

Abstract
Villin is a tissue-specific actin modifying protein that is associated with actin filaments in the microvilli and terminal web of epithelial cells. It belongs to a large family of actin-binding proteins which includes actin-capping, -nucleating and/or - severing proteins such as gelsolin, severin, fragmin, adseverin/scinderin and actin crosslinking proteins such as dematin and supervillin. Studies done in epithelial cell lines and villin knock out mice have demonstrated the function of villin in regulating actin dynamics, cell morphology, epithelial-to-mesenchymal transition, cell migration and cell survival. In addition, the ligand-binding properties of villin (F-actin, G-actin, calcium, phospholipids and phospholipase C-γ1) are mechanistically important for the crosstalk between signaling pathways and actin reorganization in epithelial cells.
Villin is an epithelial cell specific actin regulatory protein
Epithelial cells display distinct structural and functional attributes, which warrants examining their biological function independently from that of non-polarized cells. Epithelial cells form continuous mono- or multilayered sheets of tightly adherent cells. These cells have characteristic structures that determine cell-cell contacts called the tight junction complex and equally complex structure that regulates contacts with the cellular basement membranes called hemidesmosomes. During development these cells emerge as non-polarized cells and differentiate into polarized cells. One of the major functions of epithelial cells is to serve as a physiological and structural barrier. In addition, most epithelial cells participate in vectorial transport of ions, solutes and water. Epithelial cells exhibit a characteristic cell surface organization that is well adapted for these specialized functions. All these roles of the epithelia depend on the polarized distribution of proteins and lipids between the apical and basolateral membrane domains. The apical surface of epithelial cells has a distinctive morphology consisting of extensions of uniform diameter with a core of crosslinked actin filaments, called the brush border or the microvilli. The microvilli are believed to increase the cell surface area thus regulating the absorptive and secretory functions of these cells. Actin filaments in epithelial cells are localized predominantly at the apical surface. The actin cytoskeletal structure of the apical membrane consists of the microvillus core, the terminal web that underlies the apical plasma membrane and the tight junction complex that determines cell-cell adhesion. The apical cytoskeleton of epithelial cells is also unique in that it is characterized by the absence of microtubules and intermediate filaments. The exclusive expression of microfilaments at the apical surface of epithelial cells underscores the relevance of actin and actin-binding proteins in the regulation of epithelial cell function. A distinct spatial and functional polarity in the actin isoforms has also been noted in epithelial cells. For instance, in gastric parietal cells, β-actin facilitates the fusion of tubulovesicles with the apical membrane, thus regulating acid secretion by these cells [1]. There is growing evidence for the involvement of microfilaments in the organization and function of cell membranes. Likewise, membrane-bound proteins and lipids act to regulate the microfilament structure. Several actin-binding proteins of the microvillar compartments have been identified as phospholipid-binding proteins. The best examples of this include villin [2] and ezrin [3] (reviewed in [4]). It is speculated that actin-binding proteins determine the cell shape and thus plasma membrane surface available for the ion transport proteins. Alternatively, it has been suggested that the interaction of actin-binding proteins with the plasma membrane may help stabilize the lipid bilayer which contains the ion transporting molecules. This intimate interaction between the apical microfilament and the plasma membrane also regulates trafficking of vesicles to and from the apical membrane (reviewed in [5]). It is worth noting that there are several actin regulatory proteins that are solely expressed in epithelial cells, such as villin. We suggest that understanding the regulation of epithelial cell function in the context of these specific proteins is imperative to our appreciation of epithelial cell physiology and pathophysiology.
Another characteristic property of epithelial cells is their ability to undergo a reversible process called “epithelial-to-mesenchymal” transition (EMT). EMT involves the loss of an epithelial cell gene expression program coincident with the acquisition of a mesenchymal phenotype. EMT is physiologically relevant during embryogenesis and in adult tissue for wound healing. However, EMT is also associated with tumor metastasis. It is worth mentioning that greater than 90% of all tumors are carcinomas and the current thinking is that all carcinoma cells undergo either partial or complete EMT in order to become motile and invasive. The way epithelial cells move both in vitro and during tumor metastasis is itself different from non-epithelial cells such as fibroblasts. First, in vitro most epithelial cells migrate at significantly lower speeds than non-epithelial cells such as fibroblasts. Second, epithelial cells, in vitro, move as sheets maintaining cell-cell contact. Third, epithelial cells at the wound edge undergo EMT. Interestingly only the neoplastic cells at the outer edge of the epithelial tumor mass undergo EMT, confirming the role of the stroma in inducing EMT. The reversibility of EMT indicates that it is triggered by heterotypic signals originating from stromal cells, suggesting an intimate communication between the epithelial cells and the surrounding stroma. The mechanistic basis of these stromal-epithelial interactions is poorly understood, but growth factors are believed to mediate these cell-cell interactions. Normal epithelial tissue homeostasis depends on the communication between the epithelia and the stromal compartments. This interaction between the mesenchyme, epithelia and the stroma is an important regulatory mechanism required for organ development. The molecular basis of endodermal differentiation and its interaction with the mesoderm have been best studied in the development of the mature intestine. In most organs, the mesenchymal cells are found below the epithelia and the exchange of signals between these two cell compartments drives growth and differentiation during organ development. The current thinking is that signaling pathways relevant to organogenesis are recapitulated during oncogenesis. Not surprisingly, defects in epithelial function and growth control play a major role in human diseases. Most epithelial tissue also self-renew throughout adult life due to the presence of multipotent stem cells and/or unipotent progenitor cells. Epithelial stem cells are specified during development and are controlled by epithelial-mesenchymal interactions. Despite morphological and functional differences among epithelia, common signaling pathways appear to control epithelial stem cell maintenance, activation, lineage determination, and differentiation. Additionally, deregulation of these pathways can lead to diseases including cancer.
While the differences between epithelial and non-epithelial cells itself deserves a separate discussion, it is clear from these examples that epithelial morphology and function is far more complex than that of non-epithelial cells. In this review, we highlight how villin, an epithelial cell-specific actin-binding protein is uniquely adapted to regulate the epithelial cell structure and function and describe how villin regulates the characteristic features of the epithelia in which it is expressed, in molecular terms. Villin is an epithelial cell-specific actin-binding protein that has been identified in very significant amounts in the gastrointestinal, renal and urogenital epithelial cells. However, villin has also been identified in exocrine glands of endodermic lineage such as the thymus and other epithelial cells such as brush cells that line the respiratory tract, taste receptor cells and osteocytes (reviewed in [6]). Then again, villin is also expressed in cells that do not form microvilli-like structures such as crypt and M cells in the intestine. It is quite likely that villin is expressed in most epithelial cells. Most of these cells are either functionally similar or share a common embryonic origin. In the gastrointestinal tract, it has been noted that villin expression increases as the cells differentiate and move from the crypt to the tip of the villi. A similar observation has been made in intestinal cell lines in culture [7]. Villin is detected in the immature digestive tract and there is a significant increase in the expression of villin prior to the formation of the microvilli, thus villin is regarded as an early marker of committed intestinal absorptive cells. Villin is also considered as an early marker of endodermal cell lineage and as a marker of cells that arise from mesenchymal/epithelial conversion in the developing kidney. More recent studies have demonstrated that villin responds to morphogenetic cues associated with intestinal and renal development. The expression of villin is maintained in all carcinomas derived from tissue that normally express this gene. In addition, the expression of villin has also been associated with intestinal metaplasia, implicating a role for villin in the development of preneoplastic lesions [8,9].
Villin as a regulator of actin dynamics
Villin is an unusually versatile actin modifying protein in that it can both polymerize and depolymerize actin filaments. It is an actin-capping, -severing, -nucleating and -bundling protein. To the best of our knowledge, no other actin-binding protein has been identified so far that retains all the actin modifying functions like villin. Structurally and functionally villin can be compared to two groups of actin-binding proteins. The first includes proteins that nucleate, cap and/or sever actin filaments and contain repeats of a conserved domain (Fig. 1). The smallest protein of this family, severin contains three of these conserved domains (S1-S3). Villin and the homologous protein gelsolin contain six such repeats (S1-S6). This forms the “core” of the villin protein. The second group includes proteins that share a villin-like carboxyl-terminal domain called the “headpiece” and includes proteins like dematin, supervillin and advillin (Fig. 1). Most but not all proteins that share villin-like headpiece crosslink actin filaments. This unique structure of villin which includes both the core and the headpiece is responsible for its ability to nucleate, cap, sever and crosslink actin filaments (Fig. 2). Thus, understanding the molecular mechanisms of villin's function can help elucidate the functional properties of a large family of structurally and functionally related proteins.
Figure 1. Domain structure of the villin superfamily.
