

Abstract
Autotaxin (ATX) is a potent tumor cell motogen that can produce lysophosphatidic acid (LPA) from lysophosphatidylcholine. LPA is a lipid mediator that has also been shown to modulate tumor cell invasion. Autotaxin mRNA is expressed at high levels in the intestine. Likewise, LPA2 receptor levels have been shown to be elevated in colon cancers. The molecular mechanism of ATX/LPA-induced increase in intestinal cell migration however, remains poorly understood. Villin is an intestinal and renal epithelial cell specific actin regulatory protein that modifies epithelial cell migration. In this study we demonstrate that both Caco-2 (endogenous villin) and MDCK (exogenous villin) cells, which express primarily LPA2 receptors, show enhanced cell migration in response to ATX/LPA. ATX and LPA treatment results in the rapid formation of lamellipodia and redistribution of villin to these cell surface structures, suggesting a role for villin in regulating this initial event of cell locomotion. The LPA-induced increase in cell migration required activation of c-src kinase and downstream tyrosine phosphorylation of villin by c-src kinase. LPA stimulated cell motility was determined to be insensitive to pertussis toxin, but was regulated by activation of PLC-γ1. Together, our results show that in epithelial cells ATX and LPA act as strong stimulators of cell migration by recruiting PLC-γ1 and villin, both of which participate in the initiation of protrusion.
Keywords: autotaxin, LPA, villin, cell migration, actin, PLC-γ1
Introduction
LPA has emerged as a pleiotropic lipid that regulates among other things, changes in the actin cytoskeleton and cell migration which implicate LPA as a mediator of tumor cell invasion [1]. Recent studies have suggested that levels of LPA2 receptors are elevated in colon and gastric cancers [2]. Further, most human colon cancer and stomach cancer cell lines predominantly express LPA2 receptors [3]. Likewise, LPA has also been reported to enhance intestinal cell migration [4] and to ameliorate intestinal injury in a dinitrobenzene model of colitis in rats [5]. LPA interacts with at least five different heptahelical receptors (LPA1-5) which in turn activate Gi, Gq, Gs/h and G12/13 proteins to elicit multiple biological responses. However, the specific functions and cellular targets regulated by these LPA receptors in epithelial cells are not well characterized. Further, few studies have examined the LPA receptor subtype involved in LPA-induced cell motility in intestinal and renal epithelial cells. More recently, lysophospholipase D, a key enzyme in the production of LPA in human blood, was shown to be identical to autotaxin (ATX), a well-established tumor motility factor [6]. While the role of LPA/ATX in cell migration is undisputed, the molecular mechanisms regulating this function of LPA/ATX in epithelial cells remain to be examined.
Villin is an epithelial cell specific actin binding protein that regulates actin reorganization, cell morphology, epithelial-to-mesenchymal transition, cell invasion as well as cell migration indicating the significance of this protein to intestinal and renal cell function [7, 8]. The expression of villin gene is maintained in carcinomas derived from intestinal and renal epithelial cells and villin is also expressed in other adenocarcinomas even though it is absent from normal tissue such as Barrett's metaplasia, esophageal adenocarcinomas, gastric cardia adenocarcinomas and in chronic atrophic gastritis [9, 10]. Chronic Helicobacter pylori infection has also been shown to induce endogenous villin expression in the stomach [11]. These and similar studies, suggest that villin may be an important marker of preneoplastic lesions that develop in the gastrointestinal tract in response to chronic injury. In this study, we examined the effects of LPA/ATX on villin-induced cell migration. Understanding the ability of LPA/ATX to enhance epithelial cell motility provides a new dimension of knowledge linking lipid mediators, and alimentary LPA in particular, to the dysregulation of signaling in carcinomas.
Materials and Methods
Materials
1-oleoyl-LPA (18:1) was purchased from Avanti Polar Lipids Inc. Caco-2 cells clone C2BBe1 were purchased from American Type Culture Collection, Rockville, MD. MDCK Tet-Off cells were a kind gift from Dr. Keith E. Mostov (University of California-San Francisco). mCherry cloned in pRSET B was a kind gift from Dr. Roger Y Tsien (University of California at San Diego, CA). Autotaxin was a kind gift from Dr. Timothy Clair (National Cancer Institute, MD). Rac 1 inhibitor NSC23766 was purchased from Calbiochem. Autotaxin inhibitor, 3-carba analog of cyclic phosphatidic acid (16:1) has been described previously [12]. Antibodies against the following proteins were used: villin and PLC-γ1 (Transduction Laboratories); phospho-tyrosine (clone PY-20; ICN); influenza A virus haemagglutinin (HA) tag (Roche) and; c-src (UBI). Phospho-villin antibody (VP-70782) has been described previously [8]. pEGFP-actin was purchased from BD Biosciences. Pertussis toxin, U-73122, U-73343 were purchased from Calbiochem. Control and PLC-γ1 specific siRNA were purchased from Dhramacon-Upstate.
Cell culture and treatment
Caco-2 cells were cultured as described before [13]. MDCK Tet-Off cells stably transfected with full-length or mutant villin cDNA were cultured in the absence or presence of doxycycline (10 ng/ml) as described previously [14]. Cells were treated with LPA (2 μM) or ATX (1 μg/ml). Alternatively, prior to LPA treatment, cells were incubated with 2 μg/ml pertussis toxin (PTX), 10 μg/ml U-73122 or 10 μg/ml U-73343. For downregulation of PLC-γ1, cells were transfected with the RNA oligonucleotides (200 nM) using Lipofectamine 2000 reagent. Western blot analysis was performed to assess gene silencing. All experiments were performed 48 h post-transfection.
Quantitative RT-PCR
Quantitative PCR reactions were performed on 0.5 μl of the cDNA mix with 300 nmol of each primer in a final volume of 25 μl of 2 X RT2 Real-Time SYBR Green/ROX Master Mix. The following primer pairs were used for identification of LPA1-5 receptors in MDCK cells: LPA1: (forward) 5'-GTCTTCTGGGCCATTTTCAA-3' and (reverse) 5'-TCATAGTCCTCT-GGCGAACA-3'; LPA2: (forward) 5'-GAGGCCAACTCACTGGTCA-3' and (reverse) 5'-GGCGCATCTCAGCATCTC-3'; LPA3: (forward) 5'-GAAGCTAATGAAGACGGTGATGA-3' and (reverse) 5'-AGCAGGAACCACCTTTTCAC-3'; LPA4: (forward) 5'-TCTGGATCCTAGTCCTCAGTGG-3' and (reverse) 5'-CCAGACACGTTTCCCAAACG-3'; LPA5: (forward) 5'-ATCTTCCTGCTGTGCTTCGT-3' and (reverse) 5'-CAGCAGCACCATCACCAT-3'; S1P1 (forward) 5'-GCCACCACCTACAAGCTCAC-3' and (reverse) 5'-CTCCCGTTGTGGACGACATGGAGAA-3'; β-actin: (forward) 5'-AACTGGGACGACATGGAGAA-3' and (reverse) 5'-GTACATGGCTGGGGGTGTTG-3'. The transcript number of dog β-actin was quantified as an internal RNA control, and each sample was normalized on the basis of its β-actin content. The relative gene expression level of each gene was then normalized to LPA1 gene.
