial cells, these results strongly support the concept that PAR1 plays a major role mediating protective effects of APC on the vascular barrier integrity in vivo. How APC-PAR1 signaling leads to reduced permeability of the vascular barrier in vivo remains to be established. In tissue culture the endothelial barrier enhancing effect of APC requires the activity of cellular sphingosine kinase, sphingosine 1-phosphate (S1P)-receptor-1 transactivation, and activity of the small GTPase Rac1 (14, 15). Given that S1P reduces the increased vascular permeability in animal models of acute inflammatory lung injury (28), it seems likely that S1P pathway- and Rac1-dependent changes of the organization of the endothelial actin cytoskeleton and cell-cell contacts also play a role in APC’s vascular barrier protective activity in vivo.
Our results demonstrate that PAR1 plays a major role in mediating protective effects of infused APC on pulmonary fluid accumulation in a model of endotoxemia. The inflammatory response in this model of systemic inflammation includes strong upregulation of tissue factor expression on monocytic cells leading to thrombin generation (29). The reported PAR1 dependent effects could therefore either be direct by APC-PAR1 signaling or indirect by downregulation of thrombin generation and reduced thrombin-PAR1 signaling. To avoid such strong systemic responses we developed an animal model quantifying the dermal vascular barrier function. The dermis was chosen because of its easy accessibility, the high expression levels of EPCR and PAR1 within its vascular bed and its clinical relevance in protein C deficiency. VEGF acts directly on endothelial cells and mediates a well defined hyperpermeability response in vitro and in vivo (30). The finding that APC infusion protects vascular barrier integrity in both the endotoxemia and the skin model in a PAR1 dependent manner while PAR1-deficiency itself does not affect barrier integrity in either model, strongly argues that PAR1 signaling by the infused APC is beneficial. It remains to be established whether any remaining protective effects in the absence of PAR1 require APC’s anticoagulant activity or are mediated by novel non PAR1-dependent APC signaling mechanisms in endothelial cells or other cell types such as leukocytes.
Given PAR1’s role mediating vascular barrier protection by exogenously administered APC, it seems likely that endogenously generated APC also mediates protective effects through PAR1, especially in view of the finding that protein C activation on the endothelial surface is coupled to highly efficient PAR1-dependent barrier protective signaling by the generated APC in cultured endothelial cells (19). Our new data add to accumulating evidence that similar to thrombin itself the prototypical thrombin receptor PAR1 has dual-faced physiological roles, mediating both pro-inflammatory and protective effects. Originally PAR1 has been described as a receptor that mediates pro-inflammatory responses (31). Even though thrombin-PAR1 signaling is indeed pro-inflammatory in mouse models of glomerulonephritis (32), renal ischemia-reperfusion injury (33), and inflammatory bowel disease (34) PAR1 deficiency does not affect survival in mouse models of endotoxemia (6, 35, 36). Thus any beneficial effects of the absence of pro-inflammatory PAR1 signaling may be offset by the absence of cytoprotective PAR1-dependent signaling in the endotoxemia models. In a mouse sepsis model a pepducin PAR1 antagonist improved survival when the antagonist was administered immediately after surgery but not when given at later time points, whereas a PAR1 agonist was beneficial when given at late time points (37). These data indeed support the conclusion that PAR1 has opposite roles in systemic inflammation that may depend on the stage of the inflammatory response.
In conclusion, our results in complementary cell and mouse models demonstrate that PAR1 plays similar roles in endothelial barrier protective signaling by APC in humans and mice and they support the concept that APC by signaling through PAR1 maintains the integrity of the vascular barrier in systemic inflammation. The protein C pathway may have a role not only in containing the thrombotic response but also in preventing excessive spreading of vascular barrier dysfunction during inflammation.
Acknowledgments
Grant support This study was supported by National Institutes of Health grants HL 73318 (to M.R.) and HL 31950 (to J.H.G.), grants from the University of Zurich, Switzerland (Zangger-Stiftung), the Swiss Foundation for Medical-Biological Grants (SSMBS) and the Mach-Gaensslen Stiftung (to R.A.S.), and an Austrian fellowship, Erwin Schrödinger-Auslandsstipendium J2413-B13 (to C.F.).
