

Abstract
Oxidations of three porphyrin-iron(III) complexes (1) with ferric perchlorate, Fe(ClO4)3, in acetonitrile solutions at −40 °C gave metastable porphyrin-iron(IV) diperchlorate complexes (2) that isomerized to known iron(III) diperchlorate porphyrin radical cations (3) when the solutions were warmed to room temperature. The 5,10,15,20-tetraphenylporphyrin (TPP), 5,10,15,20-tetramesitylporphyrin (TMP), and 2,3,7,8,12,13,17,18-octaethylporphyrin (OEP) systems were studied by UV-visible spectroscopy. Low temperature NMR spectroscopy and effective magnetic moment measurements were possible with the TPP and TMP iron(IV) complexes. Reactions of two corrole systems, 5,10,15-tris(pentafluorophenyl)corrole (TPFC) and 5,15-bis(pentafluorophenyl)-10-p-methoxyphenylcorrole (BPFMC), also were studied. The corrole-iron(IV) chlorides reacted with silver salts to give corrole-iron(IV) complexes. The corrole-iron(IV) nitrate complexes were stable at room temperature. (TPFC)-iron(IV) toslyate, (TPFC)-iron(IV) chlorate, and (BPFMC)-iron(IV) chlorate were metastable and rearranged to their electronic isomers iron(III) corrole radical cations at room temperature. (TPFC)-iron(III) perchlorate corrole radical cation was the only product observed from reaction of the corrole-iron(IV) chloride with silver perchlorate. For the metastable iron(IV) species, the rates of isomerizations to the iron(III) macrocycle radical cation electronic isomers in dilute acetonitrile solutions were relatively insensitive to electron demands of the macrocyclic ligand but reflected the binding strength of the ligand to iron. Kinetic studies at varying temperatures and concentrations indicated that the mechanisms of the isomerization reactions are complex, involving mixed order reactivity.
Keywords: high-valent, iron, porphyrin, corrole, oxidation, isomerization, valence tautomer
Introduction
Reactions of high-valent iron complexes have attracted considerable interest because iron-oxo intermediates are formed in catalytic oxidations in the laboratory and in nature [1–4]. In a typical catalytic sequence, an iron(III) species is oxidized to an iron-oxo transient with a formal oxidation state of +5 on iron by a peroxy- or oxo-containing sacrificial oxidant, and the high-valent iron-oxo species then oxidizes a substrate in a two-electron, oxo-transfer process to return the iron(III) complex [5–7]. Synthetic porphyrin-iron complexes and related corrole-iron complexes mimic heme-containing peroxidase and cytochrome P450 enzymes, and high valent iron-oxo species with both types of macrocycles have been known for years [8,9]. The aromatic macrocycle ligands provide unique energy manifolds for high-valent iron atoms in comparison to saturated multidentate ligands because the macrocycle can be oxidized to a radical cation permitting, for example, a formal iron(V)-oxo derivative to exist in a more stable electronic configuration as a iron(IV)-oxo macrocycle radical cation [10,11].
In principle, a macrocycle-iron+n species and its analogous iron+(n−1)-macrocycle radical cation can be resonance forms of one another. When an energy barrier exists for electron transfer between the macrocycle and the iron atom, the two species will be isomers of one another, specifically electronic isomers (electromers) that are also known as valence tautomers [12]. The barriers that isolate the isomers from one another kinetically can be reasonably large due to, for example, considerable differences in bond lengths and relative atom positions. Thus, it is possible that the higher energy electromer or tautomer can react by an alternative pathway faster than it decays to the thermodynamically favored species by internal electron transfer relaxation. This reaction pathway might be important in some catalytic oxidations where, for example, a transient porphyrin-iron(V)-oxo species formed by reaction of a porphyrin-iron(III) complex with a sacrificial oxidant could be a reactive oxidant in catalytic oxidations even if not detected, whereas the better-known iron(IV)-oxo porphyrin radical cation might be a sluggish oxidant formed as a side-product of the major oxidation pathway [6,7,13].
True iron(V)-oxo species are rare. Que and co-workers reported formation of a tridentate ligand iron(V)-oxo complex [14], and evidence for another such species has been reported [15]. In the case of aromatic macrocyclic ligands, no iron(V)-oxo species has been fully characterized, but highly reactive iron-oxo transients produced in photochemical ligand cleavage reactions with both porphyrin [16] and corrole [9] macrocycles were tentatively assigned as iron(V)-oxo species. Computational studies [10,11] and thermodynamic cycles [17] suggest that macrocycle-iron(V)-oxo complexes should be energetically accessible, i.e. they should lie within several kcal/mol from their iron(IV)-oxo macrocycle radical cation electromers, but the iron(IV)-oxo macrocyclic radical cations are predicted to be the energetically favored species in most cases. Interestingly, corrolazine-iron(V)-oxo species [18] are predicted to be more stable than their iron(IV) macrocycle radical cation isomers [19].
For high-valent iron-macrocycle complexes with a formal +4 oxidation state of the metal, the energies of the iron(IV) complexes with “neutral” macrocycles and their iron(III) macrocycle radical cation electronic isomers apparently are closely matched. Understanding the position of oxidation in porphyrin-iron(III) complexes was an important objective in the chemistry of high-valent iron species [12,20], and studies of the position of oxidation in corrole-iron(III) complexes are contemporary [21]. The thermodynamically favored species in this class of complexes can switch from the iron(IV) “neutral” macrocycle to the iron(III) macrocycle radical cation as a function of the axial ligand binding strength [22,23], the pH of an aqueous solution [20], and the substitution on the macrocycle [12]. Complexes with formal +4 oxidation on iron are more stable than the more highly oxidized formal +5 iron-oxo complexes, and they provide an opportunity to study the mechanisms and barriers for interconversion of electronic isomers [16,24]. In this work, we report the production and studies of stable iron(IV) “neutral” macrocycle (porphyrin and corrole) complexes and metastable iron(IV) complexes that convert to their iron(III) macrocycle radical cation isomers. Similar behavior for porphyrin-manganese(IV) species and their manganese(III) porphyrin radical cations was reported previously [25].
Materials and Methods
Materials
HPLC grade acetonitrile (99.9%) was obtained from Sigma-Aldrich. Chloroform and methylene chloride were obtained from Fisher Scientific. All solvents were distilled prior to use. All chemical reagents were purchased from Sigma-Aldrich Co. Analyses were preformed by Atlantic Microlab, Inc.