Transfection of MDCK Tet-Off cells with full-length and mutant villin cDNA
Superenhanced yellow fluorescent protein (SEYFP)-tagged full-length villin protein (SEYFP/VIL) and SEYPF-tagged phosphorylation site mutant villin protein (SEYPF/AYFM) were cloned in pTRE-HA as described previously [8, 15]. mCherry-tagged full-length villin (mCherry/VIL) protein was prepared by cloning the mCherry cDNA into the Sal I site of full-length villin cloned in pTRE-HA to generate amino-terminal tagged mCherry/VIL using the following primers: 5'-GATAGCGTCGACGATGGTGAGCAAGGGCG-3' (forward) and 5'-GATAGCGTCGACGCTGCTCCTGCCTTGTACAGCTCGTCC-3'(reverse). Full-length SEYFP/VIL, mCherry/VIL as well as SEYFP/AYFM were stably transfected in MDCK Tet-Off cells as described previously [14, 15]. Alternatively, MDCK Tet-Off cells were stably transfected with EGFP-tagged actin (EGFP/actin) and transiently transfected with mCherry/VIL.
Adenoviral infection
Recombinant adenoviral vectors encoding either vector alone (Ad EGFP) or dominant-negative c-src kinase (Ad DN cSrc) have been described elsewhere [8]. MDCK cells were either mock infected with Ad EGFP or infected with Ad DN cSrc at a multiplicity of infection of 100 using standard protocol. Cell lysates were analyzed by Western analysis for c-src kinase expression. Caco-2 cells were infected with recombinant adenovirus expressing either vector alone or the villin phosphorylation site mutant, VIL/AYFM as described previously [15, 16]. VIL/AYFM was expressed as an HA-tagged protein, which allowed us to determine its expression in Caco-2 cells that also endogenously express villin.
Cell motility assay
Cell migration was measured as described previously with slight modification, namely wounds were generated using a sharp blade [8]. MDCK Tet-Off cells transfected with wild-type or mutant villin proteins were seeded in six-well plates and cultured in the absence (VIL +) or presence (VIL −) of doxycycline. Data were normalized to control values (which were set as 100) and expressed as percent change in migration compared to control. For Caco-2 cells, control refers to cell migration at 24 h post-wounding while for MDCK cells, control refers to villin-null cell migration 7 h post-wounding, treated with vehicle alone or transfected/infected with vector alone. Caco-2 cell migration was measured in the presence of mitomycin C (50 μg/ml). Confluent monolayers were scraped with a sharp blade across the diameter of the well to produce wounds of ∼ 1 mm width. Cells were rinsed to remove cellular debris, and images were obtained at the initial time of wounding and at various times up to 24 h post-wounding. Images were collected with a Nikon Eclipse TE2000-U inverted microscope equipped with a CoolSnap ES charge-coupled device (CCD) camera, an Optiscan motorized stage system and an Intel Pentium IV computer with Metamorph image analysis software. Images were collected by programming the X, Y, and Z coordinates of each wound location, allowing the stage to return to the precise location of the original wound. Comparisons between mean values were made using one-way repeated-measures analysis of variance and Tukey's modified t-test (Benferroni criteria).
Time-lapse microscopy
For time-lapse microscopy, cells were incubated on 35-mm glass bottom dishes. LPA (2 μM) or ATX (1 μg/ml) were added to the cells 5 min before time-lapse images were captured. Time-lapse images were acquired with a 40 × objective on a confocal microscope (LSM 5 PASCAL, Carl Zeiss, Thornwood, NY). Images were captured every 2 min for a maximum of 60 min.
Measurement of lamellipodial protrusion rate
Lamellipodia were characterized as thin regions 3-10 μm wide located at the cell margin. Using time-lapse imaging, lamellipodia extension was measured as the increase in total cell area after LPA (2 μM) treatment essentially as described before [15]. The mean relative area was calculated using Metamorph software and plotted as a function of time. Approximately 10 cells were examined and for all these cells the rate of lamellipodial extension remained constant during the 30 min of observation.
Immunoprecipitation and Western blot Analysis
MDCK Tet-Off cells transfected with full-length villin (VIL/FL) cultured in the absence or presence of doxycycline as well as Caco-2 cells which express endogenous villin, were treated with ATX (1 μg/ml) or LPA (2 μM). Cell extracts were obtained using a buffer containing 1% Triton X-100, 20 mM HEPES, 150 mM NaCl, pH 7.2. Tyrosine phosphorylated villin was immunoprecipitated from the detergent-soluble extracts and detected by Western analysis as described previously [8].
Results
Autotaxin and LPA enhance intestinal and renal epithelial cell migration
ATX is widely expressed with highest mRNA levels detected in brain, placenta, ovary and intestine [17-19]. Even though ATX mRNA is expressed at very high levels in the intestine, there are no studies so far that have examined the effects of ATX on intestinal cell motility. Likewise there are very few studies that have examined the molecular basis of LPA-induced increase in intestinal cell migration. We and others have previously reported that the epithelial cell specific cytoskeletal protein, villin, is a major regulator of intestinal cell migration [7, 8]. The role of the microfilament structure in cell locomotion as well as the motogenic effects of LPA are undisputed, hence we reasoned that a logical step would be to examine the role of villin in LPA/ATX-induced cell migration. For these studies we used the human adenocarcinoma cell line Caco-2, which endogenously express villin and the non-transformed dog kidney cell line MDCK Tet-Off which were stably transfected with full-length human villin cDNA (Fig. 1A). MDCK cells transfected with villin were cultured in the presence of doxycycline (DOX), which inhibits villin expression or in the absence of doxycycline to maintain villin expression (Fig. 1A). It has previously been reported that Caco-2 cells express primarily LPA2 receptors [3]. To the best of our knowledge there are no studies done on the LPA receptor expression in MDCK cells. Using LPA1-5 receptor primers, a control primer for sphingosine 1-phosphate 1 receptor (S1P1) and β-actin as a house-keeping gene, we determined using real-time RT-PCR that MDCK cells express primarily LPA2 receptors (Fig. 1B). Likewise, Caco-2 cells were found to have insignificant mRNA levels for LPA5 receptor (data not shown). Based on these data we conclude that both cell lines used in this study express primarily LPA2 receptors.
