

Abstract
The cardiac extracellular matrix is a dynamic structural support network that is both influenced by, and a regulator of, pathological remodeling and hypertrophic growth. In response to pathologic insults the adult heart re-expresses the secreted extracellular matrix protein periostin (Pn). Here we show that Pn is critically involved in regulating the cardiac hypertrophic response, interstitial fibrosis, and ventricular remodeling following long-term pressure overload stimulation and myocardial infarction. Mice lacking the gene encoding Pn (Postn) were more prone to ventricular rupture in the first 10 days after a myocardial infarction, but surviving mice showed less fibrosis and better ventricular performance. Pn−/− mice also showed less fibrosis and hypertrophy following long-term pressure overload, suggesting an intimate relationship between Pn and the regulation of cardiac remodeling. In contrast, inducible overexpression of Pn in the heart protected mice from rupture following myocardial infarction and induced spontaneous hypertrophy with aging. With respect to a mechanism underlying these alterations, Pn−/− hearts showed an altered molecular program in fibroblast function. Indeed, fibroblasts isolated from Pn−/− hearts were less effective in adherence to cardiac myocytes and were characterized by a dramatic alteration in global gene expression (7% of all genes). These are the first genetic data detailing the function of Pn in the adult heart as a regulator of cardiac remodeling and hypertrophy.
Keywords: Cardiac, Signaling, Hypertrophy, Remodeling, Mouse genetics
INTRODUCTION
Heart disease remains one of the most prevalent and lethal diseases in the western world, with heart failure representing the fastest growing subclass of disease that presently afflicts nearly 5 million Americans1-3. One common characteristic that underlies nearly all forms of heart failure is a remodeling of the extracellular matrix (ECM) and an associated change in ventricular geometry4,5. For example, dilated cardiomyopathy is characterized by a thinning of the ventricular walls that likely results, in part, from a “loosening” of the ECM within the heart, while a restrictive cardiomyopathy is often associated with an increase in ventricular stiffness, an over abundance of fibrosis, and inappropriate ECM expansion4,5. These heart failure-associated alterations in the cardiac ECM are likely regulated, in part, by the cardiac fibroblast through both stretch-sensitive signaling pathways and by neuroendocrine effectors such as angiotensin II and transforming growth factor β (TGFβ)6,7. In response to disease stimuli that are associated with ventricular remodeling and heart failure, the activity of myocardial fibroblasts and the molecular composition of the ECM becomes altered6,7. For example, pressure overload and myocardial infarction (MI) in both rodents and humans is associated with a dramatic re-expression of the ECM protein periostin (Pn)8-11.
Pn is a 90 kDa secreted protein involved in cell adhesion, but otherwise of relatively unknown function12,13. Pn contains 4 repetitive fasciclin domains that are similar in sequence to the insect protein fasciclin I, which is involved in neuronal cell-cell adhesion14. Pn is related to another secreted fasciclin domain containing protein βIG-H3, which like Pn is expressed in collagen-rich connective tissues or remodeling centers, and is induced by TGFβ13,15. Pn shows a dynamic expression profile, both developmentally and in adult tissues that are undergoing remodeling or active stress. Pn is expressed in the developing endocardial cushions of the heart and the mature valves16, the periosteum and periodontal ligament13, injured vessels17, tumors and metastatic cancer cells18, and in cells undergoing mesenchymal transformation18,19. With respect to celltype of expression, Pn appears to be expressed exclusively in fibroblasts, or in cells that adopt fibroblast-like characteristics following an injury event16,20-22. As a secreted ECM protein that associates with areas of fibrosis, Pn can directly interact with other ECM proteins such as fibronectin, tenascin-C, collagen I, collagen V, and heparin22-25. Pn serves as a ligand for select integrins, such as αvβ3, αvβ5, and α4β6, where it can affect the ability of cells (fibroblasts or cancer cells) to migrate and/or undergo a mesenchymal transformation in select tissues18,19,26,27.
MATERIALS AND METHODS
For an expanded Materials and Methods, please see the online data supplement available at http://circres.ahajournals.org
Animals
Pn−/− (Postn gene) mice were generated by gene targeting in embryonic stem cells, while PntTA inducible transgenic mice were generated using a tetracycline-regulated system based on the α-myosin heavy chain (α-MHC) promoter28. However, constitutive Pn expression produced no phenotypic abnormalities or enhancements at baseline, so regulated expression by doxycycline administration and withdrawal was not used. Pn−/− mice were in the C57BL/6 strain, while PntTA mice were in the FVB strain, although only strain-matched controls were used throughout (no mixing).
Echocardiography, pressure overload and MI injury
Procedures for echocardiography in mice were described previously29, as were procedures for pressure overload by transverse aortic constriction (TAC) in mice30 and MI in mice29.
Histology and immunohistochemistry
Hearts were fixed in buffered 10% formalin, embedded in paraffin and cut into 6 μm sections for H&E or Masson's trichrome staining. Immunohistochemistry for Pn, vimentin, or collagen V α3 was performed in 5 μm cryosections fixed previously in buffered 4% paraformaldehyde in 50 mM KCl, and embedded in Tissue-Tek O. C. T. compound (Sakura Finetek USA Inc.). Rabbit polyclonal anti-mouse Pn antibody was described previously18.
Western blotting and hydroxyproline content determination
Western blotting for cardiac proteins and the processing of cardiac extracts was performed as described previously29. Determination of cardiac collagen content by analysis of hydroxyproline content was also described previously31.
Myeloperoxidase (MPO) assay
The MPO assay for granulocyte content in the heart was performed as described previously32.
Fibroblast culturing and proliferation
Conditions for these assays are relatively standard and are described in detail in the supplemental methods.
Affymetrix gene expression profiling and bioinformatics
Standard generation of total RNA and cDNA, array profiling with the mouse Affymetrix MOE 430−2 gene chip set, and bioinformatics criteria were described by us previously31.
Statistics
Statistical significance was determined with a paired Student's t-test for two groups, and analysis of variance (ANOVA) was used to determine significance between multiple groups.
RESULTS
Generation of Pn−/− mice and PntTA inducible transgenic mice
To begin to understand the functional role of Pn re-expression in the heart following injury, we undertook both a genetic gain- and loss-of-function approach in the mouse. We generated Pn−/− mice by targeting the Postn gene in embryonic stem cells, replacing exons 4−10 encoding three of the four fasciclin domains, which produced a null allele (Fig 1A). Indeed, analysis of Pn protein expression in the developing feet (sites of high Pn expression) or hearts of one day-old mice showed no expression in Pn−/− compared with wildtypes (Fig 1B). No significant Pn protein is found within the ECM of the heart after postnatal development, although the valves show persistent expression throughout adulthood and low levels of expression is maintained in fibroblasts (data not shown).
