

Abstract
Tumor necrosis factor (TNF) receptor–associated factor 3 (TRAF3) regulates both innate and adaptive immunity by modulating signaling by Toll-like receptors (TLR) and TNF receptors. TRAF3 was recently identified as a tumor suppressor in human multiple myeloma, suggesting a prominent role in plasma cell homeostasis. We have generated transgenic mice expressing human TRAF3 in lymphocytes. These mice are normal at birth, but they develop over time plasmacytosis and hypergammaglobulinemia, as well as systemic inflammation and tertiary lymphoid organ formation. The analysis of the humoral responses of the TRAF3 mice demonstrated increased responses to T-dependent and T-independent antigens with increased production of antigen-specific immunoglobulin Gs (IgGs) compared with wild-type mice. Furthermore, TLR-mediated IgG production is also increased in TRAF3 B cells. In addition, TRAF3 mice develop autoimmunity and are predisposed to cancer, particularly squamous cell carcinomas of the tongue (≈ 50% incidence) and salivary gland tumors. In summary, TRAF3 renders B cells hyperreactive to antigens and TLR agonists, promoting autoimmunity, inflammation, and cancer, hereby providing a new model for studying de novo carcinogenesis promoted by B cell–initiated chronic inflammation.
Introduction
Tumor necrosis factor (TNF) receptor–associated factors (TRAFs) constitute a family of adapter proteins that interact with the cytosolic regions of multiple TNF-family receptors (TNFRs) upon their activation. TRAFs function as docking molecules for proteins involved in TNFR signaling. Furthermore, most TRAFs also catalyze ubiquitination of various target proteins via their intrinsic E3 ubiquitin ligase activity, stimulating substrate conjugation with either lysine 48- or lysine 63-linked polyubiquitin chains, with differing consequences in terms of proteasome-dependent protein degradation and protein activation, respectively.1–3
TRAF3 is among 6 members of this family of proteins in humans and mice and has been shown to interact with several members of the TNFR family.3,4 Unlike many other TRAF-family proteins that enhance nuclear factor-κB (NF-κB) activation, TRAF3 has been reported to suppress TNFR family–induced NF-κB activation5 and was identified as a negative regulator of NF-κB inducing kinase (Nik), promoting its degradation.6 Consistent with an antagonistic effect of TRAF3 on the alternative NF-κB activation pathway, p100 NF-κB2 deficiency rescues mice from lethality caused by TRAF3 gene ablation.7 Moreover, recent results indicate that mice with TRAF3 deficiency targeted to B cells develop splenomegaly and lymphadenopathy, with hyperglobulinemia and autoimmunity,8 suggesting a role for TRAF3 in B-cell homeostasis. In this regard, a tumor suppressor role for TRAF3 has been revealed in human multiple myeloma (MM). Indeed, mutations resulting in homozygous TRAF3 gene inactivation have been found in 4% to 12% of these plasma cell malignancies.9,10
TRAF3 has also been identified as a key regulator of innate immunity, by participating in Toll-like receptor (TLR)–mediated responses to pathogens.11–14 Furthermore, TLR-function is also required for B-cell responses to T cell–dependent (TD) antigens,15 as well as for germinal center formation and plasma cell differentiation, which suggests that TRAF3 may participate in the regulation of TLR-mediated B-cell responses.
In this report, we describe the generation of lymphocyte-specific TRAF3 transgenic mice. These mice overexpress TRAF3 in B cells, and develop hypergammaglobulinemia, plasmacytosis, autoimmunity, systemic inflammation, and cancer. These findings, showing a key role for TRAF3 in B-cell homeostasis, suggest that TRAF3 might indirectly promote carcinogenesis through B cell–initiated proinflammatory actions. The reported mouse model also provides the first example of solid tumors arising de novo in the setting of B cell–initiated chronic inflammation without requirement for an exogenous carcinogen, thus mimicking human conditions associated with cancer risk in the setting of chronic inflammation and providing a novel animal model for testing chemopreventive strategies for head and neck cancers.
Methods
Transgenic mice
Lymphocyte-specific TRAF3 transgenic FVB/N mice were generated by randomly inserting a cassette encompassing full-length human TRAF3 cDNA under the control of the Vh8C4 promoter and the immunoglobulin H (IgH) μ-chain enhancer (kindly provided by Dr Hitoshi Kikutani, Osaka University). Analysis of the transgenic mouse genotypes was performed by polymerase chain reaction (PCR) using primers specific for human TRAF3, and verification of the transgene expression was accomplished by immunoblotting using an anti-human TRAF3 polyclonal antibody.16 All animal procedures and protocols were approved by the Institutional Animal Care and Use Committee of the Burnham Institute for Medical Research. Euthanasia was performed according to the rules of the American Veterinarian Medical Association. Unless otherwise specified, all data shown were generated using the ϵ-line of TRAF3 transgenic mice and their normal littermate controls.
Cell isolation
Cells were isolated from spleens, lymph nodes, and bone marrow (obtained from femurs). Mononuclear cells were isolated by Ficoll density-gradient centrifugation. B cells were purified using the murine B-cell enrichment cocktail from StemCell Technologies (Vancouver, BC) following the manufacturer's specifications. T cells were purified using mouse T-cell enrichment columns (R&D Systems, Minneapolis, MN). Blood was collected from the cavernous sinus into tubes coated with heparin.
Flow cytometry
Lymphocyte suspensions were incubated with 50 μg/mL human γ-globulin to block Fc receptors. Then, 1 to 5 × 105 cells were incubated with a combination of allophycocyanin (APC)–conjugated, fluorescein isothiocyanate (FITC)–conjugated, or phycoerythrin (PE)–conjugated antibodies recognizing various surface markers (all from BD Biosciences, San Jose, CA). After a 1-hour incubation at 4°C, cells were washed and subjected to flow cytometry analysis (FACScanto; BD Biosciences).
Immunohistochemistry
Tissues and organs from mice were fixed either in Bouin (Sigma-Aldrich, St Louis, MO) or zinc-buffered (Z-fix; Anatech, Hayward, CA) solutions and embedded in paraffin. Tissue sections (5 μm) were stained with hematoxylin and eosin (H&E) or used for immunohistochemistry as described.16
Immunoblotting
Cell lysates from frozen mouse tissues were prepared in modified Laemmli buffer and subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) followed by immunoblot analysis as described.17 Anti–human TRAF316 was previously described. Antibodies against XBP-1, TRAF3 (C-20), and TRAF2 were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-IκBα was from Cell Signaling Technology (Danvers, MA). Anti–mouse Bcl-2 was from BD Biosciences. The light chain of Ig was detected using horseradish peroxidase (HRP)–labeled anti–mouse Ig antibodies (Bio-Rad Laboratories, Hercules, CA).