Fig. 1. Regulation of intestinal cell migration by ATX and LPA.

(A) Expression of villin in Caco-2 and MDCK Tet-Off cells. MDCK Tet-Off cells were stably transfected with human villin cDNA. Cells cultured in the presence of doxycycline (DOX) (10 ng/ml) do not express villin while cells cultured in the absence of doxycycline express villin. Caco-2 cells endogenously express villin. (B) Real-time RT-PCR was used to determine that MDCK cells express primarily LPA2 receptors. The transcript number of dog β-actin was quantified as an internal RNA control and each sample was normalized with this control. The relative gene expression level was then normalized to LPA1 gene. Additional controls included S1P1. (C) Villin enhances the effects of LPA on MDCK cell migration. MDCK villin-null and villin expressing cells were used to measure cell migration in the absence or presence of LPA (2 μM). Data are normalized to the control values (which was set as 100) and expressed as percent change in migration compared to control. Control refers to cell migration 7-h post-wounding in villin-null cells in the absence of LPA. The error bars are the measured S.E.M. and asterisk (*)and cross (†) denote statistically significant values (p<0.01, n=12 and p<0.001, n=12, respectively) compared to control cells. (D) LPA increases Caco-2 cell migration. Cell migration was measured in the absence or presence of LPA (2 μM). Data are normalized to the control values (which were set as 100) and expressed as percent change in migration compared to control. Control refers to cell migration 24-h post-wounding in the absence of LPA. The error bars are the measured S.E.M. and the asterisk (*) denotes statistically significant values (p<0.001, n=12) compared to control cells. (E) Villin regulates ATX-induced MDCK cell migration. MDCK Tet-Off cells cultured in the absence (VIL +) or presence (VIL −) of doxycycline were treated with ATX (1 μg/ml). The error bars are the measured S.E.M. and asterisk (*)and (†) denotes statistically significant values (p<0.01, n=6 and p<0.001, n=6, respectively) compared with villin-null cells in the absence of ATX. (F) Caco-2 cell migration was measured in the absence or presence of ATX (1 μg/ml). Data are normalized to the control values (which were set as 100) and expressed as percent change in migration compared to control. Control refers to cell migration 24 h post-wounding in the absence of ATX. The error bars are the measured S.E.M. and the asterisk (*) denotes statistically significant values (p<0.01, n=6) compared to control cells. (G) Caco-2 cell migration was measured in the absence or presence of ATX (1μg/ml) as well as in the absence or presence of ATX inhibitor, 2CCPA (16:1) (10 μM). Data were normalized to the control values (which were set as 100) and expressed as percent change in migration compared to control. Control refers to cell migration 24 h post-wounding in the absence of ATX or 2CCPA. The error bars are the measured S.E.M. and the asterisk (*) denotes statistically significant values (p<0.001, n=6) compared to control cells and cross (†) denotes statistically significant values (p<0.01, n=6) compared to ATX treated Caco-2 cells.
An in vitro wound-healing assay was used to determine the role of villin in ATX/LPA-induced epithelial cell migration. Cells were maintained in low serum (1.0% fetal bovine serum) and mitomycin (2 μg/ml) to inhibit cell proliferation and to maintain cell viability. LPA treatment increased both MDCK and Caco-2 cell migration. As shown in Fig. 1C, treatment of MDCK villin-null cells with LPA (2 μM) increased cell migration (34.28 ± 2.12 %, p<0.01, n=12 compared to untreated villin-null cells). In the presence of villin, LPA treatment further enhanced cell migration (78.0 ± 3.8%, p<0.001, n=12 compared to untreated villin-null cells) (Fig. 1C). These data then demonstrate that while villin and LPA both increase basal cell migration to comparable levels, the effect of LPA on cell migration are significantly enhanced in the presence of villin. Similar to the effects of LPA on MDCK cells, Caco-2 cells demonstrate enhanced cell migration in the presence of LPA (51% increase, p<0.001 , n=12)(Fig. 1D). Thus, epithelial cells that express villin endogenously (Caco-2) or exogenously (MDCK) migrate significantly faster than villin-null cells in the presence of LPA. It may be noted that while the mitogenic effects of LPA have been demonstrated in Caco-2 cells, no studies have examined the motogenic effects of LPA in Caco-2 cells. In concert with the LPA data, treatment of villin-null MDCK cells with ATX (1 μg/ml) significantly enhanced cell migration (21.0 ± 1.85% increase compared to villin-null cells in the absence of ATX, p<0.01, n=6; Fig. 1E). Expression of VIL/FL in MDCK cells enhanced cell migration rates, consistent with our previous observations (35.1 ±1.42% increase compared to untreated villin-null cells, p<0.01, n=6) [8]. Treatment of MDCK cells expressing full-length villin with ATX further enhanced cell migration (62.0 ± 2.8 %, p<0.001, n=6 compared to untreated villin-null cells). These data demonstrate that villin increases ATX-induced cell motility thus, acting as a stimulator of ATX-mediated cell migration. Likewise, treatment of Caco-2 cells with ATX enhanced cell migration (21.0 ± 1.52, p<0.001, n=6) (Fig. 1F). The effects of ATX were lower than LPA in these studies, because we measured cell motility in the presence of exogenous ATX but in the absence of exogenous lysophosphatidylcholine (LPC). In addition treatment of Caco-2 cells with the ATX inhibitor, 2CCPA (16:1) (10 μM) significantly inhibited ATX-induced cell migration [12](Fig. 1G). While 2CCPA did not completely block basal cell migration, these data support our hypothesis that at least a significant part of basal colon adenocarcinoma cell migration is regulated by ATX.
