

Abstract
Microglia are resident immune cells of the CNS. When stimulated by infection, tissue injury, or other signals, microglia assume an activated, “ameboid” morphology and release matrix metalloproteinases, reactive oxygen species, and other proinflammatory factors. This innate immune response augments host defenses, but it can also contribute to neuronal death. Zinc is released by neurons under several conditions in which microglial activation occurs, and zinc chelators can reduce neuronal death in animal models of cerebral ischemia and neurodegenerative disorders. Here, we show that zinc directly triggers microglial activation. Microglia transfected with a nuclear factor-κB (NF-κB) reporter gene showed a severalfold increase in NF-κB activity in response to 30 μm zinc. Cultured mouse microglia exposed to 15–30 μm zinc increased nitric oxide production, increased F4/80 expression, altered cytokine expression, and assumed the activated morphology. Zinc-induced microglial activation was blocked by inhibiting NADPH oxidase, poly(ADP-ribose) polymerase-1 (PARP-1), or NF-κB activation. Zinc injected directly into mouse brain induced microglial activation in wild-type mice, but not in mice genetically lacking PARP-1 or NADPH oxidase activity. Endogenous zinc release, induced by cerebral ischemia–reperfusion, likewise induced a robust microglial reaction, and this reaction was suppressed by the zinc chelator CaEDTA. Together, these results suggest that extracellular zinc triggers microglial activation through the sequential activation of NADPH oxidase, PARP-1, and NF-κB. These findings identify a novel trigger for microglial activation and a previously unrecognized mechanism by which zinc may contribute to neurological disorders.
Keywords: PARP-1, NF-κB, ischemia, neurodegeneration, NADPH oxidase, superoxide
Introduction
Zinc is involved in the pathogenesis of several diseases that affect the CNS (Frederickson et al., 2005). In mammalian brain, zinc is concentrated in the presynaptic vesicles of a subset of glutamatergic axon terminals (Beaulieu et al., 1992). These axon terminals are distributed throughout the forebrain, and are particularly dense in hippocampus and in cerebral cortex (Danscher et al., 1985; Frederickson, 1989). Vesicular zinc is released into the extracellular space in a calcium-dependent manner during normal neuronal activity (Assaf and Chung, 1984; Howell et al., 1984), and is massively released, along with protein-bound zinc, in many pathological conditions (Lee et al., 2003; Frederickson et al., 2005). Treatment with zinc chelators has been shown to reduce neuronal death in animal models of cerebral ischemia, trauma, hypoglycemia, and neurodegenerative disorders (Koh et al., 1996; Suh et al., 2000, 2004; Cherny et al., 2001; Calderone et al., 2004; Nguyen et al., 2005). Zinc release is thought to promote brain injury through direct neurotoxic effects (Weiss et al., 2000; Frederickson et al., 2005) and through effects on protein aggregation (Cherny et al., 2001; Nguyen et al., 2005). Here, we evaluated the possibility that extracellular zinc might also act as a direct stimulus for microglia activation.
Microglia are derived from circulating monocytes and function as resident immune cells of the CNS (Kreutzberg, 1996; Carson and Sutcliffe, 1999). Resting microglia have branched processes and maintain regular spacing throughout the brain and spinal cord. When activated by infection, tissue injury, or other signals, microglia retract these processes to form an activated, “ameboid” morphology. Activated microglia migrate to lesion foci and release matrix metalloproteinases, reactive oxygen species, and certain cytokines. This innate immune response augments host defenses to infection, but contributes to neuronal death in cerebral ischemia, trauma, and neurodegenerative disorders (Kreutzberg, 1996; Block et al., 2007).
Endogenous factors, including glutamate, ATP, and 7-ketocholesterol, have been previously identified as triggers for microglial activation (Tikka et al., 2001; Diestel et al., 2003; Farber and Kettenmann, 2006). The underlying signal transduction pathways are not well established for these signals, but the transcription factor nuclear factor-κB (NF-κB) has been identified as a key regulator of cellular immune responses, including microglia activation (Hoffmann and Baltimore, 2006; Wang et al., 2007). Many effects of NF-κB require the coactivation of poly(ADP-ribose) polymerase-1 (PARP-1) (Chang and Alvarez-Gonzalez, 2001; Ullrich et al., 2001; Chiarugi and Moskowitz, 2003; Kraus and Lis, 2003; Kauppinen and Swanson, 2005), and zinc is reported to induce PARP-1 activation (Kim and Koh, 2002; Sheline et al., 2003; Suh et al., 2007b). NADPH oxidase can also be activated by zinc (Noh and Koh, 2000), and the production of superoxide by NADPH oxidase also promotes activation of microglia (Qin et al., 2004; Mander et al., 2006). Results of the present studies suggest that extracellular zinc elevations induce microglial activation in vitro and in situ, by a mechanism involving both PARP-1 and NADPH oxidase.
Materials and Methods
These studies were performed in accordance with protocols approved by the San Francisco Veterans Affairs Medical Center. N-(6-Oxo-5,6-dihydrophenanthridin-2-yl)-N,N-dimethylacetamide (PJ-34) was obtained from Inotek Pharmaceuticals, and (E)-3-(4-methylphenylsulfonyl)-2-propenenenitrile (BAY 11-7082) was obtained from Alexis Biochemicals. All other reagents were obtained from Sigma-Aldrich except where noted.
Microglial cultures.
Cultures were prepared from PARP-1−/−, p47phox−/−, and wild-type (wt) mice as described previously (Kauppinen and Swanson, 2005). The PARP-1−/− mice were descendents of the 129S-AdprtltmlZqw strain, originally developed by Wang et al. (1995) and obtained from The Jackson Laboratory. These mice were outbred for seven generations with wt CD-1 mice, and wt CD-1 mice were used as controls for the PARP-1−/− mice. The p47phox−/− mice (The Jackson Laboratory) were of the C57BL/6 strain, and wt C57BL/6 mice were used as control cultures prepared from the C56BL/6 p47phox−/− mice. With all strains, cortices from 1-d-old mice were dissociated by mincing and incubation in papain and DNase. After centrifugation for 5 min at 500 × g, the cells were resuspended by trituration with a fire-polished Pasteur pipette, plated on six-well plates (Falcon; BD Biosciences Discovery Labware) at a density of 6.4 × 105 cells/well, and maintained in a 37°C, 5% CO2 incubator. The culture medium consisted of Eagle's Minimal Essential Medium (MEM) (Invitrogen) containing 5 mm glucose and supplemented with 10% endotoxin-free fetal bovine serum (HyClone), 2 mm glutamine, and streptomycin. The medium was changed at 3 d in vitro and once per week thereafter. This procedure produces cultures consisting of astrocytes and microglial cells. After 2 weeks in vitro, microglia were harvested by mildly shaking the cultures and collecting the floating cells. These cells were replated at a density of 5 × 105 cells/well in 24-well plates. The microglial cultures were used for experiments at days 2–3 after replating (in vitro days 16–17). Each culture well was visually inspected by phase contrast microscopy before use, and wells containing >2% contaminating astrocytes or >30% activated microglia were not used for experiments. Three hours before experiments, the culture medium was gently replaced with MEM. All drug stocks were diluted in MEM.