Fig 1.

Generation of Pn−/− and PntTA inducible transgenic mice. (A) Schematic of the targeting strategy that deletes 7 exons in the Postn gene. (B) Western blot for Pn protein from 1 day-old neonatal C57 wildtype (Wt) and Pn−/− mice from the developing bones of foot and heart. (C) Schematic of the cardiac-specific Pn expressing “responder” transgene that permits inducible expression in conjunction with the tTA “driver” transgene. (D) Western blot for Pn protein from adult hearts of the indicated genotypes in the constitutively induced state. PntTA mice contain both the responder and driver transgenes required for expression. (E) Body weight of the indicated genotype of mice at 8 weeks of age. *P<0.05 vs. C57 Wt. (F,G) Assessment of ventricular weights (VW) normalized to body weights (BW) in Pn−/−, PntTA and Wt (FVB) mice at 8 or 32 weeks of age. *P<0.05 vs. Wt. The number of mice analyzed is shown in the bar.
Pn overexpressing transgenic mice were also generated to investigate the biologic effect normally associated with induced expression in the heart. A modified α-MHC promoter was used as the responder transgene28, which when crossed with transgenic mice containing the α-MHC promoter driven tet-transactivator (tTA) protein, produces cardiac-specific expression that can be regulated, although only constitutive expression was used throughout this study (Fig 1C). While Pn expression is normally exclusive to fibroblasts, its production in cardiac myocytes here served as a surrogate to achieve heart-specificity (see Figure 2C). Cardiomyocytes do have limited expression of collagen and are capable of altering the composition of the ECM by direct expression. More importantly, cardiomyocytes efficiently produced and released Pn into the ECM in a pattern that was identical to endogenous production after stress stimulation from fibroblasts. However, a potential disadvantage, which we could not directly assess, was the possibility of differential modification of Pn in fibroblasts versus myocytes. Four independent lines of PntTA containing mice were initially characterized with robust expression, but only one line is shown given a similar overall phenotype (Fig 1D). The level of Pn overexpression in this line is approximately 5−6-fold greater than is normally observed in the heart following MI or pressure overload (data not shown).
Fig 2.

Assessment of cardiac Pn induction by pathological stimulation. (A,B) Western blot and quantitation of Pn from cardiac protein extracts of adult mice subjected to sham or TAC for the indicated periods of time. *P<0.05 vs. sham. (C) Immunohistochemistry for Pn (green) in a Wt sham heart (left panel), after 8 weeks of TAC (middle panel), and in an adult PntTA transgenic heart (right panel). Sections were co-stained for cardiac troponin I (red). (D,E) Western blot and quantitation of Pn from cardiac protein extracts of adult mice subjected to sham or MI for the indicated periods of time in days (d) or weeks (w). *P<0.05 vs. sham. (F) Immunohistochemistry for Pn (green) 1 week after a sham or MI procedure. The infarct and border zone is shown. (G) Western blot for Pn protein from the hearts of mice subjected to swimming (left panel) or wheel running (right panel) exercise for the indicated time in days or weeks. Control (con) is from a PntTA transgenic heart. (H) Confocal immunohistochemistry for Pn (green) and vimentin (red) from adult wildtype hearts 7 days after MI injury. The arrows show areas of co-localization to fibroblasts. (I) Quantitation of fibroblast content based on vimentin staining area in hearts of wildtype (C57) and Pn−/− mice after a sham or MI procedure (7 days afterwards). *P<0.05 vs. sham; #P<0.05 vs C57 MI. Tubulin is shown as a loading control throughout the figure.
The majority of Pn−/− mice survived well into adulthood, but showed smaller overall body weights (Fig 1E) and no alteration in ventricular weight normalized to body weight (Fig 1F). Deletion of Pn by 2 other groups also revealed a similar phenotype of smaller body size, as well as defective tooth development and an approximate 14% neonatal lethality25,33. Otherwise, Pn−/− mice appeared relatively normal and were fertile as adults with no alterations in cardiac morphology or ventricular performance, although hearts were slightly smaller (Supplemental Figure 1 and Supplemental Tables 1). PntTA transgenic mice were also unaffected as young adults and showed normal heart morphology, ventricular performance, and a lack of fibrosis (Fig 1G, Supplemental Figure 1 and Supplemental Table 2). However, by 24 weeks of age they showed signs of hypertrophy by echocardiography, which produced a significant increase in ventricular-weight normalized to body weight by 32 weeks of age (Fig 1G, and supplemental Table 2). Despite the hypertrophy, PntTA transgenic mice maintained normal ventricular performance up to 36 weeks of age, suggesting that Pn overexpression did not lead to decompensation.
Pn is re-expressed in cardiac fibroblasts following injury
Western blotting from adult hearts showed re-expression of Pn protein following pressure overload induced by TAC for 1 day, 1 week, 2 weeks, and 8 weeks, while no protein was observed in the ventricles of sham-operated adult mice (Fig 2A,B). Immunohistochemical analysis of Pn protein in the ventricles showed no expression in sham mice, but abundant accumulation that was localized to the interstitial space after 8 weeks of TAC in wildtype hearts (mice were 14−16 weeks of age) (Fig 2C). The same pattern of interstitial localization of Pn protein was observed in unstressed PntTA transgenic mice (10 weeks of age), showing that the transgene produced a similar overall biologic effect as endogenous re-expression after injury (Fig 2C). Pn protein re-expression was also observed in the ventricles by 4 days after a MI, and then maintained thereafter up to 8 weeks of age (Fig 2D,E). Interestingly, Pn protein appears to be relatively stable such that it persists for a number of weeks after an injury event, even after mRNA induction is lost (data not shown). Also of interest, two protein isoforms of Pn are usually observed upon induction in the heart, which may reflect differential splicing within the 23 exons comprising the gene. Indeed, at least 9 distinct Pn splice variants were detected by RT-PCR (Supplemental Fig 2). However, the full-length 23 exon-containing form of Pn is the dominantly expressed isoform, which was why it was selected for overexpression in our transgenic approach. Immunohistochemical analysis 1 week after MI injury showed massive accumulation of Pn protein within the developing scar itself, as well as between myocytes in the ECM of the peri-infarct region (Fig 2F). Interestingly, hypertrophy induced by forced swimming or voluntary wheel running exercise did not induce Pn protein expression, despite a 25% and 16% hypertrophy response, respectively (Fig 2G)30. Thus, Pn protein re-expression is likely more selective to pathological stimuli and is not involved in the hypertrophy response that accompanies exercise.
