

Abstract
The light microscopic distinction between complex atypical hyperplasia (CAH) and invasive endometrioid carcinoma (UEC) on endometrial sampling is problematic and often has significant clinical implications. Using mouse models of endometrial tumorigenesis based on two of the most common molecular alterations found in primary human UEC we sought to characterize the transition from CAH to carcinoma to identify clinically useful biomarkers. We used the previously described Pten+/-;Mlh1-/- mouse model. DNA was isolated from microdissected lesions (CAH and carcinoma) and analyzed for LOH and mutations of Pten and additional candidate genes. In order to identify novel candidate genes associated with invasion, global gene expression profiles were compared from uteri with extensive CAH and carcinoma. The majority of CAHs as well as carcinomas, arising in this model showed biallelic inactivation of Pten mediated through LOH or intragenic mutation of the wild-type allele suggesting that complete loss of Pten is insufficient for the development of carcinoma. The global gene expression studies detected increased expression of oviduct-specific glycoprotein (OGP) in carcinoma as compared to CAHs. This finding was validated using immunohistochemical staining in a collection of primary human UECs and CAHs. Our studies identify a molecular marker for invasive endometrial cancer that may have clinical significance, and highlight the usefulness of this mouse model in not only understanding the genetic underpinnings of endometrial carcinoma, but as a tool to develop clinically relevant biomarkers.
Keywords: mouse model, endometrial tumorigenesis, molecular marker of invasive disease Journal Category: Cancer Genetics
Introduction
Endometrial carcinoma is the most common malignancy of the female genital tract and the endometrioid variety constitutes the vast majority (85%) of cases. Uterine endometrioid carcinoma (UEC) arises from proliferative endometrium through a series of hyperplastic precursor lesions. The endometrium consists of both stromal and glandular elements that respond to the influence of estrogen and progesterone coordinately produced by the ovaries during the menstrual cycle. During the first half of the cycle, primarily under the influence of estrogen, the endometrium undergoes a 14-fold expansion in tissue volume due to proliferation of both the glands and stroma. After ovulation the endometrial glands and stroma differentiate in response to progesterone, and in the absence of embryo implantation menses occurs and the upper two-thirds of the endometrium is shed. However, under conditions of unopposed estrogen (endogenous or exogenous) stimulation, and/or genetic alterations, the endometrium becomes hyperplastic, defined as an increase in the gland to stroma ratio, due to unrestrained glandular proliferation. Over time the glandular architecture becomes more complex progressing from simple hyperplasia to complex hyperplasia. If the complex hyperplastic process develops cytologic atypia it is referred to as complex atypical hyperplasia (CAH). Clinicopathological and molecular studies suggest that CAH is the direct precursor of endometrioid carcinoma. Furthermore, studies have shown that 25-40% of women with a diagnosis of CAH on endometrial sampling will have endometrial carcinoma at the time of hysterectomy 1, 2. Although the discrepancy may be due, in part, to sampling errors, the ability of pathologists to reliably distinguish between CAH and carcinoma on routine microscopic evaluation is less than robust. Currently, because of this diagnostic shortcoming many women undergo hysterectomy for non-invasive disease. Not only does this lead to significant morbidity, it can also pose significant issues for younger women concerned with fertility issues. Thus, objective markers of invasion would have meaningful clinical ramifications.
Molecular genetic analyses of CAH and uterine endometroid carcinoma (UEC) have identified a number of common genetic alterations. Approximately 30-80% of UEC have intragenic mutations in the PTEN tumor suppressor gene, with a similar mutational frequency found in CAH 3-6. Interestingly, about 20% of simple and complex hyperplasia without atypia harbor mutations in PTEN, suggesting that mutations in PTEN are a relatively early event in endometrial tumorigenesis. Further evidence for an early role of PTEN inactivation comes from a Pten knock out mouse model. All female mice with a germline heterozygous deletion of Pten develop CAH, and in 20% of animals it progresses to carcinoma 7-10. The Pten mouse model, along with the human studies, establishes a central role of PTEN in the development of hyperplasia. However, its role, if any, in the progression to invasive endometrial carcinoma is not well understood.
In addition to PTEN mutations, deficiencies in DNA mismatch repair (MMR), detected as microsatellite instability (MSI), are relatively common in UEC and numerous studies have demonstrated an association between PTEN mutations and MSI in UEC 11-15. Although some studies have reported an increase in insertions and deletions in repetitive sequences in PTEN in UEC with MSI, other studies have not. Thus, the biological significance of the association remains unclear. The most common mechanism underlying MSI in sporadic endometrial carcinoma is inactivation of the MLH1 gene via promoter hypermethylation 16-18. Although the MSI phenotype has been detected in CAH with associated carcinoma, it has not been identified in lesser degrees of hyperplasia. However, methylation of the MLH1 promoter has been detected in simple and complex hyperplasia without aytpia 16 . Thus, both PTEN mutations and inactivation of MLH1 appear to occur relatively early in the neoplastic process. Previous studies from our laboratory have established that MMR deficiency accelerates endometrial tumorigenesis in Pten heterozygous mice. Pten heterozygous, Mlh1-deficient mice develop endometrial hyperplasia and carcinoma at much younger ages than Pten heterozygous mice, suggesting that the association seen in primary human tumors has biological relevance 8.
Here, we present data to show that specific mutations in Pten are a consequence of MMR deficiency in the mouse model and complete loss of Pten alone is not sufficient for the development of invasive endometrial carcinoma. Notably, by combining the gene expression profiles from endometrial lesions arising in the mouse models and immunohistochemical validation in primary human tumors, we found that the expression of oviduct-specific glycoprotein (OGP) is increased in carcinoma when compared to CAH, suggesting that it may be a molecular marker of invasive endometrial carcinoma.
Materials and Methods
Mice and genotyping
Pten heterozygous mice and Pten+/-;Mlh1-/- double mutants in C57BL6/129SvJ mixed background have been described previously8. Pten heterozygous mice in the CD-1 background were generated by backcrossing C57BL6/129SvJ Pten heterozygous male mice to wild type female CD-1 mice for 10 generations. CD-1 is an outbred strain of mice that has been used to study estrogen induced endometrial carcinoma. The Pten+/- and the Pten+/-;Mlh1-/- were sacrificed at 36-40 weeks and 14-18 weeks of age, respectively. Animals were genotyped by PCR as described previously 8. Virgin females were euthanized and uteri were dissected ensuring that all the adipose tissue was removed. All animal experiments were performed according to approved IACUC protocols. Each uterine horn was initially cut transversely into two halves. Both the top and bottom halves of each horn were further split lengthwise to obtain four uterine segments. The longitudinally split segments are expected to be mirror images of each other and accordingly, similar lesions would be present in both the segments. This was previously ascertained by histological analysis using several control mice as well as Pten heterozygote mice. One longitudinal segment was frozen for RNA extraction. The remaining half was fixed in formalin and subsequently embedded in paraffin for histological analysis and microdissection. Gene expression profiling was carried out using mice from the C57BL6/129SvJ mixed background. Eight mice with CAH (non-invasive) and four mice with stromal and/or myometrial invasion were selected for gene expression profiling.