Microglia morphology, immunostaining, and survival.
Microglia morphology was assessed in unfixed tissues by phase contrast microscopy. Microglia were scored as activated when less than two branching processes could be discerned. Immunostaining was performed with cultures fixed with 1:1 methanol:acetone at 4°C. Cultures were characterized with antibodies to glial fibrillary acidic protein (GFAP) (Millipore Bioscience Research Reagents; 1:1000 dilution) and Iba-1 (Waco; 1:1000 dilution) as previously described (Kauppinen and Swanson, 2005). Antibody binding was visualized with Alexa Fluor-conjugated anti-IgG (Invitrogen). Negative controls were prepared by omitting the primary antibodies (data not shown). For detection of poly(ADP-ribose), cultures were incubated with rabbit antibody to poly(ADP-ribose) (Trevigen) diluted 1:1000 (Kauppinen and Swanson, 2005). Microglia were counted as immunopositive for poly(ADP-ribose) when a fluorescent signal clearly above background was observed. Microglial survival was assessed by the propidium iodide exclusion method (Suh et al., 2007a). Twenty-four hours after the designated treatments, 2 μg/ml propidium iodide was added to the culture medium and the cultures were photographed with a fluorescence microscope to identify fluorescent (dead) cell nuclei. For quantifications of morphology, immunostaining, or cell survival, five randomly selected fields were photographed in each culture well and at least 100 cells were tabulated in each well.
Nitric oxide and cytokine production.
Microglial cultures were placed in 300 μl of MEM and incubated with ZnCl2 alone or with drugs as indicated. The zinc and drugs were washed out at 90 min and the microglia were placed in fresh MEM for the remainder of the experiment. Medium samples were taken at the time of zinc washout (90 min) or at 8 or 24 h later. For measurement of nitric oxide production, 50 μl of medium from each culture well was placed in triplicate wells of a 96-well plate. An equal volume of Greiss reagent (Nikodemova et al., 2006) was added to each well for 10 min, and optical densities were measured at 540 nm. The nitrite concentration was determined from a standard curve prepared by adding NaNO2 to MEM. For cytokine assays, 50 μl of the medium was evaluated with a Beadlyte mouse 21-plex cytokine detection system (Millipore) according to the manufacturer's instructions. This immunoassay method employs 21 cytokine-specific antibodies tagged with fluorescent beads. Assays were performed in duplicate, and the fluorescent signal corresponding to each cytokine was measured with a BioPlex 200 system (Bio-Rad). Values for both nitrite and cytokine assays were normalized to the protein content of each well as determined by the bicinchoninic assay (Smith et al., 1985).
Immunoblots.
Cultured were lysed in radioimmunoprecipitation assay buffer, mixed with loading buffer containing 62.5 mm Tris-HCl, pH 6.8, 10% glycerol, 4% SDS, 0.01% bromophenol blue, and 5% β-mercaptoethanol, incubated for 15 min at 65°C, electrophoresed on 10% SDS-PAGE gels, and transferred onto Immobilon-FL Transfer Membranes (Millipore). After a 1 h incubation in blocking buffer (5% BSA, 0.1% Tween in 1 mm PBS), the membranes were incubated overnight at 4°C with a 1:50 dilution of rat anti-mouse F4/80 antibody (Serotec) and 1:10,000 dilution of mouse monoclonal β-actin antibody (Sigma-Aldrich), followed by a 2 h incubation with 1:1000 dilutions of Alexa Fluor 488 anti-rat IgG and Alexa Fluor 555 anti-mouse IgG (Invitrogen). Protein bands were visualized with a Typhoon scanner (GE Healthcare). Band densities were quantified with the NIH Image J program, and F4/80 bands were normalized to densities of the control cultures after normalization to β-actin bands serving as a loading control.
Reactive oxygen species.
Dihydroethidium (DHE) (1 μm) was added to the cultures at the beginning of 90 min ZnCl2 incubations as described previously (Suh et al., 2007a). Fluorescence photographs were obtained after 90 min incubation.
NF-κB activity.
Lentivirus encoding destabilized, enhanced green fluorescence protein driven by the NF-κB promoter (Lenti-κB-dEGFP) was generously provided by Dr. Li Gan (Gladstone Institute, University of California, San Francisco, San Francisco, CA) (Chen et al., 2005). Cultures containing astrocytes and microglia were infected with Lenti-κB-dEGFP at 8–9 d in vitro. The microglia were isolated and replated 7–8 d later, and used for experiments 2–3 d after replating. Photographs were prepared at 2 or 24 h after zinc exposure. Green fluorescent protein (GFP)-expressing cells were counted in five random fields within each well and expressed as a percentage of total number of cells counted.
Intracerebral ZnCl2 injections.
wt, PARP-1−/−, and p47phox−/− mice were given a stereotaxic injection of 1 μl of ZnCl2 (100 μm) into hippocampus (anteroposterior 2.0, mediolateral 1.5, dorsoventral 2.0 mm from bregma and cortical surface), using a 10 μl Hamilton syringe. The injections were made over a 5 min period, and the needle was withdrawn after an additional 5 min. The PARP-1−/− mice were 129S-AdprtltmlZqw strain, and the p47phox−/− mice were C57BL/6 strain (The Jackson Laboratory). wt control mice were of the same congenic strains as the transgenic mice.
Transient ischemia.
wt C57BL/6 mice were anesthetized with 1–3% isoflurane in a 75:25 mixture of nitrous oxide and oxygen. Core temperature was kept at 36.5–37.5°C with a heating blanket. Forebrain ischemia was induced by clamping both common carotid arteries for a 20 min interval (Yang et al., 1997). Mice undergoing sham ischemia received the same surgical incisions and handling but without carotid artery occlusion. One group of mice was injected with CaEDTA into the right lateral ventricle at termination of ischemia. As described previously (Lee et al., 2000), a burr hole was made 0.5 mm caudal to bregma and 1.5 mm lateral to the midline, and the syringe needle tip was lowered 3.0 mm below the dura. One microliter of 100 mm CaEDTA was injected over a 5 min period.
Immunohistochemistry.