Porphyrin complexes
The macrocycles for the porphyrin (Por) complexes were 5,10,15,20-tetraphenylporphryin (TPP), 5,10,15,20-tetramesitylporphyrin (TMP), and 2,3,7,8,12,13,17,18-octaethylporphyrin (OEP). (TPP)FeIII(Cl) and (OEP)FeIII(Cl) were purchased from Sigma-Aldrich Co. The TMPH2 free ligand and the (TMP)FeIIICl complex were prepared by reported methods [26,27]. (TPP)FeIII(ClO4) (1a), (TMP)FeIII(ClO4) (1b), and (OEP)FeIII(ClO4) were prepared by reported methods [23,26–28]. Authentic samples of (TPP)+•FeIII(ClO4)2 (3a), (TMP)+•FeIII(ClO4)2 (3b), and (OEP)+•FeIII(ClO4)2 (3c) were prepared by reported methods [23,29,30].
Porphyrin-iron(IV) diperchlorate complexes (2)
In a typical reaction, solutions of 1a (20 μM in CH3CN) and Fe(ClO4)3 (0.5 mM in CH3CN) were loaded into two gas-tight syringes. The solutions (200 μL each) were simultaneously injected into a stopped-flow optical cell in the UV-visible spectrometer. The data was analyzed with Applied Photophysics software. Decay or growth traces measured in Q-band regions were well fit to single exponential functions.
Corrole complexes
The macrocycles for the corrole (Cor) complexes were 5,10,15-tris(pentafluorophenyl)corrole (TPFC) and 5,15-bis(pentafluorophenyl)-10-p-methoxyphenylcorrole (BPFMC). The free ligands (TPFC)H3 and (BPFMC)H3 were prepared according to reported methods [31,32]. (TPFC)FeIII(Et2O)2 and (TPFC)FeIVCl (4a) were prepared by the reported method [21]. The corrole (Cor) species studied in this work are shown in Chart 2. The corrole systems were 5,10,15-tris(pentafluorophenyl)corrole (TPFC) and 5,15-bis(pentafluorophenyl)-10-p-methoxyphenylcorrole (BPFMC).
Chart 2.

(BPFMC)FeIVCl (6a)
The procedure described for insertion of iron into the (TPFC) ligand [21] was used to prepare (BPFMC)FeIII(OEt2)2. Thus, 80 mg (0.1 mmol) of the free ligand and 0.25 g (2 mmol) of iron(II) chloride in DMF was heated at reflux under nitrogen for 30 min. The process was monitored by TLC (silica gel, hexanes/methylene chloride, 2:1). Evaporation of solvent followed by column chromatography (silica gel, ether) resulted in isolation of (BPFMC)FeIII(OEt2)2 (75 mg, 0.08 mmol, 80%). UV-visible (CH3CN): λ max, nm (log ε), 406 (4.75), 550 (1.58). 1H NMR (CDCl3): δ 15.67 (s, 4H), 7.15–7.92 (m, 4H), 4.07 (s, 3H), −59.13 (s, 1H), −64.91 (s, 1H), −124.50 (br s, 2H). LRMS (ESI): m/z 789.3 [M]+.
The iron(IV) complex 6a was prepared from (BPFMC)FeIII(OEt2)2 by a method similar to that used for preparation of 4a from its iron(III) complex [21]. Thus, 50 mg (0.05 mmol) of (BPFMC)FeIII(OEt2)2 was dissolved in 30 mL of CH2Cl2. The resulting mixture was washed twice with HCl (10%) and with water. During the acidic washing, the color of the mixture changed from red-brown to deep brown. The solution was dried over Na2SO4, and the solvent was removed under vacuum. Recrystallization of the precipitated solid from benzene/heptane gave 38 mg (0.045 mmol, 90 %) of (BPFMC)FeIVCl. UV-visible (CH3CN) λ max, nm (log ε), 364 (4.30), 390 (4.56). 1H NMR (CD3CN): δ 24.84 (s, 1H), 24.60 (s, 1H), 10.84 (d, 4H), 4.07 (s, 3H), −0.55 (s, 1H), −3.00 (s, 1H), −3.31 (s, 1H), −17. 33 (s, 1H), −38.58 (s, 1H). LRMS (ESI): m/z 789.3 [M-H-Cl]+.
Corrole-iron(IV) chlorates (4d and 6d) and iron(III) corrole radical cation chlorates (5d and 7d)
The chlorate complexes 4d and 6d were prepared in stiu by rapidly mixing a solution of (Cor)FeIVCl (2 × 10−5 M) with an excess amount of Ag(ClO3) (2–3 × 10−2 M) in CH3CN at 22 °C. The resulting solutions were immediately analyzed by UV-visible spectroscopy. At room temperature, complexes 4d and 6d rapidly rearranged to the corrole radical cation complexes 5d and 7d. Spectral details for these compounds are collected in Table 2.
Table 2.
Properties and magnetic moments of Iron-Corrole Complexes
| Complex | UV-vis (nm)a | 1N NMR (δ)b | μeff (μB)c |
|---|---|---|---|
| (TPFC)FeIVCl (4a) | 367 (4.31), 394 (4.60) | 3.8, −5.9, −17.1, −49.9 (−20 °C) 4.1, −2.8, −11.6, −36.8 | 2.87 |
| (TPFC)FeIV(NO3) (4b) | 396 (4.61) | −4.3, −20.4, −47.2 | 2.89 |
| (TPFC)FeIV(OTs) (4c) | 397 (4.63) | −5.0, −13.4, −45.9 | 2.85 |
| (TPFC)FeIV(ClO3) (4d) | 397 (4.65) | 2.87 | |
| (TPFC)+•FeIII(OTs) (5c) | 379 (4.61), 647 | −3.3, −21.9, −48.0, −52.8 | 2.90 |
| (TPFC)+•FeIII(ClO3) (5d) | 380 (4.55), 650 | −4.7, −9.9, −22.4, −61.2 (−20 °C) | 2.95 |
| (TPFC)+•FeIII(ClO4) (5e) | 378 (4.53) | ||
| (BPFMC)FeIVCl (6a) | 364 (4.30), 390 (4.56) | ||
| (BPFMC)FeIV(NO3) (6b) | 398 (4.64) | ||
| (BPFMC)FeIV(ClO3) (6d) | 394 (4.63) | ||
| (BPFMC)+•FeIII(ClO3) (7d) | 381 (4.52) |
In CH3CN at room temperature; log ε values are in parentheses.