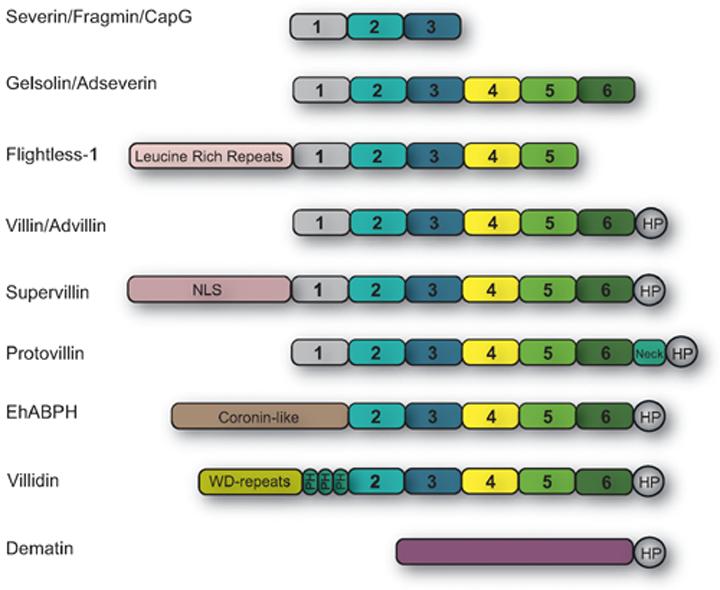
The amino-terminal homologous domains conserved between villin „core„ and other members of the villin superfamily are numbered 1-6. The smallest proteins of this family include severin, fragmin and CapG which contain three of these conserved domains (S1-S3). Villin and gelsolin contain all six of these domains (S1-S6), while EhABPH and flightless-1 do not have S1 and S6 domains respectively. In addition, villin and EhABPH contain the „headpiece„ domain (HP). The headpiece (HP) domain is also conserved in proteins like supervillin, advillin, protovillin, villidin and dematin.
Figure 2. Schematic representation of the domain structures of villin.
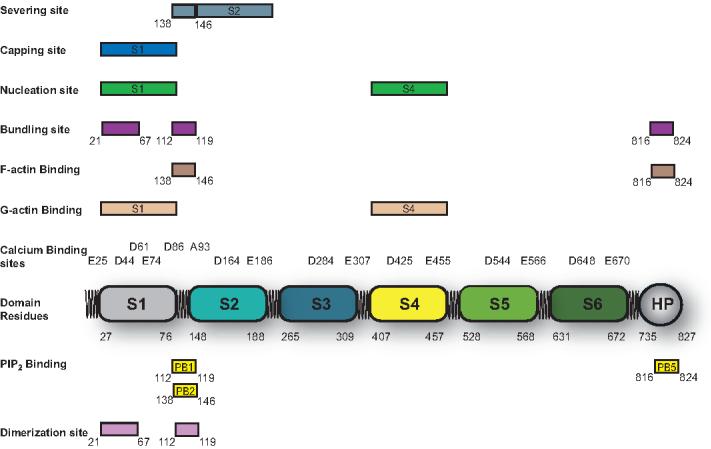
The six homologous domains conserved between villin and other proteins of its family are denoted by numbers S1-S6. Villin contains two F-actin domains one in the villin core and the second in the headpiece. The putative monomer (G-actin) binding sites in villin are shown in S1 and S4. The F-actin domain in the core regulates actin severing while the F-actin domain in the headpiece regulates actin crosslinking by villin. Actin bundling by villin also requires its self-association. The villin dimerization domain is shown in the amino-terminal region (between residues 21-67 and 112-119 in human villin). The phospholipid binding domains are indicated as PB1, PB2 and PB5. In the absence of PIP2, PB1 participates in villin dimerization. PIP2 binding to PB2 inhibits actin severing activity of villin, while PIP2 binding to PB5 enhances actin bundling by villin. Also shown in this figure are the calcium-sensitive sites identified in villin, the actin capping, actin nucleating, actin severing and actin bundling domains identified in human villin protein.
Actin binding by villin is completely dependent on Ca2+. Two F-actin binding sites, one in the core and the second in the headpiece, have been identified in villin (Fig. 2). Villin's association with F-actin is regulated by tyrosine phosphorylation of villin, which decreases the affinity of villin for F-actin [10]. In addition, other F-actin binding proteins such as tropomyosin can inhibit villin's association with F-actin [11]. Two monomer binding sites are estimated in the villin core on the basis of the monomer-binding sites identified in the homologous protein gelsolin (in domains S1 and S4). However, the identity of these sites has not been confirmed in villin, either by mutational analysis or functionally. Villin binds the barbed ends of actin filaments with very high affinity (binding constant of 1011 M-1) thus capping actin filaments [12]. In vitro, half-maximal actin capping by villin occurs at calcium concentrations between 10-30 nM [13]. In the presence of calcium, villin also binds monomeric actin thus nucleating actin assembly from the pointed ends of filaments. In vitro, at very high calcium concentrations (100-200 μM) villin also severs actin filaments [14]. Alternatively, tyrosine phosphorylated villin severs actin at nanomolar calcium concentrations [15]. This suggests that tyrosine phosphorylation rather than high calcium may be the mechanism by which villin severs actin in vivo. At an actin:villin ratio of 4:10, villin bundles actin filaments. While previous studies have suggested that both the F-actin binding sites in villin are required for it to crosslink actin filaments, we have recently reported that only the F-actin binding site in the headpiece participates in actin-bundling by villin [16]. However, villin self-associates to exhibit actin bundling using a single F-actin binding site [16]. A detailed functional analysis of villin mutants allowed us to demonstrate that the exclusive function of the F-actin binding site in the core is to sever actin filaments while the F-actin binding site in the headpiece only bundles actin filaments [16]. These studies provided a more physiologically relevant model for these morphologically distinct functions of villin in cells. The actin-capping function of villin is contained within the first (S1) domain of villin [17]. In contrast, the entire villin core is required for actin-nucleation by villin [17]. It is speculated that the actin capping activity of villin is regulated by the monomer binding site in domain S1, while the actin nucleating activity is regulated by both monomer binding sites in domains S1 and S4 (reviewed in [6]). Mutational analysis has confirmed the identity of these functional domains which were previously studied in proteolytic fragments of villin or characterized biochemically. A cluster of basic amino acid residues (Arg-138, Lys-143 and Lys-145 in human villin) have been identified as the F-actin binding residues in the villin core [18]. The identity of the F-actin binding residues in the headpiece was first described by Friederich et al [19] again as a cluster of basic amino acid residues with the 821KKEK824 motif. However, high-resolution crystal structure and NMR structures of chicken villin headpiece have led to the identification of F-actin binding residues that do not include this motif. These recent studies describe the F-actin binding site in the headpiece to include a “hydrophobic cap” consisting of Arg-787, Lys-816 and Lys-815 (in human villin) and a “crown” of positive and negative charges which includes Lys-789 [20,21]. In the same study it was demonstrated that Glu-823 and Lys-824 (in the KKEK motif) do not contribute to actin binding by the headpiece [20].
The ligand-binding properties of villin as well as post-translational modification of villin allow it to discriminate between its two disparate actin modifying functions. The actin modifying functions of villin are regulated by calcium, phosphoinositides and by tyrosine phosphorylation of the villin protein. The actin capping function of villin is regulated by calcium (increases it) and phosphatidylinositol 4,5-bisphosphate (PIP2) (decreases it) [2,22]. The actin nucleating activity of villin is inhibited by tyrosine phosphorylation and calcium is required for villin to nucleate actin filaments [10,13]. Actin severing is enhanced by calcium (at concentration between 100-200 μM) and by tyrosine phosphorylation of villin, which allows villin to sever actin at much lower calcium concentrations (nM) [13,15]. Actin severing by villin is inhibited by direct association of villin with PIP2 [2]. The actin bundling function of villin is enhanced by villin's interaction with PIP2 but is inhibited by tyrosine phosphorylation of villin [2,10].