Finally, we also confirmed previous reports that Pn expression is exclusive to fibroblasts16,20-22. Confocal microscopy for Pn and vimentin co-localization showed expression in fibroblasts within the ECM of the heart 7 days after MI (Fig 2H). A similar pattern of co-localization was also observed between Pn and DDR2, which is another fibroblast enriched marker (data not shown). There were no obvious differences in overall fibroblast numbers in the hearts of adult wildtype versus Pn−/− mice as assessed by vimentin immunohistochemistry at baseline, although after MI Pn−/− hearts had significantly less of an increase in fibroblasts in the left ventricle (Fig 2I).
Analysis of MI-mediated rupture and remodeling in Pn−/− mice
Since Pn protein is re-expressed following cardiac injury, we reasoned that it might alter the course of an injury response given its prominent deposition in the ECM. Indeed, Pn−/− mice showed a significant increase in death in the first 10 days after MI injury, associated with a 2-fold greater rate of ventricular wall rupture compared with strain-matched wildtype controls (P<0.05) (Fig 3A). However, Pn−/− mice that survived the initial scar formation phase maintained cardiac function better than did wildtype controls over the next 8 weeks, as assessed by longitudinal echocardiography (Fig 3B, Supplemental Table 3). One concern with these data is that Pn−/− mice may be inherently more protected from ischemic injury, generating a selection bias in the functional data. However Pn−/− mice showed the same infarction area (IA) normalized to area-at-risk (AAR) as wildtype mice 24 hours after ischemia-reperfusion injury (Fig 3C). The relative degree of fibrosis and the overall size of the scar itself were noticeably reduced in Pn−/− mice compared with wildtype mice, suggesting a reason underlying the improved functional profile of Pn−/− mice (Fig 3D). Indeed, Pn−/− mice also showed less inflammatory cell recruitment as assessed with a total tissue MPO activity assay or a western blot for a macrophage specific maker (CD68) following MI, consistent with the observed reduction in fibrosis and scar size (Fig 3E,F).
Fig 3.
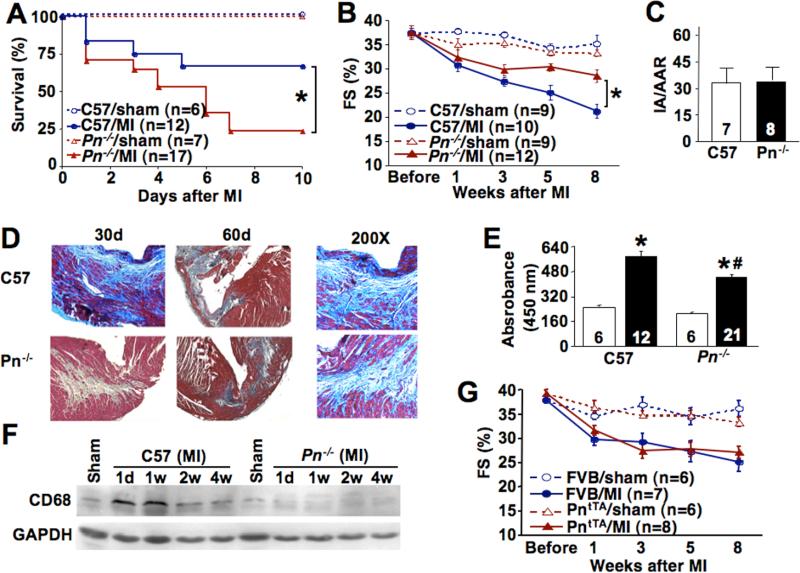
Pn−/− and PntTA mice following MI injury. (A) Survival rate of Pn−/− and C57 Wt male mice in the first 10 days after MI. *P<0.05 by Logrank test vs. C57 Wt MI mice. (B) Assessment of cardiac fractional shortening (FS, %) after sham operation or MI in the indicated groups of mice for the indicated times. *P<0.05 vs. C57 Wt MI. (C) Infarct area normalized to the area at risk (IA/AAR) in Wt and Pn−/− mice after ischemia-reperfusion injury for 24 hours. (D) Masson's trichrome staining of cardiac histological sections in the MI and border zone in Wt and Pn−/− mice for the indicted period of time. Blue staining indicates fibrosis. The 200X panels are higher magnification images to show the cellular organization and fibrosis in more detail. (E) Myeloperoxidase (MPO) assay from hearts of the indicated genotype of mice 8 weeks after a sham operation (white bar) or MI (black bar) (mice were 14−16 weeks of age). (F) Western blot for macrophage content in the heart (CD68 antibody) in Wt and Pn−/− mice after a sham operation or MI. (G) Assessment of cardiac fractional shortening (FS, %) after a sham operation or MI in Wt FVB or PntTA mice at 8 weeks of age, and followed longitudinally for the indicated time points.
While loss of Pn was protective following the later phases after an MI by reducing fibrosis and scar expansion, overexpression of Pn in PntTA mice showed no effect on these indices or the degree of functional deterioration (Fig 3G, and Supplemental Table 3). Thus, increased Pn did not increase fibrosis or scar size following infarction injury, suggesting that Pn secretion is not a rate limiting aspect of the fibrotic response itself. However, none of the expired PntTA transgenic mice showed ventricular wall rupture compared with obvious examples of rupture in the wildtype controls. Thus, Pn overexpression appears to protect from myocardial wall rupture (see discussion).
Pn−/− mice have preserved function and less hypertrophy following pressure overload
Pressure overload in the mouse induced by constricting the proximal aorta induces profound cardiac hypertrophy and ventricular remodeling associated with increased fibrosis. Since Pn is re-expressed fairly rapidly in pressure overloaded mouse hearts we reasoned that it might modulate the ensuing disease response. Indeed, while the cardiac hypertrophic response was not different between Pn−/− mice and wildtype mice in the first 2 weeks of pressure overload, Pn−/− mice showed no progression or worsening of hypertrophy after 8 weeks, compared with significantly more hypertrophy in wildtype mice (Fig 4A). As a control, pressure gradients across the aortic constriction were not different between Pn−/− mice and wildtype mice (68.51±3.82 mmHg, n=16 versus 67.67±3.46 mmHg, n=12, respectively). Consistent with the functional ramifications underlying these data, PntTA transgenic mice showed significantly greater cardiac hypertrophy 8 weeks after pressure overload compared with wildtype controls of the same strain (Fig 4B). Once again, pressure gradients across the aortic constrictions were not different between PntTA and wildtype mice (data not shown). At the cellular level, cross-sectional areas of myocytes from ventricular histological sections were significantly smaller in Pn−/− mice compared with their respective controls, while areas were significantly larger in PntTA transgenic mice compared with FVB controls following 8 weeks of pressure overload (Fig 4C,D).
Fig 4.