Mice were killed 6 h after ischemia or ZnCl2 injections. Brains were removed after transcardial perfusion with a 0.9% saline and 4% paraformaldehyde (PFA). Brains were postfixed in 4% PFA for overnight, and then cryoprotected by immersion in 20% sucrose for 24 h. Coronal 30 μm sections were prepared and immunostained as described previously (Suh et al., 2003). Rat anti-mouse F4/80 antibody (Serotec) was used at a 1:75 dilution and visualized with an Alexa Fluor 488-conjugated chicken anti-rat IgG (Invitrogen). Negative controls were prepared by omitting the primary antibodies. Digital images were acquired with a confocal laser-scanning microscope and processed identically. A grading scale for microglial activation was based on morphology and F4/80 staining (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Images were scored by an investigator who was blinded to the identification of the brain sections.
Statistical analysis.
All data are expressed as the mean ± SEM. Microglial activation scores were evaluated with the Kruskal–Wallis test followed by the Dunn's test for multiple-group comparisons. All other data were compared with ANOVA followed by either Tukey's test for multiple-group comparisons or Dunnett's test for multiple comparisons against a common control group. For studies using microglia cultures, n denotes the number of independent experiments, each performed in triplicate.
Results
Zinc induces microglia activation
The microglial cultures were uniformly immunopositive for Iba-1 and contained <2% GFAP-positive contaminating astrocytes (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Under basal conditions, >70% of the microglia had multiple branched processes characteristic of the resting morphology (Fig. 1). The addition of ZnCl2 to the medium caused these cells to retract their processes and assume the rounded morphology of activated microglia (Fig. 1), similar to that induced by lipopolysaccharide (LPS). These changes were nearly maximal within 2 h. The increase in number of activated microglia was significant at 15 μm and maximal at 60 μm ZnCl2 (Fig. 1B). Medium exchange alone (0 μm ZnCl2) typically produced a smaller, 5–10% increase over basal microglia activation, and consequently this was used as the control condition for all of the studies using microglial cultures. Microglial cell death was negligible at ZnCl2 concentrations of <60 μm (Fig. 1C).
Figure 1.

Zinc triggers microglia activation. A, Resting microglia have multiple branched processes, whereas microglia incubated for 90 min with 30 μm zinc or 50 ng/ml LPS show retracted processes and a rounded, ameboid morphology. Scale bar, 10 μm. B, ZnCl2 has a concentration-dependent effect on the fraction of microglia exhibiting the activated morphology. C, ZnCl2 effects on microglial survival, assessed 24 h after a 90 min exposure. n = 4; p < 0.01 versus control. Error bars indicate SEM.
PARP-1 activity is required for zinc-induced microglia activation
Several aspects of microglial activation require interaction between activated PARP-1 and NF-κB (Ullrich et al., 2001; Chiarugi and Moskowitz, 2003; Kauppinen and Swanson, 2005). Here, we evaluated the role of PARP-1 in zinc-induced microglial activation by using PARP-1−/− microglia and by using wild-type microglia treated with the PARP inhibitor, PJ-34 (Mabley et al., 2001). Zinc-induced microglial activation was almost completely prevented under both of these conditions (Fig. 2A,B). Blocking NF-κB activation with 5 μm BAY 11-7085 (BAY), an inhibitor of I-κB phosphorylation (Pierce et al., 1997), also prevented zinc-induced microglial activation. Immunostaining the cultures for poly(ADP-ribose) (abbreviated as PAR) confirmed that PARP-1 was activated by ZnCl2 exposure. As expected, the PAR formation was absent in cultures treated with PJ-34 or prepared from PARP-1−/− mice, but unaffected in cultures treated with BAY (Fig. 2A,C).
Figure 2.

Zinc-induced microglial activation requires PARP-1 and NADPH oxidase. A, Panels show phase-contrast images of microglial morphology (top row), immunofluorescence for poly(ADP-ribose) (PAR) (middle row), and merged images (bottom row). Wild-type microglia exhibit both morphological activation and nuclear PAR accumulation in response to 90 min incubation with 30 μm ZnCl2. Both of these responses are absent in PARP-1−/− microglia and in wild-type microglia coincubated with the PARP-1 inhibitor, PJ-34 (400 nm). Incubation with 5 μm BAY 11-70820, an inhibitor of NF-κB activation, blocks morphological activation but not PAR formation. Scale bar, 10 μm. B, C, Quantitative assessment of microglial activation and PAR formation in cultures treated as in A. D, Zinc-induced microglial activation and PAR accumulation are also absent in p47phox−/− microglia and in wild-type microglia coincubated with 500 μm of the NADPH oxidase inhibitor, apocynin. E, F, Quantitative assessment of microglial activation and PAR formation in cultures treated as in D. n = 3–5; *p < 0.01 versus ZnCl2 alone; †p < 0.01 versus wild-type control. Error bars indicate SEM.
NADPH oxidase activity is required for zinc-induced microglia activation
Superoxide production by NADPH oxidase has been proposed as an intermediate step linking zinc and PARP-1 activation (Kim and Koh, 2002). We evaluated this possibility by using p47phox −/− microglia, which are unable to assemble a functional NADPH oxidase complex (Huang et al., 2000), and by using wild-type microglia treated with the NADPH oxidase inhibitor apocynin (Stolk et al., 1994). Zinc-induced microglial activation was almost completely prevented under both of these conditions (Fig. 2D,E), indicating that NADPH oxidase plays a requisite role in this signaling pathway. PAR formation was also prevented under these conditions (Fig. 2F), suggesting that NADPH oxidase activation is upstream of PARP-1 activation.
The functional role of NADPH oxidase in microglia activation was further evaluated by assessing the production of reactive oxygen species (ROS) in zinc-treated cultures. Dihydroethidium is oxidized by superoxide and superoxide-derived ROS to fluorescent products that are trapped within cells (Zhao et al., 2003; Robinson et al., 2006; Suh et al., 2007a). Microglia exposed to zinc increased ROS production (Fig. 3A). This increase was blocked by apocynin but not by PJ-34, further indicating that NADPH oxidase acts upstream of PARP-1 activation.
Figure 3.

Role of reactive oxygen species in microglial activation. A, Superoxide formation in microglia is detected by oxidized DHE fluorescence, and microglial morphology is shown in the corresponding phase contrast images. ZnCl2 induces superoxide formation during microglial activation. Apocynin (500 μm) blocks both of these effects, whereas PJ-34 (400 nm) blocks microglial activation without blocking superoxide formation. Images are representative of four independent studies performed in triplicate. Scale bar, 40 μm. B, Zinc-induced microglial activation is blocked by PJ-34, and by the ROS scavenger Trolox (100 μm), but not by superoxide dismutase (SOD) plus catalase (each 200 U/ml). Exogenous delivery of 50 μm H2O2 to the medium also induces microglial activation that is blocked by PJ-34 or Trolox. n = 3; *p < 0.01 versus ZnCl2 or H2O2 alone; †p < 0.01 versus control. C, p47phox−/− microglia assume the activated morphology when exposed to H2O2. *p < 0.05 versus control; n = 3. Error bars indicate SEM.