During embryogenesis, villin associates with the apical surface of epithelial cells even before an actin-based cytoskeleton is established in these cells. This suggests that villin's apical distribution is not entirely determined by the microfilament density. We speculate that villin interacts with the plasma membrane and may even participate in assembling the apical microvillar core. In vitro, villin associates with phosphoinositides, which are concentrated in the inner leaflet of the plasma membrane, with the following binding affinity: PIP2>PIP>PI [2]. The dissociation constant for villin-PIP2 binding was determined to be 39.4 μM. This suggests that villin in fact has a much higher binding affinity for PIP2 than PLC-γ1 has for PIP2. In accord with this, we have demonstrated that in vitro, villin can inhibit PLC-γ1 activity by sequestering its substrate, PIP2 [23]. A direct interaction of villin with PIP2 in living cells has also been noted by us in FRET analysis using superenhanced yellow fluorescent protein (SEYFP)-tagged villin and cerulean-tagged pleckstrin homology (PH) domain of PLC-δ (George, S.P. and Khurana, S. unpublished observation). Three PIP2 binding sites have been identified in villin (PB1, PB2 and PB5 between amino acids 112-119; 138-146 and 816-824 in human villin protein; Fig. 2) [2]. The first PIP2 binding site participates in villin self association, in the absence of PIP2 [16]. The second PIP2 binding site, PB2, overlaps with the F-actin binding site in the villin core. Consistent with this observation, PIP2 binding to this site inhibits actin severing by villin [2,24]. We have reported that in vitro, PIP2 enhances actin crosslinking by villin [2]. Based on our more recent studies, we speculate that this is mediated by the association of PIP2 with the headpiece PIP2 binding domain, PB5 [16]. All three PIP2 binding sites in villin are also conserved among related actin-binding proteins such as gelsolin, scinderin, adseverin, CapG, supervillin, advillin, dematin and pervin. The three PIP2 binding sites in villin consist of a cluster of basic amino acid residues with the motif x4R/KxR/KR/K, where x is any residue. The association of villin with PIP2 does not result in global changes in the protein conformation, however in studies done with villin peptides encompassing PB1 and PB5 significant changes in the secondary structure were noted upon PIP2 binding by these peptides [2]. The only actin modifying activity of villin that is not regulated by PIP2 is actin nucleation [2]. More recently, we have also demonstrated the direct interaction of villin with the more hydrophilic phospholipid, lysophosphatidic acid (LPA) with a higher affinity than PIP2 (Kd of 22 μM) [25]. Interestingly, both PIP2 and LPA compete for the same binding sites in villin, but unlike the effects of PIP2, the association of villin with LPA inhibits all actin regulatory functions of villin [25]. In cells, association of villin with LPA could sequester LPA thus modifying not only the actin organization in these cells but also LPA-induced intracellular signaling. Alternatively, LPA could be a potent intracellular modulator of actin reorganization in epithelial cells.
Villin is a globular protein that like gelsolin exists in an autoinhibited conformation. Hesterberg and Weber [26] first suggested that villin's association with calcium releases this conformation by a “hinge mechanism.” It is also speculated that this calcium-induced conformational change exposes the F-actin binding site in the villin core, thus regulating the actin-severing function of villin. Full-length villin associates with Ca2+ with a Kd value of 80.5 μM, a stoichiometry of 6 and a hill coefficient of 1.2 [22]. In vitro studies have shown that the calcium-binding sites in villin associated with actin-severing have a much lower affinity than the calcium-binding sites regulating actin-capping by villin [13,14]. Using mutational analysis, we identified two major calcium-sensitive sites in villin that induce conformational changes in villin protein [22]. The first site (Glu-25; Asp-44 and Glu-74) regulates the actin capping and actin severing activity of villin; while the second site (Asp-86; Ala-93, Asp-61) regulates only the actin severing function of villin [22]. Some of these calcium-sensitive sites have since been confirmed functionally in cells [27].
Villin is tyrosine phosphorylated both in vitro and in vivo [23,28]. Ten tyrosine phosphorylation sites have been identified in human villin (Y-46, -60, -81, -256, -324, -461, 604 and 725) [29,30]. All ten sites are localized within the villin core. The nonreceptor tyrosine kinase c-src regulates the phosphorylation of villin both in vitro and in cells [31]. Although a recent study has suggested that Janus kinase 3 phosphorylates villin, detailed analysis from our laboratory have demonstrated that intestinal epithelial cell lines as well as epithelial cells in mouse intestine do not express Jak3, consistent with its identified limited expression in haematopoeitic cells [31,32]. RT-PCR allowed us to demonstrate unequivocally that the intestinal epithelial cell lines, HT-29/clone 19A and Caco-2 cells used to demonstrate phosphorylation of villin by Jak3, in fact, do not even express Jak3 mRNA, thus we question the validity of this report [31]. Villin's interaction with actin is sensitive to monovalent salt concentrations, suggesting that electrostatic interactions are required for this association [17]. We have speculated that tyrosine phosphorylation changes the surface charge distribution in villin thus influencing villinactin interactions. Consistent with this, we have shown a decrease in the binding affinity of phosphorylated villin for F-actin [10]. Tyrosine phosphorylation also alters villin's affinity for its other ligands including PLC-γ1 and PIP2. While phosphorylation of villin is required for its association with the SH2 domain of PLC-γ1, tyrosine phosphorylation prevents villin's association with PIP2 [23,28,33]. Based on our in vitro studies we proposed a working model (Fig. 3) to demonstrate the crosstalk between villin-regulated microfilament structure, the plasma membrane and components of the transmembrane signaling network [23]. Subsequent studies from our laboratory as well as other laboratories have confirmed this model in both epithelial cell lines as well as in animal studies [30,33-38].
Figure 3. Schematic model representing the molecular mechanism of actin remodeling and catalytic activation of PLC-γ1 by tyrosine phosphorylated villin.

In response to receptor activation (epidermal growth factor, EGF binding to its receptor, EGFR), c-src kinase and PLC-γ1 are activated. Activation of c-src results in the tyrosine phosphorylation of villin which enhances its actin depolymerizing functions. Activation of PLC-γ1 recruits it to the plasma membrane. Tyrosine phosphorylated villin dissociates from phosphatidylinositol 4,5-bisphosphate (PIP2) and associates with the SH2 domain of PLC-γ1 This results in the catalytic activation of PLC-γ1 at the plasma membrane. PLC-γ1 activation generates other second messengers, diacylglycerol (DAG) and inositol 1,4,5 trisphosphate (IP3), which results in the release of calcium from intracellular stores (endoplasmic reticulum, ER) further enhancing the actin severing activity of villin. Actin severing by villin generates new barbed ends for actin assembly at the plasma membrane, thus enhancing cell migration.
Villin as an actin-capping, actin-bundling and actin-severing protein in epithelial cells
Studies done in renal and intestinal epithelial cell lines as well as in villin-null mice have established the function of villin in regulating actin dynamics, signal transduction, cell morphology, epithelial-to-mesenchymal transition, cell migration, cell invasion and cell survival (Fig. 4) [30,33,36-39]. In addition, it has been demonstrated that villin's actin severing function is required for the invasion and dissemination of enteropathogens in the gastrointestinal tract [39]. These studies point to the significance of villin in both epithelial cell physiology as well as pathophysiology. Overexpression of villin in fibroblasts and other villin-null cells results in significant actin reorganization, which includes the loss of stress fibers and formation of microvilli-like structures on the dorsal surface of these cells [16,37,40,41]. As a result, there is a drastic change in cell morphology from a flat to a more rounded, epitheloid cell shape [37]. Over two decades ago it was demonstrated that simple mixtures of villin and F-actin form uniformly polarized bundles similar to those seen in the brush border of epithelial cells [42]. Thus, villin inherently has the ability to organize the actin filaments into ordered bundles in cells by recruiting actin filaments from other microfilament structures such as stress fibers. In epithelial cells, villin is the major protein associated with the actin filaments in the microvillar core. Some villin is also found in the rootlet area of the terminal web along with gelsolin and scinderin, two related proteins of the villin family [43]. The presence of three proteins of the same family with overlapping functions, highlights the significance of these proteins to epithelial cell function. Studies done in developing mice and chicken has demonstrated that villin is the first major actin-bundling protein that concentrates at the apical surface of enterocytes [44,45]. Thus, villin plays a major role in the initiation, organization and formation of the microvilli [46,47]. Consistent with these observations, changes in the microvillar structure have been noted in the microvilli of villin knock-out mice, which are not as well organized as their wild-type littermates [48]. More recently, the absence of villin and the concomitant structural defects in the microvilli have been associated with biliary atresia-like disorder in children [49]. In these children with progressive cholestasis and liver failure, structural abnormalities were noted within the microvilli of the bile duct canaliculi which were determined to be due to a lack of villin expression.
Figure 4. Biological functions regulated by villin in cells.
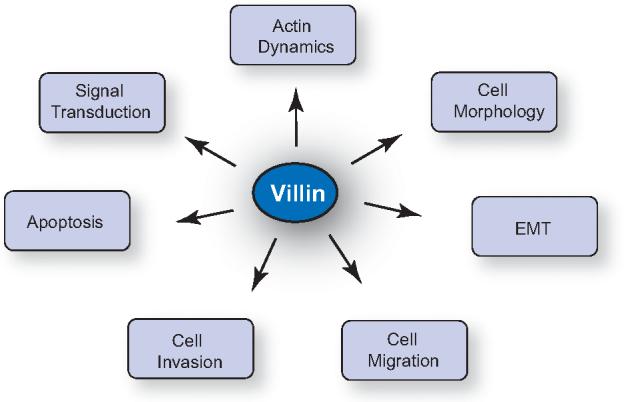
Studies done in epithelial cell lines and in villin knock out mice have identified the biological functions of villin to include regulation of actin dynamics, cell morphology, signal transduction, epithelial-tomesenchymal transition (EMT), cell migration, cell invasion and apoptosis.
