

Abstract
The neuropeptide corticotropin-releasing factor (CRF) plays a critical role in the proper functioning of the stress response system through its actions on its receptors, CRF receptor 1 (CRF1) and CRF receptor 2 (CRF2), located at multiple anatomical sites. Clinical data indicate that stress response dysfunctions, such as excessive CRF activity and hyperstimulation of CRF1, are present in a range of stress-related disorders, including depression and anxiety disorders. Our previous work along with that of other laboratories has demonstrated that mice deficient in CRF2 (CRF2−/−) display increased anxiety and depression-like behaviors. In this study we found CRF2−/− mice display increased hippocampal levels of activated (phosphorylated) mitogen-activated protein kinase (MAP kinase)/ERK kinase (MEK), extracellular signal-regulated kinases 1 and 2 (ERK1/2), and ribosomal protein S6 kinases 1 (RSK1). These changes can be explained by overactive hippocampal CRF1, in view of the finding that the application of the non-selective CRF receptor antagonist [Glu11,16] astressin ([Glu11,16]Ast) into the dorsal hippocampus of mutant mice returned the levels of the phosphorylated proteins to baseline. Moreover, inhibition of the hippocampal MEK/ERK pathway with the specific MEK inhibitor U0126, decreased depression-like behaviors in the forced swim test and tail suspension test of CRF2−/− mice. Similarly, treatment with [Glu11,16]Ast reversed depression phenotype of CRF2−/− mice without affecting the phenotype of wild-type littermates. Our results support an involvement of CRF receptors in the development of depression, such that elevated hippocampal CRF1 activity, in the absence of CRF2, produces a depression-dominated phenotype via activation of the MEK/ERK pathway.
Keywords: CRF, CRF1, hippocampus, MEK/ERK, knockout mouse, depression
Introduction
Depression is a devastating illness that affects approximately 17% of the population at some point in life, resulting in major social and economic consequences (Kessler, 1997). Evidence links stress, and the sensitivity of the individual to stressful stimuli, to the development of depression. The stress response is essential for adaptation and homeostatic balance. However, chronic stress can speed up disease, cause neural degeneration, and lead to depression or other affective disorders (Hammen, 2005).
Corticotropin releasing factor (CRF) is a 41-residue polypeptide that plays a key role in integrating the endocrine, autonomic, and behavioral responses to stress (Vale, et al 1981). CRF exerts its action through G-protein-coupled receptors (CRF1 and CRF2). CRF1 is expressed in high levels in neocortical areas, specifically, the basolateral and medial nucleus of the amygdala, anterior pituitary, hypothalamic nuclei, and cerebellar Purkinje cells among others. CRF2 has been detected in more discrete brain regions, including the dorsal raphe nucleus, lateral septum, ventromedial hypothalamus, and cortical nucleus of the amygdala (Chalmers, et al 1995; Van Pett, et al 2000).
Compelling evidence indicates that central CRF circuits are hyperactive in major depression. The most prominent evidence comes from clinical studies showing that a subset of severely depressed patients exhibit increased CRF concentrations in the cerebrospinal fluid, elevated expression of CRF in the paraventricular nucleus of the hypothalamus, increased CRF-immunoreactivity in the prefrontal and frontal cortex, locus coeruleus, and median and dorsal raphe nuclei, and decreased CRF-binding sites in the frontal cortex (Arborelius, et al 1999; Hauger, et al 2006; Schüle, 2007). These observations have been reinforced by behavioral studies in which central administration of CRF, preferentially binding to CRF1, resulted in behavioral changes including anxiety, motoric disturbances, and sleep, anorexia, and vegetative abnormalities (Heinrichs and Koob, 2004), which are cardinal symptoms of depression. Moreover, behavioral studies of transgenic mice with modified expression of CRF or CRF receptors support the notion that CRF overexpression increases anxiety-like behaviors, whereas deficiency of CRF1 (Smith, et al 1998; Timpl, et al 1998) or CRF2 genes (Bale, et al 2000; Kishimoto, et al 2000) reduces or heightens anxiety, respectively.
In addition, CRF2−/− mice tested in the forced swim test (FST) display increased immobility as an indicator of depression-like behavior (Bale and Vale, 2003; Todorovic, et al 2005). When treated with the CRF1 antagonist antalarmin the time spent immobile decreases, while swimming and climbing, i.e. active stress coping behavior increases (Bale and Vale, 2003). Although there were no controls to indicate whether antalarmin reduces depression-like behavior in wild-type mice, the effectiveness of CRF1 antagonism might be explained by the previous finding that CRF2−/− mice show increased CRF levels in the central nucleus of the amygdala and increased urocortin 1 (Ucn1) levels in the Edinger Westphal nucleus (Bale, et al 2000). The specific interaction, however, between CRF2 deletion effects and CRF1 on depression-like behavior remains unresolved. CRF1 signalling via the extracellular signal-regulated kinases 1/2 (ERK1/2)-mitogen-activated protein kinase (MAPK) cascade has been observed in neuronal, cardiac, and myometrial cells, as well as in recombinant expression systems (Hauger, et al 2006). In vivo, intracerebroventricular (i.c.v.) CRF administration increases phosphorylated ERK1/2 (pERK1/2) levels specifically in the CA3 and CA1 hippocampal subfields and basolateral complex of the amygdala, both structures related to external environmental information processing and behavioral aspects of stress (Refojo, et al 2005). Moreover, recent findings suggest that the ERK pathway in the hippocampus is involved in the molecular pathophysiology of depression and affective regulation, so ERK1/2 might be a significant target to examine the neuronal mechanisms of affective disorders induced by stress (Duman, et al 2007; Tronson, et al 2007)
Based on these considerations, the present study investigated the hypothesis that elevated phosphorylation of hippocampal ERK1/2 via enhanced activation of CRF1 might be responsible for depression-like behaviors observed in CRF2−/− mice, and that the ERK signaling pathway might be a therapeutically relevant target of the antidepressant-like actions of CRF1 antagonists.
Methods
Animals
CRF2−/− mice were generated using standard gene-targeting procedures with embryonic stem (ES) cells derived from the 129/SvJ mouse strain (Kishimoto, et al 2000). Briefly, genomic clones encompassing the CRF2 transcript were obtained from a 129/SvJ mouse library. A targeting vector was constructed in which the third intracellular loop of CRF2 was replaced by neomycin-resistant gene sequences. This knockout however does not target the soluble CRF2 α isoform, which lacks functional exons 8−14 present in the standard form that contains the region coding for the third intracellular loop targeted in the knockout (Chen, et al 2005). ES cells derived from 129/SvJ mice were electroporated with the targeting construct and selected with G418/ganciclovir using Southern-blot analysis. Heterozygous ES cell clones were injected into C57BL/6J blastocysts to generate chimeric mice. Chimeric founder males were bred to C57BL/6J females to generate F1 heterozygous offspring. Homozygous CRF2 mice were obtained from F1 intercrosses. Genotyping was performed by polymerase chain reaction-based DNA amplification using primer sets recognizing the third intracellular loop of CRF2 or the neomycin-resistance coding sequence as previously described (Kishimoto, et al 2000). Data reported here were obtained using F3-F5 generation hybrids. From the age of 8 weeks, mice were individually housed in standard Macrolon cages with free access to food and water. Mice were 10 weeks of age at the time of testing. They were kept in a 12 h light–dark cycle with lights switched on at 7 a.m. The experiments were performed during the light phase. All experimental procedures were in compliance with the European Council Directive (86/609/EEC) by permission of the Animal Section Law enforced by the District Government of Braunschweig, Lower Saxony, Germany. Experiments performed in Honolulu were approved by the University of Hawaii Animal Care Committee in accordance with National Institute of Health guidelines. All efforts were made to minimize animal suffering. For each behavioral test and molecular study different mice were used.