ROS generated by NADPH oxidase could act as an intercellular messenger if released into the extracellular space, or as an intracellular messenger if released internally (Rhee, 2006; Bedard and Krause, 2007). We evaluated these possibilities by comparing the effects of extracellular and intracellular ROS scavengers (Fig. 3B). Zinc-induced microglia activation was blocked by Trolox, which readily passes into cells, but not by the combination of superoxide dismutase plus catalase, which remain in the extracellular space. This result suggests an intracellular signaling route, but does not exclude intercellular or autaptic signaling through loculated extracellular spaces that do not communicate with the culture medium.
If ROS are important mediators of zinc-induced microglial activation, then the addition of exogenous ROS to the culture medium should also induce microglial activation. As shown in Figure 3B, H2O2 induced microglial activation by a mechanism that, like zinc-induced activation, was blocked by the PARP inhibitor PJ-34. Not only wild-type, but also p47phox−/− microglia could be activated by H2O2, thus confirming that the effect of p47phox gene deletion is mediated by reduced ROS production in these cells (Fig. 3C). Of note, the p47phox−/− microglia were found to be hyperresponsive to H2O2 relative to wild-type microglia (Fig. 3C).
Zinc induces NF-κB activation in microglia
The finding that microglial activation was prevented by the NF-κB inhibitor BAY 11-7082 (Fig. 2B) is consistent with the known role for NF-κB in many aspects of the microglial inflammatory response. More direct evidence that zinc leads to NF-κB activation was provided by studies using microglia transfected with an NF-κB-driven GFP reporter gene (Chen et al., 2005) (Fig. 4). Tumor necrosis factor α (TNFα) is known to induce a robust activation of NF-κB (Baud and Karin, 2001) and the degree of GFP expression induced by TNFα thus provides an index of the NF-κB reporter gene transfection efficiency. ZnCl2 produced a large increase in the number of microglia expressing GFP when assessed at either 2 or 24 h (Fig. 4B). This increase was severalfold greater than basal expression, but less than that induced by TNFα. PJ-34 prevented the effect of zinc at both the 2 and 24 h time points. Apocynin also prevented zinc-induced GFP expression at 2 h, but could not be tested at the 24 h time point because of toxicity associated with the longer incubation period.
Figure 4.
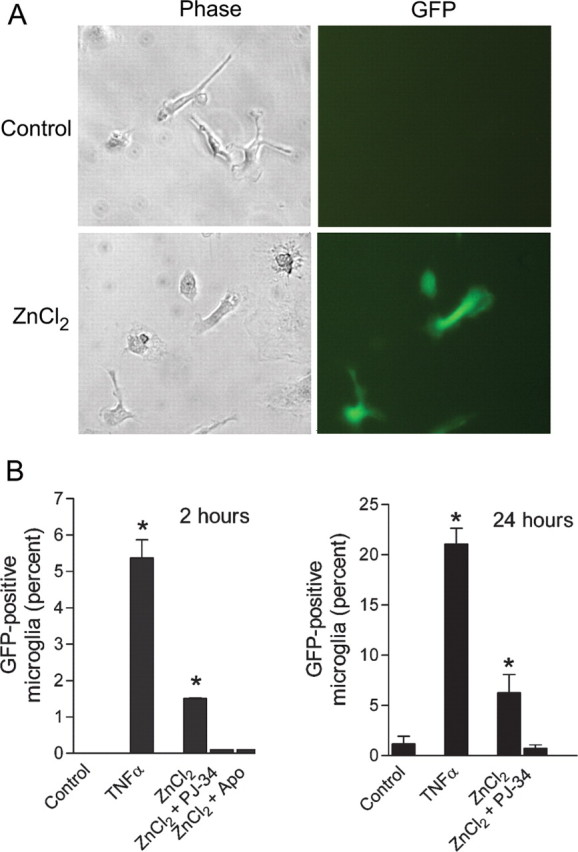
Zinc activates NF-κB. Cultures were transfected with a GFP reporter gene driven by an NF-κB promoter. A, Photomicrographs show microglial morphology (left) and GFP expression (right) in cultures treated with 0 or 15 μm zinc for 24 h. B, Quantified evaluation of NF-κB expression in microglia, assessed at 2 or 24 h after incubation with ZnCl2. The positive control, TNFα, indicates the fraction of microglia successfully transfected with the GFP reporter gene. *p < 0.05 versus control; n = 3. Error bars indicate SEM.
Inducible nitric oxide synthase activation and F4/80 expression are induced by zinc
Additional studies were performed to identify effects of zinc on aspects of microglial activation other than morphological changes. Nitric oxide production was measured by evaluating medium nitrite concentration after a 90 min incubation with ZnCl2 and after 8 or 24 h after washout of the ZnCl2. No appreciable nitrite accumulation was detected at the 90 min or 8 h time points, but by 24 h there was a 3.5-fold increase in medium nitrite content (Fig. 5A). At this time point, the nitrite concentration in the medium of control microglia ranged from 0.71 to 4.98 μm (7.45–59.80 pmol/mg protein; n = 4). The increase induced by zinc was blocked by the nitric oxide synthase inhibitor aminoguanidine and, like the microglial morphological changes, by the PARP-1 inhibitor PJ-34. Aminoguanidine did not, however, block the zinc-induced PAR formation or morphological changes (data not shown). The effect of LPS on nitric oxide production was also examined, and found to increase nitrite accumulation to a significantly greater extent than zinc (Fig. 5A).
Figure 5.