LPA and ATX enhance lamellipodia formation
To further characterize LPA-induced increase in epithelial cell migration, we elected to measure cell migration in live cells using time-lapse imaging. For these studies, we used MDCK Tet-Off cells stably transfected with SEYFP-tagged full-length human villin (SEYFP/VIL) or EGFP-tagged actin (EGFP/actin). Time-lapse images of SEYFP/VIL cells revealed that LPA enhanced the rate of lamellipodial extension in cells expressing villin within the first 30 min of LPA treatment (Fig. 2A (c)). Villin expressing cells treated with LPA formed large cell surface lamellipods which was accompanied by the rapid distribution of villin to these cell surface structures. This redistribution of villin was associated with a significant increase in the size of the lamellipodia and forward extension of the cell body, both of which are strong indicators of increased cell migration. Cells expressing EGFP-actin showed no lamellipodia formation in the absence of LPA (data not shown) or during this initial period of LPA treatment (Fig. 2A (b)). We quantified these morphological changes by measuring the rate of lamellipodial protrusion in MDCK cells expressing SEYFP/VIL as well as EGFP/actin. LPA-treated MDCK cells expressing SEYFP/VIL demonstrated a significant increase in the rate of lamellipodia formation (0.5 ± 0.008 μm/min; p<0.001 n=10) compared to untreated MDCK cells expressing villin (0.05 ± 0.004 μm/min) or LPA-treated MDCK cells expressing EGFP/actin (0.1 ± 0.003 μm/min). These data suggest that optimal lamellipodia formation and maximum increase in cell migration following LPA treatment occurred in cells expressing full-length villin protein. Likewise, treatment of SEYFP/VIL cells with ATX also resulted in rapid lamellipodia formation within 5 min of ATX treatment (Fig. 2B (b)), while treatment of MDCK cells transfected with EGFP/actin with ATX resulted in no cell protrusion during this initial period of treatment (Fig. 2B(a)). Our studies demonstrate that in the presence of villin, LPA/ATX treatment causes rapid lamellipodia formation (in less than 5 min). To further confirm these observations we performed the reverse experiment, namely MDCK cells stably transfected with EGFP/actin which do not form ruffles or lamellipodia in response to LPA treatment were transiently transfected with mCherry-tagged villin. As shown in Fig. 2C, expression of villin in these cells induced rapid membrane ruffles and lamellipodia formation in response to LPA treatment. Further these data demonstrate that both villin and F-actin are redistributed to the developing lamellipodia in response to LPA treatment.
Fig. 2. ATX and LPA cause rapid lamellipodia formation in cells expressing full-length villin.


(A) MDCK Tet-Off cells expressing SEYFP-tagged full-length villin (SEYFP/VIL (a; c)) or EGFP tagged β-actin (EGFP/actin; (b)) were treated without (a) or with (b, c) LPA (2 μM) and time-lapse images were recorded 5 min after the addition of LPA for a total of 30 min. Bars, 10 μM. Lower panel shows lamellipodia protrusion rates. Average velocity of lamellipod formation is denoted in μm/min. The error bars are the measured S.E.M. and the asterisk denotes statistically significant values (p<0.001, n=10, compared with SEYFP/VIL in the absence of LPA. (B) MDCK cells expressing SEYFP/VIL (b) or EGFP/actin (a) were treated with ATX (1 μg/ml) and time-lapse images were recorded 5 min after the addition of ATX for a total of 30 min. Bars, 10 μM. (C) MDCK Tet-Off cells stably transfected with EGFP/actin were transiently transfected with mCherry/VIL and time lapse images were recorded before and 5 min after treatment with LPA for a total of 30 min. Panel (a) shows expression of EGFP/actin, panel (b) shows expression of mCherry/VIL and panel (c) shows merged images of EGFP/actin and mCherry/VIL. Bar, 10 μM.
Tyrosine phosphorylation of villin is required for LPA/ATX-induced increase in epithelial cell migration
Activation of c-src kinase downstream of LPA receptor activation has been reported [20]. We have previously show that tyrosine phosphorylation of villin by c-src kinase regulates villin's function in cell migration [8]. We have also identified ten tyrosine phosphorylation sites in human villin that regulate villin-induced cell migration [8, 15]. We now report that both LPA and ATX result in the tyrosine phosphorylation of villin in MDCK as well as Caco-2 cells, suggesting that the effects of LPA/ATX on villin phosphorylation are not cell-type specific (Fig. 3A). To further characterize the role of tyrosine phosphorylated villin in LPA-induced cell migration, we used two approaches: first, we elected to inhibit the tyrosine phosphorylation of villin by down-regulating the endogenous c-src kinase, by either overexpressing dominant-negative c-src kinase or with the pharmacological inhibitor of c-src, PP2. The second approach was to use a phosphorylation site mutant of villin (all ten identified phosphorylation sites mutated to phenylalanine, SEYFP/AYFM). MDCK cells expressing SEYFP/VIL were infected with adenoviral constructs expressing either EGFP-tagged vector (Ad EGFP) or dominant-negative c-src (Ad DN c-src) (Fig. 3B). Expression of control vector had no effect on LPA's ability to form lamellipodia in MDCK cells expressing full-length villin (Ad EGFP SEYFP/VIL; Fig. 3C(a)). In contrast, overexpression of dominant-negative c-src significantly inhibited the ability of LPA to induce lamellipodia formation (Ad DN c-Src SEYFP/VIL; Fig. 3C(b)). There was a significant decrease in the rate of lamellipodial extension in cells expressing SEYFP/VIL and dominant-negative c-src compared to MDCK cells expressing SEYFP/VIL and infected with EGFP-vector (0.074 ± 0.004 μm/min in cells infected with dominant negative c-src versus 0.36 ±0.038 μm/min in cells infected with EGFP-vector; p<0.001, n=10). To correlate these changes with villin phosphorylation, we also examined the effect of villin phosphorylation site mutant SEYFP/AYFM on LPA-induced cell migration. MDCK cells stably transfected with the villin phosphorylation site mutant, SEYFP/AYFM showed no lamellipodia formation in response to LPA treatment and a significant inhibition of lamellipodial extension rates (0.025 ± 0.012 μm/min; p<0.001, n=10) (Fig. 3C(c)). Treatment of MDCK cells without or with the src-kinase inhibitor, PP2 (10 nM) confirmed our observation that c-src kinase activation is required for LPA induced increase in villin (+) cell migration (Fig. 3D). The inactive analog, PP3 had no effect on basal or LPA-induced cell migration (data not shown). PP2 had no effect on basal cell migration rates in untransfected MDCK cells. Interestingly, PP2 reversed both villin-induced increase in cell migration (to levels comparable to those seen with villin-null cells) as well as LPA-induced increase in migration in villin-expressing cells. This is consistent with our previous report in which we demonstrated that villin's function in basal cell migration requires tyrosine phosphorylation by c-src kinase [8]. Expression of villin mutant VIL/AYFM confirmed the significance of phosphoryable villin in MDCK (Fig. 3E) and Caco-2 cell (Fig. 3F) migration in response to LPA treatment. Overexpression of VIL/AYFM mutant in Caco-2 cells inhibited both basal as well as LPA-induced cell migration. Similarly, comparison of MDCK cells stably transfected with full-length villin or VIL/AYFM demonstrates that MDCK cells expressing villin mutant migrate at rates similar to villin-null cells.
Fig. 3. Tyrosine phosphorylation of villin is required for LPA-induced increase in epithelial cell migration.