Cytokine and autoantibody detection
Mouse cytokines were quantified using the mouse Inflammatory Bead Array kit (CBA; BD Biosciences). C-reactive protein (CRP) was detected using a mouse CRP ELISA kit (Kamiya Biomedical, Thousand Oaks, CA). Autoantibodies against double-strand (ds) DNA (Kamiya Biomed), nuclear antigens (ANA), single-strand (ss) DNA, and Sjögren syndrome A antigen (SSA)/Ro (Alpha Diagnostic International, San Antonio, TX) were quantified by enzyme-linked immunosorbent assay (ELISA).
Serum Igs
The amounts of IgM, IgG1, IgG2a, IgG2b in serum from TRAF3, and wild-type mice were determined by isotype-specific ELISA. Briefly, antibodies against mouse IgM, IgG1, IgG2a, and IgG2b (0.25 μg in 50-μL per well) were added to a 96-well plate (Nunc-Immuno plate, MaxiSorp surface; Nalge Nunc International, Roskilde, Denmark) and incubated overnight. Wells were washed with phosphate-buffered saline (PBS) and blocked with PBS containing 0.05% Tween 20. Sera dilutions were added to the wells and incubated for 3 hours. After extensive washing with PBS, alkaline phosphatase (AP)–labeled anti–mouse IgM, IgG1, IgG2a, and IgG2b antibodies recognizing different complementary epitopes were added to the wells and incubated for 2 hours. Wells were washed 3 times with PBS, then isotype-specific Igs were quantified by colorimetric assay (AP substrate kit; Bio-Rad Laboratories). All antibodies used for these experiments were from Southern Biotech (Birmingham, AL)
Antigen challenges
TRAF3 mice and wild-type littermates (3-6 months old) were inoculated either with 25 μg trinitrophenyl-conjugated bovine serum albumin (TNP-BSA) in complete Freund adjuvant (followed 7 days later with the same inoculum, except with incomplete adjuvant) or with 25 μg TNP-conjugated lipopolysaccharide (TNP-LPS). Production of anti-TNP antibodies was determined by ELISA using TNP-conjugated keyhole limpet hemocyanin (TNP-KLH; 0.25 μg/well) as a trapping antigen, and detection of isotype-specific anti-TNP antibodies was achieved using AP-labeled anti-IgM, anti-IgG1, and anti-IgG2a polyclonal antibodies (Southern Biotech). TNP antigens were kindly provided by Dr Ian Zitron (William Beaumont Hospital, Royal Oak, MI).
TLR activation and Ig production
Purified B cells were cultured in RPMI containing 10% fetal calf serum (FCS), 1 mM L-glutamine, antibiotics, oxalacetate, pyruvate, and insulin (OPI) supplement (Sigma-Aldrich), and 50 μM 2-mercaptoethanol. B cells were seeded in 12-well plates (2 × 105 cells/well), then either left untreated or activated with 2 μg/mL LPS or with 1 μM cytosine-phosphate-guanine (CpG; ODN1826; InvivoGen, San Diego, CA). Culture supernatants and cells were collected at various times. The quantification of secreted IgM, IgG1, and IgG2a was performed as described above.
Results
Generation of mice overexpressing TRAF3 in lymphocytes
We produced transgenic mice expressing full-length human TRAF3 under the control of a Vh promoter and an IgH μ-chain enhancer (Figure 1A). Three TRAF3 founders (termed α, ϵ, and μ) showing stable expression of the TRAF3 transgene were obtained (Figure S1A, available on the Blood website; see the Supplemental Materials link at the top of the online article) with the ϵ-line showing the highest levels of expression as determined by immunoblot analysis of human TRAF3 protein levels. We therefore focused our analysis on this transgenic line. Expression of the TRAF3 transgene was restricted to the lymphoid lineage (Figure 1B). TRAF3 mice express the TRAF3 transgene predominantly in B lymphocytes, although expression was also detected in a small subset of thymocytes and T lymphocytes (Figure 1C and data not shown). Analysis of total levels of TRAF3 (endogenous plus transgenic) in purified B cells from spleens of TRAF3 heterozygous mice, by immunoblotting, showed that levels of TRAF3 protein in transgenic B cells are 3- to 7-fold (average 4.8-fold) elevated compared with B cells from nontransgenic (wild-type) mice (Figure 1D). However, this increase in TRAF3 expression is within physiologic levels, since similar amounts of TRAF3 protein are produced upon B-cell activation (Figure 1E).16 Furthermore, the levels of TRAF3 in our mice are similar to those observed in B cells from a transgenic mouse model lacking functional TRAF2 (TRAF2DN; Figure 1D).18 In this regard, increased levels of TRAF3 have also been reported in Traf2-deficient B cells,19 in agreement with a role for TRAF2 in regulating TRAF3 degradation.20
Figure 1.

Generation of TRAF3 transgenic mice. (A) Representation of the TRAF3 transgenic constructs. The various components of transgenic cassette are indicated. (B) Immunoblot analysis of the hTRAF3 transgene expression in various tissues from the TRAF3 transgenic mice (ϵ-line) and wild-type littermates. hTRAF3 expression was detected using an antibody that specifically recognizes human TRAF3, but not mouse TRAF3.16 (C) Analysis of TRAF3 and IκBα expression in total splenocytes and in affinity-purified B lymphocytes and T lymphocytes isolated from TRAF3 transgenic mice and wild-type littermates. The TRAF3 antibody used recognizes only human TRAF3.16 The purity of B cells and T cells was greater than 95%. (D) Purified B cells from spleens of the various transgenic mice indicated and wild-type littermates were lysed in Laemmli buffer and sonicated. Lysates were normalized for protein content, and 25 μg were immunoblotted. Total TRAF3 and IκBα levels are shown. The TRAF3 antibody used (C-20) recognizes both human and mouse TRAF3. (E) B cells from wild-type mice were left resting or stimulated with 2 μg/mL LPS for 16 or 72 hours. TRAF3 and Bcl-2 expression were analyzed by immunoblotting. (F) Kaplan-Meier analysis of the lifespan of wild-type and TRAF3 littermates (17 mice per group). Statistical significance was calculated by log-rank analysis. (G) The weight of spleens from mice up to 8 months (TRAF3, n = 7; wild-type, n = 5) or more than 16 months old (TRAF3, n = 27; wild-type, n = 19) was determined. Statistical significance was determined by unpaired Student t test.