Cannulation and administration of drugs
The injection system (C235; Plastics One, Roanoke, VA) consisted of double-guided cannulae, dummy and a cap. Double-guided cannulae (internal, 28 gauge; guide, 22 gauge) were inserted bilaterally into the dorsal hippocampi (intrahippocampal injection; 2 mm posterior to the bregma, 2 mm lateral, and 1.6 mm below the dural surface for the CA1 region (Franklin and Paxinos, 1997), and affixed to the skull by dental cement. The animals were allowed to recover for 7−10 days before the experiments started. On the day of the experiment, the mice were exposed to a light isoflurane anesthesia, the cap and the dummy were removed, and solutions were delivered through an injector linked to two Hamilton microsyringes with plastic tubing. The drugs were administered bilaterally by a microinjector (CMA/Microdialysis, Sweden) over a 30 s period so that 0.25 μl volume was injected to each side. The cannula placement was verified for each mouse immediately after the behavior experiments by histological examination of the brains following methylene blue injection (0.25 μl/site). Only data obtained from mice with correctly inserted cannulae were included in the statistical analysis. For behavioral studies, 16 of 114 operated animals were excluded due to incorrect cannula placement. The MAPK/extracellular signal-regulated kinase (ERK) kinase (MEK) inhibitor 1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto) butadiene (U0126) (Promega, Madison, WI) was dissolved in 100% DMSO to a final stock concentration of 2 mg/ml. For behavioral testing, the drug was diluted with artificial CSF (aCSF; in mM: 120 NaCl, 2.4 KCl, 1.2 CaCl, 1.2 MgCl, 0.9 NaH2PO4, 1.4 Na2HPO4, and 0.3 ascorbic acid) to yield final concentrations of 250 μg/0.25 μl per side. We have demonstrated previously that the employed dose of this inhibitor is effective in vivo and significantly impairs the phosphorylation of its downstream targets during fear conditioning (Ahi, et al 2004). U0126 is a specific ERK/MAPK inhibitor and has no effect at a range of concentrations on other kinases, such as PKA, calcium-calmodulin kinase II, or PKC (Roberson, et al 1999). The vehicle was made as above with the omission of U1026. The highly potent non-selective CRF receptor antagonist [Glu11,16] astressin ([Glu11,16]Ast) (IC50 = 3.3 nM for CRF1 and IC50 = 1.1 nM for CRF2) (Eckart, et al 2001) was initially dissolved in 10 mM acetic acid before distilled water and 2-fold concentrated sterile aCSF was added to obtain the final dose of 120 pmol (215 ng/0.25 μl per side) of ([Glu11,16]Ast. Solution was adjusted to pH 7.4. The dose used was selected on the basis of its ability to prevent behavioral effects of the wide range of doses (0−35 pmol) of oCRF preferentially binding to CRF1 (Eckart, et al 2001). The vehicle was made as above with the omission of ([Glu11,16]Ast. The number of mice in behavioral experiments was 6−8 per group. The number of mice in each experiment is specified in the corresponding figure legends.
Tail suspension test
On the day of the tail suspension test (TST), CRF2−/− mice and their wild-type littermates were transferred to the experiment room and allowed to acclimatize for 1 h. Mice were injected intrahippocampally (i.h.) with vehicle (50% DMSO), or U0126 (250 μg/0.25 μl per side) 30 min before the TST trial. An automated tail-suspension apparatus (TSE Systems, Bad Homburg, Germany), with a tail hanger connected to a precision linear load cell, was used. One centimeter of the mouse's tail was inserted into the tail hanger and secured with non-irritating adhesive tape. Mice remained suspended by the tail, at a height of 35 cm from the tabletop, for 6 min. During this time the load cell recorded the mouse's movements and transmitted the information to a central computer, which then recorded the rate of immobility within the course of the session, and calculated total duration of immobility. Decreases in basal levels of immobility are highly predictive of antidepressant efficacy (Cryan, et al 2005).
Forced swim test
For the forced swim test (FST), a fresh group of CRF2−/− mice and their wild-type littermates were subjected to swim sessions in individual glass cylinders (height 39 cm, diameter 21.7 cm) containing water, 15-cm deep, at 23−25°C. On Day 1, all animals were placed in the cylinder for a preswim session of 12 min. On the test day 24 h later (Day 2), the mice were subjected to a testswim session for 5 min. Mice were injected on Day 1, i.h., with vehicle (50% DMSO in aCSF), or U0126 (250 μg/0.25 μl per side) 30 min before the preswim session of 12 min. Since CRF2−/− mice did not display depression-like phenotype during a testswim on Day 2, pharmacological interventions were omitted for that day. The water was changed between subjects. All sessions were recorded by a video camera positioned directly above the cylinder. A competent observer blind to treatment scored the videotapes. The behavioral measure scored was the duration of floating, defined as time spent still or only using righting movements to keep the head above water, and the latency to assume immobile posture. Increase in floating time, and decrease in latency time, were interpreted as an increase of depression-like behavior (Porsolt, et al 1978).
Locomotor activity test
Locomotor activity was measured in a Plexiglas chamber (35 cm × 20 cm × 20 cm) located in a box (58 cm × 30 cm × 27 cm) with a grey interior and a 12 W light at the ceiling. Activity (total distance travelled in centimetres) was measured during a 30 min period by an infrared beam system (detection rate 10 Hz) and analyzed by TSE software (VideoMot 2, Bad Homburg, Germany).
Immunohistochemistry of phospho-ERK1/2
CRF2−/− mice and their wild-type littermates were used to look at levels of ERK1/2 expression in the hippocampus without any prior treatment or behavioral analysis. Mice were deeply anesthetized and transcardially perfused with ice-cold phosphate-buffered saline (PBS) (pH 7.4), followed by 0.1 M phosphate buffer, pH 7.4, containing 4% paraformaldehyde (pH 7.4, 150 ml per mouse). The brains were post-fixed for 48 h in the same fixative (4% PFA) and then immersed for 24 h each in 10, 20, and 30% sucrose in PBS. The brains were cryo-sectioned (50μm slices). Immunostaining was performed using the free-floating method with mouse phospho-ERK1/2 (Thr202/Tyr204) monoclonal antibody (1:500; Cell Signaling Technology, Beverly, MA), followed by biotinylated secondary antibody and avidin-biotin complex system (Vector ABC kit, Vector Laboratories, Burlingame, CA). Diaminobenzidine (DAB) (Sigma tablet set) was used as a chromogen. For immunofluorescence analysis of pERK1/2, a similar protocol as the one used for perfusion and postfixation of mice brains was followed. Immunofluorescent labeling was performed as previously described (Sherrin, et al 2008). Briefly, sections (50μm thick) were washed with TBS before overnight incubation with mouse monoclonal anti-ERK1/2 IgG (Cell Signaling Technology) at 1:200 dilution followed by incubation with biotinylated anti-mouse secondary antibody (1:400; Vector Labs). The sections were washed, Fluorescin avidin DCS (Vector Labs) was applied, rendering green fluorescence. The sections were mounted on Superfrost Plus slides (Menzel Glaser, Braunschweig, Germany) using Vectashield mounting medium (Vector Labs) to prevent rapid photobleaching of fluorescein. An Axioimager Zeiss microscope and Zeiss Axio Cam HRM camera were used for the observations and photography of the slides, respectively.