Pn−/− and PntTA mice have an altered pressure overload response. (A) Ventricular weight (VW) normalized to body weight (BW) in Pn−/− and C57 Wt mice at 2 and 8 weeks after TAC or a sham operation. *P<0.05 vs. sham. #P<0.05 vs. C57 Wt at 8 weeks of TAC. (B) VW normalized to BW in PntTA and FVB Wt mice 8 weeks after TAC or a sham operation. *P<0.05 vs. sham. #P<0.05 vs. FVB Wt after TAC. (C,D) Assessment of myocyte cross-sectional area from left ventricular histological sections of the indicated genotypes (n=3 hearts each, with at least 300 cells counted in total). *P<0.05 vs. sham of the same genotype; #P<0.05 vs. Wt TAC. (E,F) Assessment of cardiac fractional shortening (FS, %) after a sham operation or TAC in Pn−/− and their strain-matched control mice, or PntTA and their respective control mice at 8 weeks of age, and followed longitudinally for the indicated time points. *P<0.05 vs. C57 Wt TAC. (G) Cardiac fibrosis assessed by hydroxyproline biochemical determination in the indicated mice 8 weeks after TAC. *P<0.05 vs. sham. #P<0.05 vs. C57 Wt TAC.
Pn−/− mice also maintained ventricular performance better than wildtype mice at 3, 5, and 8 weeks of pressure overload, suggesting that loss of Pn protected the heart from decompensation (Fig 4E, Supplemental Table 4). This functional improvement also correlated with significantly less collagen accumulation in the heart over 8 weeks in Pn−/− mice compared with wildtype mice (Fig 4G). However, contrary to what might be predicted based on the loss-of-function data, overexpression of Pn did not enhance functional decompensation of the heart following 8 weeks of pressure overload, nor did it increase cardiac fibrosis (Fig 4F,G, Supplemental Table 4). This lack of increased fibrosis suggests that Pn expression may not be a rate-limiting aspect in the overall fibrotic response (see discussion). It should also be noted that the FVB genetic background consistently showed little collagen accumulation following pressure overload, in contrast to a robust response in the C57 strain background, although this difference likely reflects the overall greater susceptibility of the C57 strain to decompensation following cardiac injury (Fig 4G).
Altered fibrotic gene program in Pn−/− mouse hearts
To begin to dissect the mechanistic underpinnings of the reduced fibrotic response in Pn−/− hearts following MI or long-term pressure overload, we utilized an Affymetrix gene array to identify differentially expressed mRNAs (Fig 5A). Pn−/− hearts at 8 weeks of age showed 449 genes with significantly altered expression compared with wildtype hearts of the same strain. Interestingly, a large set of these differentially expressed genes could be assigned to a biologic category involving fibrosis, cell adhesion, ECM, or that are consistent with fibroblast function. For example, Pn−/− hearts showed dramatically reduced levels of collagen Vα3, although collagen I and collagen III were expressed normally in the adult heart of those animals that survived neonatal development (Fig 5B,C). Indeed, Pn−/− hearts showed significantly less collagen V expression/accumulation in the heart following MI or TAC compared with wildtype hearts (Fig 5D,E).
Fig 5.
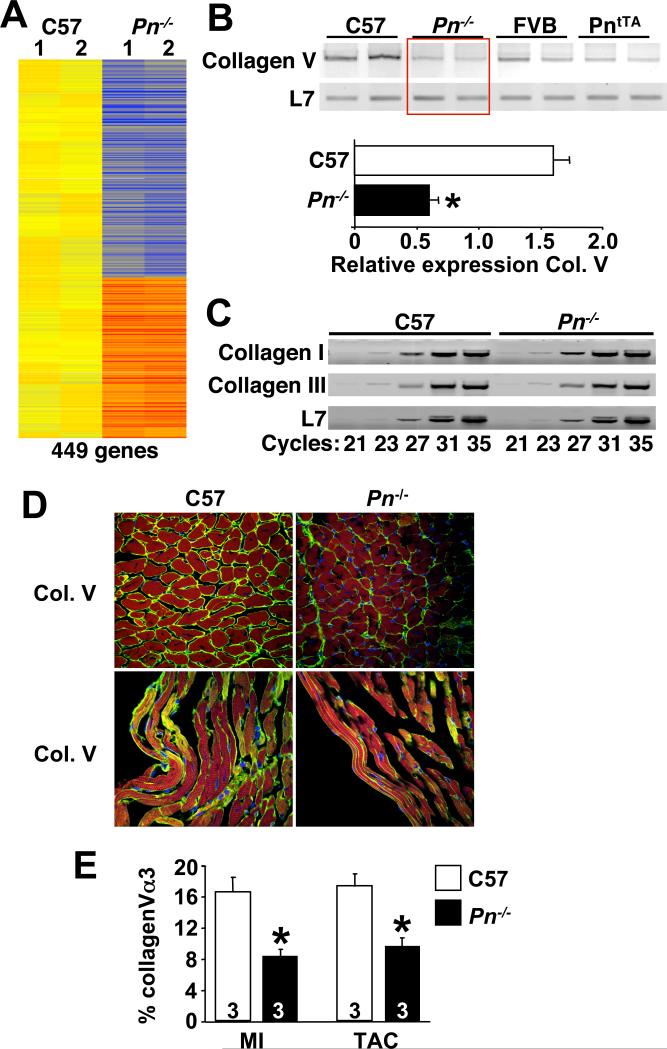
Hearts from Pn−/− mice show changes in gene expression suggestive of an altered fibroblast program. (A) Diagram of the genes that were significantly altered in expression by Affymetrix array profiling in C57 Wt and Pn−/− 8 week-old mouse hearts. Yellow is unchanged, orange and red represent increased expression, and blue represents diminished expression (n=2 hearts each). (B,C) RT-PCR for the indicted mRNA species from the hearts of the indicted genotypes. L7 was used as a control. Twenty three cycles of amplification was used in B, while panel C indicates the cycle number used. Quantitation of type V collagen α3 (Col. V) mRNA is shown in the lower panel in B. *P<0.05 vs. C57 (D,E) Immunohistochemistry and quantitiation of type V collagen α3 (Col. V) from hearts sections of Wt or Pn−/− adult mice 8 weeks after MI. Green staining in the MI border zone indicates Col. V, red indicates cardiomyocytes (troponin I), while blue indicates nuclei. Quantitation of staining in pressure overloaded hearts (TAC) is also shown in E. *P<0.05 vs. C57
The difference in gene expression in Pn−/− hearts discussed above suggested an alteration in fibroblast function. To address this issue we generated fibroblast cultures from wildtype and Pn−/− adult hearts for in vitro analysis. Fibroblast proliferation rates assessed by [3H]-thymidine incorporation or immunocytochemistry for phosphorylated histone H3 did not vary between wildtype and Pn−/− fibroblasts (Fig 6A,B). Consistent with these observations, increased expression of Pn using a recombinant adenovirus also did not alter proliferation rates in wildtype of Pn−/− fibroblast cultures (Fig 6C). There was also no difference in the percentage of myofibroblasts induced at low culture densities as quantified by staining for smooth muscle α-actin between wildtype and Pn−/− fibroblasts (Fig 6D). However, Pn−/− fibroblasts showed dramatically different adhesive properties. Plates of wildtype or Pn−/− fibroblasts were incubated overnight to form a lawn, after which rat neonatal cardiomyocytes were overlaid at a concentration of 1.25×105/well and washed with PBS to remove unattached cells after 4, 8, 16, and 48 hrs, followed by quantitation of attachment. At all time points analyzed, fewer neonatal cardiomyocytes attached to Pn−/− fibroblasts compared with wildtype fibroblasts (Fig 7A,B). The use of a mouse-rat co-culturing system between fibroblasts and cardiomyocytes has been described previously34. The reciprocal experiment was also performed in which a lawn of neonatal cardiomyocytes was used to quantify the attachment of wildtype or Pn−/− fibroblasts at 1 and 3 hrs, and the results showed a similar defect in Pn−/− fibroblasts (Fig 7C). Interestingly, addition or restoration of Pn protein in wildtype and Pn−/− fibroblasts, respectively, did not enhance adhesion in either cell culture assay (Fig 7C, and data not shown). These results suggested that Pn−/− fibroblasts might be inherently different based on their development in the absence of Pn protein. Indeed, we performed an Affymetrix array for global gene expression changes from cardiac fibroblasts isolated at 6 weeks of age from wildtype and Pn−/− mice. The results showed a significant change in approximately 7% of all expressed genes (data not shown).