Mice overexpressing TRAF3 have reduced lifespan and develop cachexia
TRAF3 mice are born with normal Mendelian frequencies, lacking any gross abnormalities at birth. However, the survival of TRAF3 mice is reduced compared with wild-type littermates (Figure 1F). Interestingly, TRAF3 mice develop an age-dependent phenotype characterized by oily hair (which becomes evident 2-3 months after birth) and cachexia (weight loss starting 8-12 months after birth), becoming progressively runted as they age. A comparison of wild-type and runted TRAF3 littermates is shown in Figure S2A,B. Weight loss affects both males and females (Figure S2C), although affecting females more severely (Figure S2D). TRAF3 mice with this phenotype lack body fat (Figure S2B), have reduced muscular mass, and show severe reduction of organ sizes and weights, including liver, kidneys, lungs, heart (not shown), and spleen (Figure 1G and Figure S2E,F). The reduction in spleen size is age-dependent, since younger TRAF3 mice did not show significant differences in spleen weight compared with wild-type littermates (Figure 1G and Figure S2E). Similar phenotypes were observed in mice bred into Fvb/n and mixed Fvb/n times BALB/c backgrounds (not shown).
TRAF3 mice have decreased B cells but develop plasmacytosis and hypergammaglobulinemia
Analyses of lymphocyte populations in spleens, lymph nodes (LNs), and bone marrows (BM) from older TRAF3 mice showed a substantial increase in the ratio of T cells versus B cells (Figure S3A). Total numbers of B lymphocytes (B220+) were reduced in older TRAF3 mice, while the number of T cells remained similar to wild-type littermates (Figure S3B). Nonetheless, the ratio of mature (IgDH) and immature (IgMH) B lymphocytes was not altered in TRAF3 mice compared with wild-type littermates (Figure S3C).
Interestingly, even in the earlier stages of the disease, TRAF3 showed significant increases in plasmablast/plasma cell populations compared with wild-type littermates (Figure 2A). This expanded plasma cell population in spleen and lymph nodes from TRAF3 mice expressed B220low-to-null/IgMlow-to-null and high levels of syndecan-1 (Figure 2A) and cytosolic IgG (not shown).
Figure 2.

TRAF3 mice develop plasmacytosis. (A) Phenotypic analysis of plasma cells. Submaxillary lymph nodes (LN) and spleen lymphoid populations were analyzed by 3-color flow cytometry in a representative 14-month-old TRAF3 transgenic mouse (TRAF3) and wild-type littermate (WT). Total nucleated cells were stained for B220, Syndecan-1 and IgM surface expression and analyzed with no gate (top) or selecting the IgM null cells (bottom). Numbers indicate the percentage of cells in that gate. (B) Analysis of plasma cells in lymph nodes. Immunohistochemical analysis of a representative lymph node from a diseased TRAF3 mouse showing severe plasmacytosis. Plasma cells were identified by cytosolic Ig staining. Bars, 100 μm. (C) Plasma cell infiltrates in tissues from TRAF3 mice. Immunohistochemical analysis of the tongue (left) and pancreas (right) of a representative diseased TRAF3 mouse (top and middle panels) and a wild-type littermate (bottom panels). Left, H&E (top and bottom) or anti-IgG (middle) staining of sections of the tongue. Right, H&E staining of infiltrating plasma cell in the pancreas. Bars, 50 μm. (D) Analysis of the expression XBP-1 and IgG in purified B cells. B-cell extracts (20 μg) were analyzed by SDS-PAGE and immunoblotting. (E) TRAF3 mice develop hypergammaglobulinemia. Serum IgG2a, IgG2b, and IgM concentrations from TRAF3 mice (ϵ-line) and wild-type littermates (10-15 months old) were determined using isotype-specific ELISA. P values are indicated in the figure. Statistical significance was determined by unpaired Student t test.
In agreement with this result, immunohistochemical analyses of spleen and lymph nodes of TRAF3 mice revealed severe plasmacytosis. Mature plasma cells, as well as less mature plasmacytoid or plasmablastic cells, were abundant in medullary cords and paracortex of nodes (Figure 2B). Furthermore, histologic analysis of various tissues and organs revealed generalized plasma cell infiltration (Figure 2C). The accumulation of plasma cells was not limited to the ϵ-line of TRAF3 transgenic mice. Although far fewer animals were analyzed, 2 other lines of TRAF3 transgenic mice derived from independent founders (α- and μ-lines) also showed evidence of plasmacytosis or plasmadyscrasias at frequencies higher than control mice. Comparison of the α-, ϵ-, and μ-lines suggests a rough correlation between levels of TRAF3 transgene expression and the frequency of animals showing abnormalities in plasma cell homeostasis (Figure S1A).
Immunoblot analysis of B cells purified from TRAF3 transgenic mice (ϵ-line) showed high levels of XBP-1 protein, typical of mature plasma cells,21 as well as increased levels of IgG (Figure 2D) in TRAF3 mice. The increase in tissue plasma cells was accompanied by hypergammaglobulinemia in TRAF3 mice, which contained increased serum levels of IgG2a and IgG2b, while serum IgM levels remained similar in both control and TRAF3 littermates (Figure 2E). Hypergammaglobulinemia was age-dependent and not observed in younger mice (not shown). Moreover, high levels of IgG2a and IgG2b were simultaneously present in TRAF3 mice, suggesting that plasmacytosis is the result of polyclonal expansions of plasma cells and not a neoplastic process. Thus, TRAF3 appears to promote plasma cell accumulation in vivo.
Immunohistochemical analysis of several tissues from the TRAF3 mice also showed infiltrating immune cells organized into follicles, sometimes with germinal centers. These ectopic lymphoid structures were frequently found in pancreas (Figure 3A), kidney, liver, lung, and in mesentery and gut serosa (not shown) of TRAF3 mice. In contrast, ectopic lymphoid structures were never found in wild-type littermates. Both B lymphocytes and T lymphocytes were abundantly represented within these ectopic lymphoid accumulations (Figure 3B,C), indicating a reactive rather than neoplastic process. Surrounding cuffs of hematopoietic cells composed of lymphocytes (Figure 3D), as well as macrophages (histiocytes), mast cells, and plasma cells were also present within or in adjacency to the lymphoid aggregates, in addition to inflammatory cells dispersed through several organs in TRAF3 mice (data not shown).
Figure 3.