Analysis of Pten and additional loci on chromosome 19
The status of the wild type Pten allele was analyzed in microdissected lesions from Pten+/-;Mlh1-/- and Pten+/- mice in both the mixed and CD-1 strains. Lesions were microdissected from paraffin- embedded 5 micron sections under light microscope visualization with a 26-gauge needle, ensuring that at least 70% of the microdissected tissue consisted of lesional cells. The DNA was extracted using the protocol described previously 5 . PCR-based LOH analysis of Pten was performed as described previously 5. Briefly, a common 5′ primer within the intron of exon 5 ( 5′ GGGATTATCTTTTTGCAACAGT 3′) and two 3′ primers ( 5′ GGGCCTCTTGTGCCTTTA 3′ and 5′ TTCCTGACTAGGGGAGGAGT 3′ ) were used to amplify Pten intragenic regions. Control DNA was prepared from either microdissected normal tissue or from tail DNA from Pten+/-;Mlh1-/-, Pten+/- and wild-type mice. PCR was performed in 50-μl reactions containing 10 mmol/L Tris-HCl (pH 9.2), 1.5 mmol/L MgCl2, 75 mmol/L KCl, 0.4 μmol/L of each 3′ primer, 0.8 μmol/L 5′ primer, 160 μmol/L each dNTP, and 2.5 U of Taq polymerase (Life Technologies, Inc., Gaithersburg, MD). Forty cycles of PCR were performed, each cycle consisted of 1 minute at 95°C, 1 minute at 57°C, and 1 minute at 72°C, followed by a single 5-minute extension at 72°C. The products were separated on 2% agarose gels, stained with ethidium bromide, and the relative intensities of the bands were visually scored. A mutant to wild-type ratio greater than 2:1 was scored as a LOH of the wild-type Pten allele. Each sample was repeated and scored separately in a blinded manner by two individuals. In addition to LOH analysis of the Pten wild type allele, the D19Mit40 and D19Mit74 loci located at 25cM and 54cM respectively on chromosome 19 were also analyzed. These dinucleotide repeat loci were amplified with alpha-32dCTP using commercially available primers (Research Genetics, Huntsville, AL) and LOH was determined on denaturing polyacrylamide gels visualized by autoradiography.
Mutational analysis
Extracted DNA from microdissected lesions was analyzed for intragenic Pten mutations in exons 1 to 9, K-ras mutations in exon 1, β-catenin mutations in exon 3, and Fas, Riz, Chk1, Axin2 and Igf2r mutations in repetitive coding sequences using intron-based, exon-specific PCR amplification. These genes were chosen because the repetitive sequences altered in primary human tumors are conserved in mice. PCR amplifications were performed in the presence of the following primers (Table 1). PCR conditions were as follows: initial denaturation at 95°C for 5 min, followed by 40 cycles of amplification (95°C for 1 min, annealing temperature for 1 min, 72°C for 1 min) and finally, 72°C for 5 min. Sequencing was carried out by the Cornell University Sequencing Facility. All lesions showing mutations were re-microdissected, re-amplified and re-sequenced to ensure reproducibility.
Table 1.
Primers used in PCR amplification
| Gene | Exon/ Repeat |
Forward primer | Reverse primer | Annealing temperature |
|---|---|---|---|---|
| Pten | exon 1 | 5′ CTCCTTTTTCTTCAGCCACA 3′ | 5′ AGAAAGCAAAGAGGAACAGC 3′ | 58°C |
| exon 2 | 5′ GCA TGCATATTTGTGTCATTG 3′ | 5′ GTTTTCGGGATCTTTTCTAAAT 3′ | 60°C | |
| exon 3 | 5′ GTTTTAGTCCTGTGCAGCAT 3′ | 5′ TCTTCAACAAAGAAATAACTTTTA 3′ | 55°C | |
| exon 4 | 5′ TCCTTCCAGTGTGAAAGGTAA 3′ | 5′ TCACCAGGCAGTAAAAGACA 3′ | 58°C | |
| exon 5 | 5′ CACTGGGATTATCTTTTTGC 3′ | 5′ CAAAGAGGGAGGAAGGAA 3′ | 58°C | |
| exon 6 | 5′ TTTTCTTTTGTCTCCCTCCTC 3′ | 5′ TCCGACACACAGACAGCTAA 3′ | 58°C | |
| exon 7 | 5′ GACATTTCTTGTGAAATGATCCTA 3′ | 5′ GGCTTTAAGCAAAAGGTCTG 3′ | 60°C | |
| exon 8 | 5′ TGTTGACTATTTGTGGTACATTTTT 3′ | 5′ TCACAATCAGAATAAAACACACC 3′ | 60°C | |
| exon 9 | 5′ TGTCTTTGCTAATACAGAACTCA 3′ | 5′ TCATGGTATTTTATCCCTCTTGA 3′ | 58°C | |
| K-ras | exon 1 | 5′ CTTGTGTGAGACATGTTCTAATTT 3′ | 5′ CTGCCGTCCTTTACAAGC 3′ | 62°C |
| β-catenin | exon 3 | 5′ CTGACCTGATGGAGTTGGAC 3′ | 5′ AGCTACTTGCTCTTGCGTGA 3′ | 62°C |
| Fas | A (9) | 5′ CTTTCTTTCTCTCTTTCTTTACTTC 3′ | 5′ GTACTCCTTCCCTTCTGTGC 3′ | 58°C |
| Riz | A (8) | 5′ GAACTCAGCAAAATGTCTCCA 3′ | 5′ CGTTCTTCGCAGATCTGTTC 3′ | 58°C |
| Chk1 | A (9) | 5′ CCCAGTGATACGTGTCAGGA 3′ | 5′ TACCCAGAGGAGCAGAATCA 3′ | 60°C |
| Axin2 | G(6) | 5′ AACCCGGGTCTTGCACTGT 3′ | 5′ GCTCACTCTCCAACATCCAC 3′ | 60°C |
| G(7) | 5′ GGATGGAGGAAAATGCCTA 3′ | 5′ GACCTTTAGGCTCCCCAGT 3′ | 60°C | |
| Igf2r | G(7) | 5′ GAAGCTGACTTTCGAAAACG 3′ | 5′ TCTGTGTAGTCCGGTCACAGT 3′ | 60°C |
Total RNA isolation and microarray analysis
Total RNA was extracted from the frozen uterine segments with extensive CAH or invasion using TRIzol reagent (Invitrogen, Carlsbad, CA) and further purified using the RNeasy Mini kit (Qiagen, Valencia, CA). RNA was quantified by measuring absorbance at 260 nm and the quality was assessed by HCHO-agarose denaturing gel electrophoresis. Five micrograms of total RNA was used for microarray analysis. The preparation and processing of labeled cRNA targets was carried out according to the manufacturer’s protocol (Affymetrix, Santa Clara, CA). Target RNA was hybridized to the GeneChip Mouse Genome 430A (Affymetrix, Santa Clara, CA). Each GeneChip contains over 22,600 probe sets representing approximately 14,000 well-characterized mouse genes. Hybridization and scanning were performed according to the GeneChip Expression Analysis Technical Manual. Raw data was analyzed using the Microarray Suite 5.0 software package (Affymetrix). All chips met the following criteria: 1) background <100, 2) noise <4, and 3) ratio of 3′/5′ hybridization <2 for β-actin, 4) percentage of present genes around 60%. Array data was normalized at the probe-level by using the Robust Multi-chip Average, with GC-content background correction algorithm (GC-RMA). Statistical analysis was performed with GeneSpring software 7.2 (Agilent, Redwood City, CA) for identifying the differentially expressed genes between non-invasive and invasive lesions.