In CD3CN at 22 °C unless noted.
Effective magnetic moment measured in CD3CN at 22 °C.
Corrole-iron(IV) nitrates (4b and 6b)
The stable nitrate complexes were prepared by stirring an equivalent amount of silver nitrate (AgNO3) (2–3 × 10−5 M) with 4a or 6a (2–3 × 10−5 M) in CH3CN for 15 min at 22 °C. The mixtures were filtered and used immediately for further studies. Spectral details for these compounds are listed in Table 2.
The (TPFC)FeIV(NO3) complex was isolated from CH3CN and analyzed: Anal.: calcd for C37H8F15FeN5O3•H2O: C 47.82, H 1.08, N 7.54; found: C 47.59, H 1.22, N 7.87.
(TPFC)FeIV(OTs) (4c) and (TPFC)+•FeIII(OTs) (5c)
The metastable tosylate complex 4c was prepared by stirring a solution of 4a (2 × 10−5 M) with an equivalent amount of silver tosylate in CH3CN at 22 °C for 15 min. Filtration gave a solution containing 4c that slowly reacted to give complex 5c. Spectral details for these compounds are in Table 2.
Instrumentation
UV-visible spectra were recorded on an Agilent 8453 diode- array spectrophotometer. Room temperature stopped-flow mixing studies were performed on an RX2000 rapid kinetics spectrometer accessory (Applied Photophysics) coupled with the above spectrometer. Variable temperature kinetics were measured on an Ocean Optics USB4000 spectrometer with a thermostated reaction cell holder that was cooled with circulating coolant from a temperature-controlled bath; temperatures were measured with a thermocouple placed adjacent to the cell in the cell holder. 1H NMR spectra were recorded in deuterated solvents on a Bruker Advance DRX-500 (500 MHz) instrument with digital sample-temperature control unit. Chemical shifts (δ) are reported in parts per million (ppm) relative to tetramethylsilane (TMS, δ = 0 ppm). Data are reported as follows: chemical shift, multiplicity (s = singlet), numbers of hydrogen atoms, and peak assignment. X-band EPR spectra were obtained at 77 K on a Varian E109E spectrometer.
EPR spectroscopic studies
EPR spectra were collected with the following conditions: sample concentration, 5 mM; central magnetic field, 4000 G; scan width, 8000 G; modulation amplitude, 4 G; gain, 1.25 × 1000 × 10; microwave frequency, 8.89 GHz; microwave power 1 mW; scan time, 240 s; sample temperature, 77 K.
For complexes 2, a solution of 40 mM Fe(ClO4)3 in CH3CN was freshly prepared. A solution of complex 2 (5 mM) in CH3CN was cooled to −40 °C, and 1.1 equivalent of Fe(ClO4)3 was added with vigorous mixing. The color changed quickly from red to brown (2a) or dark purple (2b). The mixture was rapidly transferred to a cooled EPR tube with a pre-cooled pipette, and the tube was sealed with a septum cap and freeze-quenched in liquid nitrogen. Upon warming, these solutions changed to a green color.
For corrole-iron(IV) chlorate complex 4d, a solution of 4 mM (TPFC)FeIVCl in CH3CN was prepared and cooled to −40 °C before 2–3 equivalents of AgClO3 was added with vigorous mixing. The EPR spectrum was obtained as above. Upon warming, the solution changed color from green to brown. The relatively stable corrole-iron(IV) tosylate 4c was prepared at room temperature from reaction of 4a in CH3CN (4 mM) with 1–2 equivalents of AgOTs.
NMR and magnetic susceptibility studies
For both 1H NMR and magnetic susceptibility studies, samples of 2a and 2b were prepared as described above for the EPR studies with the exceptions that 1.0 equivalents of Fe(ClO4)3 was used, CD3CN was the solvent, NMR tubes were used, and the samples were maintained at −40 °C after mixing. After the spectra of 2a and 2b were acquired, the samples were warmed to room temperature and then cooled to −40 °C again for spectral acquisition that confirmed formation of 3a and 3b. In the magnetic susceptibility studies, a coaxial tube containing neat CD3CN was used, and the magnetic susceptibility was measured for the 1H signal of CHD2CN with a narrow window (−5 to 5 ppm). NMR experiments were attempted with 2c, but the spectrum obtained at low temperature was the same as that of authentic 3c.
For corrole complexes 4b, 4c, and 5c, the stable complexes were prepared in a vial, and AgCl was removed by filtration into an NMR tube. For unstable species, 5d and 5e, the samples were prepared in an NMR tube, and the AgCl was not filtered from the solution.
Results and Discussion
Metastable porphyrin iron(IV) diperchlorate species
The porphyrin (Por) complexes studied in this work are shown in Chart 1. 5,10,15,20-Tetraphenylporphryin (TPP), 5,10,15,20-tetramesitylporphyrin (TMP), and 2,3,7,8,12,13,17,18-octaethylporphyrin (OEP) macrocycles were employed. Iron complexes of these macrocycles are well known, and (Por)FeIII(Cl) are commercially available or readily prepared. We exchanged the chloride ligand with perchlorate by reaction of the chloride complex with Ag(ClO4) to give complexes 1 so that the identity of ligands on the metal in the more highly oxidized species would not be an issue [29,30].
Chart 1.