Recent studies from our laboratory have demonstrated that villin self-association is required not only for its actin bundling function, but also for the apical targeting of villin protein and for villin-induced loss of stress fibers (presented at “The American Society for Cell Biology 47th Annual Meeting,” Abstract B86: Dimerization of villin is required for its apical localization. George, S.P., Siddiqui, M.R. and Khurana, S.). The villin dimerization site contains a conserved cysteine residue, which could regulate palmitoylation of villin protein. We have previously demonstrated that tyrosine phosphorylated villin redistributes to a Triton-soluble fraction of the plasma membrane [28,30]. Hence, we speculate that tyrosine phosphorylation of villin may target it to the plasma membrane while palmitoylation of villin may anchor it to the plasma membrane. Alternatively, palmitoylation may drive the protein to the apical surface where c-src kinase may phosphorylate villin protein. If it is determined that villin is indeed palmitoylated, it would also require us to re-examine the role of the PIP2-binding sites identified in villin and their role in determining the association of villin with the apical plasma membrane in epithelial cells. For over two decades it has been theorized that PIP2 binding domains in actin regulatory proteins, including villin, gelsolin and ezrin, determine the interaction of the microfilament structure with the plasma membrane. In that event, we would speculate that the major function of these sites would be to sequester PIP2, thus regulating phosphoinositide-mediated signal transduction pathways, including the regulation of second messengers such as PLC-γ1 which is a known ligand for both villin and its related protein, gelsolin [50]. Alternatively, the association of villin with PIP2 rich domains in the plasma membrane could lead to spatial clustering of villin in the vicinity of other ligands thus regulating cell surface functions of epithelial cells. The increase in stress fibers noted in mutant cell lines expressing the dimerization mutant of villin, may be related to villin's actin capping function. We have previously identified a calcium-sensitive site in the NH2-terminal domain of villin that regulates the actin-capping function of villin (Glu-25, Asp-44 and Glu-74) [22]. Two of these three amino acids are deleted in Δ21-67/112-119) the [16]. Based on our preliminary studies, we would suggest that loss of actin capping by villin would increase the F-actin levels in the cells, thus altering actin filament dynamics in mutant cell lines. Since overexpression of villin is associated with loss of stress fibers, we speculate that the actin capping function of villin serves to suppress excess formation of F-actin. Thus, the capping and uncapping of filament ends by villin may spatially and temporally control actin polymerization in epithelial cells. Villin expression in epithelial cells may as a result, serve to regulate the cellular levels of F-actin. The actin capping function of villin is regulated by physiological calcium concentrations (see discussion above) and may therefore be an innate function of villin, like its ability to crosslink actin filaments. Another role of villin may be to regulate the proper distribution of F-actin filaments in cells. Villin dimerization mutant localizes to the basal surface of polarized epithelial cells, increasing F-actin assembly at the ventral surface of cells. Cells expressing the villin dimerization mutant form lamellipodia like cells expressing wild-type villin protein, yet the mutant cell lines do not migrate at the same rate as cells expressing the wild-type protein. Loss of cell migration function of villin by this mutant may also be related to the loss of actin capping by villin. These studies identify potentially new and previously unrecognized functions of villin in epithelial cells.
For more than two decades, villin's physiological function was believed to be limited to actin-bundling, despite the fact that in vitro it can sever, cap and nucleate actin filaments. This idea was based on the fact that to sever actin, villin requires very high calcium concentrations (100-200 μM) that are not physiologically appropriate. Our laboratory was the first to identify the tyrosine phosphorylation of villin and to report that phosphorylation of villin enhanced the actin severing activity of villin even in the absence of high calcium [15,28]. Our in vitro studies were later confirmed in the villin-null mice which were shown to have an actin-severing defect [51]. It must be pointed out that villin is the only actin-severing protein that is expressed in the microvilli. Subsequently it was demonstrated that the actin severing activity of villin in mice required tyrosine phosphorylation of villin [36]. These studies in villin-null mice provided a temporal relationship between tyrosine phosphorylation of villin and actin severing by villin. Using villin mutants that lack the tyrosine phosphorylation sites, we provided a causal link between tyrosine phosphorylation of villin and regulation of villin's severing function in cells [30,33,37,38].
Like other proteins of its family such as gelsolin, overexpression of villin increases cell motility, irrespective of the cell type [33,37]. The absence of villin (in mice) as well as the overexpression of villin phosphorylation site mutants (in intestinal epithelial cell lines) are both associated with an inhibition of intestinal cell migration [33,36]. Nine of the ten tyrosine phosphorylation sites identified in villin are required for its function in cell migration. Three NH2-terminal residues in human villin namely Y60, -81 and -256, are required for villin's intracellular distribution at the cell surface [37]. Villin mutants that failed to localize at the cell margin, failed to increase cell migration [37]. In addition, six COOH-terminal tyrosine phosphorylation sites are required for villin's interaction with the SH2 domain of PLC-γ1 and for villin-induced increase in cell migration [30]. The direct interaction of phosphorylated villin with PLC-γ1 was demonstrated by FRET analysis in live cells [33]. Further, using PLC-γ1-/- fibroblasts, we demonstrated that tyrosine phosphorylation of villin alone is not sufficient for its function in cell migration. The interaction of phosphorylated villin with PLC-γ1 and the catalytic activation of PLC-γ1 by villin are essential to its function in cell migration and cell invasion [30,33]. The most crucial event in cell migration is rapid actin polymerization and formation of a pseudopod in the direction of cell movement. Actin polymerization can be initiated either by uncapping of barbed ends of filaments or by actin severing that generates new barbed ends. Tyrosine phosphorylation is required for both actin-severing by villin as well as regulation of cell migration by villin [10,30,37]. Tyrosine phosphorylation of villin enhances its actin severing activity thus generating new barbed ends for actin filament assembly and formation of lamellipodia. Another important step in cell locomotion is development of cell polarity. PLC-γ has been implicated in this first step of cell migration [52]. Since villin-induced cell migration also requires PLC-γ1 and since villin can regulate the catalytic activation of PLC-γ1, we have suggested that the synergistic interaction between PLC-γ1 and villin regulates the initial changes in the generation of free barbed ends, thus regulating cell migration (Fig. 5) [23,30,33].
Figure 5. Stimulated protrusion model showing the synergistic interaction of villin with PLC-γ1 in the regulation of cell migration.

(1) Under resting condition, villin is not activated. (2) The localized activation of villin generates free barbed ends. (3) Profilin bound ATP-G-actin is added to the barbed end, generating ATP actin caps, where activated Arp2/3 complex binds and initiates branch formation. (4) Rapid growth occurs at the barbed end of actin filaments which pushes the membrane forward. Translocation of PLC-γ1 to the plasma membrane initiates the early steps involved in actin polymerization and assembly by defining the directionality of a polarized motile cell.
Src proteins play a critical role in the proliferation and differentiation of normal intestinal epithelial cells [53]. Src activation is also evident in 80% of human colon cancers, particularly those that metastasize to the liver [54-56]. Despite the fact that there is considerable evidence associating aberrant Src activation with the development and progression of colorectal cancer and even though Src kinase inhibitors have entered phase I clinical trials, there are surprisingly few studies that have analyzed the consequences of Src activation on intestinal epithelial cell function. There are also no reports of complementary mechanistic studies to correlate the contribution of the different Src kinases (c-src, c-yes and c-fyn) in the regulation of intestinal epithelial cell function. One of the major functions of Src kinases is to modulate the actin cytoskeleton that controls cell migration [57]. However, the exact mechanism by which Src proteins induce remodeling of the actin cytoskeleton remains unknown. By examining the phosphorylation of villin by these three ubiquitously expressed Src kinase and correlating these with changes in cell behavior and signaling, we have provided a biochemical and cellular model that helps explain changes in epithelial cell actin dynamics by Src activation. Using the c-src, c-yes and c-fyn null fibroblast transfected individually with the ubiquitously expressed Src kinases we have recently reported that c-src kinase but not c-yes or c-fyn is required for villin-induced increase in cell migration [31]. Further, we determined that two protein tyrosine phosphatases, SHP-2 and PTP-PEST negatively regulate villin's function in cell migration by dephosphorylating c-src kinase at the catalytically active tyrosine residue Y416 within the kinase domain of c-src, thus preventing the phosphorylation of villin by c-src kinase. Both SHP-2 and PTP-PEST have been shown to reduce carcinoma invasiveness-related properties of cells in vitro [58,59]. Whether, villin-induced increase in cell migration and cell invasiveness is aberrantly regulated by c-src kinase during colon cancer progression and whether it contributes to colon tumor invasion and metastasis remains to be investigated in the future. However, we have reported that attenuation of c-src kinase activity as well as inhibition of villin phosphorylation can inhibit migration of human adenocarcinoma cells [31,33]. Caco-2 cells are human adenocarcinoma cells that endogenously express villin. Overexpression of dominant negative c-src kinase or treatment with the Src kinase inhibitor PP2; overexpression of SHP-2 or PTP-PEST; downregulation of PLC-γ1 with PLC-γ1 siRNA or the PLC-γ1 inhibitor U73122 all inhibit Caco-2 cell migration, consistent with the model described by us for villin-induced cell motility (Fig. 5) [31,33,38]. Likewise, overexpression of a phosphorylation site mutant of villin in Caco-2 cells had a dominant negative effect, inhibiting both basal as well as growth factor induced cell migration, confirming that downregulation of c-src kinase and PLC-γ1 both inhibited cell migration by inhibiting villin's function in Caco-2 cells [33]. Together, these data suggest that in colorectal carcinogenesis, elevation of c-src may promote cancer metastasis and invasion by regulating villin-induced cell migration.