TRAF3 transgenic mice develop systemic inflammation. (A) Histologic analysis. H&E staining shows lymphoid neogenesis (tertiary lymphoid organ [TLO] formation) in omentum and pancreatic parenchyma partly replaced by inflamed adipose tissue in TRAF3 mouse. (B) B cells in TLO were stained with B220 mAb. (C) T cells in TLO were stained with anti-CD3 monoclonal antibody (mAb). (D) H&E staining of an inflammatory cuff surrounding renal artery with thickened tunica media. (E) H&E staining of a joint from a representative TRAF3 mouse with arthritis. Note the distended joint spaces, pannus formation, proliferation of synoviocytes, the irregular proliferation of cartilage at the junction of cartilage bone, and synovium or cartilage erosion. Bars in panels A through E represent 100 μm. (F) Analysis of proinflammatory proteins in serum. Sera from TRAF3 transgenic mice and wild-type littermates (10-15 months old) were analyzed to determine the concentrations of inflammatory cytokines and CRP. Statistical significance was determined by unpaired Student t test. P values are indicated in figure. Hash marks indicate means.
Elevated levels of inflammatory cytokines in the serum of TRAF3 transgenic mice
Levels of inflammatory cytokines were significantly elevated (P < .05) in serum of TRAF3 transgenic mice compared with age-matched wild-type animals, including TNF, monocyte chemoattractant protein-1 (MCP-1) and interferon-γ (IFN-γ), consistent with a systemic inflammatory disorder rather than neoplasia. C-reactive protein (CRP) was also significantly higher in TRAF3 transgenic mice (Figure 3F). Serum levels of interleukin-6 (IL-6), a known growth factor for plasma cells, were also higher in TRAF3 transgenic mice (37.4 ± 3.6 pg/mL) compared with normal (29.4 ± 9.1 pg/mL), but did not reach statistical significance (P = .20).
TRAF3 mice develop autoimmunity
We measured autoantibodies in serum of mice. TRAF3 mice contained significantly higher concentrations of anti-nuclear antigens (ANA), (P = .001), anti-dsDNA antibodies (P < .001), anti-ssDNA antibodies (P = .001), and anti-SSA/Ro antibodies (P = .001), compared with wild-type littermates (Figure 4A).
Figure 4.

TRAF3 mice develop autoimmunity. (A) Analysis of circulating autoantibodies in TRAF3 mice and wild-type littermates. Blood from TRAF3 mice (12-17 months old) and from wild-type littermates was extracted for serum collection. The presence of autoantibodies in serum was determined by ELISA (anti-dsDNA, 1:150 dilution; anti-ANA, anti-ssDNA, and anti-SSA/Ro, 1:50 dilution). Statistical significance was determined by unpaired Student t test. (B) Pathologic alterations in kidneys from TRAF3 mice. Morphologic lesions encompass both glomerular (i-xi) and tubulointerstitial (i,iii) alterations, demonstrated by H&E (i-iii), Jones silver methanamine (iv,v), and Masson trichrome (vi,vii) stains. Depositions of IgG (viii,ix) and C3 complement (x,xi) were detected by immunostaining. Bars, 50 μm.
Histologic analysis of kidneys from the TRAF3 mice also showed clear evidence of autoimmune kidney disease, involving primarily glomerular lesions but also tubulointerstitial changes (Figure 4B). Although the majority of the affected glomeruli were characterized by focal segmental damage, global lesions encompassing the total glomerular tuft were occasionally noticed (Figure 4Bi). Glomerular alterations included mesangial hypercellularity, mesangial matrix expansion with Ig deposition, glomerulonephritis with necrotizing and sclerosing lesions (Figure 4Bi-iii), segmental necrosis associated with epithelial crescents, and alterations of peripheral capillary loops. Lobular accentuation and mesangialization of the peripheral capillary loops, associated with thickening of the peripheral capillary walls, were demonstrated by Jones silver methenamine (Figure 4Biv,v) and Masson trichrome (Figure 4Bvi,vii) stains. IgG (Figure 4Bviii,ix) and complement (C3; Figure 4Bx,xi) immunostainings visualized deposits confined to the mesangium or paramesangium or scattered in the subepithelial region. In addition, interstitial inflammation, fibrosis, and tubular epithelial changes were frequently present (Figure 4Biii). Pronounced renal pathology was found in 54% of TRAF3 males (14/26; 13-31 months old) and 51% of females (20/39; 7-31 months old) but no wild-type animals. In contrast, occasional wild-type littermates (5/23; 22%) had only very mild renal lesions, limited to scattered mononuclear cell infiltrations.
TRAF3 mice also develop arthritis (Figure 3E). In this regard, these mice had distended joint spaces, pannus formation, proliferation of synoviocytes, and irregular proliferation of cartilage and/or cartilage erosion at the junction of cartilage bone and synovium. In addition to joint involvement, foci of lymphocytic periductal infiltration were detected in the submaxillary and parotid glands (Figure 6F,G) of 5 of 27 male (19%) and in 1 of 30 female (3%) TRAF3 mice. These inflammatory salivary gland lesions occurred in the absence of other oral cavity disease (see below), implying they arose de novo and were not secondary to trauma or malignancy. Altogether, these findings are consistent with development of severe autoimmune disease in TRAF3 mice, probably most consistent with lupus erythematosus, with Sjögren features.
Figure 6.

Cancer development in TRAF3 transgenic mice. (A) Percentage of tumors found in wild-type mice (n = 24; 17 ± 1.5 months) and in TRAF3 mice (n = 70; 18 ± 0.8 months). (B) Graphic representation of percentage of mice bearing tumors in the tongue (squamous cell carcinoma, n = 19; papilloma, n = 2), other oral tumors (pleomorphic adenomas, cystadenomas, and adenocarcinomas), lung carcinomas (papillary bronchioloalveolar carcinoma, n = 15; adenoma, n = 2), and hepatocarcinoma (n = 7). (C) Comparative analysis of tongues from 2-month-old (bottom) and 14-month-old (top) TRAF3 mice and wild-type littermates. The arrow indicates the squamous cell carcinoma in the tongue. (D,E) Squamous cell carcinoma of the tongue (H&E). (F,G) Example of lymphoid infiltration in submaxillary gland and parotid gland, respectively, in TRAF3 mice. (H,I) H&E staining of parotid glands with pleomorphic adenoma and sarcoma, respectively. (J) Papillary bronchioloalveolar carcinoma of lung in a TRAF3 mouse. (K) H&E staining of hepatocellular carcinoma in TRAF3 mouse. Note the presence of subcapsular fat necrosis with chronic inflammatory cells. Bars (D-K), 100 μm.