Phospho-ERK1/2 density measurements
The CA1 and CA3 hippocampal subfields, the dentate gyrus, and the corpus callosum of mouse were outlined at the anatomical coordinates −1.4 to −1.6 mm posterior to the bregma (Franklin and Paxinos, 1997). All photographs were taken at 20× magnification using identical light intensity and exposure times. All images were imported into the NIH image analysis program version 1.63, and the black and white images were inverted to simulate dark-field illumination. For densitometric analysis, two separate standard area contours were drawn for each digital image. The selected contour for positive pERK immunoreactivity delineated the CA1 (encompassing dorsal stratum pyramidale, and partially strata radiatum, and oriens), CA3 hippocampal subfields (encompassing dorsal stratum pyramidale, and partially strata radiatum and lucidum containing mossy fibers from the granule cells), or dentate gyrus (encompassing dorsal deep hilus and hilar border with granule cell layer). The other contour for nonspecific DAB background lacking pERK immunoreactivity delineated corpus callosum from the same brain section. Because the corpus callosum had less variation and a low optical value reflecting less immunoreactivity, it was chosen as the reference. Mean optical density numbers were derived by subtracting the non-specific DAB background from the positive pERK-immunoreactive areas.
Protein Extraction and Western Blot Analysis
Hippocampi were dissected out and immediately homogenized at 4°C with a plastic homogenizer, in a Cell Lysis buffer (Cell Signalling, Beverly, MA) and a protease and phosphatase inhibitor cocktail (Pierce, Rockford, IL). The insoluble material was removed by centrifugation at 14 000 g for 10 min at 4°C. Protein concentrations were determined by the Bradford method (BioRad, Munich, Germany). Equal amounts of protein for each group were separated on a 10% SDS gel and transfered to an Immobilon-PVDF membrane (Millipore Corporation, Bedford, MA, USA). The blots were first probed with antibodies against the phosphorylated forms of the protein and then stripped and probed with antibodies against total proteins of same type. Antibodies were from Cell Signaling (Beverly, MA) (pMEK (Ser217/221) (1:1000), pERK1/2 (Thr202/Tyr204) (1:1000), pRSK1 (Thr359/Ser363) (1:1500), pp38 (Thr180/Tyr182) (1:2000), and pJNK1−3 (Thr183/Tyr185) (1:2000), and respective total-protein (1:1000)) Immunocomplexes were detected using the ECL method (Amersham Biosciences, Piscataway, NJ). Quantification of the immunoblots was performed by densitometric scanning of the film using NIH ImageJ software. To assess for changes in the phosphorylated kinase levels they were normalized to respective total kinase levels in hippocampal extracts. Total kinase levels did not differ significantly across genotypes. Normalized phosphorylated kinase levels in CRF2−/− mice were expressed as a percentage of those in wild-type mice (+/+).
Statistical Analysis
The data for behavioral and molecular studies are expressed as mean ± SEM, and were analyzed using a one-way ANOVA with factor Genotype and two-way ANOVA with factors Treatment and Genotype. Bonferroni/Dunn test applied, post hoc, for individual between-group comparisons at the p < 0.05 level of significance.
Results
In our first experiments, depression-like behavior of male CRF2−/− mice was measured. Bale and Vale (2003) demonstrated that male and female CRF2−/− mice show increased immobility, compared to wild-type mice, during 5 min test swimming on Day 2 in the FST as an indicator of depression. Similar to results described by Bale and Vale (2003), we observed elevated depression-like behavior for male CRF2−/− mice compared to wild-type mice. However, in our experiments the CRF2−/− mice showed increased floating (F(1,14) = 14.45; p < 0.05) and decreased latency to assume floating posture (F(1,14) = 3.12; p < 0.05) only in the 12 min preswim on Day 1 (Fig 1a, b), but not in the 5 min testswim session 24 hr later (p > 0.05 for both behavioral measures) (Day 2) (Fig 1c, d). In addition, CRF2−/− and wild-type mice were examined in the TST. CRF2−/− mice exhibited an increase in immobility compared with wild-type mice (F(1,14) = 5.15; p < 0.05) (Fig. 1e). Overall, our data confirmed and extended previous results that male CRF2−/− mice display elevated depression–like behaviors.
Figure 1. Increased depression-like behaviours of CRF2-deficient mice in the forced swim test and tail suspension test.
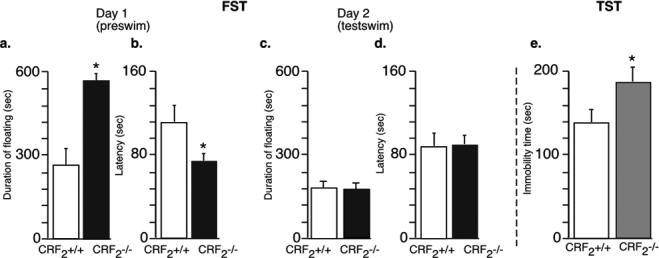
Behavior of wild-type, and CRF2−/− mice in the FST. The number of animals per group in the FST test was 8 wild-type and 8 CRF2−/− mice. CRF2−/− male mice showed an increase in time spent passively floating on the preswim on Day 1 (12 min) of the FST (a), and decreased latency to attain immobile posture (b). Statistically significant differences: *p<0.05, versus wild-type mice. CRF2−/− mice did not display any significant changes in depression-like behaviors during the testswim (5 min) on Day 2 (c, d). Behavior of wild-type, and CRF2−/− mice in the TST. CRF2−/− mice demonstrated significantly increased immobility as compared to wild-type mice (8 wild-type and 8 CRF2−/− mice) (e). Bar graphs are mean ± SEM. Statistically significant differences: * p < 0.05, versus wild-type littermates.
We next investigated whether the increased depression-like behavior of male CRF2−/− mice is caused by differences in activation (phosphorylation) of hippocampal ERK1/2, its upstream activator MEK, and substrate RSK1 implicated in mood regulation (Duman, et al 2007). Therefore, we determined the levels of phosphorylated MEK, ERK1/2, and RSK1 under baseline conditions and 30 min after exposure to the FST on Day 1. In the hippocampal formation, a significant increase in pMEK (F(1,6) = 14.15; p < 0.05) pERK1, (F(1,6) = 6.82; p < 0.05), pERK2 (F(1,6) = 4.44; p < 0.05), and pRSK1 (F(1,6) = 9.56; p < 0.05) was detected in CRF2−/− mice as compared with the wild-type mice under baseline conditions (Fig. 2a, b). Two other subfamilies of MAPKs, c-Jun amino-terminal kinases1−3 and p38 MAPKs did not differ between genotypes (p > 0.05). In the control study, Western blot analysis did not reveal significant changes in MEK, ERK1/2, and RSK1 protein levels in the somatosensory cortex and striatum, indicating relative region specific alteration of the MEK/ERK pathway in the mutant mice (data not shown). Exposure to the FST for 12 min on Day 1 did not change hippocampal pERK1 and pERK2 activation of CRF2−/− mice and their wild-type littermates (p > 0.05) (Fig. 2c). Immunohistochemical analysis revealed subfield-specific phosphorylation of ERK1/2 within the hippocampus (Fig. 3a), as in the CA3 (F(1,8) = 12.98; p < 0.05) (Fig. 3b, Fig. 3e left panel), and CA1 (F(1,8) = 8.22; p < 0.05) (Fig. 3c, Fig. 3e right panel), hippocampal subfields, but not the dentate gyrus (p > 0.05), pERK1/2 was significantly elevated (Fig. 3a-e).