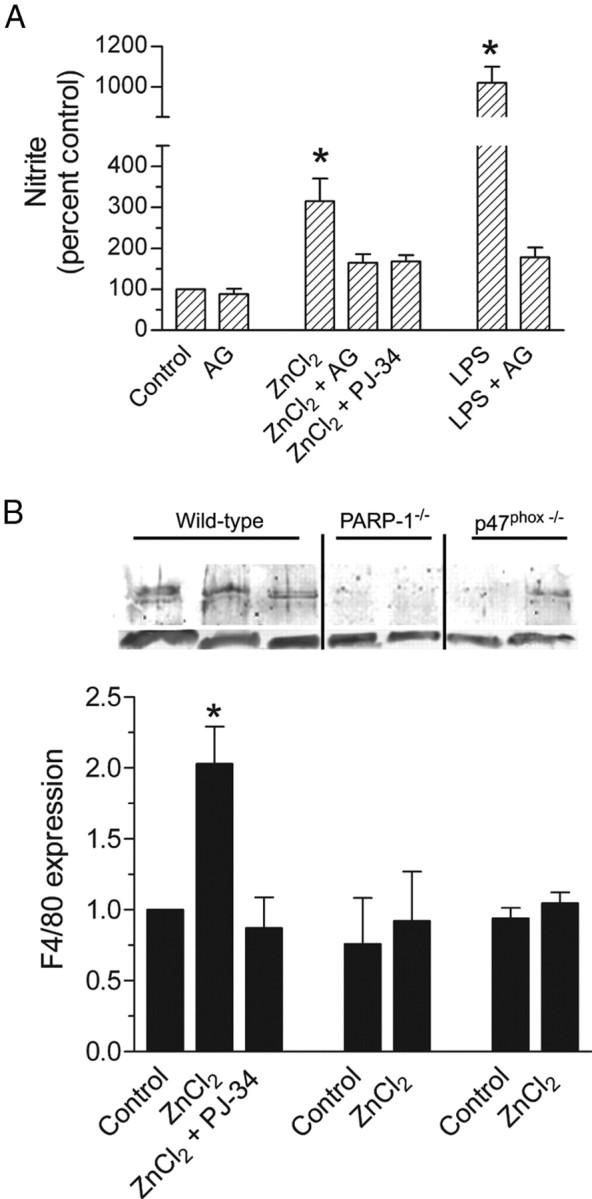
Zinc increases nitric oxide synthase (NOS) activation and F4/80 expression. A, Nitrite was measured 24 h after 90 min incubations under the designated conditions. ZnCl2, 30 μm; aminoguanidine (AG), 1 mm; PJ-34, 400 nm; LPS, 50 ng/ml. n = 4; *p < 0.05. B, Western blot shows F4/80 expression at 160 kDa (top) and β-actin loading control (bottom). The vertical lines denote independently prepared blots. Cultures were prepared from wild-type, PARP-1−/−, or p47phox−/− microglia, after treatment as denoted on the bar graph. Abbreviations are as in A. Graph indicates quantified data from n = 3, normalized to expression in control, wild-type cells. *p < 0.05 versus wild-type control. Error bars indicate SEM.
F4/80 is a surface molecule on microglia and macrophages that is upregulated during immune responses (Austyn and Gordon, 1981). Cultured microglia exposed to 30 μm ZnCl2 for 90 min exhibited a twofold increase in F4/80 expression, assessed 24 h later. This increase was blocked by PJ-34 and blocked by PARP-1 or p47phox gene deficiency (Fig. 5B). We used a multiplex cytokine assay to determine whether ZnCl2 exposure also induced changes in cytokine expression patterns. Of the 21 cytokines evaluated, interleukin-1α (IL-1α), IL-1β, IL-3, and granulocyte–macrophage colony-stimulating factor were found to be modestly increased (1.5- to 3-fold) in culture media harvested 24 h after a 90 min incubation with 30 μm ZnCl2. These increases were absent in cultures treated with PJ-34 and in cultures derived from PARP-1−/− or p47phox−/− mice (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Zinc-induced microglial activation in vivo
To determine whether extracellular zinc can induce microglial activation in situ, we injected ZnCl2 solutions directly into mouse hippocampus. ZnCl2 injection induced soma enlargement, process retraction, and increased F4/80 expression typical of microglial activation (Fig. 6; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). These changes were evident within 6 h of the ZnCl2 injections and were restricted to an area ∼500 μm from the injection needle track. In contrast, mice injected with vehicle (saline) alone showed microglial activation only in the immediate vicinity of the needle track. Consistent with the cell culture results, ZnCl2 injected into either PARP-1−/− or p47phox−/− mouse hippocampi produced substantially less microglial activation than ZnCl2 injected into wild-type mice of the corresponding background strains (Fig. 6).
Figure 6.
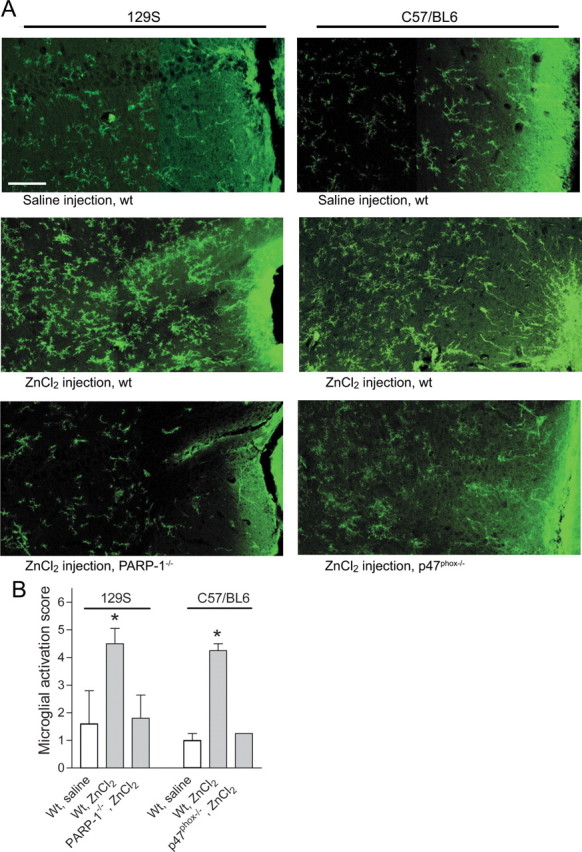
Microglial activation in brain is induced by ZnCl2 injection. A, Photomicrographs show microglial morphology and F4/80 expression (green) in mouse hippocampus after stereotaxic injection of ZnCl2 or saline vehicle. The needle track is visible at the right-hand edge of each composite image. Microglia in wild-type mice of both the C57BL/6 and 129S strains showed increased F4/80 immunoreactivity and activated morphology after ZnCl2 injections. These changes were attenuated in C57BL/6 p47phox−/− mice and the 129S PARP-1−/− mice. Scale bar, 50 μm. Quantified data are shown in B. n = 3–5; *p < 0.05 versus saline control. Error bars indicate SEM.
Endogenous zinc is released during ischemia–reperfusion and in other conditions in which microglial activation occurs (Frederickson et al., 2005). The effects of extracellular zinc can be blocked with the zinc chelator, CaEDTA (Koh et al., 1996). Thus, to evaluate the role of endogenous zinc release in microglia activation, we examined the effect of intracerebroventricular injection of CaEDTA on microglial activation after brain ischemia–reperfusion. At 6 h after ischemia, there was a robust microglial response throughout hippocampus and other brain regions (Fig. 7). These changes were almost completely absent in mice receiving intracerebroventricular CaEDTA immediately after the ischemic interval.
Figure 7.