Quantitative real-time PCR
The regulation of genes identified by microarray analysis was verified using TaqMan real-time PCR. First-strand cDNA was synthesized from 4μg of total uterine RNA with oligo(dT) primers and SuperScript III reverse transcriptase as per the instructions provided with the First-Strand cDNA Synthesis Kit (Invitrogen, Grand Island, NY). Primer sets and probe for OGP were ordered from Assays-on-Demand gene expression products (Applied Biosystems, Foster City, CA). Primer F (5′GCCAACCGTGAAAAGATGA3′) and Primer R (5′GCCTGGATGGCTACGTACATG3′) were the forward and reverse primers for β-actin respectively, while the sequence 6FAMCATGTTTGAGACCTTCAACMGBNFQ was used as the TaqMan MGB probe. Primers for each gene were in adjacent exons. Real-time PCR reactions were carried out in ABI PRISM 7700 Sequence Detection System (Applied Biosystems). All samples were run in triplicate. The threshold line was determined as 10 times the SD of the baseline fluorescence signal, and threshold cycle (Ct) was defined as cycle number at this point. Real time PCR was performed for all the samples and individual Ct values were obtained. The average Ct value for invasive lesions and CAHs was calculated. Fold change in OGP expression between non-invasive and invasive lesions was calculated by the comparative Ct method. The mean values with SD for four invasive lesions and eight CAH were calculated (ΔCt(inv)= 6.495±0.494; ΔCt(CAH)=9.59±2.08). Next, we calculated ΔΔCt using the formula ΔΔCt=ΔCt(Inv)-ΔCt(CAH), which was found to be -3.095.□□ The fold change between invasion and CAH was calculated as: Fold Change = -ΔΔCt(log 2)□ = -3.095(log2) = 8.54. β-actin was used as an internal control.
Human specimen collection
Two separate series of paraffin-embedded endometrial tissue samples were selected retrospectively from the surgical pathology files of New York Presbyterian Hospital and Johns Hopkins Hospital. The first series includes 25 cases of CAHs, while the second series includes 21 cases of UECs. These cases were reviewed by three gynecological pathologists (L.H.E., L.I. & B.M.R.) and the protocol was approved by the IRB at both institutions.
Immunohistochemistry
Five-μm sections were prepared from formalin-fixed, paraffin-embedded tissue. The immunostaining for OGP was carried out using Ultra Vison Detection System (Lab Vision Corporation, UK) according to manufacturer’s protocol. Antigen retrieval was performed in 10mM citrate buffer (pH 6.0) for 15 min. Sections were incubated in rabbit polyclonal anti-HuOGP antibody 19 at a dilution of 1:1000 for 1.5 hours at room temperature. Scores for OGP expression were assigned semiquantitatively according to the percentage of cells stained (0 for no positive cells; 1 for 1-25%; 2 for 26%-50%; 3 for 51-75%; 4 for 76-100%) and the intensity of staining (0 for no staining; 1 for weak staining; 2 for moderate staining; 3 for strong staining). The two scores were then multiplied to get the staining index a method commonly used in reporting immunohistochemical staining 20. The cases were considered positive with a score of 1 and above and the scores ranged from 0-12. Mann-Whitney U rank sum nonparametric test was used for statistical analysis using SPSS, version1 1.0 software.
Results
LOH of Pten in endometrial lesions and at additional loci on chromosome 19
To determine the status of the wild-type Pten allele, loss of heterozygosity (LOH) analysis was carried out on DNA isolated from microdissected lesions (both CAH and invasion, Figure 1A) from 6 Pten+/-;Mlh1-/- and 7 Pten+/-;Mlh1+/+ littermates in a C57Bl6/129SvJ background and 7 Pten+/- mice in a CD-1 background. Since all mice developed scattered, physically distinct lesions, more than one lesion was microdissected from all but one animal. In Pten+/-;Mlh1-/- double mutants 17 of 31 (54.8%) microdissected lesions, including both invasive lesions, showed LOH of the wild-type Pten allele, while 21 of 37 (56.8%) and 23 of 40 (57.5%, all CAH) lesions in Pten+/- (C57Bl6/129SvJ) and Pten+/- (CD-1) mice displayed LOH, respectively (Table 2 & Figure 1B). Both invasive lesions in Pten+/- mice showed LOH. Clearly, there is a similar frequency of LOH despite the age, genotype or the background strain. Interestingly, lesions in close proximity showed different LOH patterns suggesting that individual lesions are clonal.
Figure 1.

LOH and mutation analysis of Pten in Pten+/- and Pten+/-;Mlh1-/- mice. A) Representative photomicrographs of microdissected invasive (1) and CAH (2) lesions. B) PCR based analysis of LOH of Pten in endometrial lesions from CD-1 Pten+/- mice. Lane M, 1kb ladder. Lane 1, amplification of DNA from microdissected normal tissue from a Pten+/- mouse showing both mutant (*) and wild-type (arrow head) alleles. Lane 2, amplification of DNA from microdissected tissue from a wild-type mouse. Lane 3 and 6, amplification of DNA from two microdissected lesions without LOH. Lane 4, 5 and 7, amplification of DNA from three microdissected lesions showing LOH of the wild-type Pten allele. C) LOH at microsatellite loci D19Mit40 and D19Mit71 at Chromosome 19 in the CAH with LOH of Pten from a CD-1 Pten+/- mouse. Lane N, microdissected normal tissue showing two alleles. Lane C, microdissected CAH showing LOH at both loci tested. D) A frameshift mutation in exon 5 of Pten from a Pten+/-;Mlh1-/- lesion without LOH of Pten. A 1-bp deletion (T) at codon 146 is detected (arrow), which leads to a stop at codon 146.
Table 2.
Incidence of LOH of Pten and mutations in Pten, K-ras, β-catenin, Fas, Riz, Chk1, Axin2, Igf2r in the endometrial lesions from Pten+/-;Mlh1-/- and Pten+/- mice
| LOH (%) |
Pten mutation (in lesions w/o LOH) |
K-ras (exon 1) (in lesions w/o biallelic inactivation of Pten) |
β-catenin (exon 3) |
Fas A(9) |
Riz A(8) |
Chk1 A(9) |
Axin2 G(6)&G(7) |
Igf2r G(7) |
|
|---|---|---|---|---|---|---|---|---|---|
|
Pten+/-
;M lh1-/- |
54.8 (17/31)1 |
35.7 (5/14) |
0/9 | 0/20 | 0/21 | 0/15 | 0/6 | 0/6 | 0/6 |
|
Pten+/- (mixed) |
56.8 (21/37)2 |
0 (0/16) | 0/16 | 0/5 | |||||
|
Pten+/- (CD-1) |
57.5 (23/40)3 |
5.89 (1/17) |
0/13 |
Includes one invasive lesion and 30 CAH. The one invasive lesion had LOH. The additional nvasive lesion could not be submitted to LOH analysis due to stromal contamination.
Included two invasive lesions and 35 CAH. Both the invasive lesions had LOH.
All 40 lesions were CAH.
To further understand the mechanism of LOH, we analyzed the extent of LOH on chromosome 19 using microsatellite markers D19Mit40 and D19Mit71. All informative, microdissected CAHs with LOH of Pten showed loss of heterozygosity at both microsatellite loci (Figure 1C) demonstrating that LOH of Pten is not due to small intrachromosomal deletions.
