

Abstract
A preclinical humanized mouse model of β thalassemia major or Cooley anemia (CA) was generated by targeted gene replacement of the mouse adult globin genes in embryonic stem cells. The mouse adult α and β globin genes were replaced with adult human α globin genes (α2α1) and a human fetal to adult hemoglobin (Hb)–switching cassette (γHPFHδβ0), respectively. Similar to human infants with CA, fully humanized mice survived postnatally by synthesizing predominantly human fetal Hb, HbF (α2γ2), with a small amount of human minor adult Hb, HbA2 (α2δ2). Completion of the human fetal to adult Hb switch after birth resulted in severe anemia marked by erythroid hyperplasia, ineffective erythropoiesis, hemolysis, and death. Similar to human patients, CA mice were rescued from lethal anemia by regular blood transfusion. Transfusion corrected the anemia and effectively suppressed the ineffective erythropoiesis, but led to iron overload. This preclinical humanized animal model of CA will be useful for the development of new transfusion and iron chelation regimens, the study of iron homeostasis in disease, and testing of cellular and genetic therapies for the correction of thalassemia.
Introduction
Hemoglobin (Hb) is the oxygen carrier in red blood cells (RBCs). Hb is a tetramer composed of 2 α-like and 2 β-like globin chains. Thalassemia is a common genetic blood disorder caused by an imbalance in the production of α and β globin chains. The most severe form of β thalassemia (β thalassemia major, β0 thalassemia, or Cooley anemia [CA]) is marked by the absence of adult β globin chains, resulting in clinically severe anemia that requires regular blood transfusion. Because human RBCs contain high HbF levels at birth, CA patients do not develop severe anemia until after their fetal to adult Hb switch is completed during the first year of life.
In the absence of β globin chains in thalassemia major, excess α globin chains precipitate forming inclusions that can cause the premature intramedullary apoptosis of erythroblasts in the bone marrow.1–3 The anemia triggered by this ineffective erythropoiesis results in erythroid hyperplasia in the marrow and extramedullary erythropoiesis in tissues like the liver. Thalassemic erythroblasts that escape early destruction can complete their maturation to reticulocytes and exit the bone marrow into the peripheral blood. However, these damaged erythrocytes have a shortened lifespan in the circulation, and their hemolysis leads to severe anemia.4,5
Without treatment, β thalassemia major is lethal in early childhood. Treatment consists of lifelong blood transfusions to correct the anemia and suppress ineffective erythropoiesis. Iron overload, a negative consequence of chronic blood transfusion, can lead to heart failure, liver disease, and endocrinopathies.6 Hemosiderosis is managed with iron chelation therapy. The availability of a preclinical animal model would enable the development of novel therapies for the clinical treatment of thalassemia major.
The lack of a mouse model that recapitulates the perinatal timing of human fetal to adult Hb switching has hindered the postnatal study of CA in vivo. Because mice have no fetal globin gene equivalent,7,8 early models of β thalassemia major were all embryonic lethal.9–14 One adult CA model that is generated by lethal irradiation of wild-type mice, followed by transplantation with fetal liver cells collected from homozygous mouse β globin knockout embryos, survives until their endogenous wild-type RBCs senesce.15 However, unlike CA patients, this interesting model is nonheritable; has a late adult onset; and lacks reticulocytosis, hemolysis, and Hb switching; and the donor erythroblasts exhibit little apoptosis or the capacity for maturation.15,16
Recently, we generated a humanized CA mouse model by targeted gene replacement of the mouse adult β globin genes with a human γβ0 globin gene-switching cassette.17 Similar to humans, these mice undergo 2 β-like Hb switches during development, from murine embryonic to human fetal and then finally to a nonfunctional adult human β0 globin gene. Although human HbF levels are high enough for the mice to survive to birth, the mice soon expire from severe anemia upon completion of the human γ to β0 Hb switch.17
In this report, novel humanized CA mice are produced that more closely mimic the disease in humans by increasing the postnatal levels of human HbF and HbA2. Fully humanized CA mice are born alive, surviving solely upon human HbF and minor amounts of HbA2. CA mice become progressively anemic upon completion of the fetal to adult Hb switch, and the majority died before weaning age. Blood transfusions can rescue CA mice from their lethal anemia with a concomitant rise in tissue iron stores. This preclinical humanized animal model of CA will be useful for the development of new transfusion and iron chelation regimens, the study of iron homeostasis in disease, and testing of cellular and genetic therapies for the correction of thalassemia.