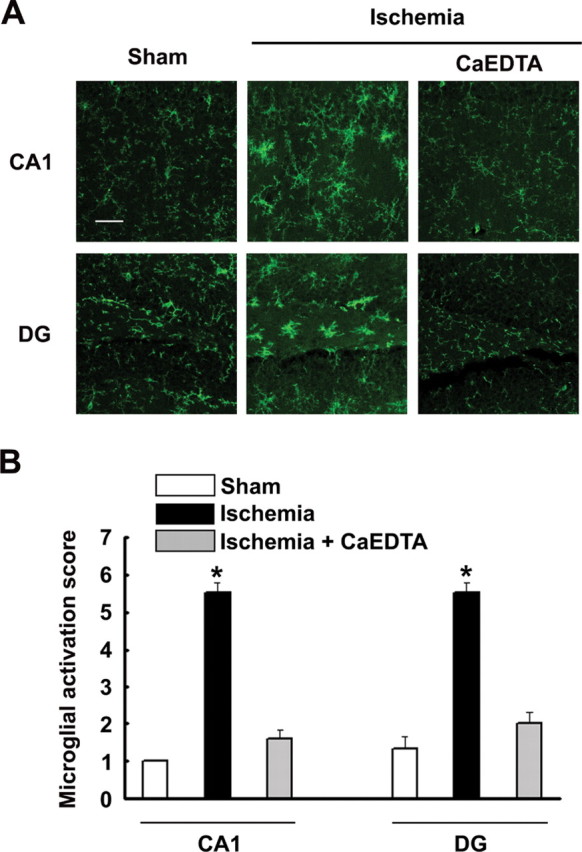
Microglia activation after cerebral ischemia is blocked by a zinc chelator. A, Images show microglia morphology and F4/80 expression (green) in the CA1 and dentate gyrus (DG) regions of hippocampus after ischemia, sham ischemia, and ischemia followed by intracerebroventricular injection of the zinc chelator, CaEDTA (100 μm in 1 μl volume). Scale bar, 50 μm. Quantified data are shown in B. n = 5; *p < 0.05. Error bars indicate SEM.
Discussion
Activated microglia promote neuronal death in both cerebral ischemia and neurodegenerative disorders (Dheen et al., 2007). Zinc has also been found to promote injury in these disorders, through direct neurotoxicity and effects on protein aggregation (Frederickson et al., 2005). The present findings suggest that extracellular zinc can function as a signal for microglial activation, and that this signal is mediated by the sequential activation of NADPH oxidase, PARP-1, and NF-κB. These results identify a novel mechanism by which zinc may contribute to neurological disorders.
The production of ROS by microglial NADPH oxidase is an innate response to infection and other stimuli. NADPH oxidase and ROS also promote microglial activation in a feedforward manner (Min et al., 2004; Mander et al., 2006). The signaling mechanisms linking ROS production and microglial activation have not been established, but PARP-1 activation is a candidate intermediary step because PARP-1 is activated during oxidative stress (Kauppinen et al., 2006; Schreiber et al., 2006) and required for both NF-κB transcriptional activity and microglial activation. In the present studies, zinc induced activation of both NADPH oxidase and PARP-1. Inhibition or genetic disruption of NADPH oxidase blocked both PARP-1 activation and microglial activation, indicating that ROS production by NADPH oxidase is upstream of PARP-1 in this signaling pathway. Moreover, microglial activation was induced by exogenous H2O2 in the absence of added zinc, and this microglial activation also was blocked by PARP-1 inhibition. The reason for the markedly increased sensitivity of p47phox−/− microglia to H2O2 was not explored in this study, but may result from reduced ROS scavenging or other compensatory changes in response to reduced NADPH oxidase activity in these cells.
A limitation of studies with cultured microglia is that these cells are not in a true resting state, as evidenced here by the substantial fraction of microglia with retracted processes under basal conditions. This partial activation may be attributable in part to the absence of normal astrocyte influences which dampen microglial activation (Giulian et al., 1995). To confirm that zinc can activate microglia in situ, we performed parallel studies to evaluate the effects of zinc injected directly into brain. These studies showed the same pattern of responses as the cell culture studies: zinc injection induced microglial activation, and this activation was attenuated in mice deficient in PARP-1 or NADPH oxidase activity. Of note, the rate of zinc-induced microglial activation in situ was considerably slower than in cell culture. The reason for this difference is not established here, but may be related to the basal, partial activation of microglia in culture.
Microglial response is a graded phenomenon (Kreutzberg, 1996). Process retraction appears to be universal manifestation of the immune response in these cells (Giulian et al., 1995), but other responses are stimulus dependent. Different stimuli induce different patterns of gene expression, proliferation, and protein secretion in cultured microglia (Giulian and Ingeman, 1988; Ganter et al., 1992; Kauppinen and Swanson, 2005; McKimmie et al., 2006). Here, we found that zinc caused, in addition to morphological changes, an increase in nitric oxide release and an increase in F4/80 expression, but only modest elevations in proinflammatory cytokines. Thus, zinc induces an attenuated and more restricted microglial activation response than that induced by LPS or TNFα in cell culture. The response of microglia to zinc in situ may, however, differ from that observed in culture as a result of interactions with other secreted factors (Farber and Kettenmann, 2006; Lund et al., 2006).
Although the role of zinc in neurological disorders is well established, the actual concentrations of zinc in brain extracellular space remain uncertain. Zinc is released from both synaptic vesicles and protein binding sites during ischemia–reperfusion (Lee et al., 2003; Bossy-Wetzel et al., 2004; Frederickson et al., 2005). Ischemia-induced zinc release has been calculated to raise extracellular zinc concentrations up to 300 μm, which exceeds the 30 μm zinc used in the cell culture studies here. However, direct measures of free zinc in brain extracellular space during ischemia have identified elevations to only the nanomolar range (Frederickson et al., 2006). Because of these uncertainties, we also evaluated the effect of endogenous zinc, released during ischemia–reperfusion, on microglial activation using the zinc chelator CaEDTA, which has been shown to prevent ischemic neuronal death (Koh et al., 1996; Calderone et al., 2004). The near-complete suppression of microglial activation in brains treated with CaEDTA in these studies suggests that endogenous zinc release is an early signal triggering microglial activation after ischemia. An alternative possibility, that microglial activation is reduced because of reduced zinc-induced neuronal death in the CaEDTA-treated mice, is unlikely because neuronal death occurs well after microglial activation in this ischemic model (Lee et al., 2004). It is therefore possible that the effect of CaEDTA on neuronal survival after ischemia may be attributable, at least in part, to reduced microglial activation.
Zinc has also been implicated in neurodegenerative disorders, specifically Huntington's disease and Alzheimer's disease. Zinc chelators slow progression in animal models of these diseases (Cherny et al., 2001; Nguyen et al., 2005), an effect that has been attributed to reduced abnormal protein synthesis or aggregation. However, microglial activation is also a prominent and early event in these disorders (Monsonego and Weiner, 2003; Tai et al., 2007). The present findings suggest that interventions targeting elevated brain zinc concentrations may act in part by attenuating this innate immune response.
Footnotes
This work was supported by National Institutes of Health Grant NS145434 (R.A.S.) and by Department of Veterans Affairs. We thank Dr. Lynn Pulliam for assistance with the cytokine assays and Dr. Li Gan for the NF-κB reporter gene lentivirus.