Mice treated with pristane (2,6,10,14-tetramethyl-pentadecane) develop lymphoid neogenesis and autoimmunity,22 as well as plasmacytoma on susceptible strain backgrounds.23 Treating TRAF3 mice with pristane resulted in exacerbated inflammation, necrosis, and ectopic lymphoid organ formation compared with untreated aged-matched TRAF3 mice and to pristane-treated wild-type littermates (not shown). However, pristane-treated TRAF3 mice bred onto the pristane-sensitive BALB/c background did not develop myeloma, indicating that TRAF3 overexpression in B cells predisposes to inflammation but not myleoma or other B-cell malignancies.
Enhanced humoral responses in TRAF3 mice
Because TRAF3 mice have hypergammaglobulinemia and produce autoantibodies, we functionally assessed humoral immune responses in TRAF3 mice. For these experiments, TRAF3 mice and wild-type littermates were challenged with either TNP-BSA (T-dependent antigen) or TNP-LPS (T-independent antigen). IgM-specific responses to these antigens were similar in TRAF3 and wild-type mice (Figure 5A). However, IgG1 and particularly IgG2 responses to these antigens were significantly enhanced in TRAF3 mice. This enhanced antibody response was particularly evident in TRAF3 mice challenged with LPS, resulting in significantly higher amounts of antigen-specific IgG2a and IgG2b antibodies compared with wild-type littermates (Figure 5A). Thus, TRAF3 improves humoral responses to both TD and T cell–independent (TI) antigens.
Figure 5.

TRAF3 mice exhibit enhanced humoral responses to TD and TI antigens. (A) Mice were challenged with TNP-BSA (T-dependent antigen) in complete Freund adjuvant or with TNP-LPS (T-independent antigen). Blood was collected before and after injection of TRAF3 mice (◇) and wild-type littermates (■) with antigens at the days indicated. Titers of anti-TNP IgM and anti-TNP IgG2 were determined by ELISA using TNP-KLH–coated plates. Data represent mean (± standard error of the mean [SEM]; n = 4). (B) B-cell antibody responses to TLR agonists. B cells were cultured in 12-well plates (2 × 105 cells/mL) and either untreated or treated with 2 μg/mL LPS or 1 μM CpG (ODN1826). Culture supernatants were collected at various days, and secreted IgM, IgG1, and IgG2a were quantified. Data represent mean (± SEM) for TRAF3 mice (n = 5,  ) and wild-type littermates (n = 4, ■).
) and wild-type littermates (n = 4, ■).
TRAF3 B cells are hyperresponsive to TLR activation
A role for TRAF3 in innate immune responses has been previously suggested.11,12 Growing evidence indicates that inappropriate activation of TLRs may be involved in development of lupus erythematosus and other autoimmune diseases.24 Given that TRAF3 mice exhibited enhanced humoral responses to TLR agonists and develop autoimmunity, we assessed Ig production by B cells activated with the TLR agonists LPS and CpG. TRAF3 B cells activated with either LPS or CpG showed a striking increase in IgG1 and IgG2a production compared with wild-type B cells (Figure 5B). In contrast, LPS- and CpG-induced IgM production was similar for TRAF3 and wild-type B cells.
To test whether hyperresponses to TLR were the result of accelerated B-cell proliferation, purified B cells from TRAF3 and wild-type mice were culture in the presence of a variety of TLR agonists. TRAF3 and wild-type B cells proliferate at similar rates in response to TLR activation (Figure S4A). These results suggest that TRAF3 does not affect TLR-mediated B-cell proliferation but rather regulates TLR-mediated B-cell differentiation.
TRAF3 overexpression reduces Nik, but does not alter CD40- or BAFF-R–mediated B-cell proliferation and signaling
TRAF3 has been shown to interact with and regulate CD40 and BAFF-R.4 Therefore, to determine whether TRAF3 overexpression alters the normal responses to these receptors, we first assessed whether TRAF3 overexpression in B cells altered their proliferation in response to CD40 and BAFF-R activation, but found that TRAF3 B cells proliferate at similar rates compared with wild-type B cells in response to these agents (Figure S4B,C), either alone or in combination with B-cell receptor (BCR) activation (Figure S4D).
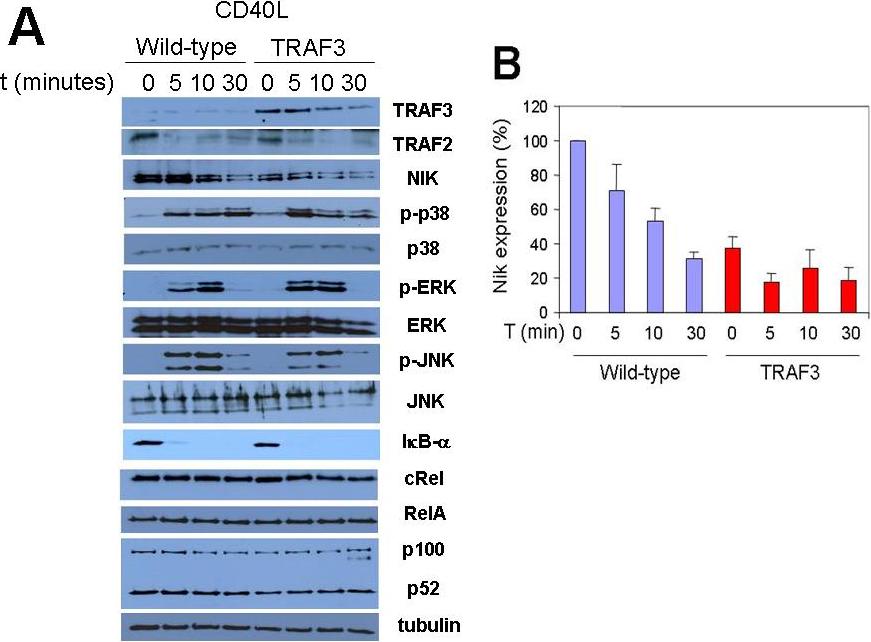
As expected, TRAF3 overexpression reduced endogenous levels of Nik (Figure S5A,B), in agreement with the previously described role of TRAF3 in controlling Nik degradation.6,25 The average levels of Nik expression in TRAF3 mice were 37% (± 7%) of wild-type mice. While starting with lower basal levels of Nik, stimulation with either CD40L or BAFF reduced Nik protein levels in B cells from TRAF3 mice, similar to the wild-type B cells (Figure S5A,B). TRAF3 expression in B cells had no discernible effect on MAP3K activation (JNK, p38, and ERK), IκB degradation, p100 processing, p52 levels, or TRAF2 down-regulation in response to activation either with CD40L (Figure S5A) or BAFF (not shown) in TRAF3 B cells compared with wild-type B cells within the time frame examined here.