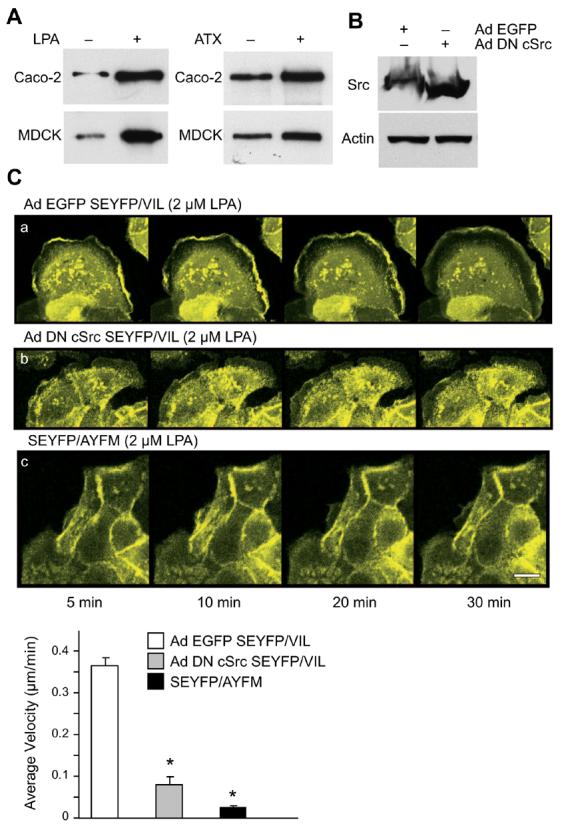

(A) Cell extracts from MDCK Tet-Off cells stably transfected with full-length villin as well as Caco-2 cell extracts were immunoprecipitated with phospho-villin antibody (VP-70782) and Western analysis was done with a phosphotyrosine antibody (PY-20) as described in Methods. (B) MDCK cells expressing full-length villin were infected with recombinant adenovirus expressing either EGFP (Ad EGFP) or dominant negative c-src (Ad DN c-src) for 4 h at a multiplicity of infection of 100. Virus-containing media were then removed and cells were cultured for an additional 18 h to allow for expression of transgenic proteins. This Western blot is representative of three experiments with similar results. A quantitative Western with actin was run in parallel. (C) MDCK cells expressing SEYFP/VIL (a, b) were treated with LPA (2 μM) in the presence of dominant negative c-src kinase (Ad DN c-src; (b)) or EGFP-tagged vector (Ad EGFP; (a)). MDCK cells expressing SEYFP/AYFM (c) were also treated with LPA (2 μM). Time-lapse images were collected essentially as described under Fig. 2. Lower panel shows lamellipodia protrusion rates. Average velocity of lamellipod formation is denoted in μm/min. The error bars are the measured S.E.M. and the asterisk (*) denotes statistically significant values (p<0.001, n=10, compared with SEYFP/VIL infected with Ad EGFP. (D) MDCK villin (−) and villin (+) cells n were used to measure cell migration in the absence or presence of LPA (2 μM) and the src kinase inhibitor, PP2 (10 nM). Data are normalized to the control values (which was set as 100) and expressed as percent change in migration compared to control. Control refers to cell migration 7-h post-wounding in villin (−) cells in the absence of LPA and PP2. The error bars are the measured S.E.M. and asterisk (*) denotes statistically significant values compared to untreated control cells (p<0.01, n=12) while (†) denotes statistically significant values compared to LPA-treated villin (+) cells (p<0.001, n=12). (E) MDCK cells stably transfected with wild-type villin (VIL/FL) and villin mutant VIL/AYFM were treated without or with LPA (2 μM) and cell migration measured essentially as described above. The error bars are the measured S.E.M. and asterisk (*) denotes statistically significant values compared to control cells (untreated villin-null cells; p<0.01, n=12) and cross (†) denotes statistically significant values (p<0.001, n=12) compared to untreated villin-null cells. (F) Caco-2 cells were infected with adenovirus to overexpress HA-tagged mutant villin, VIL/AYFM. The expression of the mutant villin was determined by Western analysis using HA monoclonal antibodies. Cell migration was measured in Mock infected and VIL/AYFM infected Caco-2 cells in the absence or presence of LPA. The error bars are the measured S.E.M. and asterisk (*) denotes statistically significant values compared to control cells (Mock infected (VIL/AYFM (−)), untreated cells; p<0.01, n=12) and cross (†) denotes statistically significant values (p<0.001, n=12) compared to LPA treated Caco-2 cells.
LPA-induced epithelial cell migration is regulated by activation of PLC-γ1
LPA appears to work through different G protein-coupled cascades to activate the GTPase RhoA or PLC, both of which are known to remodel the actin cytoskeleton during cell migration [21, 22]. Based on these observations, we elected to characterize the villin-induced increase in cell migration in response to LPA treatment in the absence or presence of PLC-γ1 inhibitors U-73122 and its inactive analog, U-73343 and a Gαi inhibitor, pertussis toxin (PTX). As shown in Fig. 4A, LPA treatment enhanced cell migration in MDCK cells expressing full-length villin (43.35 % compared to untreated control cells, n=12, p<0.01). PTX (2 μg/ml) pretreated cells migrated as effectively as LPA treated VIL/FL cells (39.43 % increase in cell migration, n=12, p<0.01 compared to untreated control cells). However, pretreatment of MDCK cells expressing VIL/FL with the PLC-γ1 inhibitor, U-73122 (10 μg/ml) resulted in a statistically significant inhibition of cell migration (p<0.001, n=12 compared to LPA-treated VIL/FL cells). While cells treated with the inactive analog, U-73343 did not inhibit LPA-stimulated villin-induced cell migration (40.0% increase compared to untreated control cells, p<0.01, n=12 compared to untreated villin expressing cells). Additional studies were done using the Rac 1 inhibitor, NSC23766 and overexpression of constitutively active Rac1 (Rac L61 mutant), neither of which had any effect on basal, LPA or ATX induced cell migration in either villin-null or villin expressing MDCK cells (data not shown).
Fig. 4. LPA-induced epithelial cell migration is regulated by PLC-γ1.