References
- Assaf SY, Chung SH. Release of endogenous Zn2+ from brain tissue during activity. Nature. 1984;308:734–736. doi: 10.1038/308734a0. [DOI] [PubMed] [Google Scholar]
- Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol. 1981;11:805–815. doi: 10.1002/eji.1830111013. [DOI] [PubMed] [Google Scholar]
- Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11:372–377. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- Beaulieu C, Dyck R, Cynader M. Enrichment of glutamate in zinc-containing terminals of the cat visual cortex. NeuroReport. 1992;3:861–864. doi: 10.1097/00001756-199210000-00010. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Talantova MV, Lee WD, Scholzke MN, Harrop A, Mathews E, Gotz T, Han J, Ellisman MH, Perkins GA, Lipton SA. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004;41:351–365. doi: 10.1016/s0896-6273(04)00015-7. [DOI] [PubMed] [Google Scholar]
- Calderone A, Jover T, Mashiko T, Noh KM, Tanaka H, Bennett MV, Zukin RS. Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J Neurosci. 2004;24:9903–9913. doi: 10.1523/JNEUROSCI.1713-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson MJ, Sutcliffe JG. Balancing function vs. self defense: the CNS as an active regulator of immune responses. J Neurosci Res. 1999;55:1–8. doi: 10.1002/(SICI)1097-4547(19990101)55:1<1::AID-JNR1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Chang WJ, Alvarez-Gonzalez R. The sequence-specific DNA binding of NF-kappa B is reversibly regulated by the automodification reaction of poly (ADP-ribose) polymerase 1. J Biol Chem. 2001;276:47664–47670. doi: 10.1074/jbc.M104666200. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhou Y, Mueller-Steiner S, Chen LF, Kwon H, Yi S, Mucke L, Gan L. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J Biol Chem. 2005;280:40364–40374. doi: 10.1074/jbc.M509329200. [DOI] [PubMed] [Google Scholar]
- Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron. 2001;30:665–676. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- Chiarugi A, Moskowitz MA. Poly(ADP-ribose) polymerase-1 activity promotes NF-kappaB-driven transcription and microglial activation: implication for neurodegenerative disorders. J Neurochem. 2003;85:306–317. doi: 10.1046/j.1471-4159.2003.01684.x. [DOI] [PubMed] [Google Scholar]
- Danscher G, Howell G, Perez-Clausell J, Hertel N. The dithizone, Timm's sulphide silver and the selenium methods demonstrate a chelatable pool of zinc in CNS. A proton activation (PIXE) analysis of carbon tetrachloride extracts from rat brains and spinal cords intravitally treated with dithizone. Histochemistry. 1985;83:419–422. doi: 10.1007/BF00509203. [DOI] [PubMed] [Google Scholar]
- Dheen ST, Kaur C, Ling EA. Microglial activation and its implications in the brain diseases. Curr Med Chem. 2007;14:1189–1197. doi: 10.2174/092986707780597961. [DOI] [PubMed] [Google Scholar]
- Diestel A, Aktas O, Hackel D, Hake I, Meier S, Raine CS, Nitsch R, Zipp F, Ullrich O. Activation of microglial poly(ADP-ribose)-polymerase-1 by cholesterol breakdown products during neuroinflammation: a link between demyelination and neuronal damage. J Exp Med. 2003;198:1729–1740. doi: 10.1084/jem.20030975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber K, Kettenmann H. Purinergic signaling and microglia. Pflügers Arch. 2006;452:615–621. doi: 10.1007/s00424-006-0064-7. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ. Neurobiology of zinc and zinc-containing neurons. Int Rev Neurobiol. 1989;31:145–238. doi: 10.1016/s0074-7742(08)60279-2. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci. 2005;6:449–462. doi: 10.1038/nrn1671. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Giblin LJ, Krezel A, McAdoo DJ, Muelle RN, Zeng Y, Balaji RV, Masalha R, Thompson RB, Fierke CA, Sarvey JM, de Valdenebro M, Prough DS, Zornow MH. Concentrations of extracellular free zinc (pZn)e in the central nervous system during simple anesthetization, ischemia and reperfusion. Exp Neurol. 2006;198:285–293. doi: 10.1016/j.expneurol.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Ganter S, Northoff H, Mannel D, Gebicke-Harter PJ. Growth control of cultured microglia. J Neurosci Res. 1992;33:218–230. doi: 10.1002/jnr.490330205. [DOI] [PubMed] [Google Scholar]
- Giulian D, Ingeman JE. Colony-stimulating factors as promoters of ameboid microglia. J Neurosci. 1988;8:4707–4717. doi: 10.1523/JNEUROSCI.08-12-04707.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Li J, Bartel S, Broker J, Li X, Kirkpatrick JB. Cell surface morphology identifies microglia as a distinct class of mononuclear phagocyte. J Neurosci. 1995;15:7712–7726. doi: 10.1523/JNEUROSCI.15-11-07712.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev. 2006;210:171–186. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- Howell GA, Welch MG, Frederickson CJ. Stimulation-induced uptake and release of zinc in hippocampal slices. Nature. 1984;308:736–738. doi: 10.1038/308736a0. [DOI] [PubMed] [Google Scholar]
- Huang CK, Zhan L, Hannigan MO, Ai Y, Leto TL. P47(phox)-deficient NADPH oxidase defect in neutrophils of diabetic mouse strains, C57BL/6J-m db/db and db/+ J Leukoc Biol. 2000;67:210–215. doi: 10.1002/jlb.67.2.210. [DOI] [PubMed] [Google Scholar]
- Kauppinen TM, Swanson RA. Poly(ADP-ribose) polymerase-1 promotes microglial activation, proliferation, and matrix metalloproteinase-9-mediated neuron death. J Immunol. 2005;174:2288–2296. doi: 10.4049/jimmunol.174.4.2288. [DOI] [PubMed] [Google Scholar]
- Kauppinen TM, Chan WY, Suh SW, Wiggins AK, Huang EJ, Swanson RA. Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc Natl Acad Sci USA. 2006;103:7136–7141. doi: 10.1073/pnas.0508606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Koh JY. The role of NADPH oxidase and neuronal nitric oxide synthase in zinc-induced poly(ADP-ribose) polymerase activation and cell death in cortical culture. Exp Neurol. 2002;177:407–418. doi: 10.1006/exnr.2002.7990. [DOI] [PubMed] [Google Scholar]
- Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996;272:1013–1016. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- Kraus WL, Lis JT. PARP goes transcription. Cell. 2003;113:677–683. doi: 10.1016/s0092-8674(03)00433-1. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Lee JY, Cole TB, Palmiter RD, Koh JY. Accumulation of zinc in degenerating hippocampal neurons of ZnT3-null mice after seizures: evidence against synaptic vesicle origin. J Neurosci. 2000;20(RC79):1–5. doi: 10.1523/JNEUROSCI.20-11-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Kim JH, Palmiter RD, Koh JY. Zinc released from metallothionein-iii may contribute to hippocampal CA1 and thalamic neuronal death following acute brain injury. Exp Neurol. 2003;184:337–347. doi: 10.1016/s0014-4886(03)00382-0. [DOI] [PubMed] [Google Scholar]