Figure 2. Increased expression of activated proteins in the MEK/ERK pathway in CRF2−/− mice under basal conditions.

Western blot analyses of hippocampal extracts for phosphorylated MEK, ERK1/2, RSK1, JNK1−3, p38 and their respective non-phosphorylated protein forms were carried out using specific antibodies. Hippocampal lysates from CRF2−/− mice and wild-type littermates were obtained from untreated, naive mice (a). Quantification by densitometry of the corresponding X-ray films. Activated kinase levels in CRF2−/− mice (−/−) were expressed as a percentage of those in wild-type mice (+/+). Before that calculation, activated kinase levels were normalized to total kinase levels. Differences between wild-type littermates (+/+) (n= 4) and CRF2−/− (n= 4) mice were all statistically significant (ANOVA) in the MEK/ERK pathway; *p < 0.05 relative to the wild-type (+/+) littermates. Other members of MAPK subfamilies, p38 MAPK and c-Jun NH2-terminal protein kinase1−3 (JNK1−3) did not differ between genotypes (b). Twelve minute (Day 1) exposure to the FST did not induce elevation of pERK1/2 in hippocampal lysates (wild-type, n = 4; CRF2−/−, n= 4) (c). No changes in total protein amounts were observed. Statistically significant differences: * p < 0.05, versus wild-type littermates.
Figure 3. Increased phosphorylation of ERK1/2 in CRF2−/− mice in the CA1 and CA3 hippocampal subfields under baseline conditions.

Representative photomicrographs of baseline pERK1/2 labelling in the hippocampus of wild-type (+/+) and CRF2−/− mice (a). Summarized diagram showing pERK1/2-labeled cells in CRF2−/− mice and their wild-type (+/+) controls. The data are expressed as the relative optical density of pERK1/2-immunoreactivity (mean ± SEM) in the DG, CA1, and CA3 of the hippocampus (n= 5 mice per genotype) Bar graphs are mean ± SEM. Statistically significant differences: *p < 0.05, versus wild-type littermates (b). High magnification showing pERK1/2 immunostaining of mossy fibers and CA3 pyramidal cells of wild-type (+/+) (left) and CRF2−/− mice (right). Black arrows indicate immunostained fibers, and white arrows, nuclear staining (diaminobenzidine was used as a chromogen) (c). High magnification of pERK1/2 immunostaining of CA1 pyramidal cells of wild-type (+/+) (left) and CRF2−/− mice (right). Black arrows indicate immunostained fibres of stratum radiatum, and white arrows, nuclear staining (diaminobenzidine was used as a chromogen) (d). Representative images of pERK1/2 fluorescence immunohistochemistry of CA3 (left panel) and CA1 (right panel) subregions of the hippocampus in CRF2−/− mice (lower panel) and wild-type (+/+) littermates (upper panel) (magnification 20 x) (e). Abbreviations: CA1 subregion of hippocampus (CA1); CA3 subregion of hippocampus (CA3); dentate gyrus (DG). (Scale bar, 110 μm (a); 400μm (c-e)).
In order to determine whether MEK/ERK pathway hyperphosphorylation is functionally related to elevated depression-like behavior observed in CRF2−/− mice, we tested the effects of U0126, a specific MEK inhibitor (Roberson, al 1999), on depression-like behaviors in the FST and TST. The U0126 injections were applied i.h. 30 min before the preswim on Day 1 for the FST, and 30 min before the TST. Since no differences between genotypes were observed during the testswim on Day 2 of the FST, injections of U0126 were not employed for Day 2. Two-way ANOVA (treatment X genotype) revealed that an i.h. injection of U0126 (250 μg/0.25 μl per side) significantly decreased floating time (treatment, F(1,26) =7.46; p < 0.05; genotype, F(1,26) = 2.35; p > 0.05; treatment x genotype, F(1,26) = .67; p > 0.05), increased latency time (treatment, F(1,26) = 11.2; p < 0.05; genotype, F(1,26) = 3.12; p > 0.05; treatment x genotype, F(1,26) = 1.67; p > 0.05) in the FST (Fig. 4a, b), and reduced immobility time (treatment, F(1,26) = 5.2; p < 0.05; genotype, F(1,26) = 1.89; p > 0.05; treatment x genotype, F(1,26) = .77; p > 0.05) in the TST (Fig. 4d) irrespective of genotype (Bonferroni/Dunn test; #p > 0.05 compared to vehicle treated mice; *p > 0.05 compared to vehicle treated wild-type littermates). To confirm that i.h. administration of the MEK inhibitor U0126 results in inhibition of ERK phosphorylation, we injected mice with U0126 (250 μg/0.25 μl per side), and 30 min later, hippocampal tissue was collected for determination of pERK levels. As seen in Figure 4c, 250 μg of U0126 significantly decreased basal levels of pERK1/2 in the hippocampus without altering the amount of total ERK (Bonferroni/Dunn test; *p < 0.05 compared to vehicle treated groups). We next examined the locomotor responses to pharmacological inhibition of the hippocampal MEK/ERK pathway. CRF2−/− mice and their wild-type littermates received i.h. injection of vehicle or U0126 (250 μg/0.25 μl per side), and were exposed to the locomotor activity test 30 min later. As revealed by two-way ANOVA (treatment X genotype) (treatment, F(1,12) = .46; p > 0.05; genotype, F(1,12) = 1.1; p > 0.05; treatment x genotype, F(1,12) = .62; p > 0.05), U0126 did not affect locomotor activity in the CRF2−/− and control mice (Fig. 4e). Overall, these data suggest that the blockade of the MEK/ERK pathway in the hippocampus induces selective antidepressant-like behavioral effects.
Figure 4. Inhibition of the MEK/ERK pathway in the hippocampus has antidepressant properties in both wild-type and CRF2−/− mice in the forced swim test and tail suspension test.

Intrahippocampal (i.h.) administration of MEK inhibitor U0126 (250 μg/0.25 μl per side) 30 min before 12 min FST (Day 1) (a, b), or 6 min TST (d), reversed elevated depression-like behaviors of CRF2−/− mice and showed antidepressant-like properties in the wild-type mice. Locomotor activity as indicated by total distance travelled (cm) was not affected by i.h. U0126 (250 μg/0.25 μl per side) treatment (e). The number of animals per group in the FST and TST tests was 7 for CRF2 −/−, and 8 for CRF2+/+. For locomotor activity test, number of mice per group was 4. Different mice were used for separate tests. Representative blots (left), and mean ± SEM percent of pERK1 and pERK2 immunoreactivity (right) from hippocampal extracts taken from wild-type mice given i.h. infusions of 50% DMSO (vehicle) (n= 4 mice), or 1 μg/μl of U0126 (n= 4 mice) (a, b, d). The immunoblots were obtained from individual hippocampal lysates isolated 30 min after injection of inhibitor or vehicle. Normalized activated kinase levels in CRF2−/− mice were expressed as a percentage of those in wild-type mice (+/+) (c). Statistically significant differences: # p > 0.05 compared to vehicle treated mice of the same genotype; * p > 0.05 compared to vehicle treated wild-type littermates.