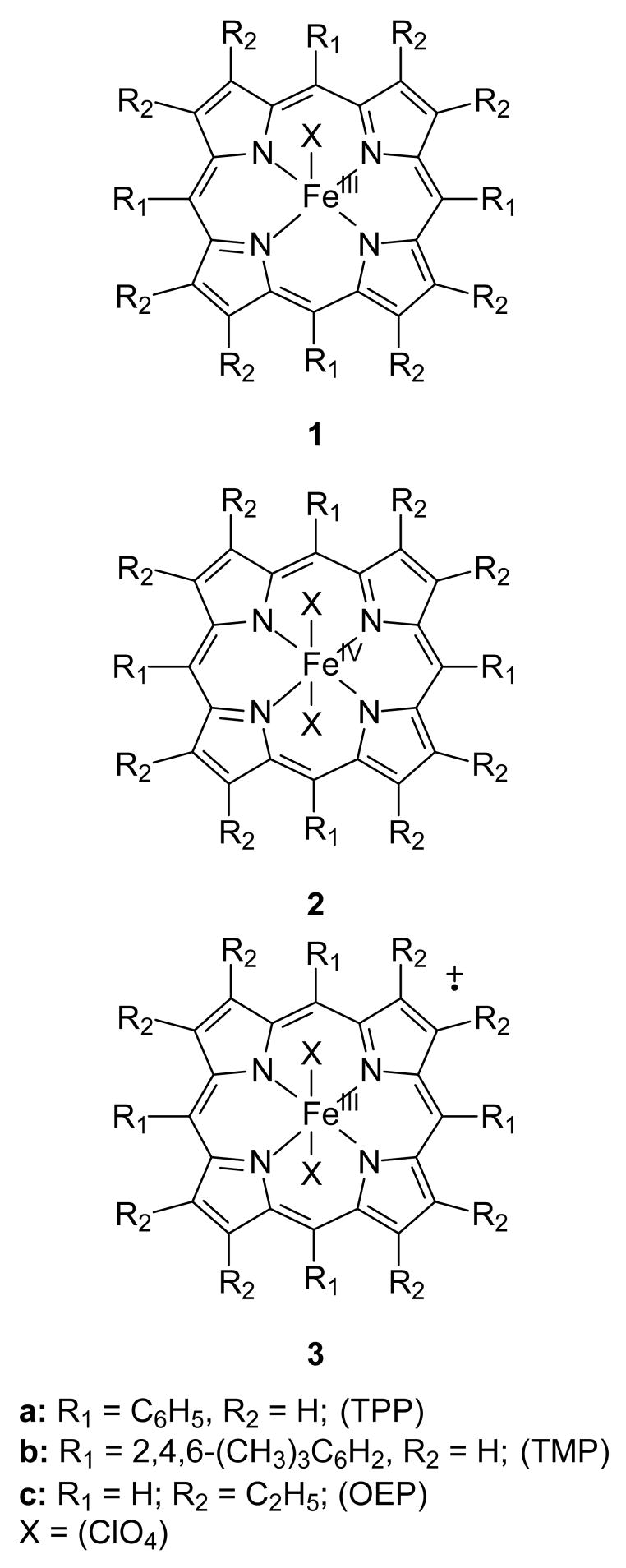
Oxidation of complexes 1 can occur on the metal to give the iron(IV) “neutral” porphyrins 2 or the iron(III) porphyrin radical cations 3. For each of the systems studied here, oxidation in methylene chloride solution with ferric perchlorate, Fe(ClO4)3, has been reported to give the iron(III) radical cations 3 [23,29,30]. Ferric perchlorate is more soluble in acetontirile than in methylene chloride. When oxidation reactions were conducted in acetonitrile solvent at low temperature, the first-formed product was the iron(IV) porphyrin species 2. Compounds 2 were metastable and rapidly rearranged at room temperature to give the known products 3. For the TPP and TMP systems, transients 2a and 2b were characterized by spectroscopic methods (UV-visible, EPR and 1H NMR spectroscopies) and magnetic susceptibility studies. The OEP derivative 2c appeared to convert to 3c at 22 °C with a rate similar to those for conversions of 2a and 2b as determined by kinetic UV-visible spectroscopy, but our attempts to detect 2c by NMR spectroscopy at low temperature were not successful.
Isolations of metastable species 2 were not possible because the species rearranged rapidly, often in seconds, at low temperatures. Dissociation and reassociation of a perchlorate anion in 2 is expected, and we cannot exclude the possibility that species 2 predominantly exists as a charged complex with iron bound to only one perchlorate group. Nonetheless, the when the samples of 2 were warmed gradually, the diperchlorate complexes 3 were the only species detected from reactions of 2.
In UV-visible spectral studies, solutions of compounds 1 in CH3CN were mixed rapidly at room temperature with CH3CN solutions containing an excess of Fe(ClO4)3. The spectra of compounds 1–3 are collected in Table 1. Spectra of the iron(III) porphyrin complexes 1 and iron(III) porphyrin radical cations 3 in methylene chloride or chloroform were previously reported [23,29,30], and those for species 1 and 3 in acetonitrile were similar. The spectra of the iron(IV) complexes 2 differed in a predictable manner. The Soret bands for compounds 2 were sharp peaks, and the molar extinction coefficients were greater than those of 1, whereas the Soret bands for 3 were broader and less intense. The Q-bands were characteristic, especially the Q-band for the porphyrin radical cation complexes 3, which had a broad long wavelength absorbance in the range 600–750 nm. Figure 1 shows the spectra for the TMP system.
Table 1.
Properties of iron-porphyrin complexes.
| Complex | UV-visible (nm)a | 1H NMR (δ)b | μeff (μB)c |
|---|---|---|---|
| (TPP)FeIII(ClO4) (1a) | 398 (5.11), 532 | 74.6, 18.3, 11.9, 11.7 | |
| (TMP)FeIII(ClO4) (1b) | 399 (5.07), 530 | 8.4, 3.6, 2.6, −26 | |
| (OEP)FeIII(ClO4) (1c) | 376 (4.92), 503, 553, 634, 559 | 47.0, 8.3 | |
| (TPP)FeIV(ClO4)2 (2a) | 396 (5.14), 532 | 69.6, 31.6, 16.9 | 2.96 |
| (TMP)FeIV(ClO4)2 (2b) | 397 (5.12), 530 | 66, 32, 14 | 3.03 |
| (OEP)FeIV(ClO4)2 (2c) | 391 (5.02), 495, 618 | ||
| (TPP)+•FeIII(ClO4)2 (3a) | 393 (5.03), 535, 602, 682 | 61.8, 32.8, −11.1, −14.2 | 5.50 |
| (TMP)+•FeIII(ClO4)2 (3b) | 392 (4.98), 498, 605, 680 | 92, 71.7, 31.4, 20.8 | 5.95 |
| (OEP)+•FeIII(ClO4)2 (3c) | 374 (4.85), 502, 595, 648 | 69.8, 26.9 | 5.20 |
UV-visible spectral absorbances in CH3CN; log ε of the Soret band in parentheses.