Pten mutations in endometrial lesions without LOH
Pten mutation analysis of all 9 exons was done on DNA microdissected from every lesion without LOH. In Pten+/-;Mlh1-/- mice 5 of 14 (35.7%) CAHs without Pten LOH showed mutations in the remaining allele of Pten, whereas no mutations were identified in 16 CAHs from Pten+/- mice also in the C57Bl6/129SvJ background (Table 3). This difference is statistically significant (two tail p=0.014, Fisher’s Exact Test) suggesting that Pten mutations occur more commonly in lesions with underlying DNA mismatch repair deficiency. Of note, only 1 of 17 (5.89%) CAHs in Pten+/- (CD-1) mice had a mutation in Pten. Together with the LOH findings, biallelic inactivation of Pten (LOH or intragenic mutation) was present in 71%, 56.8% and 60% of the lesions from Pten+/-;Mlh1-/-, Pten+/- (C57Bl6/129SvJ) and Pten+/- (CD-1) mice, respectively. Sixty percent of CAHs lost Pten through either LOH or intragenic mutation, while all five invasive lesions showed biallelic inactivation of Pten via LOH. In addition, only two Pten+/-;Mlh1-/- and three Pten+/- (C57Bl6/129SvJ) mice developed invasive endometrial carcinomas in the 20 mice we studied. These results suggest that complete loss of Pten is not sufficient for the development of invasive endometrial carcinoma.
Table 3.
Mutations in Pten exons in LOH-negative lesions
| Animal | Lesion No. |
Genotype | Pten mutation |
|---|---|---|---|
| LP12 | 8 | Pten+/-;Mlh1-/- | Exon 6, Cod 184, Del(A) |
| LP12 | 9 | Pten+/-;Mlh1-/- | Exon 8, Cod 323, Del(A) |
| LP186 | 1 | Pten+/-;Mlh1-/- | Exon 6, Cod 184, Del(A) |
| LP54 | 2 | Pten+/-;Mlh1-/- | Exon 5, Cod 146, Del(A) |
| LP54 | 5 | Pten+/-;Mlh1-/- | Exon 6, Cod 184, Del(A) |
| CD203 | 1 | Pten+/-(CD-1) | Exon 5, E92K |
As described above, 35.7% of CAHs without Pten LOH arising in Pten+/-;Mlh1-/- mice showed mutations in the remaining allele of Pten. In contrast, only 5.89% and 0% of CAHs in Pten+/-(CD-1) and Pten+/- (C57Bl6/129SvJ) mice had Pten mutations, respectively. Furthermore, in Pten+/-;Mlh1-/- mice, all of the intragenic Pten mutations were deletions in poly(A/T) tracts at codons 146, 184 and 323, which are expected to result in truncation of the Pten protein (Figure 1D). We have previously identified the identical codon 323 mutation in a primary sporadic human endometrial carcinoma and complex atypical hyperplasia 21. In Pten+/- (CD-1) mice the single mutation was a missense mutation at codon 92, a region that is commonly mutated in primary human tumors 4, 22. Thus, the higher frequency of Pten mutations at repetitive sequences in Pten+/-;Mlh1-/- mice compared to those in Pten+/- mice (two tail p value=0.004, Fisher’s Exact Test) indicates that specific mutations in Pten are a consequence of MMR deficiency in this setting.
Mutational analysis of additional genes
To investigate molecular differences between CAH and UEC we analyzed Kras and Ctnnb1, two genes known to be mutated in a significant percentage of primary human UECs 15, 23-25. We also analyzed portions of the Fas, Riz, Chk1, Axin2 and Igf2r genes as they have been reported to accumulate mutations in specific repetitive coding sequences in MMR deficient primary human tumors and the repetitive sequences are conserved in mice 26, 27. The primer sequences for all the genes have been listed (Table 1). Sequencing of PCR products amplified from DNA extracted from microdissected lesions did not reveal mutations in any of these genes (Table 2).
Microarray analysis of CAH and UECs and Real-time RT-PCR validation
To identify genes differentially expressed in non-invasive and invasive endometrial lesions, we performed global gene expression analysis on 8 mice uteri with extensive CAH and 4 mice uteri with carcinoma having either stromal or myometrial invasion (Table 4). The labeled cRNA from each animal was hybridized to GeneChip Mouse Genome 430A from Affymetrics. Fifty-eight genes with statistically significant differences of at least 2-fold were selected and analyzed using hierarchical clustering for gene pattern study (Table 5). Out of 54, 25 were over-expressed in invasive lesions while 34 were down-regulated.
Table 4.
Genotypes and ages of mice used for microarray analysis
| Sample No. | Genotype | Age (in weeks) |
|---|---|---|
| CAH1 | Pten+/-;Mlh1+/+ | 36-40 |
| CAH2 | Pten+/-;Mlh1+/+ | 36-40 |
| CAH3 | Pten+/;;Mlh1+/+ | 36-40 |
| CAH4 | Pten+/-;Mlh1+/+ | 36-40 |
| CAH5 | Pten+/-;Mlh1-/- | 14-16 |
| CAH6 | Pten+/-;Mlh1-/- | 14-16 |
| CAH7 | Pten+/-;Mlh1-/- | 14-16 |
| CAH8 | Pten+/-;Mlh1-/- | 14-16 |
| INV1 | Pten+/-;Mlh1+/+ | 36-40 |
| INV2 | Pten+/-;Mlh1+/+ | 36-40 |
| INV3 | Pten+/-;Mlh1-/- | 14 |
| INV4 | Pten+/-;Mlh1-/- | 16 |
Table 5.
List of genes differentially expressed with at least 2-fold change between non-invasive and invasive endometrial lesions arising in mouse models
| Affymetrix probe set |
Symbol | Gene | Genebank | Fold Change a |
|---|---|---|---|---|
| 1425233_at | 2210407C18Rik | RIKEN cDNA 2210407C18 gene | BC019553 | 13 |
| 1418872_at | Abcb1b | ATP-binding cassette, sub-family B (MDR/TAP), member 1B |
NM_011075 | 5.937 |
| 1426526_s_at | Ovgp1 | oviductal glycoprotein 1 | AU017520 | 3.711 |
| 1420771_at | Sprr2d | small proline-rich protein 2D | NM_011470 | 3.665 |
| 1451537_at | Chi3l1 | chitinase 3-like 1 | BC005611 | 3.657 |
| 1419674_a_at | Dpep1 | dipeptidase 1 (renal) | NM_007876 | 3.416 |
| 1449578_at | Supt16h | suppressor of Ty 16 homolog (S. cerevisiae) |
AW536705 | 2.829 |
| 1416468_at | Aldh1a7 | aldehyde dehydrogenase family 1, subfamily A1 |
NM_013467 | 2.683 |
| 1448680_at | Serpina1a | serine (or cysteine) proteinase inhibitor, clade A, member 1a |
NM_009245 | 2.615 |
| 1435943_at | Dpep1 | dipeptidase 1 (renal) | AI647687 | 2.602 |
| 1419323_ at | Padi1 | peptidyl arginine deiminase, type I | NM_011059 | 2.582 |
| 1424938_at | Steap | six transmembrane epithelial antigen of the prostate |
AF297098 | 2.411 |
| 1451006_at | Xdh | xanthine dehydrogenase | AV286265 | 2.365 |