TRAF3 mice are predisposed to cancer
Most TRAF3 mice (47 of 70; 67%) developed solid tumors with aging. In contrast, only a minority of wild-type mice (3 of 24; 13%) developed malignancies over the same time frame (Figure 6A). The most frequent tumor developed by the TRAF3 mice was squamous cell carcinoma of the tongue (Figure 6B,C), which was found in 50% of TRAF3 mice (19 of 38; 40% of TRAF3 males [n = 15] and 57% of TRAF3 females [n = 23]). Examples of these cancers are shown in Figures 6D,E. Tongue tumors were invaded by inflammatory cells, predominantly granulocytes and macrophages, with occasional plasma cells. Mice with tongue tumors uniformly had large reactive submandibular lymph nodes. No tongue tumors were found in wild-type mice (n = 11). Furthermore, tumors were also found in 43% of the parotid glands derived from TRAF3 females (6/14), but only in 1 of 11 of TRAF3 males (9%; Figure 6H,I). Parotide tumors included pleiomorphic adenomas, cystadenomas, and spindle cell carcinomas. Lymphoid infiltration in the parotid glands was also evident (Figure 6G). Thus, TRAF3 mice are predisposed to developing tumors of the oral cavity and salivary glands.
Lung cancers (Figure 6J) were found in 27% of TRAF3 mice (17 of 63; 39% of TRAF3 males [n = 28] and 17% of TRAF3 females [n = 35]), comprising 15 papillary bronchioloalveolar carcinomas (Figure 6J) and 2 adenomas, whereas only 14% (n = 22) of wild-type littermates developed lung cancers. Interestingly, inflammatory lesions such as perivascular cuffing were frequently found in the vicinity of these solid tumors in TRAF3 mice. Moreover, peribronchial mononuclear cell infiltrations, lymphocytic interstitial pneumonitis, and occasional pleuritis were also commonly observed in TRAF3 mice (not shown). Altogether, inflammatory pulmonary lesions were present in 38% (n = 63) of TRAF3 mice (18% [n = 28] of TRAF3 males and 54% [n = 35] of TRAF3 females), whereas lung tumors were present in 27% (n = 63) of these mice, suggesting that lung inflammation commonly accompanies the development of lung cancer in these mice.
Hepatocellular carcinoma (Figure 6K) was found in 18% (n = 38) of female TRAF3 mice, but none of the TRAF3 males (n = 27) or wild-type littermates (n = 22). Inflammatory changes, such as portal/periportal or periseptal inflammation with lymphocyte aggregates (chronic hepatitis), focal lytic necrosis, and peliosis hepatis were found in 22% (n = 27) of TRAF3 males and 44% (n = 34) of TRAF3 females (Figure 6H). In contrast, the only liver alteration found in wild-type mice was microgranuloma (9%; n = 22), which is common in mice and not necessarily associated with liver pathology.
Discussion
TRAF3 was recently described as a tumor suppressor candidate in human MM, implying an important role for this adapter protein in B-cell and plasma cell biology. As many as 4% to 12% of MM specimens and 17% of MM cell lines harbor homozygous deletions or inactivating mutations in their traf3 genes.9,10 Mice lacking TRAF3 (traf3−/−) in B cells through tissue-specific gene ablation develop splenomegaly and lymphadenopathy, as well as hyperglobulinemia and autoimmunity,8 consistent with the idea that elimination of TRAF3 removes an obstacle to B-cell expansion in vivo. In contrast to mice with traf3−/− B cells (ϵ-line), our animals overexpressing TRAF3 in B cells developed hypotrophy of spleen with aging and decreased numbers of B cells in vivo. Remarkably, TRAF3 transgenic mice develop hypergammaglobulinemia and severe plasmacytosis in spleen and lymph nodes, with frequent infiltration of plasmablasts and plasma cells in tissues. Thus, despite TRAF3's potential to inhibit B-cell accumulation in vivo, plasma cell expansion is enhanced in TRAF3 transgenic mice, which seems at odds with a tumor suppressor role for TRAF3 in myeloma. These results strongly suggest a role for TRAF3 in promoting plasma cell differentiation, where the overexpression of TRAF3 in B cells drives these cells toward a plasma cell phenotype. Conversely, however, the absence of TRAF3 does not prevent B cells from differentiating into antibody-producing plasma cells, as indicated by development of hyperglobulinemia in mice harboring TRAF3-deficient B cells.8 Thus, TRAF3 is not required for B cells to differentiation into plasma cells, although it may enhance this differentiation. Importantly, we have previously reported that endogenous TRAF3 is prominently expressed in normal plasma cells in both humans and mice,16 supporting a physiologic role for TRAF3 in plasma cell differentiation.
The apparent paradox that both TRAF3 overexpression and TRAF3 knockout cause hyperglobulinemia and autoimmunity in vivo could reflect different roles for TRAF3 that result in a similar phenotype, but for different reasons. For example, TRAF3 deficiency may cause plasma cell expansion (and thus hyperglobulinemia) because it prolongs plasma cell survival,8 while TRAF3 overexpression may cause plasma cell expansion because it drives B-cell differentiation. In this regard, traf3−/− B cells reportedly have increased survival ex vivo, which might account for in vivo plasma cell accumulation in traf3−/− mice. Conversely, we can eliminate a role for TRAF3 in controlling B-cell proliferation, in that B cells from our TRAF3 transgenic mice exhibit normal proliferative responses in culture compared with wild-type B cells when stimulated with several different stimuli, including CD40L, BAFF, and TLR agonists, which is in agreement to what has been described for traf3−/− B cells.8 Thus, TRAF3 seems to have complex effects on B-cell survival and differentiation, which should be considered in the context of TRAF3's putative role as a tumor suppressor.