Villin as an anti-apoptotic protein
Some of the most comprehensible data supporting links between actin and apoptosis in eukaryotic cells come from studies using drugs that affect actin turnover. Actin and actin binding proteins are also substrates of activated caspases, although it remains to be established if such changes are important for the progression of the apoptotic process. In yeast, however, it has been clearly demonstrated that changes in actin turnover caused by direct mutations in actin, addition of actin-binding drugs or by mutations in actin-binding proteins changes cell fate [60]. Yeast with reduced actin dynamics exhibit accumulation of F-actin, loss of mitochondrial permeability and increased cell death. The precise mechanisms by which alterations in actin dynamics regulate apoptosis in higher eukaryotes remains to be established. However, several recent studies clearly support a role for actin dynamics in cell survival. It has been suggested that actin filaments could regulate mitochondrial structure and function by regulating mitochondrial motility (such as is seen in neurons), mitochondrial morphology (fission and fusion) and/or transport of pro-apoptotic cargo to the mitochondria [61-66].
The best studied actin regulatory protein that is also associated with apoptosis is gelsolin, a homologous protein of the villin family. The roles of gelsolin however, are disputed in terms of its involvement in contrasting activities such as oncogenic [67,68], anti-oncogenic [69,70], pro-apoptotic [71] and anti-apoptotic [72,73]. Overexpression of gelsolin has been shown to inhibit caspase-3 activation indicating its role as an anti-apoptotic protein [74,75]. At the same time overexpression of gelsolin has also been shown to fail to regulate lymphocyte apoptosis [76]. To explain this disparity, it has been reported that human gelsolin but not mouse gelsolin inhibits apoptosis, although there are studies done with mouse gelsolin and its anti-apoptotic effects [73,77]. The most convincing data come from 2 studies that have examined neurons and liver tissue from gelsolin-null mice, and both these studies suggest that gelsolin functions as an anti-apoptotic protein [78,79]. Our own studies with the villin knock out mouse and the villin and gelsolin-double knock out mice suggest that in vivo gelsolin functions as an anti-apoptotic protein [80]. Gelsolin is also cleaved by caspase-3 and the NH2-termianl cleaved fragment (S1-S3) has been shown to enhance apoptosis while the COOH-terminal fragment (S4-S6) has been shown to inhibit apoptosis [71,81]. Again, the physiological relevance of these findings is unclear. Both full-length gelsolin and fragment 5 (S5) of gelsolin have been shown to inhibit apoptosis by directly binding to the voltage-dependent anion channels (VDAC) [72,81]. However, even this finding is undermined by a recent study by Baines and colleagues that demonstrates unequivocally that VDAC's do not regulate mitochondrial apoptosis [82]. To further complicate this picture, gelsolin in complex with PIP2 has been shown to directly inhibit caspase-9 and -3 as well as prevent apoptotic progression in a cell free system [73]. At the same time gelsolin also inhibits Fas-triggered death signals, which suggests that gelsolin prevents apoptosis by acting upstream of caspase activation. We believe our studies with villin knock out mice and with the villin-gelsolin double knock out mice provide an opportunity to not only understand epithelial cell survival but also to introduce a molecular basis for gelsolin's anti-apoptotic function in the gastrointestinal epithelium that could be extended to other tissues.
The homeostatic balance between proliferation and apoptosis is essential for the intestinal epithelium to function as a physiological and structural barrier. Intestinal epithelial cells have a high rate of cell turnover accompanied by an equally high rate of apoptosis [83]. This normal apoptosis is essential for the hierarchical organization of the intestinal epithelium and apoptotic epithelial cells have been detected both at the base of the crypt as well as the villus tips of the small and large intestine [84,85]. Defects in apoptosis are also associated with several gastrointestinal disorders such as villus atrophy, epithelial hyperplasia, loss of normal absorptive functions and increased risk of tumorigenesis. Abnormalities in apoptotic function contribute both to the pathogenesis of colorectal cancer as well as its resistance to chemotherapy and radiotherapy [86,87]. Surprisingly, there are few data available on whether apoptosis is an important mechanism of action in vivo in the gastrointestinal epithelium. We have recently reported that the villin-null mice have higher levels of apoptosis compared to their wild-type littermates that correlates with the severity of colitis induced in dextran sodium sulfate-treated villin-null mice [88]. Further in vitro experiments allowed us to characterize villin as an epithelial cell specific anti-apoptotic protein. These studies provided a molecular mechanism for villin's role in regulating epithelial cellular plasticity related to cell injury [88]. We also determined from these studies that villin is cleaved late in the apoptotic cycle (Wang, Y. and Khurana, S., unpublished observation) and we have previously reported that an NH2-terminal fragment of villin is pro-apoptotic [30]. Based on ongoing research in our laboratory, we speculate that full-length villin is anti-apoptotic. However, as the epithelial cells migrate from the crypt to the tip and are ready to be shed into the lumen, villin may be cleaved to generate the pro-apoptotic S1-S3 fragment thus enhancing and/or assisting the process of cell extrusion from the gastrointestinal epithelium. To examine the role of villin and its related proteins in the regulation of epithelial cell survival, we have generated the villin-gelsolin double knock-out mice [80]. The double knock-out mice have higher levels of apoptotic cells compared to their wild-type littermates [80]. Deletion of both genes resulted in increased susceptibility of these mice to both γ-radiation as well as DSS-colitis induced apoptosis and injury [80]. Transmission electron micrographs of the small intestine showed a dramatic change in the mitochondrial morphology in these mice, but not in wild-type or the single knock out mice. The mitochondria were swollen and irregular with significant loss of cristae structures and electron dense inclusions were noted within the inner membrane. These mice had a large number of abnormal mitochondria undergoing autophagy and a significant increase in the accumulation of lipofuscin [80]. All these findings are indicative of perturbed mitochondrial morphology and function and predictive of lower ATP levels. These studies suggest that in vivo, both villin and gelsolin function as anti-apoptotic proteins. Further, these studies suggest that these proteins may have overlapping functions in epithelial cell survival, since the phenotype of the villin-gelsolin double knock out mouse is more severe than that of either single knock out mice [80,88]. A correlation between absence of villin and increased apoptosis has been noted in several inflammatory diseases of the gastrointestinal tract. A decrease in the levels of villin expression in enterocytes from patients with inflammatory bowel disease relative to healthy controls has been reported [89]. Evidence from animal and human studies have demonstrated that increased apoptosis in the intestinal epithelium contributes to the tissue damage and the severity of colonic inflammatory response [85]. Decreased villin expression has also been associated with other inflammatory diseases such as pancreatitis, a disease believed to be related to abnormal apoptosis/necrosis [90].
Changes in intracellular calcium, changes in actin cytoskeleton as well as activation of PI3-kinase all have been demonstrated to regulate apoptosis in eukaryotic cells. Villin is an actin-nucleating, -capping, -severing and -crosslinking protein that also associates with Ca2+, PIP2 and PLC-γ1. Any of these properties of villin could thus regulate its anti-apoptotic functions. We have used wild-type and mutant villin proteins lacking each of these actin modifying or ligand-binding sites and examined the effects on epithelial cell survival. Our studies allowed us to demonstrate unequivocally, that the actin-severing function of villin was required for its anti-apoptotic function [91]. Overexpression of either villin or gelsolin in epithelial cell lines delayed apoptosis by maintaining the actin dynamics [91]. In contrast, in the absence of these proteins or in the presence of actin-severing mutants of these proteins, there was significant increase in the F-actin content of cells accompanied by a significant decrease in the G-actin content of cells [91]. Thus, our studies allow us to conclude that both villin and gelsolin function as anti-apoptotic proteins by regulating actin dynamics in cells. Further, the actin-severing functions of both these proteins prevent the accumulation of F-actin in cells subjected to apoptotic injury. It may be noted that both in yeast as well as in eukaryotic cells, reduced actin dynamics and accumulation of F-actin has been linked to increased cell death [60,92].