Fig 6.
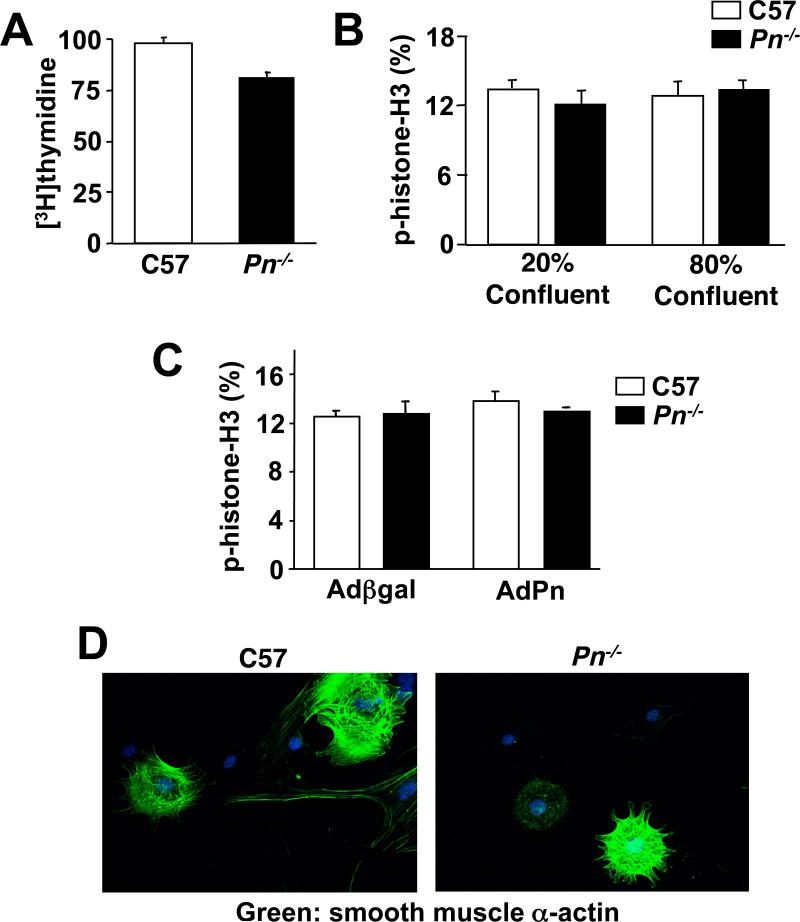
Assessment of cardiac fibroblasts from Pn−/− mice. (A) Assessment of cardiac fibroblast proliferation by [3H]-thymidine incorporation in Wt or Pn−/− fibroblasts in culture. (B,C) Assessment of cardiac fibroblast proliferation by immunocytochemistry for phosphorylated-histone H3 in Wt or Pn−/− cultures at the indicated confluence. Panel C also shows the effect of Pn overexpression by adenoviral infection with AdPn, or a control, Adβgal. (D) Florescent images of smooth muscle α-actin (green) in wildtype (C57) and Pn−/− fibroblasts at low density to show myofibroblast content. Nuclei are shown in blue.
Fig 7.

Assessment of fibroblast-cardiomyocyte adhesion in vitro. (A) The number of adherent rat neonatal cardiomyocyte (CM) normalized to an area containing 100 Wt or Pn−/− cardiac fibroblasts after 4 and 8 hrs of incubation. *P<0.05 vs. C57 Wt fibroblasts. (B) Representative immunocytochemistry for cardiomyocyte adherence on pre-plated Wt or Pn−/− cardiac fibroblasts at 16 and 48 hours. Pn−/− fibroblasts attach fewer cardiomyocytes. Red staining indicates cardiac troponin I (to show myocytes), and blue indicates nuclei. (C) Quantity of wildtype or Pn−/− fibroblasts that adhered to pre-plated neonatal cardiomyocytes cultures at 1 and 3 hrs (20 fields each). AdPn was used to generate conditioned media from Pn overexpressing myocytes and incubated on the experimental cells for 24 hrs prior to the attachment assay. Adβgal infection was used as a control. *P<0.05 vs. C57 Wt fibroblasts.
DISCUSSION
Here we observed that Pn contributes to the cardiac hypertrophic response, an observation that was unanticipated given its primary function as an ECM cellular adhesion protein. Overexpression of Pn produced a concentric hypertrophy response in mice as they aged beyond young adulthood, while unaffected young adult mice showed a significantly greater hypertrophy response after pressure overload stimulation. Consistent with these data, Pn−/− mice showed less progression of hypertrophy and dilation between 2 and 8 weeks of pressure overload stimulation, collectively suggesting a potential signaling role in the cardiac growth response and/or decompensation. That a cellular adhesion protein might affect the cardiac hypertrophic response is not without precedence, especially given the known importance of integrins in regulating cardiac reactive signaling35,36 and given Pn's known role as a substrate for multiple combinations of integrins. For example, both focal adhesion kinase (FAK) and integrin-linked kinase (ILK), which are intracellular signaling components of the integrin complex, function as important regulators of the cardiac hypertrophy response37,38. Indeed, mice lacking the protein osteopontin, which is another cardiac inducible and secreted ECM protein that binds integrins, showed less pressure overload hypertrophy compared with wildtype mice39. Based on these results it is tempting to speculate that Pn alters the cardiac hypertrophic response by affecting signaling within cardiac myocytes and fibroblasts through alterations in their engagement of integrins or other fasciclin domain containing proteins (membrane bound versions). The lack of Pn in the developing heart also appears to dramatically alter the molecular program within fibroblasts themselves, so that adult Pn−/− fibroblasts show dramatic changes in gene expression (7% of all genes). This inherent alteration in fibroblasts in Pn−/− hearts might also impact the hypertrophic response (as well as the fibrotic response), although a specific mechanism was not identified.