Role of TRAF3 in the control of humoral responses
TRAF3 loss-of-function and gain-of-function through genetic manipulations in B cells result in increased in vivo Ig production and development of autoimmunity, as described above, suggesting that TRAF3 has a critical role in the control of tolerance. These seemingly contradictory results might underline 2 different mechanisms controlled by TRAF3. While lack of TRAF3 increases survival of B cells and consequently dysregulates B-cell homeostasis, which might result in the expansion of autoreactive B cells and autoimmunity,8 TRAF3 overexpression in B cells enhances humoral responses against TD and TI antigens, essentially improving antibody generating activity, presumably by promoting B-cell differentiation. Consistent with the hypothesis that different mechanisms underlie these seemingly similar phenotypes of TRAF3 gain- and loss-of-function, the time-course with which traf3−/− and TRAF3 transgenic mice manifest their antibody-mediated pathologies differs. Thus, mice lacking TRAF3 in B cells develop hyperglobulinemia and autoimmunity at young age, consistent with a role of TRAF3 in the control of B-cell homeostasis with immediate effects on Ig production. In contrast, mice overexpressing TRAF3 in B cells develop hypergammaglobulinemia and autoimmune pathology at older ages (> 1 year), with younger TRAF3 transgenic mice showing no evidence of disease, consistent with a cumulative effect of TRAF3 overexpression in enhancing humoral responses during a lifetime of antigenic exposure. Consistent with this interpretation, the ability of TRAF3 B cells to mount enhanced humoral responses to antigens in vivo, and upon TLR activation in vitro, was already evident in young (3-6 months old) asymptomatic mice.
Another striking difference in traf3−/− and TRAF3 transgenic mice is found in the primary (IgM) versus secondary (IgG) antibody responses. In this regard, TRAF3 transgenic mice that develop hyperglobulinemia have similar IgM titers compared with wild-type littermates. Moreover, IgM production in response to both TI and TD antigens in vivo and to TLR activation in vitro is similar in both TRAF3 and wild-type mice. In contrast to IgM, it is IgG titers that rise in TRAF3 mice with aging, and production of IgG, in particular IgG2a and IgG2b, is greatly enhanced upon immunization, especially with TI antigens and upon activation with TLR agonists. These results suggest that TRAF3 overexpression in the B-cell lineage does not regulate the initiation of the humoral response, but rather controls later stages of B-cell differentiation or the duration of antibody responses. In contrast, mice lacking TRAF3 in B cells show increased Ig production in response to TI antigens, including IgM and IgG isotypes, which is more consistent with heightened antibody responses as a result of increased survival of antigen-challenged B cells, rather than an alteration in B cell differentiation,8 as elaborated above. Altogether, these results support the idea that TRAF3 has different functions at different stages of B-cell development.
Recent data suggest that TRAF3 is a key regulator of innate immunity, by participating in the control of TLR-mediated signal transduction pathways activated by pathogens.11,12 Our results show that TRAF3 is indeed involved in TLR-mediated humoral responses and that its overexpression in B cells greatly enhances humoral responses to both TI and TD antigens. Furthermore these results are also generally in agreement with the recently described role for TLRs in the control of B cell–dependent responses to TD antigens,15 which showed that TLR signaling is required for germinal center formation and plasma cell differentiation. Thus, we favor the idea that TLR-mediated signaling (rather than BCR-mediated signaling) is the principal context in which TRAF3 overexpression in B cells leads to heightened antibody responses and plasma cell expansion.
B cells, chronic inflammation, and cancer
TRAF3 mice develop systemic inflammation, manifested by high levels of inflammatory cytokines in serum, elevated CRP, white blood cell infiltration into tissues, and ectopic lymphoid organ formation. Furthermore, TRAF3 mice also develop cancer with high incidence, in particular squamous cell carcinomas of the oral cavity. Although we cannot exclude a gene expression–modifying role of transgene insertion into the mouse genome, we hypothesize that the frequent development of cancers in TRAF3 transgenic mice results from chronic inflammation.
Abundant data have established that cancer is promoted by chronic inflammation.26,27 In particular, it is well documented that chronic inflammation, irrespective of the underlying etiology, can lead to the development of squamous cell carcinomas.28,29
Interestingly, De Visser and coworkers30 using a transgenic model of enforced expression of human papillomavirus type 16 (HPV16) early-region genes in keratinocytes, have recently shown that B lymphocytes are required for establishing chronic inflammatory states that promote de novo carcinogenesis. Indeed, malignant transformation of epithelium seems to require the participation of B cells and Ig depositions in premalignant lesions. The inflammatory alterations and cancer developed by the B cell–specific TRAF3 mice may have a similar etiology. However, we have failed to consistently observe Ig depositions in the squamous cell carcinomas developed by the TRAF3 mice (not shown). While further research will be required to assess the role of Ig deposition in inflammation and cancer development in the TRAF3 transgenic mice, it is worth noting that these mice require not exogenous carcinogens or oncoproteins to develop tumors, representing the first model of de novo carcinogenesis involving B cell–initiated inflammation and providing a novel model for testing chemopreventive therapies aimed at oral cancers, a setting where premalignant lesions progress to full-blown malignancy with the assistance of chronic inflammation.
Supplementary Material
Acknowledgments
We are indebted to S. Hourmezian, M. Thomas, X.-S. Huang, B. Charbono, and J. Groos. We thank Dr Craig Franklin (University of Missouri, Columbia, MO), and Drs Herbert C. Morse III and Jerrold M. Ward (National Institute of Allergy and Infectious Diseases, Rockville, MD) for expert pathology opinion.
This work was supported by National Institutes of Health grants AI-069356 and CA-69381, Programa Ramón y Cajal, SAF2004-7675, and a fellowship from the Spanish Ministerio de Sanidad y Consumo (FI05/00 191).
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: D.L. and M.K. contributed equally to this work; J.M.Z. contributed to all scientific aspects of the manuscript and wrote the paper; M.K. contributed the immunohistochemical analyses; D.L. designed and performed research and analyzed data; S.L. and C.L.K. performed research; and J.C.R. contributed by designing research, analyzing data, and writing the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current address for Dr Llobet is Hospital Universitari Arnau de Vilanova, Servei Anatomia Pathològica-IRB, 25198 Lleida, Spain. The current address for Dr Lefebvre is 236 rue du Trianon, 34400 Lunel, France.
Correspondence: Juan M. Zapata, Centro de Biología Molecular Severo Ochoa, UAM/CSIC, Cantoblanco, Madrid 28049, Spain; e-mail: jmzapata@cbm.uam.es; or John C. Reed, Burnham Institute for Medical Research, 10901 N Torrey Pines Rd, La Jolla, CA 92037; e-mail: reedoffice@burnham.org.