We were also interested to examine if increased hippocampal CRF1 activity in the absence of CRF2 was the possible cause of MEK/ERK pathway hyperphosphorylation, and the increased depression-like behaviors in CRF2−/− mice. Figure 5a shows that i.h. administration of 120 pmol ([Glu11,16]Ast (215 ng/0.25 μl per side), resulted in normalization of hippocampal pMEK and pERK1/2 levels in CRF2−/− mice 30 min afterwards without changing the amount of total proteins. Moreover, the same treatment did not change pMEK and pERK1/2 levels in the wild-type littermates (Bonferroni/Dunn test; p > 0.05 compared to vehicle treated groups) (Fig. 5a). Based on that finding, in the next experiment, 120 pmol [Glu11,16]Ast (215 ng/0.25 μl per side) >was applied i.h. to CRF2−/− mice and wild-type littermates 30 min before Day 1 test of the FST (12 min) and the TST (6 min). Two-way ANOVA did not reveal a significant difference in floating (treatment, F(1,20) = 1.82; p > 0.05; genotype, F(1,20) = 2.43; p > 0.05; treatment x genotype, F(1,20) = 1.67; p > 0.05), and latency time (treatment, F(1,20) = 1.22; p > 0.05; genotype, F(1,20) = 2.17; p > 0.05; treatment x genotype, F(1,20) = 1.33; p > 0.05) in the FST of male [Glu11,16]Ast-treated CRF2-mutant mice compared to vehicle-treated mutant mice (Bonferroni/Dunn test; p > 0.05) (Fig. 5b, c). Similarly, the level of immobility detected in the [Glu11,16]Ast -treated mutant male mice was similar to that found in vehicle treated wild-type littermates in the TST (Bonferroni/Dunn test; p > 0.05) (Fig. 5d). ). These results demonstrated that administration of CRF1/CRF2 non-specific antagonist [Glu11,16]Ast in CRF2−/− mice resulted in a balanced activation of the MEK/ERK pathway and attenuated ERK-mediated depression-like phenotype, suggesting a role for CRF1 in these processes. Non-specific blockade of CRF receptors in the hippocampus did not result in altered depression-like behavior, unrelated to mouse genotype.
Figure 5. Blockade of CRF1 in the hippocampus normalizes MEK/ERK pathway and reverses depression-like behaviors of CRF2−/− mice.
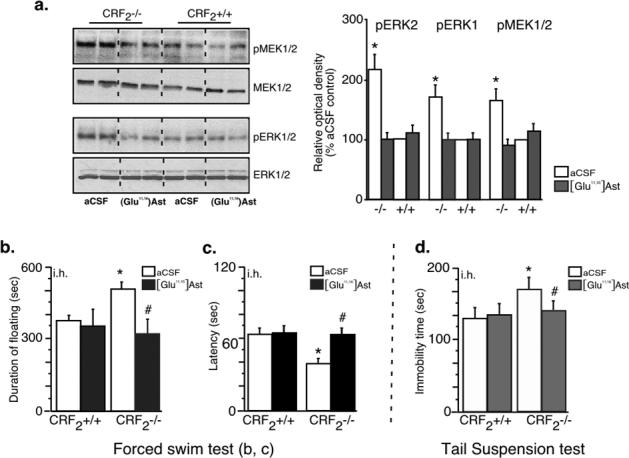
Intrahippocampal (i.h.) administration of CRF receptor non-selective antagonist ([Glu11,16]Ast (120 pmol (215 ng/0.25 μl per side) normalized hippocampal levels of pMEK and pERK1/2 in CRF2−/− mice. Differences between wild-type (+/+) (n= 3) and CRF2 −/− mice (n= 3) were not statistically significant: p> 0.05. The immunoblots were obtained from individual hippocampal lysates obtained 30 min after injection of antagonist or vehicle (a). The same treatment with 120 pmol [Glu11,16]Ast (215 ng/0.25 μl per side, injected i.h.) 30 min before 12 min FST on Day 1 (b, c), or 6 min TST (d), returned depression-like behaviors of CRF2−/− mice to baseline levels, without affecting behavior of wild-type mice. The number of animals per group in the FST and TST tests was 6 for CRF2 −/−, and 6 for wild-type mice (+/+). Different mice were used for separate tests. Statistically significant differences: #p > 0.05 compared to vehicle treated mice of the same genotype; *p > 0.05 compared to vehicle treated wild-type littermates.
Discussion
In these studies, male CRF2−/− mice showed increased depression-like behaviors in both FST and TST, the most commonly used animal models of depression (Cryan, et al 2002). Male CRF2−/− mice demonstrated both increased floating time, as well as decreased latency to attain floating posture, compared with their wild-type male littermates in the FST. In addition, CRF2−/− mice showed a significant increase in immobility time during TST. These behavioral differences between genotypes were functionally coupled with the hyperphosphorylation of the hippocampal MEK/ERK pathway found in the CRF2−/− mice. This overactivation of the MEK/ERK pathway is hypothesised to result from the unimpeded activation of hippocampal CRF1. In support of this interpretation, i.h. application of the CRF receptor subtype-nonselective antagonist [Glu11,16]Ast reversed the observed molecular and behavioral differences between genotypes. Additionally, i.h. blockade of the MEK/ERK pathway had antidepressant effects independent of the genotype.
Overall, these results support the prevailing hypothesis that increased CRF1 activity results in increased susceptibility for the development of depression, since previous studies have demonstrated an involvement of enhanced CRF1 activity or increased production of CRF with the development of depression-like behaviors in rodents and humans (Arborelius, et al 1999; Reul and Holsboer, 2002). Similar to our results, it has been previously reported that male and female CRF2−/− mice show increased immobility in the FST, and that i.c.v. treatment of CRF2−/− mice with the CRF1 antagonist antalarmin decreases immobility time and increases swim time in both sexes (Bale and Vale, 2003). The same mutants display increased CRF mRNA in the central nucleus of the amygdala, Ucn1 mRNA in the Edinger-Westphal nucleus, and Urocortin3 mRNA levels in the lateral perifornical area (Bale, et al 2000; Bale and Vale, 2003). Taking into account our present finding that i.h. application of CRF receptor subtype-nonselective antagonist [Glu11,16]Ast reduced depression-like behaviors of the CRF2−/− mice, it is conceivable that deregulation of a circuitry involving the central nuclei of the amygdala and the hippocampal CRF receptors may result in a predisposition for developing the depression-dominated phenotype (Swanson and Cowan, 1977). It is worth noting, that i.h. application of [Glu11,16]Ast did not affect depression-like behaviors of wild-type mice. Since CRF1 antagonists are most effective in producing an antidepressant effect when applied to animals bred for high innate anxiety or previously exposed to stress, more often than not, CRF1 antagonists do not affect baseline depression-related behaviors when rodents are tested in the standard or modified FST or TST. (Keck, et al 2001; Hauger, et al 2006). As we recently suggested, antagonism of CRF1 may be especially effective in treating individuals who are genetically predisposed to develop anxiety and affective disorders, or who have been exposed to stress by means of ‘priming’ sensitizing them to subsequent stress challenges (Sherrin, et al 2008).