NMR spectra in CD3CN solution at −40 °C.
Effective magnetic moment determined in CD3CN at −40 °C.
Figure 1.

UV-visible spectra for (TMP)FeIV(ClO4)2 (2b) (solid line) and (TMP)+•FeIII(ClO4)2 (3b) (dashed line) in acetonitrile.
The 1H NMR spectra in the TPP and TMP series also suggested the identity of the iron(IV) species. 1H NMR spectra in CD2Cl2 or CDCl3 solutions were reported previously for compounds 1a [33], 3a [34], 1b [23], 3b [23], 1c [28] and (OEP)+•FeIII(Cl)(ClO4) [29]. We confirmed the identity of compounds 1 and 3 by obtaining 1H NMR spectra in CD2Cl2 solutions and in CD3CN solutions, which were similar (Table 1). For the TPP and TMP series, NMR spectra of the metastable complexes 2a and 2b also were obtained. When the iron(III) porphyrin complexes in CD3CN solutions cooled to −40 °C were treated with 1 equivalent of Fe(ClO4)3, new NMR signals formed that were attributed to transients 2a and 2b. When the samples were warmed to room temperature and again cooled to −40 °C, the NMR spectra of complexes 3a and 3b were observed. The characteristic features of these spectra were that compounds 1 had relatively sharp signals, the signals from compounds 2 were quite broad, and the signals from compounds 3 were again relatively sharp. Figure 2 shows the results for the (TMP) complexes 1b, 2b, and 3b. We specifically note that species 2a and 2b were expected to have very small, upfield signals in their 1H NMR spectra at δ ≈ −30 for pyrrole protons (see ref [23] for an example). Unfortunately, the NMR spectra of 2a and 2b at −40 °C contained only broad signals (cf. Figure 2), and we could not detect any upfield signals.
Figure 2.

1H NMR spectra in CD3CN at −40 °C of (TMP)FeIII(ClO4) (1b), (TMP)FeIV(ClO4)2 (2b), and (TMP)+•FeIII(ClO4)2 (3b). The relative intensities of the spectra are the same.
For EPR spectroscopic studies, samples of compounds 2a and 2b were prepared in CH3CN at −40 °C, and the samples were cooled to 77 K. The EPR spectra contained small signals from residual starting compounds 1, but no new signals for compounds 2 were observed. Integration of the signals from compounds 1 indicated that more than 80% of the material had been converted to EPR silent species. The iron(IV) species 2 were expected to be EPR silent because they have S = 1 [35].
Magnetic susceptibility studies also were performed with samples of 2a and 2b using the Evans NMR method [36,37]. The samples were prepared in CD3CN at −40 °C in NMR tubes containing a coaxial tube with neat CD3CN solvent. The chemical shift differences in the signals from CHD2CN in the solvent were measured, and the effective magnetic moment (μeff) was calculated from the chemical shift differences [37]. At −40 °C, the effective magnetic moments of 2a and 2b were μeff = 2.96 μB and μeff = 3.03 μB, respectively, where we estimate the errors to be ±10%. These values are close to the value expected for an S = 1 species (μeff = 2.83 μB) [38]. When the samples were warmed, the μeff values increased as increasing amounts of species 3 were formed. The samples of 3 were cooled to −40 °C again, and NMR spectra were obtained to determine the values of μeff under the same conditions as used for the samples of 2, which gave μeff = 5.50 μB for 3a and μeff = 5.95 μB for 3b, values consistent with uncoupled spin-admixed iron(III) (S = 3/2, 5/2) and porphyrin radical (S = 1) [38]. Note that any contribution to the magnetic susceptibility from the by-products of the ferric perchlorate will be constant in these measurements, and the difference in values before and after warming results only from the isomerization of 2 to 3.
Corrole iron(IV) species
The corrole (Cor) species studied in this work are shown in Chart 2. The corrole systems were 5,10,15-tris(pentafluorophenyl)corrole (TPFC) and 5,15-bis(pentafluorophenyl)-10-p-methoxyphenylcorrole (BPFMC). Isomers 4 and 6 are iron(IV) species with “neutral” corrole ligands (actually corrole tri-anions), and isomers 5 and 7 are iron(III) species with corrole “radical cations” (actually corrole di-anion radicals). Note that neutral corroles have three acidic protons on nitrogen atoms whereas porphyrins have two; therefore fully deprotonated corrole-iron complexes with the metal in a formal +4 oxidation state have a single associated anion and are inherently more stable than porphyrin species with the metal in the +4 oxidation state.
The starting points for the corrole-iron species were the corrole-iron(IV) chlorides 4a and 6a, which were formed by air oxidation of the corresponding corrole-iron(III) complexes in the presence of HCl [39]. The chloride counterions in 4a and 6a were exchanged by reaction with various silver salts to give nitrate, tosylate, chlorate, and perchlorate derivatives [9].
The structures of formal +4 iron-corrolates have been an issue in recent years [21,40]. Either the iron(IV) trianionic corrole structures or the iron(III) corrole dianion radical structures are possible. The former is an S = 1 species, and the latter is an S = 3/2 iron-center that could have the macrocycle radical antiferromagnetically coupled to the unpaired electrons on iron [38]. As such, both structures could be EPR silent, and both could have similar effective magnetic moments of μeff ≈ 3 μB. Differentiation between the two structures is possible from UV-visible and NMR spectral data and from computational results.
In our studies, we relied primarily on UV-visible spectra with support from 1H NMR spectroscopy for assignment of the corrole-iron species electronic structures. Figure 3 shows typical results for the chlorate isomers in both series of corroles studied. The iron(IV) neutral corrole species have a relatively sharp Soret absorbance and quite weak or absent visible-band absorbances [39,41]. The iron(III) corrole radical cations have broader, less intense Soret bands that are blue-shifted from the iron(IV) species and weak but obvious long wavelength absorbances (λmax ≈ 650 nm) that are characteristic of radicals in highly conjugated systems.
Figure 3.