Summary
Villin is an extraordinarily versatile actin regulatory protein that organizes, integrates and regulates multiple epithelial cell functions such as cell morphology, cell motility and cell death. To perform these diverse biological functions villin's actin modifying activities are tightly regulated both spatially and temporally. Villin responds to extracellular stimuli as well as interacts with several second messengers including PIP2, PLC-γ1 and calcium, it is also a substrate of c-src kinase and yet unidentified phosphatases. Thus, villin serves to converge signals at the cell surface to regulate cell migration but also intracellulary to regulate cell survival. Understanding the molecular mechanisms of villin's function in cells allows us to appreciate the synergy between actin-binding proteins and signal transduction pathways in the regulation of biological processes. While many potential drug targets are available to treat human diseases, development of cytoskeleton-based strategies to treat or prevent human disease is essentially untapped. We believe our studies identify the actin cytoskeleton as a target to protect from apoptosis and modify during metastasis and thus may have implications for therapeutic approaches to digestive and renal diseases.
Acknowledgements
The work described in this review was supported by NIDDK Grants DK-65006 and DK-54755 from the National Institutes of Health (to S.K.).
Abbreviations
- DSS
dextran sodium sulfate
- EMT
epithelial-to-mesenchymal transition
- FRET
fluorescence resonance energy transfer
- LPA
lysophosphatidic acid
- PH
pleckstrin homology
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PLC-γ1
phospholipase C-gamma 1
- SEYFP
super-enhanced yellowed fluorescent protein
- SH2
Src homology 2
- VDAC
voltage-dependent anion channel
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Yao X, Chaponnier C, Gabbiani G, Forte JG. Biochemical characterization of ezrin-actin interaction. Mol Biol Cell. 1995;6:541–57. doi: 10.1091/mbc.6.5.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kumar N, Zhao P, Tomar A, Galea CA, Khurana S. Association of villin with phosphatidylinositol 4,5-bisphosphate regulates the actin cytoskeleton. J Biol Chem. 2004;279:3096–110. doi: 10.1074/jbc.M308878200. [DOI] [PubMed] [Google Scholar]
- [3].Hirao M, Sato N, Kondo T, Yonemura S, Monden M, Sasaki T, Takai Y, Tsukita S. Regulation mechanism of ERM (ezrin/radixin/moesin) protein/plasma membrane association: possible involvement of phosphatidylinositol turnover and Rho-dependent signaling pathway. J Cell Biol. 1996;135:37–51. doi: 10.1083/jcb.135.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Niggli V. Lipid interactions of cytoskeletal proteins in: Advances in molecular and cell biology. In: Khurana S, editor. Aspects of the cytoskeleton. Vol. 37. Elsevier; 2006. pp. 221–250. [Google Scholar]
- [5].Doctor RB. The actin cytoskeleton in the apical domain of epithelial cells in: Advances in molecular and cell biology. In: Khurana S, editor. Aspects of the cytoskeleton. Vol. 37. Elsevier; 2006. pp. 25–47. [Google Scholar]
- [6].Khurana S. Structure and function of villin in: Aspects of the cytoskeleton. In: Khurana S, editor. Advances in molecular and cell biology. Vol. 37. Elsevier; 2006. pp. 89–115. [Google Scholar]
- [7].Dudouet B, Robine S, Huet C, Sahuquillo-Merino C, Blair L, Coudrier E, Louvard D. Changes in villin synthesis and subcellular distribution during intestinal differentiation of HT29-18 clones. J Cell Biol. 1987;105:359–69. doi: 10.1083/jcb.105.1.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rieder G, Tessier AJ, Qiao XT, Madison B, Gumucio DL, Merchant JL. Helicobacter-induced intestinal metaplasia in the stomach correlates with Elk-1 and serum response factor induction of villin. J Biol Chem. 2005;280:4906–12. doi: 10.1074/jbc.M413399200. [DOI] [PubMed] [Google Scholar]
- [9].Regalado SP, Nambu Y, Iannettoni MD, Orringer MB, Beer DG. Abundant expression of the intestinal protein villin in Barrett's metaplasia and esophageal adenocarcinomas. Mol Carcinog. 1998;22:182–9. [PubMed] [Google Scholar]
- [10].Zhai L, Zhao P, Panebra A, Guerrerio AL, Khurana S. Tyrosine phosphorylation of villin regulates the organization of the actin cytoskeleton. J Biol Chem. 2001;276:36163–7. doi: 10.1074/jbc.C100418200. [DOI] [PubMed] [Google Scholar]
- [11].Burgess DR, Broschat KO, Hayden JM. Tropomyosin distinguishes between the two actin-binding sites of villin and affects actin-binding properties of other brush border proteins. J Cell Biol. 1987;104:29–40. doi: 10.1083/jcb.104.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Walsh TP, Weber A, Higgins J, Bonder EM, Mooseker MS. Effect of villin on the kinetics of actin polymerization. Biochemistry. 1984;23:2613–21. doi: 10.1021/bi00307a012. [DOI] [PubMed] [Google Scholar]
- [13].Northrop J, Weber A, Mooseker MS, Franzini-Armstrong C, Bishop MF, Dubyak GR, Tucker M, Walsh TP. Different calcium dependence of the capping and cutting activities of villin. J Biol Chem. 1986;261:9274–81. [PubMed] [Google Scholar]
- [14].Walsh TP, Weber A, Davis K, Bonder E, Mooseker M. Calcium dependence of villin-induced actin depolymerization. Biochemistry. 1984;23:6099–102. doi: 10.1021/bi00320a030. [DOI] [PubMed] [Google Scholar]
- [15].Kumar N, Khurana S. Identification of a functional switch for actin severing by cytoskeletal proteins. J Biol Chem. 2004;279:24915–8. doi: 10.1074/jbc.C400110200. [DOI] [PubMed] [Google Scholar]
- [16].George SP, Wang Y, Mathew S, Srinivasan K, Khurana S. Dimerization and actin-bundling properties of villin and its role in the assembly of epithelial cell brush borders. J Biol Chem. 2007;282:26528–41. doi: 10.1074/jbc.M703617200. [DOI] [PubMed] [Google Scholar]
- [17].Janmey PA, Matsudaira PT. Functional comparison of villin and gelsolin. Effects of Ca2+, KCl, and polyphosphoinositides. J Biol Chem. 1988;263:16738–43. [PubMed] [Google Scholar]
- [18].de Arruda MV, Bazari H, Wallek M, Matsudaira P. An actin footprint on villin. Single site substitutions in a cluster of basic residues inhibit the actin severing but not capping activity of villin. J Biol Chem. 1992;267:13079–85. [PubMed] [Google Scholar]
- [19].Friederich E, Vancompernolle K, Huet C, Goethals M, Finidori J, Vandekerckhove J, Louvard D. An actin-binding site containing a conserved motif of charged amino acid residues is essential for the morphogenic effect of villin. Cell. 1992;70:81–92. doi: 10.1016/0092-8674(92)90535-k. [DOI] [PubMed] [Google Scholar]
- [20].Meng J, Vardar D, Wang Y, Guo HC, Head JF, McKnight CJ. High-resolution crystal structures of villin headpiece and mutants with reduced F-actin binding activity. Biochemistry. 2005;44:11963–73. doi: 10.1021/bi050850x. [DOI] [PubMed] [Google Scholar]
- [21].Vermeulen W, et al. Solution structures of the C-terminal headpiece subdomains of human villin and advillin, evaluation of headpiece F-actin-binding requirements. Protein Sci. 2004;13:1276–87. doi: 10.1110/ps.03518104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kumar N, Tomar A, Parrill AL, Khurana S. Functional dissection and molecular characterization of calcium-sensitive actin-capping and actindepolymerizing sites in villin. J Biol Chem. 2004;279:45036–46. doi: 10.1074/jbc.M405424200. [DOI] [PubMed] [Google Scholar]
- [23].Panebra A, Ma SX, Zhai LW, Wang XT, Rhee SG, Khurana S. Regulation of phospholipase C-gamma(1) by the actin-regulatory protein villin. Am J Physiol Cell Physiol. 2001;281:C1046–58. doi: 10.1152/ajpcell.2001.281.3.C1046. [DOI] [PubMed] [Google Scholar]
- [24].Janmey PA, Lamb J, Allen PG, Matsudaira PT. Phosphoinositide-binding peptides derived from the sequences of gelsolin and villin. J Biol Chem. 1992;267:11818–23. [PubMed] [Google Scholar]
- [25].Khurana S, Tomar A, Mathew S, George SP. 2008. Submitted.