Our data indicating that CRF2−/− mice showed increased immobility only during the preswim, but not in the testswim session 24 hours later (Days 1 and 2, respectively) are seemingly at odds with the previous study in which the CRF2−/− mice display increased immobility during testswim session (Day 2) (Bale and Vale, 2003). Certain procedural differences between the studies merit mentioning. Firstly, the latter study did not measure depression-like behaviors on Day 1. Secondly, while in the latter study mice were placed in the water for a preswim for 5 min on Day 1 and then monitored during a 5 min test 24 hr after the preswim, totalling 10 min of the forced swimming (Bale and Vale, 2003), duration of the forced swimming in our study totalled 12 min already on Day 1 (‘preswim’), the time frame in which Bale and Vale (2003) observed the differences between the genotypes. The absence of the differences between genotypes on Day 2, observed in our study, might be put in the framework of a series of findings showing that CRF1 and CRF2 regulate the dorsal raphe nucleus (DRN) serotonergic system in an opposite manner (Price, et al 2002: Hammack, et al 2003). In short, studies utilizing FST, demonstrate that CRF1 mediates inhibition of the DRN serotonergic system which facilitates short-term, active defensive responses that can be quickly terminated, while CRF2 mediates excitation of this system which promotes long-term, passive defensive responses (Valentino and Commons, 2005). It is possible, therefore, that in the absence of CRF2, CRF1-facilitated short-term defensive responses dominated, whereas CRF2-mediated passive long-term, defensive responses, characteristic of learned helplessness, were absent on Day 2 of the FST in CRF2−/− mice. In such case, the balance between innate and learned depression-related behavioral tendencies would favor the loss of hippocampally-driven depressant phenotype observed on Day 1.
As noted, i.c.v. injection of h/rCRF induces strong ERK1/2 activation in the CA3 and CA1 hippocampal subfields and basolateral complex of the amygdala. However, CRF-induced phosphorylation of ERK1/2 is absent in low-anxiety mice with a conditional knockout of forebrain and limbic CRF1 (Refojo, et al 2005). Hence, CRF1 signaling via the MEK/ERK pathway may play a role in anxiogenesis and depression. In accordance, we found that CRF2−/− mice exhibit increased levels of pERK1/2 activator pMEK, pERK1/2, and pERK1/2 substrate pRSK1, in the CA1 and CA3 subfields of the hippocampus. These results suggest that overactivation of CRF1 in these areas might stimulate activity of the MEK/ERK pathway. This suggestion was supported by the finding that local injection of [Glu11,16]Ast returned the MEK/ERK pathway activity to the level observed in wild-type mice. In short, these data point to the possibility that hippocampal CRF1 overdrive stimulates the MEK/ERK pathway, which causes depression-like behaviors in CRF2−/− mice. In line with this, i.h. injection of the selective MEK inhibitor U0126, dramatically decreased the floating and immobility time in the FST and TST, respectively, both in wild-type and mutant mice. Broadly, enhanced CRF1 activity might be attributable to a deficiency in desensitization of receptor, either to mechanisms that switch CRF1 signaling from one mode of G-protein signaling to another, or to intracellular mechanisms regulating the magnitude and duration of CRF1 signal in CRF2−/− mice (Steckler and Dautzenberg, 2005). The exact mechanism of the observed molecular changes induced by CRF2-deficiency/CRF1 overdrive is the subject of present studies in our laboratory.
Our results need to be discussed in the context of data demonstrating that chronic administration of antidepressants which increase the synaptic concentration of serotonin and/or norepinephrine, appear to activate the MEK/ERK signaling pathway in certain brain structures (Nestler, et al 2002; Coyle and Duman, 2003; Huang and Lin, 2006; Gourley, et al 2008). In line with these findings, systemic acute blockade of MEK/ERK signaling produces a depressive-like phenotype and blocks behavioral actions of antidepressants in the mouse models of depression (Duman, et al 2007). Using acute treatments as employed here, it has recently been reported that activation of ERK1/2 also acts as a depressant within the amygdala and prefrontal cortex (Huang and Lin, 2006; Qi, et al 2006). Those findings may appear inconsistent with our results at first glance. However, taking into consideration that acute and subacute (i.e. three doses over a 24-hour period) systemic MEK inhibition produce opposite effects on FST behavior (Duman, et al 2007), it remains to be elucidated whether multiple treatments or the use of different doses would consistently show depressant effects in the hippocampus, amygdala and prefrontal cortex. Also, a growing body of data suggests that region-specific manipulation of the MEK/ERK pathway may bring about quite distinct affective behavioral phenotypes (Newton, et al 2002; Einat, et al 2003). In particular, a recent finding that i.h. blockade of the MEK/ERK pathway in mice lacking ERK1 reduces the depressive-like phenotype caused by overactivation of ERK2 (Tronson, et al 2007), suggests that ERK mediates depressant actions via the hippocampus (Fumagalli, et al 2005: Tronson, et al 2007), and antidepressant actions via other brain sites like the prefrontal cortex (Qi, et al 2006).
We have previously demonstrated that CRF2−/− mice do not show alterations of locomotor activity, which would be a potential confound variable for the assessment of floating behavior, latency in the FST, and immobility time in the TST (Kishimoto, et al 2000). In addition, locomotor effects of i.h. U0126 were avoided by employing a dose that did not increase locomotion in the locomotor activity test, but instead selectively diminished the depressive-like phenotype of CRF2−/− mice.
It should also be mentioned, that recently, based on the FST, we described that the potent CRF1-selective agonist cortagine produces antidepressant-like effects similar to the CRF1 antagonists (Tezval, et al 2004). This is in apparent contrast to FST studies that found antidepressant-like effects by central CRF1 antagonism (Overstreet and Griebel, 2004; Hodgson, et al 2007), as well as with results of the present study. In addition, cortagine decreases the distance traveled in the elevated plus maze (EPM), and so reduces mobility (Tezval, et al 2004). Based on this decrease of mobility in the EPM, it would not be sufficient to suggest that the effects of cortagine in the FST are simply locomotor effects (Swiergiel, et al 2008). Attempting to reconcile these results, Farrokhi et al., (2008) recently noted that interpretation of behaviors might depend on the model employed. They suggest that anxiety-like or defensive responses may be affected by the options available for dealing with a threat source (Blanchard, et al 1997). A more potent stress source with limited options for responding such as the FST allows two options: increase the swim rate to attempt escaping, or become immobile. The EPM provides more options, and the alternative of retreat to an area of relative safety rather than struggling to escape. Thus, comparing the measure of mobility in both tests as an index for emotionality may not be appropriate. While the FST may be a useful screening model for antidepressant effects of drugs (Lucki, 1997), it may be a weak indicator of emotionality with compounds like cortagine that also produce locomotor effects or high levels of anxiety (Farrokhi, et al 2008). Further preclinical research is necessary to determine the exact role of CRF1 in depression-related behaviors.
This is of particular importance, given that the concept that abnormally enhanced central CRF/CRF1 signaling contributes to the pathophysiology of major depression was recently questioned, after clinical trials employing small molecule CRF1 receptor antagonists in patients with major depression yielded inconsistent results (Zobel et al, 2000; Binneman et al., 2008). Although in a preliminary open-label clinical trial, oral administration of the CRF1 receptor antagonist NBI-30775 decreases depression and anxiety scores in patients with major depression, presumably by blocking the activity of hyperactive CRF1 receptors in brain regions mediating symptoms of depression (Zobel et al, 2000), recent randomized, placebo controlled trial finds that another CRF1 antagonist CP-316,311 fails to demonstrate efficacy in the treatment of major depression (Binneman et al., 2008). Even though it is difficult to reconcile such apparent inconsistency, it should be kept in mind that probably only in a certain subpopulation of patients altered CRF /CRF1 signaling might account for the etiology and symptoms of major depression. This renders heterogeneity among trials in clinical setting highly probable.