(A) MDCK Tet-Off cells stably transfected with villin were used to measure cell migration in the absence or presence of LPA (2 μM), pertussis toxin (2 μg/ml), PLC-γ1 inhibitor U-73122 (10 μg/ml) or its inactive analog, U-73343 (10 μg/ml). Data are normalized to the control values (which was set as 100) and expressed as percent change in migration compared to control. Control refers to cell migration 7-h post-wounding in the absence of LPA. The error bars are the measured S.E.M. and asterisk (*) denotes statistically significant values compared to control untreated cells (p<0.01, n=12) while (†) denotes statistically significant values compared to cells in the presence of LPA (p<0.001, n=12). (B) MDCK Tet-Off cells were stably transfected with SEYFP/VIL and were pre-treated with either PTX (2 μg/ml; (a)), U-73343 (10 μg/ml; (b)) or U-73122 (10 μg/ml; (c)) followed by LPA treatment (2 μM) and time-lapse images were recorded 5 min after the addition of LPA for a total of 30 min. Bars, 10 μM. Lower panel shows lamellipodia protrusion rates. Average velocity of lamellipod formation is denoted in μm/min. The error bars are the measured S.E.M. and the asterisk denotes statistically significant values (p<0.01, n=10, compared with SEYFP/VIL in the presence of PTX). (C) MDCK villin expressing cells were transfected with PLC-γ1 or control siRNA. Gene silencing was determined 48 h post-transfection. Using Western analysis it was determined that greater than 90% of endogenous PLC-γ1 expression was downregulated by PLC-γ1 siRNA but not control siRNA (determined by densitometric analysis; n=3). Cell migration was measured in villin expressing untransfected, transfected with control siRNA and transfected with PLC-γ1 siRNA MDCK cells in the absence or presence of LPA (2 μM). Data are normalized to the control values (which was set as 100) and expressed as percent change in migration compared to control. Control refers to cell migration 7-h post-wounding in villin expressing cells transfected with control siRNA in the absence of LPA. The error bars are the measured S.E.M. and asterisk (*) denotes statistically significant values compared to control untreated cells (p<0.01, n=12) while (†) denotes statistically significant values compared to control siRNA transfected villin (+) cells in the presence of LPA (p<0.001, n=12).
Time-lapse images demonstrated that PLC-γ1 regulated LPA-induced cell motility by promoting the rapid lamellipodia formation in villin expressing cells. MDCK cells expressing SEYFP/FL and treated with LPA in the presence of PTX had no effect on the rate of lamellipodia formation. These cells extended membrane ruffles and lamellipodia (0.56 ± 0.038 μm/min) (Fig. 4B, 4C). Likewise, cells expressing SEYFP-tagged villin formed rapid lamellipodia in response to LPA treatment and U-73343 had no statistically significant effect on the rate of lamellipodia formation (0.413 ± 0.008 μm/min). In contrast, cells treated with the PLC-γ1 inhibitor, U-73122 showed a significant reduction in the rate of lamellipodia formation and extension (0.07 ± 0.012 μm/min; Fig. 3B, 3C). Additional studies were done using PLC-γ1 specific siRNA and a control siRNA transiently transfected in MDCK cells expressing full-length villin (Fig. 4C). Downregulation of PLC-γ1 (approximately 90%, determined by densitometric analysis) inhibited LPA-induced increase in villin (+) MDCK cells while the control siRNA had no effect on LPA-induced cell migration in villin (+) cells. Collectively, these data demonstrate that villin and PLC-γ1, in concert, participate in the early events associated with lamellipodia formation in response to LPA/ATX treatment, thus determining the increased rate of cell migration.
Discussion
Despite the fact that there are several lines of evidences that convincingly link LPA/ATX to more aggressive and invasive carcinomas as well as cell migration, few studies have examined the effects of LPA/ATX on intestinal cell migration and little is known about the intracellular signaling mechanisms of ATX and LPA in intestinal epithelial cells. In this study we demonstrate that ATX/LPA enhanced cell migration in both Caco-2 cells that endogenously express villin and in MDCK cells that ectopically express villin. While both villin as well as LPA/ATX enhanced basal cell migration to a similar rate (∼35-43% increase compared to untreated villin-null cells), cells expressing villin and treated with either LPA or ATX on the other hand demonstrated significantly higher rates of migration (∼65-78% increase compared to untreated villin-null cells). The effects of LPA/ATX on MDCK cells expressing villin was significantly higher than in villin-null cells, suggesting that villin enhanced the effects of LPA/ATX on epithelial cell motility. The effects of LPA/ATX on Caco-2 cells were modestly lower than the effect of these motogens on MDCK cells. We believe this is consistent with the current thinking that tumor cells migrate faster than non-transformed cells and because Caco-2 are transformed colon adenocarcinoma cells, their basal rate of migration may be higher than the non-transformed MDCK cells. The role of villin in ATX and LPA-induced cell migration was further supported by observation that cells expressing mutant villin protein failed to demonstrate enhanced cell migration in response to either LPA or ATX treatment in both Caco-2 and MDCK cells. An important step in cell locomotion is the development of a polarized phenotype with the formation of a protrusion in the direction of cell movement. During cell locomotion extracellular signals are communicated to the microfilament structure to generate a protrusion in the direction of the haptotactic and/or chemotactic signal. It has been suggested that rapid actin polymerization during this first step of cell migration, can determine the rate of cell migration [23]. Malignant tumor cells often demonstrate excessive cell protrusive activity [24]. Previous studies have demonstrated that LPA induces rapid lamellipodia formation with higher persistence time and slower retraction [25]. Our studies then suggest that villin in response to LPA/ATX activation could determine the direction of cell protrusion as well as the generation of this early increase in barbed end formation.
It has been shown previously that LPA induces a transient increase in Src kinase activity [26] and rapid tyrosine phosphorylation of several proteins involved in cell migration and adhesion [27]. It is also known that in late-stage ovarian cancers, for instance, that both the levels of LPA as well as Src family kinases are elevated [28]. In this study we demonstrate that in intestinal cells, at least one of the substrates of c-src kinase activation in response to LPA/ATX treatment is an actin-binding protein that regulates both cell migration and cell invasion, namely, villin. Overexpression of dominant negative c-src as well as treatment of cell with the pharmacological inhibitor of c-src, PP2, prevented villin-induced increase in cell migration in response to LPA treatment. These data were confirmed by demonstrating that the phosphorylation site mutant of villin, VIL/AYFM, failed to respond to LPA.
Our studies show that LPA induced increase in intestinal cell migration was pertussis toxin insensitive suggesting that Rho GTPases do not play a significant role in the initial development of protrusion in intestinal epithelial cells. This is consistent with previous reports which have demonstrated that the effects of Rho family proteins on cell migration depend on the cell type and environment specific migration [29, 30]. Studies done with epithelial cells have reported either no effect of RhoA on cell migration or have reported inhibition of epithelial cell migration by RhoA [31, 32]. Likewise, we found no effect of Rac inhibition or of overexpression of constitutively active Rac 1 on either basal or LPA-induced epithelial cell migration. In contrast, LPA induced cell migration was efficiently and significantly inhibited by the PLC-γ1 inhibitor U-73122 and by downregulation of endogenous PLC-γ1 using siRNA. There is considerable data supporting a role for PLC-γ activation in promoting rapid lamellipodium extension in response to growth factor treatment and for orientation of cell movement during chemotaxis thus, suggesting that PLC-γ determines the directionality of the protrusion [33]. It has previously been shown that PLC-γ is an important regulator of both growth factor mediated cell motility as well as for invasiveness of cancer cells [34, 35]. We suggest that ATX/LPA induced cell motility is determined by localized signaling events which recruit villin and PLC-γ1 to extend protrusions, thus regulating the initial polarization of a migrating cell.