In addition to altering the hypertrophic response, loss of Pn compromised the fibrotic response of the heart following pressure overload stimulation and MI. This reduction in fibrosis initially promoted greater ventricular rupture following MI, although long-term it improved cardiac function in the surviving animals. The mechanism for increased rupture is reminiscent of corticosteroids and anti-inflammatory treatment regimens used in humans to reduce fibrosis following MI, but with the secondary consequence of increasing rupture rates40. Even more germane, mice null for the angiotensin II type 2 receptor were characterized by significantly less fibrosis following MI injury, and a 63% death incidence due to rupture in the first 7 days, compared with a rate of 24% in wildtype mice41. These results suggest that while inhibition of cardiac fibrosis benefits the long-term remodeling process and preserves cardiac function to a greater extent, in the short term it can predispose to rupture by compromising scar formation.
Two other groups have previously attempted to manipulate expression of Pn or an alternately spliced isoform of Pn in the heart10,21. Katsuragi et al attempted to overexpress Pn in the rat heart by transfection of a plasmid mixed with liposomes coated with the hemagglutinating virus of Japan21. This same group attempted to knock-down Pn expression with a one time infusion of antisense oligonucleotides into the rat heart, after which function was assessed 43 days later21. The main concern with this prior study was that protein expression afforded by either manipulation was not examined, making it very difficult to interpret the validity of the reported results, notwithstanding the implausibility that a single infusion of antisense oligonucleotides imparted a functional effect on cardiac remodeling 6 weeks later. For example, Katsuragi reported that transfection of a Pn expressing plasmid into the rat heart caused ventricular dilation and a reduction in cardiac function, although our PntTA transgenic mice showed no such effect, even at high levels of sustained expression (for more than 7−8 months). In another study, Litvin et al injected an adenovirus encoding an alternatively spliced isoform of Pn into the heart, referred to as periostin-like factor (PLF), which after 7 days caused no dilation but instead produced a minor increase in cardiac hypertrophy10. However, our PntTA transgenic mice did not show cardiac hypertrophy until 6−8 months of age with sustained overexpression, suggesting that the viral infection protocol may have had a secondary influence in the Litvin et al study, or that the alternately spliced isoform functioned differently.
With respect to cardiac remodeling, it was interesting that overexpression of Pn did not lead to increased cardiac fibrosis or otherwise affect remodeling following pressure overload and MI injury. These observations indicate that Pn itself does not nucleate the fibrotic response, and that it is not a limiting component in the pathway to fibrotic deposition. However, loss of Pn compromised the efficiency of fibrosis development and ventricular remodeling, collectively suggesting that Pn is necessary but not sufficient in the remodeling and fibrotic response. Another consideration is that while Pn overexpression did not alter cardiac fibrosis, it still enhanced the cardiac hypertrophic response and protected hearts from rupture. We believe that the mechanistic role of Pn in altering the hypertrophy response is distinct from its potentially more passive role in affecting the fibrotic response (see below).
The alterations in cardiac remodeling and hypertrophy associated with Pn deletion are hypothesized to arise by two distinct, but potentially overlapping mechanisms. Pn might simply regulate the integrity of the ECM through its ability to bind multiple ECM proteins, such as tenascin-C, fibronectin, collagen V, collagen I, and heparin22-25, thus affecting collagen synthesis and maturation through a complex series of architectural interactions. Indeed, as part of a collaborative study we determined that collagen fibrils from Pn−/− mice were reduced in size, slightly disorganized, and less efficiently cross-linked23. Functionally, skin from Pn−/− mice exhibited lower tensile strength with a difference in elasticity23. Thus, by regulating the integrity or composition of the ECM, Pn secondarily impacts the structural properties of the heart, which could alter the hypertrophic response by simply modifying the stretch characteristics of the tissue, impacting signaling as an indirect consequence. Another aspect of this passive regulation is that Pn appears to be a more pliable substrate compared with fibronectin and vitronectin, possibly affecting how mesenchymal cells (fibroblasts, inflammatory cells, stem cells) migrate within the heart following injury18. Indeed, transformed cells preferentially secrete Pn to facilitate invasiveness and metastatic activity18,26,27. This same static feature of ECM pliability may also underlie invasiveness of mesenchymal cells into endocardial cushions in the presence of Pn in the developing heart19.
A second possibility is that Pn is a more active participant in the remodeling and hypertrophic response by altering the phenotype of fibroblasts and/or cardiomyocytes due to attachment-dependent signaling (which could also be an epigenetic developmental effect that is maintained in adulthood). Pn is an integrin binding protein and its attachment with fibroblasts may impart a signal that affects how these cells perceive their environment and respond to stress and strain. Pn supports αvβ5 integrin signaling to facilitate epithelial-mesenchymal transition and metastatic activity18,27, as well as for movement of cells in the developing bone centers or periodontal ligaments20,25,33. In the same manner, Pn secretion in the heart might alter the movement or adherence of fibroblasts or inflammatory cells into areas of injured myocardium where Pn is abundantly expressed and secreted8-11.
Supplementary Material
Fig. Histological analysis of wildtype, Pn−/−, and PntRA transgenic mice. (A) Gross histological profiles show relatively normal heart geometries. (B) Immunohistochemistry for troponin I (red) and periostin (green) in ventricular sections of the indicated genotypes. Only the PntTA transgenic show expression (green interstitial signal). Nuclei are blue.
Fig. RT-PCR analysis of Pn mRNA splicing variants from the hearts of sham or MI mice. Primer pairs that span exons 1−8, 8−15, 15−19, and 19−23 were used, which covers the entire gene. At least 9 different splice products were detected, some of which were altered in content following MI (e.g., the exon 1−8 product at 260 bp was reduced in abundance).
Acknowledgments
This work was supported by the National Institutes of Health (to J.D.M., G.W.D., and J.R.). This work was also supported by an international grant in heart failure research for the Fondation Leducq (J.D.M.). J.D.M. is an Established Investigator of the American Heart Association.