In summary, our results have confirmed previous results (Bale and Vale, 2003; Todorovic, et al 2005) that show a significant increase in depression-like behaviors in CRF2−/− mice. Presumably chronically elevated CRF and Ucn1 levels acting on the hippocampal CRF1 led to overactivation of the MEK/ERK pathway that might be contributing to the detected depressive-like phenotype of mutants. Additional studies examining the involvement of CRF family members, as well as other distinct neurotransmitter systems, will provide necessary information concerning the genetic and neurobiological basis for the complex relationship between stress and depression.
Acknowledgments
This work was supported by Max Planck Society, NIH grant 5U54NS039406-08, and NMRC grant (NMRC/0754/2003).
Footnotes
Disclosure Conflict of Interest: We declare no conflict of interest.
References
- Ahi J, Radulovic J, Spiess J. The role of hippocampal signaling cascades in consolidation of fear memory. Behav Brain Res. 2004;149:17–31. doi: 10.1016/s0166-4328(03)00207-9. [DOI] [PubMed] [Google Scholar]
- Arborelius L, Owens MJ, Plotsky PM, Nemeroff CB. The role of corticotropin-releasing factor in depression and anxiety disorders. J Endocrinol. 1999;160:1–12. doi: 10.1677/joe.0.1600001. [DOI] [PubMed] [Google Scholar]
- Arzt E, Holsboer F. CRF signaling: molecular specificity for drug targeting in the CNS. Trends Pharmacol Sci. 2006;27:531–538. doi: 10.1016/j.tips.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Bale TL, Contarino A, Smith GW, Chan R, Gold LH, Sawchenko PE, Koob GF, Vale WW, Lee KF. Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behaviour and are hypersensitive to stress. Nat Genet. 2000;24:410–414. doi: 10.1038/74263. [DOI] [PubMed] [Google Scholar]
- Bale TL, Vale WW. Increased depression-like behaviors in corticotropin-releasing factor receptor-2-deficient mice: sexually dichotomous responses. J Neurosci. 2003;23:5295–5301. doi: 10.1523/JNEUROSCI.23-12-05295.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binneman B, Feltner D, Kolluri S, Shi Y, Qiu R, Stiger T. A 6-week randomized, placebo-controlled trial of CP-316,311 (a selective CRH1 antagonist) in the treatment of major depression. Am J Psychiatry. 2008;165:617–620. doi: 10.1176/appi.ajp.2008.07071199. [DOI] [PubMed] [Google Scholar]
- Blanchard RJ, Griebel G, Henrie JA, Blanchard DC. Differentiation of anxiolytic and panicolytic drugs by effects on rat and mouse defense test batteries. Neurosci Biobehav Rev. 1997;21:783–789. doi: 10.1016/s0149-7634(96)00062-0. [DOI] [PubMed] [Google Scholar]
- Chalmers DT, Lovenberg TW, De Souza EB. Localization of novel corticotropin-releasing factor receptor (CRF-2) mRNA expression to specific subcortical nuclei in rat brain: Comparison with CRF-1 receptor mRNA expression. J Neurosci. 1995;15:6340–6350. doi: 10.1523/JNEUROSCI.15-10-06340.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AM, Perrin MH, Digruccio MR, Vaughan JM, Brar BK, Arias CM, Lewis KA, Rivier JE, Sawchenko PE, Vale WW. A soluble mouse brain splice variant of type 2alpha corticotropin-releasing factor (CRF) receptor binds ligands and modulates their activity. Proc Natl Acad Sci U S A. 2005;102:2620–2625. doi: 10.1073/pnas.0409583102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Duman RS. Finding the intracellular signaling pathways affected by mood disorder treatments. Neuron. 2003;38:157–160. doi: 10.1016/s0896-6273(03)00195-8. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Mombereau C, Vassout A. The tail suspension test as a model for assessing antidepressant activity: review of pharmacological and genetic studies in mice. Neurosci Biobehav Rev. 2005;29:571–625. doi: 10.1016/j.neubiorev.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Markou A, Lucki I. Assessing antidepressant activity in rodents: recent developments and future needs. Trends Pharmacol Sci. 2002;23:238–245. doi: 10.1016/s0165-6147(02)02017-5. [DOI] [PubMed] [Google Scholar]
- Duman CH, Schlesinger L, Kodama M, Russell DS, Duman RS. A role for MAP kinase signaling in behavioral models depression and antidepressant treatment. Biol Psychiatry. 2007;61:661–670. doi: 10.1016/j.biopsych.2006.05.047. [DOI] [PubMed] [Google Scholar]
- Eckart K, Jahn O, Radulovic J, Tezval H, van Werven L, Spiess J. A single amino acid serves as an affinity switch between the receptor and the binding protein of corticotropin-releasing factor: implications for the design of agonists and antagonists. Proc Natl Acad Sci USA. 2001;98:11142–11147. doi: 10.1073/pnas.211424998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einat H, Yuan P, Gould TD, Li J, Du J, Zhang L, et al. The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J Neurosci. 2003;23:7311–7316. doi: 10.1523/JNEUROSCI.23-19-07311.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrokhi CB, Tovote P, Blanchard RJ, Blanchard DC, Litvin Y, Spiess J. Cortagine: behavioral and autonomic function of the selective CRF receptor subtype 1 agonist. CNS Drug Rev. 2007;13:423–443. doi: 10.1111/j.1527-3458.2007.00027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin K, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. Academic Press; San Diego: 1997. [Google Scholar]
- Fumagalli F, Molteni R, Calabrese F, Frasca A, Racagni G, Riva MA. Chronic fluoxetine administration inhibits extracellular signal-regulated kinase 1/2 phosphorylation in rat brain. J Neurochem. 2005;93:1551–1560. doi: 10.1111/j.1471-4159.2005.03149.x. [DOI] [PubMed] [Google Scholar]
- Gourley SL, Wu FJ, Kiraly DD, Ploski JE, Kedves AT, Duman RS, Taylor JR. Regionally specific regulation of ERK MAP kinase in a model of antidepressant-sensitive chronic depression. Biol Psychiatry. 2008;63:353–359. doi: 10.1016/j.biopsych.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammack SE, Schmid MJ, LoPresti ML, Der-Avakian A, Pellymounter MA, Foster AC, Watkins LR, Maier SF. Corticotropin releasing hormone type 2 receptors in the dorsal raphe nucleus mediate the behavioral consequences of uncontrollable stress. J Neurosci. 2003;23:1019–1025. doi: 10.1523/JNEUROSCI.23-03-01019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauger RL, Risbrough V, Brauns O, Dautzenberg FM. Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: new molecular targets. CNS Neurol Disord Drug Targets. 2006;5:453–479. doi: 10.2174/187152706777950684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammen C. Stress and depression. Annu Rev Clin Psychol. 2005;1:293–319. doi: 10.1146/annurev.clinpsy.1.102803.143938. [DOI] [PubMed] [Google Scholar]