It may be noted that colon cancer cell lines including Caco-2, as well as MDCK cells used in this study both express primarily LPA2 receptors. In this study we confirm previous suggestions that unlike LPA1, LPA2 receptor mediated cell migration is induced by activation of PLC-γ1. It has also been noted previously that LPA2 does not contribute significantly to the activation of RhoA in the absence of LPA1 expression, consistent with our findings [36]. LPA2 expression is increased in several cancers including colon, uterus, rectum, testis and lung, suggesting that increased levels of LPA2 may be a commonly occurring effect in cancers [2, 3]. It is also noteworthy that several sequence variants in the 3' URT of human LPA2 gene that may lead to a loss of normal regulation of LPA2 gene expression have been found in cancerous cells [37].
In summary, in this study we identify an actin cytoskeletal protein that is uniquely expressed in intestinal and renal epithelial cells that mediates the effects of LPA/ATX on epithelial cell migration. Our results reinforce previous work that has identified the actin cytoskeleton as one of the major targets of the LPA-induced signaling pathways. Further, we emphasize that lamellipodia formation represents an early facet of LPA/ATX-induced increase in intestinal cell migration which is regulated by villin and PLC-γ1 activation. These studies suggest that LPA as well as actin regulatory proteins such as villin could be potential targets for treatment of carcinomas.
Acknowledgments
This work has been supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases grants DK-65006 and DK-54755 (to S.K.) and by a grant from the National Cancer Institute CA-92160 (to G.T.).
Abbreviations
- ATX
autotaxin
- LPA
lysophosphatidic acid
- PLC-γ1
phospholipase C-γ1
- PTX
pertussis toxin
- VIL/FL
full-length human villin
- VIL/AYFM
tyrosine phosphorylation site mutant of human villin
- DOX
doxycycline
- S1P1
sphingosine 1 phosphate 1
- SEYFP
superenhanced yellow fluorescent protein
- mCherry
m-Cherry tagged fluorescent protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3:582–91. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 2.Shida D, Watanabe T, Aoki J, Hama K, Kitayama J, Sonoda H, Kishi Y, Yamaguchi H, Sasaki S, Sako A, Konishi T, Arai H, Nagawa H. Aberrant expression of lysophosphatidic acid (LPA) receptors in human colorectal cancer. Lab Invest. 2004;84:1352–62. doi: 10.1038/labinvest.3700146. [DOI] [PubMed] [Google Scholar]
- 3.Yun CC, Sun H, Wang D, Rusovici R, Castleberry A, Hall RA, Shim H. LPA2 receptor mediates mitogenic signals in human colon cancer cells. Am J Physiol Cell Physiol. 2005;289:C2–11. doi: 10.1152/ajpcell.00610.2004. [DOI] [PubMed] [Google Scholar]
- 4.Hines OJ, Ryder N, Chu J, McFadden D. Lysophosphatidic acid stimulates intestinal restitution via cytoskeletal activation and remodeling. J Surg Res. 2000;92:23–8. doi: 10.1006/jsre.2000.5941. [DOI] [PubMed] [Google Scholar]
- 5.Sturm A, Sudermann T, Schulte KM, Goebell H, Dignass AU. Modulation of intestinal epithelial wound healing in vitro and in vivo by lysophosphatidic acid. Gastroenterology. 1999;117:368–77. doi: 10.1053/gast.1999.0029900368. [DOI] [PubMed] [Google Scholar]
- 6.Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, Yamori T, Mills GB, Inoue K, Aoki J, Arai H. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J Cell Biol. 2002;158:227–33. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Athman R, Louvard D, Robine S. Villin enhances hepatocyte growth factor-induced actin cytoskeleton remodeling in epithelial cells. Mol Biol Cell. 2003;14:4641–53. doi: 10.1091/mbc.E03-02-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomar A, Wang Y, Kumar N, George S, Ceacareanu B, Hassid A, Chapman KE, Aryal AM, Waters CM, Khurana S. Regulation of cell motility by tyrosine phosphorylated villin. Mol Biol Cell. 2004;15:4807–17. doi: 10.1091/mbc.E04-05-0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robine S, Huet C, Moll R, Sahuquillo-Merino C, Coudrier E, Zweibaum A, Louvard D. Can villin be used to identify malignant and undifferentiated normal digestive epithelial cells? Proc Natl Acad Sci U S A. 1985;82:8488–92. doi: 10.1073/pnas.82.24.8488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Regalado SP, Nambu Y, Iannettoni MD, Orringer MB, Beer DG. Abundant expression of the intestinal protein villin in Barrett's metaplasia and esophageal adenocarcinomas. Mol Carcinog. 1998;22:182–9. [PubMed] [Google Scholar]
- 11.Rieder G, Tessier AJ, Qiao XT, Madison B, Gumucio DL, Merchant JL. Helicobacter-induced intestinal metaplasia in the stomach correlates with Elk-1 and serum response factor induction of villin. J Biol Chem. 2005;280:4906–12. doi: 10.1074/jbc.M413399200. [DOI] [PubMed] [Google Scholar]
- 12.Baker DL, Fujiwara Y, Pigg KR, Tsukahara R, Kobayashi S, Murofushi H, Uchiyama A, Murakami-Murofushi K, Koh E, Bandle RW, Byun HS, Bittman R, Fan D, Murph M, Mills GB, Tigyi G. Carba analogs of cyclic phosphatidic acid are selective inhibitors of autotaxin and cancer cell invasion and metastasis. J Biol Chem. 2006;281:22786–93. doi: 10.1074/jbc.M512486200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hermiston ML, Gordon JI. Inflammatory bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science. 1995;270:1203–7. doi: 10.1126/science.270.5239.1203. [DOI] [PubMed] [Google Scholar]
- 14.George S, Wang Y, Mathew S, Kamalakkannan S, Khurana S. Dimerization and actin-bundling properties of villin and its role in the assembly of epithelial cell brush borders. doi: 10.1074/jbc.M703617200. (Submitted) [DOI] [PubMed] [Google Scholar]