| 1426981_at | Pace4 | paired basic amino acid cleaving system 4 | BI157485 | 2.319 |
| 1419549_at | Arg1 | arginase 1, liver | NM_007482 | 2.311 |
| 1424953_at | BC021614 | cDNA sequence BC021614 | BC021614 | 2.268 |
| 1451191_at | Crabp2 | cellular retinoic acid binding protein II | BC018397 | 2.264 |
| 1422425_at | Sprr2k | small proline-rich protein 2K | NM_011477 | 2.206 |
| 1455618_x_at | 1300010A20Rik | RIKEN cDNA 1300010A20 gene | BI440178 | 2.182 |
| 1450007_at | 1500003O03Rik | RIKEN cDNA 1500003O03 gene | NM_019769 | 2.141 |
| 1451608_a_at | 1300010A20Rik | RIKEN cDNA 1300010A20 gene | BC024685 | 2.132 |
| 1439016_x_at | Sprr2a |
AV371678 RIKEN full-length enriched, adult male colon Mus musculus cDNA clone 9030418F15 3′ |
AV371678 | 2.077 |
| 1420832_at | Qscn6 | quiescin Q6 | AK004880 | 2.068 |
| 1448303_at | Gpnmb | glycoprotein (transmembrane) nmb | NM_053110 | 2.038 |
| 1451532_s_at | Steap | six transmembrane epithelial antigen of the prostate |
AF297098 | 2.011 |
| 1422814_at | Calmbp1 | calmodulin binding protein 1 | NM_009791 | 0.492 |
| 1423775_s_at | Prc1 | protein regulator of cytokinesis 1 | BC005475 | 0.489 |
| 1423341_at | Cspg4 | chondroitin sulfate proteoglycan 4 | BB377873 | 0.485 |
| 1417910_at | Ccna2 | cyclin A2 | X75483 | 0.48 |
| 1426767_at | 3230401M21Rik | RIKEN cDNA 3230401M21 gene | AK014328 | 0.473 |
| 1422723_at | Stra6 | stimulated by retinoic acid gene 6 | NM_009291 | 0.461 |
| 1435321_at | 3732412D22Rik | Adult male corpora quadrigemina cDNA, RIKEN full-length enriched library, clone:B230216K11 |
BM117827 | 0.461 |
| 1455195_at | Rps24 | ribosomal protein S24 | BM119287 | 0.455 |
| 1451411_at | Gprc5b | G protein-coupled receptor, family C, group 5, member B |
BC020004 | 0.448 |
| 1438034_at | 6430576D04Rik |
BB115225 RIKEN full-length enriched, adult male urinary bladder Mus musculus cDNA clone 9530049K06 3′ |
BB115225 | 0.445 |
| 1451567_a_at | Ifi203 | interferon activated gene 203 | BC008167 | 0.445 |
| 1424893_at | Ndel1 | nuclear distribution gene E-like homolog 1 (A. nidulans) |
BC021434 | 0.442 |
| 1436780_at | Ogt | O-linked N-acetylglucosamine (GlcNAc) transferase |
BG065325 | 0.439 |
| 1426712_at | Slc6a15 | solute carrier family 6 (neurotransmitter transporter), member 15 |
BB129409 | 0.435 |
| 1419898_s_at | Zc3hdc7 | RIKEN cDNA A430104C18 gene | AW556219 | 0.433 |
| 1452536_s_at | Igk-V1 | immunoglobulin kappa chain variable 1 (VI) |
Z95478 | 0.433 |
| 1424713_at | Calml4 | calmodulin-like 4 | AY061807 | 0.428 |
| 1429171_a_at | 5730507H05Rik | Adult male hypothalamus cDNA, RIKEN full-length enriched library, clone:A230105F24 product:hypothetical protein |
BB702347 | 0.426 |
| 1452195_s_at | 2310047I15Rik | RIKEN cDNA 2310047I15 gene | AW744519 | 0.422 |
| 1424289_at | BC010311 | cDNA sequence BC010311 | BB817847 | 0.413 |
| 1420512_at | Dkk2 | dickkopf homolog 2 (Xenopus laevis) | NM_020265 | 0.399 |
| 1416713_at | 2700055K07Rik | RIKEN cDNA 2700055K07 gene | NM_026481 | 0.397 |
| 1427257_at | Cspg2 | chondroitin sulfate proteoglycan 2 | BM251152 | 0.395 |
| 1427256_at | Cspg2 | chondroitin sulfate proteoglycan 2 | BM251152 | 0.39 |
| 1450276_a_at | Scin | scinderin | NM_009132 | 0.342 |
| 1417504_at | Calb1 | calbindin-28K | BB246032 | 0.323 |
| 1427161_at | Lek1 | leucine, glutamic acid, lysine family 1 protein |
BE848253 | 0.322 |
| 1448755_at | Col15a1 | procollagen, type XV | AF011450 | 0.281 |
| 1423597_at | Atp8a1 | ATPase, aminophospholipid transporter (APLT), class I, type 8A, member 1 |
BB303874 | 0.276 |
| 1416957_at | Pou2af1 | POU domain, class 2, associating factor 1 | NM_011136 | 0.237 |
| 1420719_at | Tex15 | testis expressed gene 15 | NM_031374 | 0.178 |
| 1448738_at | Calb1 | calbindin-28K | BB246032 | 0.0989 |
| 1449526_a_at | 1110015E22Rik | RIKEN cDNA 1110015E22 gene | NM_024228 | 0.0234 |
Fold change represents invasion versus non-invasion.
The expression of genes identified by the microarray analysis was further validated by Real-time RT-PCR analysis on the same RNA samples. Of particular interest was the Ogp gene which encodes a protein that is normally expressed in the oviduct, but has been reported to be over-expressed in human endometrial hyperplasias and carcinomas 28. Compared with the CAHs, Ogp expression in the carcinomas was up regulated by 8.54-fold, confirming the microarray observations (Figure 2).
Figure 2.
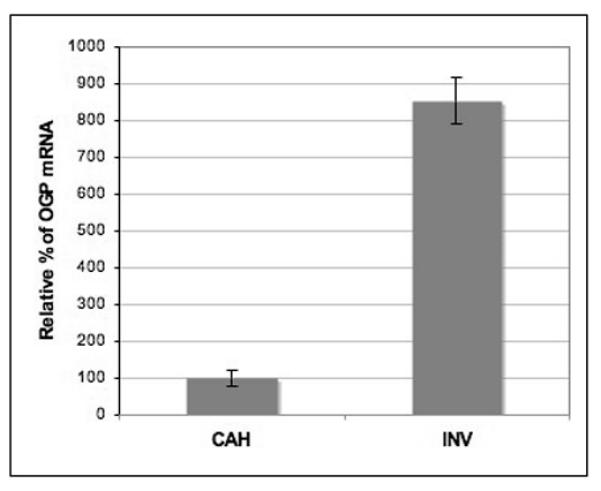
Real time PCR analysis of Ogp expression in CAH and invasion. Relative expression is determined as compared to actin mRNA. The increased expression of Ogp in invasive lesions validates the microarray analysis.
OGP as a potential molecular marker of human invasive endometrial cancer
Having identified Ogp as a potential marker up-regulated in invasive lesions in the mouse model, we next determined if the same was true in primary human tissue. Twenty-five cases with confirmed diagnosis of CAH (Figure 3C, H&E) and 21 UEC (Figure 3A, H&E) cases were analyzed for expression of OGP by immunostaining (Figure 3). OGP localized to the cytoplasm and appeared to exhibit stronger staining in the apical region of epithelial cells. The expression was quantified by assigning staining indices to all the samples, as described in materials and methods. All the UEC cases (Figure 3B) were strongly positive for OGP, with average staining index of 7.3 and more than 50% of cases with scores of 8 or greater. Of the CAH (Figure 3D) cases, 13 were positive for OGP, with average staining index of 1.4. The difference in the staining indices of the two groups was statistically significant (P=0.000, Table 6). These results indicate that OGP is a potential molecular marker of invasive endometrial cancer and further validates the relevance of the mouse model to human disease. Of note, all the cases had been previously analyzed for PTEN mutations and PTEN immunohistochemical staining (data not shown) and no correlation with OGP staining was found 21. In addition, 3 cases each of proliferative and secretory endometrium failed to show appreciable staining for OGP.