- Heinrichs SC, Koob GF. Corticotropin-releasing factor in brain: a role in activation, arousal, and affect regulation. J Pharmacol Exp Ther. 2004;311:427–440. doi: 10.1124/jpet.103.052092. [DOI] [PubMed] [Google Scholar]
- Hodgson RA, Higgins GA, Guthrie DH, Lu SX, Pond AJ, Mullins DE, Guzzi MF, Parker EM, Varty GB. Comparison of the V1b antagonist, SSR149415, and the CRF1 antagonist, CP-154,526, in rodent models of anxiety and depression. Pharmacol Biochem Behav. 2007;86:431–440. doi: 10.1016/j.pbb.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Huang TY, Lin CH. Role of amygdala MAPK activation on immobility behavior of forced swim rats. Behav Brain Res. 2006;173:104–111. doi: 10.1016/j.bbr.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Keck ME, Welt T, Wigger A, Renner U, Engelmann M, Holsboer F, Landgraf R. The anxiolytic effect of the CRH(1) receptor antagonist R121919 depends on innate emotionality in rats. Eur J Neurosci. 2001;13:373–380. doi: 10.1046/j.0953-816x.2000.01383.x. [DOI] [PubMed] [Google Scholar]
- Kessler RC. The effects of stressful life events on depression. Annu Rev Psychol. 1997;48:191–214. doi: 10.1146/annurev.psych.48.1.191. [DOI] [PubMed] [Google Scholar]
- Kishimoto T, Radulovic J, Radulovic M, Lin CR, Schrick C, Hooshmand F, Hermanson O, Rosenfeld MG, Spiess J. Deletion of Crhr2 reveals an anxiolytic role for corticotropin-releasing hormone receptor-2. Nat Genet. 2000;24:415–419. doi: 10.1038/74271. [DOI] [PubMed] [Google Scholar]
- Lucki I. The forced swim test as a model for core and component behavioral effects of antidepressant drugs. Behav Pharmacol. 1997;8:523–532. doi: 10.1097/00008877-199711000-00010. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- Newton SS, Thome J, Wallace TL, Shirayama Y, Schlesinger L, Sakai N, Chen J, Neve R, Nestler EJ, Duman RS. Inhibition of cAMP response element-binding protein or dynorphin in the nucleus accumbens produces an antidepressant-like effect. J Neurosci. 2002;22:10883–10890. doi: 10.1523/JNEUROSCI.22-24-10883.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet DH, Griebel G. Antidepressant-like effects of CRF1 receptor antagonist SSR125543 in an animal model of depression. Eur J Pharmacol. 2004;497:49–53. doi: 10.1016/j.ejphar.2004.06.035. [DOI] [PubMed] [Google Scholar]
- Porsolt RD, Bertin A, Jalfre M. ‘Behavioural despair’ in rats and mice: strain differences and the effects of imipramine. Eur Pharmacol. 1978;51:291–294. doi: 10.1016/0014-2999(78)90414-4. [DOI] [PubMed] [Google Scholar]
- Price ML, Kirby LG, Valentino RJ, Lucki I. Evidence for corticotropin-releasing factor regulation of serotonin in the lateral septum during acute swim stress: adaptation produced by repeated swim. Psychopharmacology. 2002;162:406–414. doi: 10.1007/s00213-002-1114-2. [DOI] [PubMed] [Google Scholar]
- Qi X, Lin W, Li J, Pan Y, Wang W. The depressive-like behaviors are correlated with decreased phosphorylation mitogen-activated protein kinases in rat brain following chronic forced swim stress. Behav Brain Res. 2006;175:233–240. doi: 10.1016/j.bbr.2006.08.035. [DOI] [PubMed] [Google Scholar]
- Refojo D, Echenique C, Müller MB, Reul JM, Deussing JM, Wurst W, Sillaber I, Paez-Pereda M, Holsboer F. Corticotropin-releasing hormone activates ERK1/2 MAPK in specific brain areas. Proc Natl Acad Sci U S A. 2005;102:6183–6188. doi: 10.1073/pnas.0502070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reul JM, Holsboer F. Corticotropin-releasing factor receptors 1 and 2 in anxiety and depression. Curr Opin Pharmacol. 2002;2:23–33. doi: 10.1016/s1471-4892(01)00117-5. [DOI] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüle C. Neuroendocrinological mechanisms of actions of antidepressant drugs. J Neuroendocrinol. 2007;19:213–226. doi: 10.1111/j.1365-2826.2006.01516.x. [DOI] [PubMed] [Google Scholar]
- Sherrin T, Todorovic C, Zeyda T, Tan CH, Hon PW, Zhu YZ, Spiess J. Chronic stimulation of corticotropin-releasing factor receptor 1 enhances the anxiogenic response of the cholecystokinin system. Mol Psychiatry. 2008 doi: 10.1038/sj.mp.4002121. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Smith GW, Chen R, Marchuk Y, Hauser C, Bentley CA, Sawchenko PE, Koob GF, Vale W, Lee KF. Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron. 1998;20:1093–1102. doi: 10.1016/s0896-6273(00)80491-2. [DOI] [PubMed] [Google Scholar]
- Steckler T, Dautzenberg FM. Corticotropin-releasing factor receptor antagonists in affective disorders and drug dependence-- an update. CNS Neurol Disord Drug Targets. 2006;5:147–165. doi: 10.2174/187152706776359619. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Cowan WM. An autoradiographic study of the organization of the efferent connections of the hippocampal formation in the rat. J Comp Neurol. 1977;172:49–84. doi: 10.1002/cne.901720104. [DOI] [PubMed] [Google Scholar]
- Swiergiel AH, Leskov IL, Dunn AJ. Effects of chronic and acute stressors and CRF on depression-like behavior in mice. Behav Brain Res. 2008;186:32–40. doi: 10.1016/j.bbr.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Tezval H, Jahn O, Todorovic C, Sasse A, Eckart K, Spiess J. Cortagine, a specific agonist of corticotropin-releasing factor receptor subtype 1, is anxiogenic and antidepressive in the mouse model. Proc Natl Acad Sci USA. 2004;101:9468–9473. doi: 10.1073/pnas.0403159101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JM, Stalla GK, Blanquet V, Steckler T, Holsboer F, Wurst W. Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet. 1998;19:162–166. doi: 10.1038/520. [DOI] [PubMed] [Google Scholar]
- Todorovic C, Jahn O, Tezval H, Hippel C, Spiess J. The role of CRF receptors in anxiety and depression: implications of the novel CRF1 agonist cortagine. Neurosci Biobehav Rev. 2005;29:1323–1333. doi: 10.1016/j.neubiorev.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Tronson NC, Schrick C, Fischer A, Sananbenesi F, Pagès G, Pouysségur J, Radulovic J. Regulatory Mechanisms of Fear Extinction and Depression-Like Behavior. Neuropsychopharmacology. 2007 Aug 22; doi: 10.1038/sj.npp.1301550. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale WW, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- Valentino RJ, Commons KG. Peptides that fine-tune the serotonin system. Neuropeptides. 2005;39:1–8. doi: 10.1016/j.npep.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Zobel AW, Nickel T, Kunzel HE, Ackl N, Sonntag A, Ising M, Holsboer F. Effects of the high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major depression: the first 20 patients treated. J Psychiatr Res. 2000;34:171–181. doi: 10.1016/s0022-3956(00)00016-9. [DOI] [PubMed] [Google Scholar]