Time-resolved UV-visible spectra of corrole-iron chlorates. (A) (TPFC)FeIV(ClO3) (4d) converting to (TPFC)+•FeIII(ClO3) (5d) at ambient temperature; the time slices are at 0.3, 15, 20, 31, 40, and 60 s. (B) (BPFMC)FeIV(ClO3) (6d) converting to (BPFMC)+•FeIII(ClO3) (7d) at ambient temperature; the time slices are at 0.3, 7, 18, 23, and 60 s.
1H NMR spectra could be obtained for some of the (TPFC)-iron derivatives, and these provided supporting evidence for the structure assignments. NMR chemical shifts in corrole-iron(IV) derivatives have been shown to be highly variable for different ligands on iron [42], but the NMR spectra we obtained demonstrated similarities for the corrole-iron(IV) species (three upfield peaks) and for the corrole-iron(III) radical cations (four upfield peaks) as listed in Table 2. The NMR spectra also clearly demonstrate when an isomerization occurs as shown in Figure 4, which contains the spectra of the initially-formed metastable (TPFC)-iron(IV) tosylate (4c) and its isomerization product 5c.
Figure 4.

1H NMR spectra of 4c and 5c in CD3CN recorded at 22 °C.
The formal +4 iron-(TPFC) chloride complex 4a was initially prepared by Gross who assigned the structure 4a [39]. In our preparative reactions, we employed 4a in CH3CN solutions. The species was EPR silent, and the effective magnetic moment measured in CD3CN solution at 22 °C by the Evans method [36] was μeff = 2.87 μB; both features are consistent with structure 4a, which has S = 1, or structure 5a (S = 3/2) if there is antiferromagnetic coupling of the radical with the iron electrons to give S = 1. The 1H NMR spectrum in CD3CN and UV-visible spectrum in CH3CN were similar to those reported [39] for 4a in CD2Cl2 and CH2Cl2 solutions, respectively (Table 2). Reactions of complex 4a with silver salts to exchange the anion gave different results as a function of the new counterion that was exchanged for chloride. The primary methods for structure assignments for the products of these reactions were UV-visible spectra and 1H NMR spectra (Figures 3 and 4); both types of spectra showed characteristic differences for the two electronic isomers as discussed above.
Reaction of 4a with Ag(NO3) at 22 °C gave a species that we assign as structure 4b on the basis of its UV-visible (Table 2) and 1H NMR spectra (Figure 4). Importantly, the UV-visible spectrum lacked the highly characteristic long wavelength absorbance due to a radical in the corrole macrocycle [39,41]. Species 4b was stable when warmed to room temperature.
Reactions of complex 4a with Ag(ClO3) at −20 °C and with AgOTs (Ts = tosyl = p-CH3C6H4SO2) at 22 °C in CH3CN gave metastable species that rearranged at room temperature. The spectral properties of both isomers are listed in Table 2. In both cases, the first-formed species had UV-visible spectra with sharper, more intense Soret bands than their isomers and lacked a long wavelength Q-band absorbance at ca. 650 nm, whereas both isomers that formed upon warming displayed long wavelength Q-band absorbances. From these spectral properties, we assign the first-formed species as iron(IV) corrolates 4c and 4d, and the species that formed upon warming as iron(III) corrole radical cations 5c and 5d. The 1H NMR spectra of the tosylate derivatives support the structural assignment (Figure 4). For the iron(IV) species 4c, the 1H NMR spectrum was similar to that of the iron(IV) chloride complex 4a. For the isomerized compounds 5c and 5d, the 1H NMR spectra displayed four distinct upfield signals for the four unique corrole ring protons.
When chloride complex 4a was allowed to react with Ag(ClO4) at −20 °C, the initially formed species was stable when warmed to room temperature. In this case, the UV-visible spectrum indicated that the product was the iron(III) corrole radical cation 5e (Table 2). If iron(IV) correlate 4e was formed in the initial exchange reaction, it isomerized to 5e too rapidly for detection in our method.
The chemistry of the (BPFMC)-iron species 6 and 7 was similar to that of the (TPFC)-iron species as deduced from UV-visible spectra. The iron(IV) chloride complex 6a was prepared by air oxidation of the iron(III) corrole complex in the presence of HCl. Reaction of chloride complex 6a with AgNO3 gave the iron(IV) complex 6b that did not rearrange on standing. Reaction of 6a with Ag(ClO3) at low temperature gave the iron(IV) complex 6d that rearranged to the iron(III) corrole radical cation 7d when warmed to room temperature.
The thermodynamically favored electronic isomers for the corrole complexes studied apparently are determined by the counterion binding strengths [43,44]. The iron(IV) corroles are favored with the relatively strong binding chloride and nitrate ligands (4a, 4b, 6a, 6b). For the weaker binding ligands OTs, ClO3, and ClO4, the iron(III) radical cations are the thermodynamically favored species (5c, 5d, 5e, 7d).
Isomerizations of iron(IV) macrocyles to iron(III) macrocycle radical cations
In total, six of the iron(IV) complexes prepared in this work isomerized to their iron(III) macrocycle radical cations, the three porphyrin-iron(IV) diperchlorates (2a, 2b, and 2c), (TPFC)FeIV(OTs) (4c), (TPFC)FeIV(ClO3) (4d), and (BPFMC)FeIV(ClO3) (6d). For these metastable iron(IV) species, the barriers for isomerizations to the thermodynamically favored iron(III) macrocycle radical cation species are of special interest because similar barriers might be involved in the isomerizations of macrocycle-iron(V)-oxo species to iron(IV)-oxo macrocycle radical cations.
Isomerizations of the iron(IV) species to their iron(III) macrocycle radical cations were readily observed by UV-visible spectroscopy. For example, the time-resolved spectra in Figure 3 show conversions of corrole-iron(IV) chlorates 4d and 6d to the corrole radical cation iron(III) chlorates 5d and 7d, respectively. Nonetheless, the kinetics of the isomerizations reactions could not be analyzed in a straightforward manner because the mechanisms of the isomerizations reactions appear to be highly complex. The difficulty in analyzing the results is illustrated in Figure 5, where, for demonstration purposes, we solved the kinetic data for the isomerization reactions as first-order processes. Figure 5A shows variable temperature results for reaction of the corrole-iron(IV) chlorate 4d. The data appears to fit an analysis in the context of Arrhenius theory, but the apparent activation parameters are nonsensical. Specifically, the reaction appears to have a very low activation energy of Ea = 5.6 kcal/mol, which is possible, but also an extraordinarily negative entropy term, log A = 3, which gives ΔS‡ ≈ −50 eu. Even more problematical from a mechanistic standpoint, when the initial concentrations of iron(IV) species were varied in a series of reactions at constant temperature, the apparent first-order rate constants for isomerizations increased as a fractional order of the iron reactant. Figure 5B shows apparent first-order rate constants for conversion of corrole-iron(IV) species 4d to 5d and for conversion of porphyrin-iron(IV) species 2b to 3b in log-log format. If the reactions were first-order in iron species, the slopes of these plots would be zero. Instead, both plots have slopes of ca. 0.5, indicating that the iron species are reacting with apparent molecular order of 1.5.