Figure 3.

Immunohistochemical analysis of OGP expression in primary human tumors. A) H&E staining of a uterine endometrioid carcinoma. B) Increased OGP expression in the lesions of an adjacent serial section from the same UEC case. C) H&E staining of a complex atypical hyperplasia. D) No OGP expression tested in the lesions of an adjacent serial section from the same CAH case. original magnifications, X10.
Table 6.
Immunohistochemical analysis of OGP expression in primary human tumors
| Tumor | n | No. (%) of positive cases |
No. (%) of cases with staining index>=8 |
P-value (staining index) |
|---|---|---|---|---|
| CAH | 25 | 13/25 (52) | 1/25 (4) | 0.000 |
| UEC | 21 | 21/21 (100) | 12/21 (57.1) |
Discussion
To understand the mechanisms responsible for the accelerated phenotype, and to shed light on the biological significance of the association of PTEN mutations and MMR deficiency in primary human tumors and its possible role in invasion, we analyzed lesions arising in Pten+/-;Mlh-/- mice and Pten+/- mice. Lesions from Pten+/-;Mlh1-/- and Pten+/- mice exhibited LOH at the Pten locus with equal frequency despite the difference in ages of the mice (14 weeks and 32-40 weeks respectively). All the lesions with LOH at the Pten locus also exhibited LOH at the microsatellite markers D19Mit40 and D19Mit71 on chromosome 19, suggesting that the loss of wild type Pten is not due to small chromosomal deletions, but rather is due to loss of heterozygosity of a significant portion of chromosome 19, either by deletion or recombination. In contrast, the Pten+/-;Mlh-/- mice had a statistically significant increase in intragenic mutations over the Pten+/- mice, 37.5% and 5.9%, respectively. Interestingly, the five mutations detected in Pten+/-;Mlh-/- were deletion mutations in repetitive sequences, whereas the single mutation identified in a Pten+/- mouse was a missense mutation. These findings suggest that mutations in Pten are more frequent in MMR deficient mice and that the mutations are due to the absence of DNA mismatch repair. Similar mutations have been found in MMR deficient and proficient human endometrial carcinoma with some studies reporting an increase in their frequency in MMR deficient tumors 29. Our results support that MMR deficiency results in an increase in Pten mutations that may contribute to the accelerated phenotype in the mouse model and explain, at least in part, the association between the two genetic alterations in primary human tumors 3, 4, 22, 29, 30. Of note, Pten+/+;Mlh-/- littermates did not develop endometrial lesions indicating that MMR deficiency alone is not sufficient for the development of hyperplasia or carcinoma during a similar time frame 8. Interestingly, it has been shown that MMR deficient mice treated with DES (Diethylstilbestrol) develop both hyperplasia and carcinoma 31. Perhaps PTEN alterations and DES treatment provide an environment (e.g., increased proliferation) that allows MMR deficiency to exert its effect on the neoplastic process.
In humans, MMR deficient endometrial carcinomas accumulate mutations in the repetitive sequences of coding regions of several genes, although the incidence of such mutations is low in UECs as compared to gastrointestinal MMR deficient tumors 26. In order to determine if any of these genes may be targets of MMR deficiency in the Pten+/-;Mlh1-/- mice, we analyzed the conserved repetitive target sequences in several genes (Fas, Riz, Chk1, Axin2 and Igf2r). The presence of mutations in these genes might, in concert with loss of Pten, lead to progression of CAH to UEC. No mutations were identified in the repeat sequences of these genes suggesting that they are not frequent targets of MMR deficiency in the mouse model. However, given the low incidence of such mutations in primary human tumors and the lack of reported mutations in CAH, it is quite possible that analysis of additional invasive lesions might reveal a low incidence of mutations. Another gene commonly altered in both hyperplasias and carcinomas in primary human tissues is the KRAS protooncogene. In published reports from several laboratories, mutations have been detected in approximately 26% of UECs. This is of obvious interest given the fact that activation of KRAS can lead to phosphorylation of AKT. Although KRAS and PTEN mutations are mutually exclusive in some tumor types, they are not in endometrial carcinoma 15. However, in a set of carcinomas analyzed in our laboratory, KRAS mutations were more commonly found in tumors with only a single intragenic PTEN mutation 15. Therefore, the Kras gene was sequenced in mouse lesions that lacked biallelic inactivation of Pten, but no mutations were identified. All of the lesions, hyperplasias and carcinomas, arising in the mouse model lacked expression of Pten by immunohistochemical staining suggesting that complete inactivation of Pten occurs even in lesions in which biallelic inactivation has not been detected. Consequently, it is possible that Kras mutations may not be selected for in this setting.
The high frequency of alterations in the wild-type Pten allele in CAH, in conjunction with the lack of staining for Pten by immunohistochemistry, suggests that the development of endometrial hyperplasia in Pten+/- mice is associated with complete loss of Pten. In other words, endometrial neoplasia did not develop in the context of haploinsufficiency in contrast to reports in mouse models of prostate neoplasia. This may, in part, explain why women with Cowden disease are at increased risk for endometrial carcinoma, but still show a relatively low disease penetrance. These data also indicate that alterations in other genes, in addition to complete loss of Pten, are required for the development of carcinoma. These findings are of interest in light of our recent study that found that human CAH and UEC have a similar frequency of PTEN mutations, but UEC has a statistically significant increase in PIK3CA mutations over CAH 21.
Although the above studies have provided insights into the relationship between PTEN alterations and MMR deficiency, they indicate that biallelic inactivation of Pten and mutations in additional candidate genes altered in human endometrioid carcinoma are not commonly responsible for the transition from CAH to carcinoma in the mouse model. In order to further elucidate the changes involved in this transition and identify objective markers of invasion we turned to a global gene expression profiling approach. Among the 58 genes, 24 were up-regulated in invasive lesions while 34 were down-regulated. Pace4, a member of proprotein convertases family, has been implicated in tumor cell invasion by activating matrix metalloproteinases (MT-MMPs) that results in collagenase type IV activation and collagen type IV degradation 32. Collagen type IV degradation leads to disruption and breakdown of the normal basement membrane architecture, a key process in the initiation of tumor microinvasion into the connective tissue. The over-expression of Dpep1, a membrane bound dipeptidase, was previously reported in colorectal carcinomas 28. Though the role of Dpep1 in cancer progression is currently unknown, it may be involved in the degradation of surrounding extracellular matrix components, a mechanism that would aid in the ability of tumor cells to migrate from the primary site through the extracellular matrix 28. Among the 34 down-regulated genes, Dkk2 is a negative regulator of Wnt signaling pathway, consistent with Wnt pathway abnormalities implicated in endometrial tumorigenesis 33-35. Though these genes are promising, we focused initially on Ogp since it was the most highly expressed gene that had potential relevance in human cancer. Exploration of the role of other genes will be a focus of future studies.
Recent reports have suggested that increased OGP expression is associated with ovarian and endometrial tumorigenesis 28. In addition, a recent report has described an intracellular form of OGP that is associated with tight junctions, suggesting a role for OGP in the maintenance of cell polarity and adhesion 36. Finally, an antibody was available that showed specificity in primary human tissue. OGP is responsive to estrogen in most species 37-41 and contains several half EREs in the promoter region and in primates progesterone down regulates OGP expression 38. These data contributed to OGP as an attractive candidate for further analysis. The up-regulation of OGP was first validated by real time RT-PCR analysis in which it was found to be up-regulated by 8.5 fold in the invasive lesions, corroborating the microarray analysis.