- [26].Hesterberg LK, Weber K. Demonstration of three distinct calcium-binding sites in villin, a modulator of actin assembly. J Biol Chem. 1983;258:365–9. [PubMed] [Google Scholar]
- [27].Revenu C, Courtois M, Michelot A, Sykes C, Louvard D, Robine S. Villin severing activity enhances actin-based motility in vivo. Mol Biol Cell. 2007;18:827–38. doi: 10.1091/mbc.E06-05-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Khurana S, Arpin M, Patterson R, Donowitz M. Ileal microvillar protein villin is tyrosine-phosphorylated and associates with PLC-gamma1. Role of cytoskeletal rearrangement in the carbachol-induced inhibition of ileal NaCl absorption. J Biol Chem. 1997;272:30115–21. doi: 10.1074/jbc.272.48.30115. [DOI] [PubMed] [Google Scholar]
- [29].Zhai L, Kumar N, Panebra A, Zhao P, Parrill AL, Khurana S. Regulation of actin dynamics by tyrosine phosphorylation: identification of tyrosine phosphorylation sites within the actin-severing domain of villin. Biochemistry. 2002;41:11750–60. doi: 10.1021/bi0263762. [DOI] [PubMed] [Google Scholar]
- [30].Tomar A, George S, Kansal P, Wang Y, Khurana S. Interaction of phospholipase C-gamma1 with villin regulates epithelial cell migration. J Biol Chem. 2006;281:31972–86. doi: 10.1074/jbc.M604323200. [DOI] [PubMed] [Google Scholar]
- [31].Mathew S, George SP, Wang Y, Siddiqui MR, Srinivasan K, Tan L, O'Shea JJ, Khurana S. c-Src kinase activation is required for intestinal migration. 2008. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kumar N, Mishra J, Narang VS, Waters CM. Janus kinase 3 regulates interleukin 2-induced mucosal wound repair through tyrosine phosphorylation of villin. J Biol Chem. 2007;282:30341–5. doi: 10.1074/jbc.C600319200. [DOI] [PubMed] [Google Scholar]
- [33].Wang Y, Tomar A, George SP, Khurana S. Obligatory role for phospholipase C-gamma(1) in villin-induced epithelial cell migration. Am J Physiol Cell Physiol. 2007;292:C1775–86. doi: 10.1152/ajpcell.00420.2006. [DOI] [PubMed] [Google Scholar]
- [34].Lange K, Brandt U. Calcium storage and release properties of F-actin evidence for the involvement of F-actin in cellular calcium signaling. FEBS Lett. 1996;395:137–42. doi: 10.1016/0014-5793(96)01025-3. [DOI] [PubMed] [Google Scholar]
- [35].Papakonstanti EA, Emmanouel DS, Gravanis A, Stournaras C. PLC-gamma1 signaling pathway and villin activation are involved in actin cytoskeleton reorganization induced by Na+/Pi cotransport up-regulation. Mol Med. 2000;6:303–18. [PMC free article] [PubMed] [Google Scholar]
- [36].Athman R, Louvard D, Robine S. Villin enhances hepatocyte growth factor-induced actin cytoskeleton remodeling in epithelial cells. Mol Biol Cell. 2003;14:4641–53. doi: 10.1091/mbc.E03-02-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tomar A, et al. Regulation of cell motility by tyrosine phosphorylated villin. Mol Biol Cell. 2004;15:4807–17. doi: 10.1091/mbc.E04-05-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Khurana S, Tomar A, George SP, Wang Y, Siddiqui MR, Guo H, Tigyi G, Mathew S. Autotaxin and lysophosphatidic acid stimulate intestinal cell motility by redistribution of the actin modifying protein villin to the developing lamellipodia. Exp Cell Res. 2007 doi: 10.1016/j.yexcr.2007.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Athman R, Fernandez MI, Gounon P, Sansonetti P, Louvard D, Philpott D, Robine S. Shigella flexneri infection is dependent on villin in the mouse intestine and in primary cultures of intestinal epithelial cells. Cell Microbiol. 2005;7:1109–16. doi: 10.1111/j.1462-5822.2005.00535.x. [DOI] [PubMed] [Google Scholar]
- [40].Franck Z, Footer M, Bretscher A. Microinjection of villin into cultured cells induces rapid and long-lasting changes in cell morphology but does not inhibit cytokinesis, cell motility, or membrane ruffling. J Cell Biol. 1990;111:2475–85. doi: 10.1083/jcb.111.6.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Friederich E, Huet C, Arpin M, Louvard D. Villin induces microvilli growth and actin redistribution in transfected fibroblasts. Cell. 1989;59:461–75. doi: 10.1016/0092-8674(89)90030-5. [DOI] [PubMed] [Google Scholar]
- [42].Coluccio LM, Bretscher A. Reassociation of microvillar core proteins: making a microvillar core in vitro. J Cell Biol. 1989;108:495–502. doi: 10.1083/jcb.108.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lueck A, Brown D, Kwiatkowski DJ. The actin-binding proteins adseverin and gelsolin are both highly expressed but differentially localized in kidney and intestine. J Cell Sci. 1998;111:3633–43. doi: 10.1242/jcs.111.24.3633. [DOI] [PubMed] [Google Scholar]
- [44].Ezzell RM, Chafel MM, Matsudaira PT. Differential localization of villin and fimbrin during development of the mouse visceral endoderm and intestinal epithelium. Development. 1989;106:407–19. doi: 10.1242/dev.106.2.407. [DOI] [PubMed] [Google Scholar]
- [45].Shibayama T, Carboni JM, Mooseker MS. Assembly of the intestinal brush border: appearance and redistribution of microvillar core proteins in developing chick enterocytes. J Cell Biol. 1987;105:335–44. doi: 10.1083/jcb.105.1.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fath KR, Burgess DR. Microvillus assembly. Not actin alone. Curr Biol. 1995;5:591–3. doi: 10.1016/s0960-9822(95)00117-5. [DOI] [PubMed] [Google Scholar]
- [47].Heintzelman MB, Mooseker MS. Assembly of the intestinal brush border cytoskeleton. Curr Top Dev Biol. 1992;26:93–122. doi: 10.1016/s0070-2153(08)60442-1. [DOI] [PubMed] [Google Scholar]
- [48].Pinson KI, Dunbar L, Samuelson L, Gumucio DL. Targeted disruption of the mouse villin gene does not impair the morphogenesis of microvilli. Dev Dyn. 1998;211:109–21. doi: 10.1002/(SICI)1097-0177(199801)211:1<109::AID-AJA10>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- [49].Phillips MJ, et al. Abnormalities in villin gene expression and canalicular microvillus structure in progressive cholestatic liver disease of childhood. Lancet. 2003;362:1112–9. doi: 10.1016/S0140-6736(03)14467-4. [DOI] [PubMed] [Google Scholar]
- [50].Banno Y, Nakashima T, Kumada T, Ebisawa K, Nonomura Y, Nozawa Y. Effects of gelsolin on human platelet cytosolic phosphoinositide-phospholipase C isozymes. J Biol Chem. 1992;267:6488–94. [PubMed] [Google Scholar]
- [51].Ferrary E, et al. In vivo, villin is required for Ca(2+)-dependent F-actin disruption in intestinal brush borders. J Cell Biol. 1999;146:819–30. doi: 10.1083/jcb.146.4.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Piccolo E, Innominato PF, Mariggio MA, Maffucci T, Iacobelli S, Falasca M. The mechanism involved in the regulation of phospholipase Cgamma1 activity in cell migration. Oncogene. 2002;21:6520–9. doi: 10.1038/sj.onc.1205821. [DOI] [PubMed] [Google Scholar]
- [53].Cartwright CA, Mamajiwalla S, Skolnick SA, Eckhart W, Burgess DR. Intestinal crypt cells contain higher levels of cytoskeletal-associated pp60c-src protein tyrosine kinase activity than do differentiated enterocytes. Oncogene. 1993;8:1033–9. [PubMed] [Google Scholar]
- [54].Talamonti MS, Roh MS, Curley SA, Gallick GE. Increase in activity and level of pp60c-src in progressive stages of human colorectal cancer. J Clin Invest. 1993;91:53–60. doi: 10.1172/JCI116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Irby RB, et al. Activating SRC mutation in a subset of advanced human colon cancers. Nat Genet. 1999;21:187–90. doi: 10.1038/5971. [DOI] [PubMed] [Google Scholar]
- [56].Aligayer H, Boyd DD, Heiss MM, Abdalla EK, Curley SA, Gallick GE. Activation of Src kinase in primary colorectal carcinoma: an indicator of poor clinical prognosis. Cancer. 2002;94:344–51. doi: 10.1002/cncr.10221. [DOI] [PubMed] [Google Scholar]