REFERENCES
- 1.Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 2.Esmon CT. The protein C pathway. Chest. 2003;124:26S–32S. doi: 10.1378/chest.124.3_suppl.26s. [DOI] [PubMed] [Google Scholar]
- 3.Dahlback B, Villoutreix BO. Regulation of blood coagulation by the protein C anticoagulant pathway: novel insights into structure-function relationships and molecular recognition. Arterioscler Thromb Vasc Biol. 2005;25:1311–1320. doi: 10.1161/01.ATV.0000168421.13467.82. [DOI] [PubMed] [Google Scholar]
- 4.Warren BL, Eid A, Singer P, et al. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. Jama. 2001;286:1869–1878. doi: 10.1001/jama.286.15.1869. [DOI] [PubMed] [Google Scholar]
- 5.Abraham E, Reinhart K, Opal S, et al. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. Jama. 2003;290:238–247. doi: 10.1001/jama.290.2.238. [DOI] [PubMed] [Google Scholar]
- 6.Kerschen EJ, Fernandez JA, Cooley BC, et al. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. The Journal of experimental medicine. 2007;204:2439–2448. doi: 10.1084/jem.20070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 8.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 9.Riewald M, Petrovan RJ, Donner A, et al. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880–1882. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- 10.Guo H, Liu D, Gelbard H, et al. Activated protein C prevents neuronal apoptosis via protease activated receptors 1 and 3. Neuron. 2004;41:563–572. doi: 10.1016/s0896-6273(04)00019-4. [DOI] [PubMed] [Google Scholar]
- 11.Bae JS, Yang L, Rezaie AR. Lipid raft localization regulates the cleavage specificity of protease activated receptor 1 in endothelial cells. J Thromb Haemost. 2008;6:954–961. doi: 10.1111/j.1538-7836.2008.02924.x. [DOI] [PubMed] [Google Scholar]
- 12.Robriquet L, Collet F, Tournoys A, et al. Intravenous administration of activated protein C in Pseudomonas-induced lung injury: impact on lung fluid balance and the inflammatory response. Respir Res. 2006;7:41. doi: 10.1186/1465-9921-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abraham E. Effects of recombinant human activated protein C in human models of endotoxin administration. Proc Am Thorac Soc. 2005;2:243–247. doi: 10.1513/pats.200501-004AC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105:3178–3184. doi: 10.1182/blood-2004-10-3985. [DOI] [PubMed] [Google Scholar]
- 15.Finigan JH, Dudek SM, Singleton PA, et al. Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. J Biol Chem. 2005;280:17286–17293. doi: 10.1074/jbc.M412427200. [DOI] [PubMed] [Google Scholar]
- 16.McVerry BJ, Garcia JG. Endothelial cell barrier regulation by sphingosine 1-phosphate. J Cell Biochem. 2004;92:1075–1085. doi: 10.1002/jcb.20088. [DOI] [PubMed] [Google Scholar]
- 17.Peng X, Hassoun PM, Sammani S, et al. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. American journal of respiratory and critical care medicine. 2004;169:1245–1251. doi: 10.1164/rccm.200309-1258OC. [DOI] [PubMed] [Google Scholar]
- 18.Feistritzer C, Lenta R, Riewald M. Protease-activated receptors-1 and -2 can mediate endothelial barrier protection: role in factor Xa signaling. J Thromb Haemost. 2005;3:2798–2805. doi: 10.1111/j.1538-7836.2005.01610.x. [DOI] [PubMed] [Google Scholar]
- 19.Feistritzer C, Schuepbach RA, Mosnier LO, et al. Protective signaling by activated protein C is mechanistically linked to protein C activation on endothelial cells. J Biol Chem. 2006;281:20077–20084. doi: 10.1074/jbc.M600506200. [DOI] [PubMed] [Google Scholar]