We subsequently tested the expression of OGP on human endometrial specimens by immunostaining. A set of CAHs and UECs was selected based on independent review by three gynecologic pathologists. OGP was found to be significantly over-expressed in UECs compared to CAH. Our results differ from the previous published results that suggested that OGP expression was greater in CAH than UEC 42. The reason for the discrepancy is unclear and suggests that further studies on larger sample sizes may be required. However, our results were highly significant (P=0.000), correlated with the expression studies in the mouse model, and suggest that high OGP expression is associated with invasive lesions in the endometrium. Hence, the functional significance of OGP overexpression in invasive endometrial lesions requires further investigation. It is likely that a single marker will not be sufficient to reliably distinguish carcinoma from CAH, however, it is possible that future studies analyzing the additional genes identified using the mouse model, in concert with studies on primary human tissue samples, will ultimately lead to a panel of objective markers that can be used in the clinical arena.
Acknowledgements
This work was supported by grants from the National Cancer Institute R01 CA095427 and the Bohmfalk Award.
Footnotes
Highlights: Biallelic inactivation of Pten is not sufficient for development of carcinoma Ogp could be a potential molecular marker to distinguish CAH from invasive carcinoma
References
- 1.Kurman RJ, Kaminski PF, Norris HJ. The behavior of endometrial hyperplasia. A long-term study of “untreated” hyperplasia in 170 patients. Cancer. 1985;56:403–12. doi: 10.1002/1097-0142(19850715)56:2<403::aid-cncr2820560233>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 2.Trimble CL, Kauderer J, Zaino R, Silverberg S, Lim PC, Burke JJ, 2nd, Alberts D, Curtin J. Concurrent endometrial carcinoma in women with a biopsy diagnosis of atypical endometrial hyperplasia: a Gynecologic Oncology Group study. Cancer. 2006;106:812–9. doi: 10.1002/cncr.21650. [DOI] [PubMed] [Google Scholar]
- 3.Simpkins SB, Peiffer-Schneider S, Mutch DG, Gersell D, Goodfellow PJ. PTEN mutations in endometrial cancers with 10q LOH: additional evidence for the involvement of multiple tumor suppressors. Gynecol Oncol. 1998;71:391–5. doi: 10.1006/gyno.1998.5208. [DOI] [PubMed] [Google Scholar]
- 4.Kong D, Suzuki A, Zou TT, Sakurada A, Kemp LW, Wakatsuki S, Yokoyama T, Yamakawa H, Furukawa T, Sato M, Ohuchi N, Sato S, et al. PTEN1 is frequently mutated in primary endometrial carcinomas. Nat Genet. 1997;17:143–4. doi: 10.1038/ng1097-143. [DOI] [PubMed] [Google Scholar]
- 5.Tashiro H, Blazes MS, Wu R, Cho KR, Bose S, Wang SI, Li J, Parsons R, Ellenson LH. Mutations in PTEN are frequent in endometrial carcinoma but rare in other common gynecological malignancies. Cancer Res. 1997;57:3935–40. [PubMed] [Google Scholar]
- 6.Mutter GL, Lin MC, Fitzgerald JT, Kum JB, Baak JP, Lees JA, Weng LP, Eng C. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J Natl Cancer Inst. 2000;92:924–30. doi: 10.1093/jnci/92.11.924. [DOI] [PubMed] [Google Scholar]
- 7.Lu TL, Chang JL, Liang CC, You LR, Chen CM. Tumor spectrum, tumor latency and tumor incidence of the Pten-deficient mice. PLoS ONE. 2007;2:e1237. doi: 10.1371/journal.pone.0001237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang H, Douglas W, Lia M, Edelmann W, Kucherlapati R, Podsypanina K, Parsons R, Ellenson LH. DNA mismatch repair deficiency accelerates endometrial tumorigenesis in Pten heterozygous mice. Am J Pathol. 2002;160:1481–6. doi: 10.1016/S0002-9440(10)62573-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R. Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci U S A. 1999;96:1563–8. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stambolic V, Tsao MS, Macpherson D, Suzuki A, Chapman WB, Mak TW. High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in pten+/- mice. Cancer Res. 2000;60:3605–11. [PubMed] [Google Scholar]
- 11.Helland A, Borresen-Dale AL, Peltomaki P, Hektoen M, Kristensen GB, Nesland JM, de la Chapelle A, Lothe RA. Microsatellite instability in cervical and endometrial carcinomas. Int J Cancer. 1997;70:499–501. doi: 10.1002/(sici)1097-0215(19970304)70:5<499::aid-ijc1>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 12.Gurin CC, Federici MG, Kang L, Boyd J. Causes and consequences of microsatellite instability in endometrial carcinoma. Cancer Res. 1999;59:462–6. [PubMed] [Google Scholar]
- 13.Levine RL, Cargile CB, Blazes MS, van Rees B, Kurman RJ, Ellenson LH. PTEN mutations and microsatellite instability in complex atypical hyperplasia, a precursor lesion to uterine endometrioid carcinoma. Cancer Res. 1998;58:3254–8. [PubMed] [Google Scholar]
- 14.Burks RT, Kessis TD, Cho KR, Hedrick L. Microsatellite instability in endometrial carcinoma. Oncogene. 1994;9:1163–6. [PubMed] [Google Scholar]
- 15.Lax SF, Kendall B, Tashiro H, Slebos RJ, Hedrick L. The frequency of p53, K-ras mutations, and microsatellite instability differs in uterine endometrioid and serous carcinoma: evidence of distinct molecular genetic pathways. Cancer. 2000;88:814–24. [PubMed] [Google Scholar]
- 16.Esteller M, Xercavins J, Reventos J. Advances in the molecular genetics of endometrial cancer (Review) Oncol Rep. 1999;6:1377–82. doi: 10.3892/or.6.6.1377. [DOI] [PubMed] [Google Scholar]
- 17.Salvesen HB, MacDonald N, Ryan A, Iversen OE, Jacobs IJ, Akslen LA, Das S. Methylation of hMLH1 in a population-based series of endometrial carcinomas. Clin Cancer Res. 2000;6:3607–13. [PubMed] [Google Scholar]
- 18.Parc YR, Halling KC, Burgart LJ, McDonnell SK, Schaid DJ, Thibodeau SN, Halling AC. Microsatellite instability and hMLH1/hMSH2 expression in young endometrial carcinoma patients: associations with family history and histopathology. Int J Cancer. 2000;86:60–6. doi: 10.1002/(sici)1097-0215(20000401)86:1<60::aid-ijc9>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 19.Rapisarda JJ, Mavrogianis PA, O’Day-Bowman MB, Fazleabas AT, Verhage HG. Immunological characterization and immunocytochemical localization of an oviduct-specific glycoprotein in the human. J Clin Endocrinol Metab. 1993;76:1483–8. doi: 10.1210/jcem.76.6.8501154. [DOI] [PubMed] [Google Scholar]