Figure 5.

Kinetic plots. (A) Temperature-dependent rate constants for reaction of corrole-iron(IV) chlorate 4d in acetonitrile; the kinetic values used are apparent first-order rate constants for reactions of 1 × 10−5 M solutions. (B) Concentration dependence of apparent first-order rate constants for reactions at room temperature for corrole-iron(IV) chlorate 4d (solid circles) and porphyrin-iron(IV) perchlorate 2b (open circles).
Various mechanistic schemes might be envisioned to rationalize the kinetic results, but it is apparent that detailed studies will be required to understand the reactions well. Until such information is available, we believe it is prudent to discuss the kinetics in terms of half-lives for reactions under similar conditions as opposed to rate constants. The half-lives for isomerizations of the iron(IV) complexes at ca. 1 × 10−5 M in acetonitrile at room temperature are listed in Table 3. For the porphyrin-iron(IV) species, the reactivity is 2b > 2a ≈ 2c, but all of the reactions had similar rates, with half-lives of about 10 s at room temperature corresponding to a barrier of ΔG‡ ≈ 19 kcal/mol. For the corrole-iron(IV) species, chlorate 4d reacted much faster than tosylate 4c, which had a barrier to isomerization of ΔG‡ ≈ 22 kcal/mol, indicating an expected trend that the iron(IV) species is increasingly favored by stronger π-donating ligands [43,44]. Interestingly, the changes in electron demand of the macrocycles for the two corrole-iron(IV) chlorates 4d and 6d had only a slight effect on the rates of isomerization, which is similar to the small macrocycle effect observed with the porphyrin-iron(IV) species.
Table 3.
Observed half-lives for isomerizations of iron(IV) complexes in acetonitrile.a
| Complex | Temp (°C) | t1/2 (s) |
|---|---|---|
| (TPP)FeIV(ClO4)2 (2a) | 22 | 13 |
| 9 | 20 | |
| 1 | 28 | |
| (TMP)FeIV(ClO4)2 (2b) | 22 | 10 |
| (OEP)FeIV(ClO4)2 (2c) | 22 | 13 |
| (TPFC)FeIV(OTs) (4c) | 22 | 2500 |
| (TPFC)FeIV(ClO3) (4d) | 23 | 15 |
| (BPFMC)FeIV(ClO3) (6d) | 22 | 18 |
For isomerizations of ca. 1 × 10−5 M solutions.
Conclusion
Oxidation of porphyrin-iron(III) complexes by ferric perchlorate occurs initially on the metal ion to give metastable porphyrin-iron(IV) diperchlorates, and stable and metastable corrole-iron(IV) complexes can be produced from the corrole-iron(IV) chloride by ligand exchange. Macrocycle iron(IV) complexes with oxygen-bound ligands such as nitrate, chlorate and perchlorate are attractive intermediates because they are photo-labile and react to give high-valent iron-oxo derivatives [9,16], and the methods studied here are expected to be generally useful for preparation of precursors for such photochemical studies. Rearrangements of the metastable iron(IV) species to their iron(III) macrocycle radical cation isomers are fast in some cases, but the lifetimes of the iron(IV) species often are long enough to permit some spectroscopic studies. The rates of rearrangement were found to be relatively insensitive to changes in the macrocyclic ligand substitution but strongly sensitive to the binding strength of the “fifth” ligand to iron. The isomerization rates for the iron(IV) species studied here might be related to the rates of isomerizations of higher valent iron-oxo species and suggest that appreciable barriers for electronic isomerizations of formal +5 iron species are possible.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (GM-48722).
Abbreviations
- Cor
corrole
- Por
porphyrin
- TPP
5,10,15,20-tetraphenylporphyrin or -porphyrinato
- TMP
5,10,15,20-tetramesitylporphyrin or -porphyrinato
- OEP
2,3,7,8,12,13,17,18-octaethylporphyrin or -porphyrinato
- TPFC
5,10,15-tris(pentafluorophenyl)corrole or -corroloto
- BPFMC
5,15-bis(pentafluorophenyl)-10-p-methoxyphenylcorrole or -corroloto
- LRMS
low resolution mass spectrometry
- ESI
electrospray ionization
- TMS
tetramethylsilate
References
- 1.Meunier B, editor. Metal-Oxo and Metal-Peroxo Species in Catalytic Oxidations. Springer-Verlag; Berlin: 2000. [Google Scholar]
- 2.Sheldon RA. Metalloprophyrins in Catalytic Oxidations. Marcel Dekker; New York: 1994. [Google Scholar]
- 3.Ortiz de Montellano PR, editor. Cytochrome P450 Structure, Mechanism, and Biochemistry. 3. Kluwer; New York: 2005. [Google Scholar]
- 4.Sono M, Roach MP, Coulter ED, Dawson JH. Chem Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 5.Groves JT, Watanabe Y. J Am Chem Soc. 1986;108:507–508. doi: 10.1021/ja00263a026. [DOI] [PubMed] [Google Scholar]