- [57].Moissoglu K, Gelman IH. v-Src rescues actin-based cytoskeletal architecture and cell motility and induces enhanced anchorage independence during oncogenic transformation of focal adhesion kinase-null fibroblasts. J Biol Chem. 2003;278:47946–59. doi: 10.1074/jbc.M302720200. [DOI] [PubMed] [Google Scholar]
- [58].Guo HB, Randolph M, Pierce M. Inhibition of a specific N-glycosylation activity results in attenuation of breast carcinoma cell invasiveness-related phenotypes: inhibition of epidermal growth factor-induced dephosphorylation of focal adhesion kinase. J Biol Chem. 2007;282:22150–62. doi: 10.1074/jbc.M611518200. [DOI] [PubMed] [Google Scholar]
- [59].Hatakeyama M. The role of Helicobacter pylori CagA in gastric carcinogenesis. Int J Hematol. 2006;84:301–8. doi: 10.1532/IJH97.06166. [DOI] [PubMed] [Google Scholar]
- [60].Gourlay CW, Ayscough KR. The actin cytoskeleton: a key regulator of apoptosis and ageing? Nat Rev Mol Cell Biol. 2005;6:583–9. doi: 10.1038/nrm1682. [DOI] [PubMed] [Google Scholar]
- [61].Puthalakath H, Villunger A, O'Reilly LA, Beaumont JG, Coultas L, Cheney RE, Huang DC, Strasser A. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 2001;293:1829–32. doi: 10.1126/science.1062257. [DOI] [PubMed] [Google Scholar]
- [62].De Vos KJ, Allan VJ, Grierson AJ, Sheetz MP. Mitochondrial function and actin regulate dynamin-related protein 1-dependent mitochondrial fission. Curr Biol. 2005;15:678–83. doi: 10.1016/j.cub.2005.02.064. [DOI] [PubMed] [Google Scholar]
- [63].Imoto M, Tachibana I, Urrutia R. Identification and functional characterization of a novel human protein highly related to the yeast dynamin-like GTPase Vps1p. J Cell Sci. 1998;111(Pt 10):1341–9. doi: 10.1242/jcs.111.10.1341. [DOI] [PubMed] [Google Scholar]
- [64].Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–25. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- [65].Morris RL, Hollenbeck PJ. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J Cell Biol. 1995;131:1315–26. doi: 10.1083/jcb.131.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Boldogh I, Vojtov N, Karmon S, Pon LA. Interaction between mitochondria and the actin cytoskeleton in budding yeast requires two integral mitochondrial outer membrane proteins, Mmm1p and Mdm10p. J Cell Biol. 1998;141:1371–81. doi: 10.1083/jcb.141.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Shieh DB, Godleski J, Herndon JE, 2nd, Azuma T, Mercer H, Sugarbaker DJ, Kwiatkowski DJ. Cell motility as a prognostic factor in Stage I nonsmall cell lung carcinoma: the role of gelsolin expression. Cancer. 1999;85:47–57. doi: 10.1002/(sici)1097-0142(19990101)85:1<47::aid-cncr7>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- [68].Ahn JS, Jang IS, Kim DI, Cho KA, Park YH, Kim K, Kwak CS, Chul Park S. Aging-associated increase of gelsolin for apoptosis resistance. Biochem Biophys Res Commun. 2003;312:1335–41. doi: 10.1016/j.bbrc.2003.11.061. [DOI] [PubMed] [Google Scholar]
- [69].Kuzumaki N, Fujita H, Tanaka M, Sakai N, Ohtsu M. 1998. pp. 121–131.
- [70].Asch HL, Head K, Dong Y, Natoli F, Winston JS, Connolly JL, Asch BB. Widespread loss of gelsolin in breast cancers of humans, mice, and rats. Cancer Res. 1996;56:4841–5. [PubMed] [Google Scholar]
- [71].Kothakota S, et al. Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science. 1997;278:294–8. doi: 10.1126/science.278.5336.294. [DOI] [PubMed] [Google Scholar]
- [72].Koya RC, Fujita H, Shimizu S, Ohtsu M, Takimoto M, Tsujimoto Y, Kuzumaki N. Gelsolin inhibits apoptosis by blocking mitochondrial membrane potential loss and cytochrome c release. J Biol Chem. 2000;275:15343–9. doi: 10.1074/jbc.275.20.15343. [DOI] [PubMed] [Google Scholar]
- [73].Azuma T, Koths K, Flanagan L, Kwiatkowski D. Gelsolin in complex with phosphatidylinositol 4,5-bisphosphate inhibits caspase-3 and -9 to retard apoptotic progression. J Biol Chem. 2000;275:3761–6. doi: 10.1074/jbc.275.6.3761. [DOI] [PubMed] [Google Scholar]
- [74].Ohtsu M, et al. Inhibition of apoptosis by the actin-regulatory protein gelsolin. Embo J. 1997;16:4650–6. doi: 10.1093/emboj/16.15.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kamada S, Kusano H, Fujita H, Ohtsu M, Koya RC, Kuzumaki N, Tsujimoto Y. A cloning method for caspase substrates that uses the yeast two-hybrid system: cloning of the antiapoptotic gene gelsolin. Proc Natl Acad Sci U S A. 1998;95:8532–7. doi: 10.1073/pnas.95.15.8532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Posey SC, Martelli MP, Azuma T, Kwiatkowski DJ, Bierer BE. Failure of gelsolin overexpression to regulate lymphocyte apoptosis. Blood. 2000;95:3483–8. [PubMed] [Google Scholar]
- [77].Kusano H, Shimizu S, Koya RC, Fujita H, Kamada S, Kuzumaki N, Tsujimoto Y. Human gelsolin prevents apoptosis by inhibiting apoptotic mitochondrial changes via closing VDAC. Oncogene. 2000;19:4807–14. doi: 10.1038/sj.onc.1203868. [DOI] [PubMed] [Google Scholar]
- [78].Harms C, et al. Neuronal gelsolin prevents apoptosis by enhancing actin depolymerization. Mol Cell Neurosci. 2004;25:69–82. doi: 10.1016/j.mcn.2003.09.012. [DOI] [PubMed] [Google Scholar]
- [79].Leifeld L, Fink K, Debska G, Fielenbach M, Schmitz V, Sauerbruch T, Spengler U. Anti-apoptotic function of gelsolin in fas antibody-induced liver failure in vivo. Am J Pathol. 2006;168:778–85. doi: 10.2353/ajpath.2006.050323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Khurana S, Kwiatowski DJ, Gumucio DL, Wang Y. Increased autophagy/apoptosis in villin-gelsolin double knock out mouse. Gastroenterology. 2005;128(4):A–604. Suppl 2. [Google Scholar]
- [81].Sakurai N, Utsumi T. Posttranslational N-myristoylation is required for the anti-apoptotic activity of human tGelsolin, the C-terminal caspase cleavage product of human gelsolin. J Biol Chem. 2006;281:14288–95. doi: 10.1074/jbc.M510338200. [DOI] [PubMed] [Google Scholar]
- [82].Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–5. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Hall PA, Coates PJ, Ansari B, Hopwood D. Regulation of cell number in the mammalian gastrointestinal tract: the importance of apoptosis. J Cell Sci. 1994;107(Pt 12):3569–77. doi: 10.1242/jcs.107.12.3569. [DOI] [PubMed] [Google Scholar]
- [84].Bullen TF, et al. Characterization of epithelial cell shedding from human small intestine. Lab Invest. 2006;86:1052–63. doi: 10.1038/labinvest.3700464. [DOI] [PubMed] [Google Scholar]
- [85].Ellis RE, Yuan JY, Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol. 1991;7:663–98. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- [86].Bedi A, et al. Inhibition of apoptosis during development of colorectal cancer. Cancer Res. 1995;55:1811–6. [PubMed] [Google Scholar]
- [87].Pasricha PJ, et al. The effects of sulindac on colorectal proliferation and apoptosis in familial adenomatous polyposis. Gastroenterology. 1995;109:994–8. doi: 10.1016/0016-5085(95)90411-5. [DOI] [PubMed] [Google Scholar]
- [88].Wang Y, Srinivasan K, Siddiqui MR, George SP, Tomar A, Khurana S. A novel role for villin in intestinal epithelial cell survival and homeostasis. J Biol. Chem. 2007 doi: 10.1074/jbc.M707962200. [DOI] [PubMed] [Google Scholar]
- [89].Kersting S, et al. Antigen transport and cytoskeletal characteristics of a distinct enterocyte population in inflammatory bowel diseases. Am J Pathol. 2004;165:425–37. doi: 10.1016/S0002-9440(10)63308-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Elsasser HP, Kloppel G, Mannherz HG, Flocke K, Kern HF. Immunohistochemical demonstration of villin in the normal human pancreas and in chronic pancreatitis. Histochemistry. 1991;95:383–90. doi: 10.1007/BF00266966. [DOI] [PubMed] [Google Scholar]
- [91].Wang Y, et al. 2007.
- [92].Mannherz HG, Gonsior SM, Gremm D, Wu X, Pope BJ, Weeds AG. Activated cofilin colocalises with Arp2/3 complex in apoptotic blebs during programmed cell death. Eur J Cell Biol. 2005;84:503–15. doi: 10.1016/j.ejcb.2004.11.008. [DOI] [PubMed] [Google Scholar]