- Lee SR, Tsuji K, Lo EH. Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J Neurosci. 2004;24:671–678. doi: 10.1523/JNEUROSCI.4243-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund S, Christensen KV, Hedtjarn M, Mortensen AL, Hagberg H, Falsig J, Hasseldam H, Schrattenholz A, Porzgen P, Leist M. The dynamics of the LPS triggered inflammatory response of murine microglia under different culture and in vivo conditions. J Neuroimmunol. 2006;180:71–87. doi: 10.1016/j.jneuroim.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Mabley JG, Jagtap P, Perretti M, Getting SJ, Salzman AL, Virag L, Szabo E, Soriano FG, Liaudet L, Abdelkarim GE, Hasko G, Marton A, Southan GJ, Szabo C. Anti-inflammatory effects of a novel, potent inhibitor of poly (ADP-ribose) polymerase. Inflamm Res. 2001;50:561–569. doi: 10.1007/PL00000234. [DOI] [PubMed] [Google Scholar]
- Mander PK, Jekabsone A, Brown GC. Microglia proliferation is regulated by hydrogen peroxide from NADPH oxidase. J Immunol. 2006;176:1046–1052. doi: 10.4049/jimmunol.176.2.1046. [DOI] [PubMed] [Google Scholar]
- McKimmie CS, Roy D, Forster T, Fazakerley JK. Innate immune response gene expression profiles of N9 microglia are pathogen-type specific. J Neuroimmunol. 2006;175:128–141. doi: 10.1016/j.jneuroim.2006.03.012. [DOI] [PubMed] [Google Scholar]
- Min KJ, Pyo HK, Yang MS, Ji KA, Jou I, Joe EH. Gangliosides activate microglia via protein kinase C and NADPH oxidase. Glia. 2004;48:197–206. doi: 10.1002/glia.20069. [DOI] [PubMed] [Google Scholar]
- Monsonego A, Weiner HL. Immunotherapeutic approaches to Alzheimer's disease. Science. 2003;302:834–838. doi: 10.1126/science.1088469. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Hamby A, Massa SM. Clioquinol down-regulates mutant huntingtin expression in vitro and mitigates pathology in a Huntington's disease mouse model. Proc Natl Acad Sci USA. 2005;102:11840–11845. doi: 10.1073/pnas.0502177102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikodemova M, Duncan ID, Watters JJ. Minocycline exerts inhibitory effects on multiple mitogen-activated protein kinases and IkappaBalpha degradation in a stimulus-specific manner in microglia. J Neurochem. 2006;96:314–323. doi: 10.1111/j.1471-4159.2005.03520.x. [DOI] [PubMed] [Google Scholar]
- Noh KM, Koh JY. Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. J Neurosci. 2000;20(RC111):1–5. doi: 10.1523/JNEUROSCI.20-23-j0001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, Gerritsen ME. Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272:21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279:1415–1421. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci USA. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- Sheline CT, Wang H, Cai AL, Dawson VL, Choi DW. Involvement of poly ADP ribosyl polymerase-1 in acute but not chronic zinc toxicity. Eur J Neurosci. 2003;18:1402–1409. doi: 10.1046/j.1460-9568.2003.02865.x. [DOI] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- Suh SW, Chen JW, Motamedi M, Bell B, Listiak K, Pons NF, Danscher G, Frederickson CJ. Evidence that synaptically-released zinc contributes to neuronal injury after traumatic brain injury. Brain Res. 2000;852:268–273. doi: 10.1016/s0006-8993(99)02095-8. [DOI] [PubMed] [Google Scholar]
- Suh SW, Aoyama K, Chen Y, Garnier P, Matsumori Y, Gum E, Liu J, Swanson RA. Hypoglycemic neuronal death and cognitive impairment are prevented by poly(ADP-ribose) polymerase inhibitors administered after hypoglycemia. J Neurosci. 2003;23:10681–10690. doi: 10.1523/JNEUROSCI.23-33-10681.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh SW, Garnier P, Aoyama K, Chen Y, Swanson RA. Zinc release contributes to hypoglycemia-induced neuronal death. Neurobiol Dis. 2004;16:538–545. doi: 10.1016/j.nbd.2004.04.017. [DOI] [PubMed] [Google Scholar]
- Suh SW, Gum ET, Hamby AM, Chan PH, Swanson RA. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J Clin Invest. 2007a;117:910–918. doi: 10.1172/JCI30077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh SW, Aoyama K, Alano CC, Anderson CM, Hamby AM, Swanson RA. Zinc inhibits astrocyte glutamate uptake by activation of poly(ADP-ribose) polymerase-1. Mol Med. 2007b;13:344–349. doi: 10.2119/2007-00043.Suh. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, Piccini P. Microglial activation in presymptomatic Huntington's disease gene carriers. Brain. 2007;130:1759–1766. doi: 10.1093/brain/awm044. [DOI] [PubMed] [Google Scholar]
- Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci. 2001;21:2580–2588. doi: 10.1523/JNEUROSCI.21-08-02580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich O, Diestel A, Eyupoglu IY, Nitsch R. Regulation of microglial expression of integrins by poly(ADP-ribose) polymerase-1. Nat Cell Biol. 2001;3:1035–1042. doi: 10.1038/ncb1201-1035. [DOI] [PubMed] [Google Scholar]
- Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol. 2007;184:53–68. doi: 10.1016/j.jneuroim.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZQ, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M, Wagner EF. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995;9:509–520. doi: 10.1101/gad.9.5.509. [DOI] [PubMed] [Google Scholar]
- Weiss JH, Sensi SL, Koh JY. Zn2+: a novel ionic mediator of neural injury in brain disease. Trends Pharmacol Sci. 2000;21:395–401. doi: 10.1016/s0165-6147(00)01541-8. [DOI] [PubMed] [Google Scholar]
- Yang G, Kitagawa K, Matsushita K, Mabuchi T, Yagita Y, Yanagihara T, Matsumoto M. C57BL/6 strain is most susceptible to cerebral ischemia following bilateral common carotid occlusion among seven mouse strains: selective neuronal death in the murine transient forebrain ischemia. Brain Res. 1997;752:209–218. doi: 10.1016/s0006-8993(96)01453-9. [DOI] [PubMed] [Google Scholar]
- Zhao H, Kalivendi S, Zhang H, Joseph J, Nithipatikom K, Vasquez-Vivar J, Kalyanaraman B. Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide. Free Radic Biol Med. 2003;34:1359–1368. doi: 10.1016/s0891-5849(03)00142-4. [DOI] [PubMed] [Google Scholar]