- 6.Pan Z, Zhang R, Newcomb M. J Inorg Biochem. 2006;100:524–532. doi: 10.1016/j.jinorgbio.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 7.Zhang R, Nagraj N, Lansakara-P DSP, Hager LP, Newcomb M. Org Lett. 2006;8:2731–2734. doi: 10.1021/ol060762k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Groves JT, Haushalter RC, Nakamura M, Nemo TE, Evans BJ. J Am Chem Soc. 1981;103:2884–2886. [Google Scholar]
- 9.Harischandra DN, Zhang R, Newcomb M. J Am Chem Soc. 2005;127:13776–13777. doi: 10.1021/ja0542439. [DOI] [PubMed] [Google Scholar]
- 10.Ogliaro F, de Visser SP, Groves JT, Shaik S. Angew Chem, Int Ed. 2001;40:2874–2878. [PubMed] [Google Scholar]
- 11.Dey A, Ghosh A. J Am Chem Soc. 2002;124:3206–3207. doi: 10.1021/ja012402s. [DOI] [PubMed] [Google Scholar]
- 12.Weiss R, Bulach V, Gold A, Terner J, Trautwein AX. J Biol Inorg Chem. 2001;6:831–845. doi: 10.1007/s007750100277. [DOI] [PubMed] [Google Scholar]
- 13.Newcomb M, Zhang R, Chandrasena REP, Halgrimson JA, Horner JH, Makris TM, Sligar SG. J Am Chem Soc. 2006;128:4580–4581. doi: 10.1021/ja060048y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Oliveira FT, Chanda A, Banerjee D, Shan XP, Mondal S, Que L, Bominaar EL, Munck E, Collins TJ. Science. 2007;315:835–838. doi: 10.1126/science.1133417. [DOI] [PubMed] [Google Scholar]
- 15.Lee SH, Han JH, Kwak H, Lee SJ, Lee EY, Kim HJ, Lee JH, Bae C, Lee SN, Kim Y, Kimal C. Chem Eur J. 2007;13:9393–9398. doi: 10.1002/chem.200700513. [DOI] [PubMed] [Google Scholar]
- 16.Pan Z, Zhang R, Fung LWM, Newcomb M. Inorg Chem. 2007;46:1517–1519. doi: 10.1021/ic061972w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koppenol WH. J Am Chem Soc. 2007;129:9686–9690. doi: 10.1021/ja071546p. [DOI] [PubMed] [Google Scholar]
- 18.Kerber WD, Ramdhanie B, Goldberg DR. Angew Chem Int Ed. 2007;46:3718–3721. doi: 10.1002/anie.200605156. [DOI] [PubMed] [Google Scholar]
- 19.Wasbotten I, Ghosh A. Inorg Chem. 2006;45:4910–4913. doi: 10.1021/ic0602493. [DOI] [PubMed] [Google Scholar]
- 20.Wolak M, van Eldik R. Chem Eur J. 2007;13:4873–4883. doi: 10.1002/chem.200601148. [DOI] [PubMed] [Google Scholar]
- 21.Simkhovich L, Goldberg I, Gross Z. Inorg Chem. 2002;41:5433–5439. doi: 10.1021/ic020118b. [DOI] [PubMed] [Google Scholar]
- 22.Groves JT, Quinn R, McMurry TJ, Lang G, Boso B. J Chem Soc, Chem Commun. 1984:1455–1456. [Google Scholar]
- 23.Groves JT, Quinn R, McMurry TJ, Nakamura M, Lang G, Boso B. J Am Chem Soc. 1985;107:354–360. [Google Scholar]
- 24.Nam W, Park SE, Lim IK, Lim MH, Hong JK, Kim J. J Am Chem Soc. 2003;125:14674–14675. doi: 10.1021/ja0368204. [DOI] [PubMed] [Google Scholar]
- 25.Kaustov L, Tal ME, Shames AI, Gross Z. Inorg Chem. 1997;36:3503–3511. doi: 10.1021/ic961207p. [DOI] [PubMed] [Google Scholar]
- 26.Lindsey JS, Wagner RW. J Org Chem. 1989;54:828–836. [Google Scholar]
- 27.Adler AD, Longo FR, Kampas F, Kim J. J Inorg Nucl Chem. 1970;32:2443–2445. [Google Scholar]
- 28.Ogoshi H, Watanabe E, Yoshida Z. Chem Lett. 1973:989–992. [Google Scholar]
- 29.Phillippi MA, Goff HM. J Am Chem Soc. 1982;104:6026–6034. [Google Scholar]
- 30.Gans P, Buisson G, Duee E, Marchon JC, Erler BS, Scholz WF, Reed CA. J Am Chem Soc. 1986;108:1223–1234. [Google Scholar]
- 31.Gross Z, Galili N, Saltsman I. Angew Chem Int Ed. 1999;38:1427–1429. doi: 10.1002/(SICI)1521-3773(19990517)38:10<1427::AID-ANIE1427>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 32.Gryko DT, Koszarna B. Synthesis. 2004:2205–2209. [Google Scholar]
- 33.Goff H, Shimomura E. J Am Chem Soc. 1980;102:31–37. [Google Scholar]
- 34.Buisson G, Deronzier A, Duee E, Gans P, Marchon JC, Regnard JR. J Am Chem Soc. 1982;104:6793–6796. [Google Scholar]
- 35.Walke FA. In: Porphyrin Handbook, vol. 5, Proton NMR and EPR Spectroscopy of Paramagnetic Metalloporphyrins. Kadish KM, Smith KM, Guilard R, editors. Academic; New York: 2000. pp. 81–183. [Google Scholar]
- 36.Evans DF. J Chem Soc. 1959:2003–2005. [Google Scholar]
- 37.Sur SK. J Magn Reson. 1989;82:169–173. [Google Scholar]
- 38.Walker FA. Inorg Chem. 2003;42:4526–4544. doi: 10.1021/ic026245p. [DOI] [PubMed] [Google Scholar]
- 39.Simkhovich L, Mahammed A, Goldberg I, Gross Z. Chem Eur J. 2001;7:1041–1055. doi: 10.1002/1521-3765(20010302)7:5<1041::aid-chem1041>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 40.Walker FA, Licoccia S, Paolesse R. J Inorg Biochem. 2006;100:810–837. doi: 10.1016/j.jinorgbio.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 41.Kadish KM. Prog Inorg Chem. 1986;34:435–605. [Google Scholar]
- 42.Simkhovich L, Gross Z. Inorg Chem. 2004;43:6136–6138. doi: 10.1021/ic0491332. [DOI] [PubMed] [Google Scholar]
- 43.Gross Z, Nimri S. Inorg Chem. 1994;33:1731–1732. [Google Scholar]
- 44.Czarnecki K, Nimri S, Gross Z, Proniewicz LM, Kincaid JR. J Am Chem Soc. 1996;118:2929–2935. [Google Scholar]