- 20.Schuepbach RA, Feistritzer C, Brass LF, et al. Activated protein C-cleaved protease activated receptor-1 is retained on the endothelial cell surface even in the presence of thrombin. Blood. 2007;111:2667–2673. doi: 10.1182/blood-2007-09-113076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fernandez JA, Xu X, Liu D, et al. Recombinant murine-activated protein C is neuroprotective in a murine ischemic stroke model. Blood Cells Mol Dis. 2003;30:271–276. doi: 10.1016/s1079-9796(03)00034-2. [DOI] [PubMed] [Google Scholar]
- 22.Li W, Zheng X, Gu J, et al. Overexpressing endothelial cell protein C receptor alters the hemostatic balance and protects mice from endotoxin. J Thromb Haemost. 2005;3:1351–1359. doi: 10.1111/j.1538-7836.2005.01385.x. [DOI] [PubMed] [Google Scholar]
- 23.Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci U S A. 1983;80:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riewald M, Ruf W. Protease-activated receptor-1 signaling by activated protein C in cytokine-perturbed endothelial cells is distinct from thrombin signaling. J Biol Chem. 2005;280:19808–19814. doi: 10.1074/jbc.M500747200. [DOI] [PubMed] [Google Scholar]
- 25.Cantwell AM, Di Cera E. Rational design of a potent anticoagulant thrombin. J Biol Chem. 2000;275:39827–39830. doi: 10.1074/jbc.C000751200. [DOI] [PubMed] [Google Scholar]
- 26.Gruber A, Fernandez JA, Bush L, et al. Limited generation of activated protein C during infusion of the protein C activator thrombin analog W215A/E217A in primates. J Thromb Haemost. 2006;4:392–397. doi: 10.1111/j.1538-7836.2006.01760.x. [DOI] [PubMed] [Google Scholar]
- 27.Cheng T, Liu D, Griffin JH, et al. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. 2003;9:338–342. doi: 10.1038/nm826. [DOI] [PubMed] [Google Scholar]
- 28.McVerry BJ, Garcia JG. In vitro and in vivo modulation of vascular barrier integrity by sphingosine 1-phosphate: mechanistic insights. Cell Signal. 2005;17:131–139. doi: 10.1016/j.cellsig.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 29.Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 30.Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature. 2005;437:497–504. doi: 10.1038/nature03987. [DOI] [PubMed] [Google Scholar]
- 31.Coughlin SR, Camerer E. PARticipation in inflammation. J Clin Invest. 2003;111:25–27. doi: 10.1172/JCI17564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cunningham MA, Rondeau E, Chen X, et al. Protease-activated receptor 1 mediates thrombin-dependent, cell-mediated renal inflammation in crescentic glomerulonephritis. The Journal of experimental medicine. 2000;191:455–462. doi: 10.1084/jem.191.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sevastos J, Kennedy SE, Davis DR, et al. Tissue factor deficiency and PAR-1 deficiency are protective against renal ischemia reperfusion injury. Blood. 2007;109:577–583. doi: 10.1182/blood-2006-03-008870. [DOI] [PubMed] [Google Scholar]
- 34.Vergnolle N, Cellars L, Mencarelli A, et al. A role for proteinase-activated receptor-1 in inflammatory bowel diseases. J Clin Invest. 2004;114:1444–1456. doi: 10.1172/JCI21689. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Pawlinski R, Pedersen B, Schabbauer G, et al. Role of tissue factor and protease-activated receptors in a mouse model of endotoxemia. Blood. 2004;103:1342–1347. doi: 10.1182/blood-2003-09-3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Camerer E, Cornelissen I, Kataoka H, et al. Roles of protease-activated receptors in a mouse model of endotoxemia. Blood. 2006;107:3912–3921. doi: 10.1182/blood-2005-08-3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaneider NC, Leger AJ, Agarwal A, et al. ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol. 2007;8:1303–1312. doi: 10.1038/ni1525. [DOI] [PMC free article] [PubMed] [Google Scholar]