Footnotes
Conflict of interest: None
REFERENCES
- 1.Levy D, Kenchaiah S, Larson MG, Benjamin EJ, Kupka MJ, Ho KK, Murabito JM, Vasan RS. Long-term trends in the incidence of and survival with heart failure. N Engl J Med. 2002;347:1397–1402. doi: 10.1056/NEJMoa020265. [DOI] [PubMed] [Google Scholar]
- 2.Haldeman GA, Croft JB, Giles WH, Rashidee A. Hospitalization of patients with heart failure: National Hospital Discharge Survey, 1985 to 1995. Am Heart J. 1999;137:352–360. doi: 10.1053/hj.1999.v137.95495. [DOI] [PubMed] [Google Scholar]
- 3.Lloyd-Jones DM, Larson MG, Leip EP, Beiser A, D'Agostino RB, Kannel WB, Murabito JM, Vasan RS, Benjamin EJ, Levy D. Lifetime risk for developing congestive heart failure: the Framingham Heart Study. Circulation. 2002;106:3068–3072. doi: 10.1161/01.cir.0000039105.49749.6f. [DOI] [PubMed] [Google Scholar]
- 4.Manso AM, Elsherif L, Kang SM, Ross RS. Integrins, membrane-type matrix metalloproteinases and ADAMs: potential implications for cardiac remodeling. Cardiovasc Res. 2006;69:574–584. doi: 10.1016/j.cardiores.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 5.Deschamps AM, Spinale FG. Matrix modulation and heart failure: new concepts question old beliefs. Curr Opin Cardiol. 2005;20:211–216. doi: 10.1097/01.hco.0000162397.44843.83. [DOI] [PubMed] [Google Scholar]
- 6.Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–87. doi: 10.1146/annurev.pharmtox.45.120403.095802. [DOI] [PubMed] [Google Scholar]
- 7.Baudino TA, Carver W, Giles W, Borg TK. Cardiac fibroblasts: friend or foe? Am J Physiol Heart Circ Physiol. 2006;291:H1015–H1026. doi: 10.1152/ajpheart.00023.2006. [DOI] [PubMed] [Google Scholar]
- 8.Stanton LW, Garrard LJ, Damm D, Garrick BL, Lam A, Kapoun AM, Zheng Q, Protter AA, Schreiner GF, White RT. Altered patterns of gene expression in response to myocardial infarction. Circ Res. 2000;86:939–945. doi: 10.1161/01.res.86.9.939. [DOI] [PubMed] [Google Scholar]
- 9.Wang D, Oparil S, Feng JA, Li P, Perry G, Chen LB, Dai M, John SW, Chen YF. Effects of pressure overload on extracellular matrix expression in the heart of the atrial natriuretic peptide-null mouse. Hypertension. 2003;42:88–95. doi: 10.1161/01.HYP.0000074905.22908.A6. [DOI] [PubMed] [Google Scholar]
- 10.Litvin J, Blagg A, Mu A, Matiwala S, Montgomery M, Berretta R, Houser S, Margulies K. Periostin and periostin-like factor in the human heart: possible therapeutic targets. Cardiovasc Pathol. 2006;15:24–32. doi: 10.1016/j.carpath.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Johnatty SE, Dyck JR, Michael LH, Olson EN, Abdellatif M. Identification of genes regulated during mechanical load-induced cardiac hypertrophy. J Mol Cell Cardiol. 2000;32:805–815. doi: 10.1006/jmcc.2000.1122. [DOI] [PubMed] [Google Scholar]
- 12.Takeshita S, Kikuno R, Tezuka K, Amann E. Osteoblast-specific factor 2: cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I. Biochem J. 1993;294:271–278. doi: 10.1042/bj2940271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horiuchi K, Amizuka N, Takeshita S, Takamatsu H, Katsuura M, Ozawa H, Toyama Y, Bonewald LF, Kudo A. Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta. J Bone Miner Res. 1999;14:1239–1249. doi: 10.1359/jbmr.1999.14.7.1239. [DOI] [PubMed] [Google Scholar]
- 14.Zinn K, McAllister L, Goodman CS. Sequence analysis and neuronal expression of fasciclin I in grasshopper and Drosophila. Cell. 1988;53:577–587. doi: 10.1016/0092-8674(88)90574-0. [DOI] [PubMed] [Google Scholar]
- 15.LeBaron RG, Bezverkov KI, Zimber MP, Pavelec R, Skonier J, Purchio AF. Beta IG-H3, a novel secretory protein inducible by transforming growth factor-beta, is present in normal skin and promotes the adhesion and spreading of dermal fibroblasts in vitro. J Invest Dermatol. 1995;104:844–849. doi: 10.1111/1523-1747.ep12607024. [DOI] [PubMed] [Google Scholar]
- 16.Kruzynska-Frejtag A, Machnicki M, Rogers R, Markwald RR, Conway SJ. Periostin (an osteoblast-specific factor) is expressed within the embryonic mouse heart during valve formation. Mech Dev. 2001;103:183–188. doi: 10.1016/s0925-4773(01)00356-2. [DOI] [PubMed] [Google Scholar]
- 17.Li G, Oparil S, Sanders JM, Zhang L, Dai M, Chen LB, Conway SJ, McNamara CA, Sarembock IJ. Phosphatidylinositol-3-kinase signaling mediates vascular smooth muscle cell expression of periostin in vivo and in vitro. Atherosclerosis. 2006;188:292–300. doi: 10.1016/j.atherosclerosis.2005.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gillan L, Matei D, Fishman DA, Gerbin CS, Karlan BY, Chang DD. Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(V)beta(3) and alpha(V)beta(5) integrins and promotes cell motility. Cancer Res. 2002;62:5358–5364. [PubMed] [Google Scholar]
- 19.Butcher JT, Norris RA, Hoffman S, Mjaatvedt CH, Markwald RR. Periostin promotes atrioventricular mesenchyme matrix invasion and remodeling mediated by integrin signaling through Rho/PI 3-kinase. Dev Biol. 2007;302:256–266. doi: 10.1016/j.ydbio.2006.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilde J, Yokozeki M, Terai K, Kudo A, Moriyama K. The divergent expression of periostin mRNA in the periodontal ligament during experimental tooth movement. Cell Tissue Res. 2003;312:345–351. doi: 10.1007/s00441-002-0664-2. [DOI] [PubMed] [Google Scholar]
- 21.Katsuragi N, Morishita R, Nakamura N, Ochiai T, Taniyama Y, Hasegawa Y, Kawashima K, Kaneda Y, Ogihara T, Sugimura K. Periostin as a novel factor responsible for ventricular dilation. Circulation. 2004;110:1806–1813. doi: 10.1161/01.CIR.0000142607.33398.54. [DOI] [PubMed] [Google Scholar]
- 22.Takayama G, Arima K, Kanaji T, Toda S, Tanaka H, Shoji S, McKenzie AN, Nagai H, Hotokebuchi T, Izuhara K. Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J Allergy Clin Immunol. 2006;118:98–104. doi: 10.1016/j.jaci.2006.02.046. [DOI] [PubMed] [Google Scholar]