- 20.Tashiro H, Isacson C, Levine R, Kurman RJ, Cho KR, Hedrick L. p53 gene mutations are common in uterine serous carcinoma and occur early in their pathogenesis. Am J Pathol. 1997;150:177–85. [PMC free article] [PubMed] [Google Scholar]
- 21.Hayes MP, Wang H, Espinal-Witter R, Douglas W, Solomon GJ, Baker SJ, Ellenson LH. PIK3CA and PTEN mutations in uterine endometrioid carcinoma and complex atypical hyperplasia. Clin Cancer Res. 2006;12:5932–5. doi: 10.1158/1078-0432.CCR-06-1375. [DOI] [PubMed] [Google Scholar]
- 22.Risinger JI, Hayes K, Maxwell GL, Carney ME, Dodge RK, Barrett JC, Berchuck A. PTEN mutation in endometrial cancers is associated with favorable clinical and pathologic characteristics. Clin Cancer Res. 1998;4:3005–10. [PubMed] [Google Scholar]
- 23.Esteller M, Garcia A, Martinez-Palones JM, Xercavins J, Reventos J. The clinicopathological significance of K-RAS point mutation and gene amplification in endometrial cancer. Eur J Cancer. 1997;33:1572–7. doi: 10.1016/s0959-8049(97)00154-8. [DOI] [PubMed] [Google Scholar]
- 24.Fukuchi T, Sakamoto M, Tsuda H, Maruyama K, Nozawa S, Hirohashi S. Beta-catenin mutation in carcinoma of the uterine endometrium. Cancer Res. 1998;58:3526–8. [PubMed] [Google Scholar]
- 25.Saegusa M, Hashimura M, Yoshida T, Okayasu I. beta-Catenin mutations and aberrant nuclear expression during endometrial tumorigenesis. Br J Cancer. 2001;84:209–17. doi: 10.1054/bjoc.2000.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duval A, Hamelin R. Mutations at coding repeat sequences in mismatch repair-deficient human cancers: toward a new concept of target genes for instability. Cancer Res. 2002;62:2447–54. [PubMed] [Google Scholar]
- 27.Vassileva V, Millar A, Briollais L, Chapman W, Bapat B. Genes involved in DNA repair are mutational targets in endometrial cancers with microsatellite instability. Cancer Res. 2002;62:4095–9. [PubMed] [Google Scholar]
- 28.McIver CM, Lloyd JM, Hewett PJ, Hardingham JE. Dipeptidase 1: a candidate tumor-specific molecular marker in colorectal carcinoma. Cancer Lett. 2004;209:67–74. doi: 10.1016/j.canlet.2003.11.033. [DOI] [PubMed] [Google Scholar]
- 29.Bussaglia E, del Rio E, Matias-Guiu X, Prat J. PTEN mutations in endometrial carcinomas: a molecular and clinicopathologic analysis of 38 cases. Hum Pathol. 2000;31:312–7. doi: 10.1016/s0046-8177(00)80244-0. [DOI] [PubMed] [Google Scholar]
- 30.Maxwell GL, Risinger JI, Alvarez AA, Barrett JC, Berchuck A. Favorable survival associated with microsatellite instability in endometrioid endometrial cancers. Obstet Gynecol. 2001;97:417–22. doi: 10.1016/s0029-7844(00)01165-0. [DOI] [PubMed] [Google Scholar]
- 31.Kabbarah O, Sotelo AK, Mallon MA, Winkeler EL, Fan MY, Pfeifer JD, Shibata D, Gutmann DH, Goodfellow PJ. Diethylstilbestrol effects and lymphomagenesis in Mlh1-deficient mice. Int J Cancer. 2005;115:666–9. doi: 10.1002/ijc.20918. [DOI] [PubMed] [Google Scholar]
- 32.Bassi DE, De Cicco R Lopez, Cenna J, Litwin S, Cukierman E, Klein-Szanto AJ. PACE4 expression in mouse basal keratinocytes results in basement membrane disruption and acceleration of tumor progression. Cancer Res. 2005;65:7310–9. doi: 10.1158/0008-5472.CAN-05-1213. [DOI] [PubMed] [Google Scholar]
- 33.Risinger JI, Maxwell GL, Chandramouli GV, Aprelikova O, Litzi T, Umar A, Berchuck A, Barrett JC. Gene expression profiling of microsatellite unstable and microsatellite stable endometrial cancers indicates distinct pathways of aberrant signaling. Cancer Res. 2005;65:5031–7. doi: 10.1158/0008-5472.CAN-04-0850. [DOI] [PubMed] [Google Scholar]
- 34.Shedden KA, Kshirsagar MP, Schwartz DR, Wu R, Yu H, Misek DE, Hanash S, Katabuchi H, Ellenson LH, Fearon ER, Cho KR. Histologic type, organ of origin, and Wnt pathway status: effect on gene expression in ovarian and uterine carcinomas. Clin Cancer Res. 2005;11:2123–31. doi: 10.1158/1078-0432.CCR-04-2061. [DOI] [PubMed] [Google Scholar]
- 35.Moreno-Bueno G, Hardisson D, Sanchez C, Sarrio D, Cassia R, Garcia-Rostan G, Prat J, Guo M, Herman JG, Matias-Guiu X, Esteller M, Palacios J. Abnormalities of the APC/beta-catenin pathway in endometrial cancer. Oncogene. 2002;21:7981–90. doi: 10.1038/sj.onc.1205924. [DOI] [PubMed] [Google Scholar]
- 36.Kadam KM, D’Souza SJ, Natraj U. Identification of cellular isoform of oviduct-specific glycoprotein: role in oviduct tissue remodeling? Cell Tissue Res. 2007;330:545–56. doi: 10.1007/s00441-007-0489-0. [DOI] [PubMed] [Google Scholar]
- 37.Abe H, Satoh T, Hoshi H. Primary modulation by oestradiol of the production of an oviduct-specific glycoprotein by the epithelial cells in the oviduct of newborn golden hamsters. J Reprod Fertil. 1998;112:157–63. doi: 10.1530/jrf.0.1120157. [DOI] [PubMed] [Google Scholar]
- 38.Verhage HG, Mavrogianis PA, Boomsma RA, Schmidt A, Brenner RM, Slayden OV, Jaffe RC. Immunologic and molecular characterization of an estrogen-dependent glycoprotein in the rhesus (Macaca mulatta) oviduct. Biol Reprod. 1997;57:525–31. doi: 10.1095/biolreprod57.3.525. [DOI] [PubMed] [Google Scholar]
- 39.Murray MK. Biosynthesis and immunocytochemical localization of an estrogen-dependent glycoprotein and associated morphological alterations in the sheep ampulla oviduct. Biol Reprod. 1992;47:889–902. doi: 10.1095/biolreprod47.5.889. [DOI] [PubMed] [Google Scholar]
- 40.Jaffe RC, Arias EB, O’Day-Bowman MB, Donnelly KM, Mavrogianis PA, Verhage HG. Regional distribution and hormonal control of estrogen-dependent oviduct-specific glycoprotein messenger ribonucleic acid in the baboon (Papio anubis) Biol Reprod. 1996;55:421–6. doi: 10.1095/biolreprod55.2.421. [DOI] [PubMed] [Google Scholar]
- 41.Buhi WC, Ashworth CJ, Bazer FW, Alvarez IM. In vitro synthesis of oviductal secretory proteins by estrogen-treated ovariectomized gilts. J Exp Zool. 1992;262:426–35. doi: 10.1002/jez.1402620409. [DOI] [PubMed] [Google Scholar]
- 42.Woo MM, Alkushi A, Verhage HG, Magliocco AM, Leung PC, Gilks CB, Auersperg N. Gain of OGP, an estrogen-regulated oviduct-specific glycoprotein, is associated with the development of endometrial hyperplasia and endometrial cancer. Clin Cancer Res. 2004;10:7958–64. doi: 10.1158/1078-0432.CCR-04-1261. [DOI] [PubMed] [Google Scholar]