References
- 1.Zapata JM. TRAF3: B cells and beyond. Mod Asp Immunobiol. 2002;2:236–240. [Google Scholar]
- 2.Zapata JM, Martinez-Garcia V, Lefebvre S. Phylogeny of the TRAF/MATH domain. Adv Exp Med Biol. 2007;597:1–24. doi: 10.1007/978-0-387-70630-6_1. [DOI] [PubMed] [Google Scholar]
- 3.He JQ, Oganesyan G, Saha SK, Zarnegar B, Cheng G. TRAF3 and its biological function. Adv Exp Med Biol. 2007;597:48–59. doi: 10.1007/978-0-387-70630-6_4. [DOI] [PubMed] [Google Scholar]
- 4.Zapata JM, Lefebvre S, Reed JC. Targeting TRAfs for therapeutic intervention. Adv Exp Med Biol. 2007;597:188–201. doi: 10.1007/978-0-387-70630-6_15. [DOI] [PubMed] [Google Scholar]
- 5.Hauer J, Puschner S, Ramakrishnan P, et al. TNF receptor (TNFR)-associated factor (TRAF) 3 serves as an inhibitor of TRAF2/5-mediated activation of the noncanonical NF-κB pathway by TRAF-binding TNFRs. Proc Natl Acad Sci U S A. 2005;102:2874–2879. doi: 10.1073/pnas.0500187102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liao G, Zhang M, Harhaj EW, Sun SC. Regulation of the NF-κB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. 2004;279:26243–26250. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- 7.He JQ, Zarnegar B, Oganesyan G, et al. Rescue of TRAF3-null mice by p100 NF-κ B deficiency. J Exp Med. 2006;203:2413–2418. doi: 10.1084/jem.20061166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie P, Stunz LL, Larison KD, Yang B, Bishop GA. Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity. 2007;27:253–267. doi: 10.1016/j.immuni.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keats JJ, Fonseca R, Chesi M, et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Annunziata CM, Davis RE, Demchenko Y, et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oganesyan G, Saha SK, Guo B, et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2005;439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- 12.Hacker H, Redecke V, Blagoev B, et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2005;439:204–207. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- 13.Saha SK, Pietras EM, He JQ, et al. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and cardif. EMBO J. 2006;25:3257–3263. doi: 10.1038/sj.emboj.7601220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kayagaki N, Phung Q, Chan S, et al. DUBA: a deubiquitinase that regulates type I interferon production. Science. 2007;318:1628–1632. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- 15.Pasare C, Medzhitov R. Control of B-cell responses by Toll-like receptors. Nature. 2005;438:364–368. doi: 10.1038/nature04267. [DOI] [PubMed] [Google Scholar]
- 16.Krajewski S, Zapata JM, Krajewski M, et al. Immunohistochemical analysis of in vivo patterns of TRAF-3 expression, a member of the TNF receptor-associated factor family. J Immunol. 1997;159:5841–5852. [PubMed] [Google Scholar]
- 17.Zapata JM, Takahashi R, Salvesen GS, Reed JC. Granzyme release and caspase activation in activated human T-lymphocytes. J Biol Chem. 1998;273:6916–6920. doi: 10.1074/jbc.273.12.6916. [DOI] [PubMed] [Google Scholar]
- 18.Lee SY, Reichlin A, Santana A, Sokol KA, Nussenzweig MC, Choi Y. TRAF2 is essential for JNK but not NF-κB activation and regulates lymphocyte proliferation and survival. Immunity. 1997;7:703–713. doi: 10.1016/s1074-7613(00)80390-8. [DOI] [PubMed] [Google Scholar]
- 19.Grech AP, Amesbury M, Chan T, Gardam S, Basten A, Brink R. TRAF2 differentially regulates the canonical and noncanonical pathways of NF-κB activation in mature B cells. Immunity. 2004;21:629–642. doi: 10.1016/j.immuni.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 20.Hostager BS, Haxhinasto SA, Rowland SL, Bishop GA. Tumor necrosis factor receptor-associated factor 2 (TRAF2)-deficient B lymphocytes reveal novel roles for TRAF2 in CD40 signaling. J Biol Chem. 2003;278:45382–45390. doi: 10.1074/jbc.M306708200. [DOI] [PubMed] [Google Scholar]
- 21.Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol. 2005;5:230–242. doi: 10.1038/nri1572. [DOI] [PubMed] [Google Scholar]
- 22.Nacionales DC, Kelly KM, Lee PY, et al. Type I interferon production by tertiary lymphoid tissue developing in response to 2,6,10,14-tetramethyl-pentadecane (pristane). Am J Pathol. 2006;168:1227–1240. doi: 10.2353/ajpath.2006.050125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Potter M, Morrison S, Wiener F, Zhang XK, Miller FW. Induction of plasmacytomas with silicone gel in genetically susceptible strains of mice. J Natl Cancer Inst. 1994;86:1058–1065. doi: 10.1093/jnci/86.14.1058. [DOI] [PubMed] [Google Scholar]
- 24.Krieg AM, Vollmer J. Toll-like receptors 7, 8, and 9: linking innate immunity to autoimmunity. Immunol Rev. 2007;220:251–269. doi: 10.1111/j.1600-065X.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- 25.He JQ, Saha SK, Kang JR, Zarnegar B, Cheng G. Specificity of TRAF3 in its negative regulation of the noncanonical NF-κ B pathway. J Biol Chem. 2007;282:3688–3694. doi: 10.1074/jbc.M610271200. [DOI] [PubMed] [Google Scholar]
- 26.Tan TT, Coussens LM. Humoral immunity, inflammation and cancer. Curr Opin Immunol. 2007;19:209–216. doi: 10.1016/j.coi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 27.Chen K, Huang J, Gong W, Iribarren P, Dunlop NM, Wang JM. Toll-like receptors in inflammation, infection and cancer. Int Immunopharmacol. 2007;7:1271–1285. doi: 10.1016/j.intimp.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 28.Carlson JA, Ambros R, Malfetano J, et al. Vulvar lichen sclerosus and squamous cell carcinoma: a cohort, case control, and investigational study with historical perspective; implications for chronic inflammation and sclerosis in the development of neoplasia. Hum Pathol. 1998;29:932–948. doi: 10.1016/s0046-8177(98)90198-8. [DOI] [PubMed] [Google Scholar]
- 29.Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncology (Williston Park) 2002;16:217–226. 229. discussion 230–212. [PubMed] [Google Scholar]
- 30.de Visser KE, Korets LV, Coussens LM. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell. 2005;7:411–423. doi: 10.1016/j.ccr.2005.04.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}