- 15.Tomar A, George S, Kansal P, Wang Y, Khurana S. Interaction of phospholipase C-gamma1 with villin regulates epithelial cell migration. J Biol Chem. 2006;281:31972–86. doi: 10.1074/jbc.M604323200. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Tomar A, George SP, Khurana S. Obligatory role for phospholipase C-{gamma}1 in villin-induced epithelial cell migration. Am J Physiol Cell Physiol. 2007;292:C1775–86. doi: 10.1152/ajpcell.00420.2006. [DOI] [PubMed] [Google Scholar]
- 17.Fuss B, Baba H, Phan T, Tuohy VK, Macklin WB. Phosphodiesterase I, a novel adhesion molecule and/or cytokine involved in oligodendrocyte function. J Neurosci. 1997;17:9095–103. doi: 10.1523/JNEUROSCI.17-23-09095.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee HY, Murata J, Clair T, Polymeropoulos MH, Torres R, Manrow RE, Liotta LA, Stracke ML. Cloning, chromosomal localization, and tissue expression of autotaxin from human teratocarcinoma cells. Biochem Biophys Res Commun. 1996;218:714–9. doi: 10.1006/bbrc.1996.0127. [DOI] [PubMed] [Google Scholar]
- 19.Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, Orth AP, Vega RG, Sapinoso LM, Moqrich A, Patapoutian A, Hampton GM, Schultz PG, Hogenesch JB. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci U S A. 2002;99:4465–70. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai YJ, Chen CS, Lin WC, Lin FT. c-Src-mediated phosphorylation of TRIP6 regulates its function in lysophosphatidic acid-induced cell migration. Mol Cell Biol. 2005;25:5859–68. doi: 10.1128/MCB.25.14.5859-5868.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kranenburg O, Poland M, van Horck FP, Drechsel D, Hall A, Moolenaar WH. Activation of RhoA by lysophosphatidic acid and Galpha12/13 subunits in neuronal cells: induction of neurite retraction. Mol Biol Cell. 1999;10:1851–7. doi: 10.1091/mbc.10.6.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan J, Slice LW, Gu J, Rozengurt E. Cooperation of Gq, Gi, and G12/13 in protein kinase D activation and phosphorylation induced by lysophosphatidic acid. J Biol Chem. 2003;278:4882–91. doi: 10.1074/jbc.M211175200. [DOI] [PubMed] [Google Scholar]
- 23.Piccolo E, Innominato PF, Mariggio MA, Maffucci T, Iacobelli S, Falasca M. The mechanism involved in the regulation of phospholipase Cgamma1 activity in cell migration. Oncogene. 2002;21:6520–9. doi: 10.1038/sj.onc.1205821. [DOI] [PubMed] [Google Scholar]
- 24.Wang W, Goswami S, Lapidus K, Wells AL, Wyckoff JB, Sahai E, Singer RH, Segall JE, Condeelis JS. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 2004;64:8585–94. doi: 10.1158/0008-5472.CAN-04-1136. [DOI] [PubMed] [Google Scholar]
- 25.Jourquin J, Yang N, Kam Y, Guess C, Quaranta V. Dispersal of epithelial cancer cell colonies by lysophosphatidic acid (LPA) J Cell Physiol. 2006;206:337–46. doi: 10.1002/jcp.20470. [DOI] [PubMed] [Google Scholar]
- 26.Luttrell LM, Hawes BE, van Biesen T, Luttrell DK, Lansing TJ, Lefkowitz RJ. Role of c-Src tyrosine kinase in G protein-coupled receptor- and Gbetagamma subunit-mediated activation of mitogen-activated protein kinases. J Biol Chem. 1996;271:19443–50. doi: 10.1074/jbc.271.32.19443. [DOI] [PubMed] [Google Scholar]
- 27.Seufferlein T, Rozengurt E. Lysophosphatidic acid stimulates tyrosine phosphorylation of focal adhesion kinase, paxillin, and p130. Signaling pathways and cross-talk with platelet-derived growth factor. J Biol Chem. 1994;269:9345–51. [PubMed] [Google Scholar]
- 28.Wiener JR, Windham TC, Estrella VC, Parikh NU, Thall PF, Deavers MT, Bast RC, Mills GB, Gallick GE. Activated SRC protein tyrosine kinase is overexpressed in late-stage human ovarian cancers. Gynecol Oncol. 2003;88:73–9. doi: 10.1006/gyno.2002.6851. [DOI] [PubMed] [Google Scholar]
- 29.Nakayama M, Amano M, Katsumi A, Kaneko T, Kawabata S, Takefuji M, Kaibuchi K. Rho-kinase and myosin II activities are required for cell type and environment specific migration. Genes Cells. 2005;10:107–17. doi: 10.1111/j.1365-2443.2005.00823.x. [DOI] [PubMed] [Google Scholar]
- 30.Ridley AJ, Comoglio PM, Hall A. Regulation of scatter factor/hepatocyte growth factor responses by Ras, Rac, and Rho in MDCK cells. Mol Cell Biol. 1995;15:1110–22. doi: 10.1128/mcb.15.2.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reuters I, Weber M, Schulze-Lohoff E. Rho/Rho kinase pathway regulates maintenance of the differentiated tubular epithelial cell phenotype on laminin-1. Nephron Physiol. 2006;104:p95–p106. doi: 10.1159/000094573. [DOI] [PubMed] [Google Scholar]
- 32.Ridley AJ, Comoglio PM, Hall A. Regulation of scatter factor/hepatocyte growth factor responses by Ras, Rac, and Rho in MDCK cells. Mol Cell Biol. 1995;15:1110–22. doi: 10.1128/mcb.15.2.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mouneimne G, Soon L, DesMarais V, Sidani M, Song X, Yip SC, Ghosh M, Eddy R, Backer JM, Condeelis J. Phospholipase C and cofilin are required for carcinoma cell directionality in response to EGF stimulation. J Cell Biol. 2004;166:697–708. doi: 10.1083/jcb.200405156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Falasca M, Logan SK, Lehto VP, Baccante G, Lemmon MA, Schlessinger J. Activation of phospholipase C gamma by PI 3-kinase-induced PH domain- mediated membrane targeting. Embo J. 1998;17:414–22. doi: 10.1093/emboj/17.2.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kassis J, Radinsky R, Wells A. Motility is rate-limiting for invasion of bladder carcinoma cell lines. Int J Biochem Cell Biol. 2002;34:762–75. doi: 10.1016/s1357-2725(01)00173-x. [DOI] [PubMed] [Google Scholar]
- 36.Hama K, Aoki J, Fukaya M, Kishi Y, Sakai T, Suzuki R, Ohta H, Yamori T, Watanabe M, Chun J, Arai H. Lysophosphatidic acid and autotaxin stimulate cell motility of neoplastic and non-neoplastic cells through LPA1. J Biol Chem. 2004;279:17634–9. doi: 10.1074/jbc.M313927200. [DOI] [PubMed] [Google Scholar]
- 37.Contos JJ, Chun J. Genomic characterization of the lysophosphatidic acid receptor gene, lp(A2)/Edg4, and identification of a frameshift mutation in a previously characterized cDNA. Genomics. 2000;64:155–69. doi: 10.1006/geno.2000.6122. [DOI] [PubMed] [Google Scholar]