- 23.Norris RA, Damon B, Mironov V, Kasyanov V, Ramamurthi A, Moreno-Rodriguez R, Trusk T, Potss JD, Goodwin RL, Davis J, Hoffman S, Wen X, Sugi Y, Kern CB, Mjaatvedt CH, Turner DK, Oka T, Conway SJ, Molkentin JD, Forgacs G, Markwald RR. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J Cell Biochem. 2007 Jan 16; doi: 10.1002/jcb.21224. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugiura T, Takamatsu H, Kudo A, Amann E. Expression and characterization of murine osteoblast-specific factor 2 (OSF-2) in a baculovirus expression system. Protein Expr Purif. 1995;6:305–311. doi: 10.1006/prep.1995.1040. [DOI] [PubMed] [Google Scholar]
- 25.Kii I, Amizuka N, Minqi L, Kitajima S, Saga Y, Kudo A. Periostin is an extracellular matrix protein required for eruption of incisors in mice. Biochem Biophys Res Commun. 2006;342:766–772. doi: 10.1016/j.bbrc.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 26.Baril P, Gangeswaran R, Mahon PC, Caulee K, Kocher HM, Harada T, Zhu M, Kalthoff H, Crnogorac-Jurcevic T, Lemoine NR. Periostin promotes invasiveness and resistance of pancreatic cancer cells to hypoxia-induced cell death: role of the beta(4) integrin and the PI3k pathway. Oncogene. 2007;26:2082–2094. doi: 10.1038/sj.onc.1210009. [DOI] [PubMed] [Google Scholar]
- 27.Yan W, Shao R. Transduction of a mesenchyme-specific gene periostin into 293T cells induces cell invasive activity through epithelial-mesenchymal transformation. J Biol Chem. 2006;281:19700–19708. doi: 10.1074/jbc.M601856200. [DOI] [PubMed] [Google Scholar]
- 28.Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J. Reengineering inducible cardiac-specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res. 2003;92:609–616. doi: 10.1161/01.RES.0000065442.64694.9F. [DOI] [PubMed] [Google Scholar]
- 29.Kaiser RA, Liang Q, Bueno O, Huang Y, Lackey T, Klevitsky R, Hewett TE, Molkentin JD. Genetic inhibition or activation of JNK1/2 protects the myocardium from ischemia-reperfusion-induced cell death in vivo. J Biol Chem. 2005;280:32602–32608. doi: 10.1074/jbc.M500684200. [DOI] [PubMed] [Google Scholar]
- 30.Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 31.Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837–845. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki K, Murtuza B, Smolenski RT, Sammut IA, Suzuki N, Kaneda Y, Yacoub MH. Overexpression of interleukin-1 receptor antagonist provides cardioprotection against ischemia-reperfusion injury associated with reduction in apoptosis. Circulation. 2001;104:I308–I313. doi: 10.1161/hc37t1.094871. [DOI] [PubMed] [Google Scholar]
- 33.Rios H, Koushik SV, Wang H, Wang J, Zhou HM, Lindsley A, Rogers R, Chen Z, Maeda M, Kruzynska-Frejtag A, Feng JQ, Conway SJ. periostin null mice exhibit dwarfism, incisor enamel defects, and an early-onset periodontal disease-like phenotype. Mol Cell Biol. 2005;25:11131–11144. doi: 10.1128/MCB.25.24.11131-11144.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kizana E, Ginn SL, Smyth CM, Boyd A, Thomas SP, Allen DG, Ross DL, Alexander IE. Fibroblasts modulate cardiomyocyte excitability: implications for cardiac gene therapy. Gene Ther. 2006;13:1611–1615. doi: 10.1038/sj.gt.3302813. [DOI] [PubMed] [Google Scholar]
- 35.Manso AM, Elsherif L, Kang SM, Ross RS. Integrins, membrane-type matrix metalloproteinases and ADAMs: potential implications for cardiac remodeling. Cardiovasc Res. 2006;69:574–584. doi: 10.1016/j.cardiores.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 36.Brancaccio M, Hirsch E, Notte A, Selvetella G, Lembo G, Tarone G. Integrin signalling: the tug-of-war in heart hypertrophy. Cardiovasc Res. 2006;70:422–433. doi: 10.1016/j.cardiores.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 37.Lu H, Fedak PW, Dai X, Du C, Zhou YQ, Henkelman M, Mongroo PS, Lau A, Yamabi H, Hinek A, Husain M, Hannigan G, Coles JG. Integrin-linked kinase expression is elevated in human cardiac hypertrophy and induces hypertrophy in transgenic mice. Circulation. 2006;114:2271–2279. doi: 10.1161/CIRCULATIONAHA.106.642330. [DOI] [PubMed] [Google Scholar]
- 38.DiMichele LA, Doherty JT, Rojas M, Beggs HE, Reichardt LF, Mack CP, Taylor JM. Myocyte-restricted focal adhesion kinase deletion attenuates pressure overload-induced hypertrophy. Circ Res. 2006;99:636–645. doi: 10.1161/01.RES.0000240498.44752.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie Z, Singh M, Singh K. Osteopontin modulates myocardial hypertrophy in response to chronic pressure overload in mice. Hypertension. 2004;44:826–831. doi: 10.1161/01.HYP.0000148458.03202.48. [DOI] [PubMed] [Google Scholar]
- 40.Silverman HS, Pfeifer MP. Relation between use of anti-inflammatory agents and left ventricular free wall rupture during acute myocardial infarction. Am J Cardiol. 1987;59:363–364. doi: 10.1016/0002-9149(87)90817-4. [DOI] [PubMed] [Google Scholar]
- 41.Ichihara S, Senbonmatsu T, Price E, Jr, Ichiki T, Gaffney FA, Inagami T. Targeted deletion of angiotensin II type 2 receptor caused cardiac rupture after acute myocardial infarction. Circulation. 2002;106:2244–2249. doi: 10.1161/01.cir.0000033826.52681.37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. Histological analysis of wildtype, Pn−/−, and PntRA transgenic mice. (A) Gross histological profiles show relatively normal heart geometries. (B) Immunohistochemistry for troponin I (red) and periostin (green) in ventricular sections of the indicated genotypes. Only the PntTA transgenic show expression (green interstitial signal). Nuclei are blue.
Fig. RT-PCR analysis of Pn mRNA splicing variants from the hearts of sham or MI mice. Primer pairs that span exons 1−8, 8−15, 15−19, and 19−23 were used, which covers the entire gene. At least 9 different splice products were detected, some of which were altered in content following MI (e.g., the exon 1−8 product at 260 bp was reduced in abundance).