Methods
Generation of humanized γHPFHδβ0 knockin mice
A targeting construct, γHPFHδβ0, was built in a pBluescript vector (Stratagene, La Jolla, CA) containing from 5′ to 3′ a phosphoglycerate kinase (pgk) promoter driving thymidine kinase, 1.7 kb of mouse homology upstream of the mouse βmajor(maj) globin gene (HindIII fragment), 4.7-kb human Aγ globin gene fragment (Accession U01317: 39 013-43 728), 2.9-kb human δ globin gene fragment (U01317: 54 336-57 279), 4.1-kb human β globin gene fragment (U01317: 61 320-65 426), a pgk promoter driving hygromycin (pgk-hyg) resistance gene flanked by 2 loxP sites, and 7 kb of mouse homology downstream of the mouse βminor(min) globin gene (BamHI fragment). The hereditary persistence of fetal Hb (HPFH) mutation, guanine (G) to adenine (A) transition at position −117 of the human Aγ globin gene, and the human β0 globin gene mutation, G to A transition in the first base of intervening sequence 1 (IVS1.1 G to A), were introduced by polymerase chain reaction (PCR). The plasmid was linearized by NotI digestion before electroporation into the mouse embryonic stem (ES) cell line. The ES cells were selected in hygromycin B (125 μg/mL) and ganciclovir (1 mM) in ES cell media (Dulbecco modification of Eagle medium, 15% fetal bovine serum [HyClone, Logan, UT], 1× nucleosides, 2 mM l-glutamine, 1× nonessential amino acids, 50 IU/mL penicillin, 50 μg/mL streptomycin, 0.1 mM β-mercaptoethanol, and 1000 U/mL leukemia inhibitory factor). After selection, DNA from drug-resistant ES cell colonies was screened by PCR to identify the homologous recombinants. Mice were cloned by injecting the ES cells into 8-cell stage C57BL/6J (The Jackson Laboratory, Bar Harbor, ME) blastocysts18,19 that were transferred into the uteri of outbred CD1 (Charles River Laboratories, Wilmington, MA) pseudopregnant recipient mice. The cloned mice were interbred to hCMV-Cre transgenic mice20 and human α2α1 globin knockin (KI) mice (T.M.R., unpublished data, December 2004) and to delete the hygromycin marker gene and generate doubly heterozygous γHPFHδβ0 and α2α1 KI mice, respectively. Fully humanized CA mice were produced by breeding 2 heterozygous KI mice. All procedures were approved by the University of Alabama at Birmingham (UAB) Institutional Animal Care and Use Committee.
High-performance liquid chromatography analysis of Hb chains
Hemolysates are prepared by lysing washed RBCs in hemolysate buffer (5 mM phosphate, 0.5 mM EDTA [ethylenediaminetetraacetic acid], pH 7.4), adding NaCl to 1%, and removing RBC membranes by centrifugation. A linear gradient of increasing acetonitrile with 0.1% trifluoroacetic acid at a 1.0 mL/minutes flow rate was used to separate human and mouse globin chains on a reverse-phase C4 column (Vydac, Hesperia, CA) on a Surveyor high-performance liquid chromatography (HPLC) instrument (Thermo Scientific, Waltham, MA).
Hematologic indices and histopathology
Peripheral blood was collected from anesthetized mice into Microtainer EDTA collection tubes (BD Biosciences, Franklin Lakes, NJ). Blood smears were stained by Dip-Quick staining kit (Jorgensen Laboratories, Loveland, CO). RBC counts and RBC distribution widths (RDW) were measured on a HemaVet 1700 (Drew Scientific, Waterbury, CT) hematology analyzer. Packed cell volume (PCV) was measured in a JorVet J503 (Jorgensen Laboratories) microhematocrit centrifuge. Hb concentrations were determined after conversion to cyanmethemoglobin by lysing RBCs in Drabkin reagent (Sigma-Aldrich, St Louis, MO), removing insoluble RBC membranes by centrifugation, measuring the absorbance at 540 nm on a spectrophotometer, and comparing with Hb standards. Reticulocyte counts were determined by flow cytometry after staining with thiazole orange.21 Tissues were fixed in 70% alcoholic formalin, embedded in paraffin, sectioned, and stained with hematoxylin-eosin by standard methods at the UAB Comparative Pathology Laboratory (CPL). Liver iron was stained by Prussian Blue. All slides were analyzed on Nikon Eclipse TE2000-U microscope (Nikon, Tokyo, Japan). Images were taken on Nikon Coolpix E990 digital camera and processed by Adobe Photoshop CS version 8.0 imaging software (Adobe Systems, San Jose, CA).
Blood transfusion and tissue iron quantification
Weekly transfusion therapy began 2 days after birth. Peripheral blood from green fluorescent protein (GFP) transgenic mice (C57BL/6-transgenic (UBC-GFP) 30Scha/J, stock no. 004353; The Jackson Laboratory) was collected from anesthetized mice into Microtainer EDTA collection tubes (BD Biosciences). Enucleated GFP+ RBCs were collected after Ficoll gradient separation, followed by 3 washes in cold PBS. The first 2 transfusions were through superficial temporal (facial) vein and intraperitoneal cavity, respectively. Mice older than 3 weeks were transfused by retro-orbital injection. For hypertransfusion, 300 to 400 μL blood was transfused, and half dose was used for hypotransfusion. Mice were killed at 8 weeks of age. Livers and hearts were preserved in 30% formalin and dried, and the nonheme iron concentrations were determined colorimetrically.22
Results
Establishment of humanized γHPFHδβ0 mouse model of CA
Previous studies showed that a humanized mouse model of CA that contained a human fetal to adult Hb-switching cassette could survive to birth solely upon human fetal Hb, but died shortly thereafter due to severe anemia.17 By incorporating an HPFH γ globin gene allele to further delay the fetal to adult Hb switch and including the human δ globin gene for the production of minor adult HbA2, the postnatal survival of humanized CA mice was extended to make them more amenable for study. Humanized γHPFHδβ0 CA mice were generated by targeted gene replacement of the adult mouse β globin genes, βmaj and βmin, with human fetal γHPFH globin and human adult δ and β0 globin genes by homologous recombination in ES cells (Figure 1A). The human γHPFH globin allele contains the Greek nondeletional HPFH promoter mutation at nucleotide position −117 shown to elevate γ globin gene expression levels in both humans and transgenic mice.23–26 The human adult globin alleles contain a wild-type δ globin gene and a mutant β0 globin allele found in the Mediterranean population.27,28 This nonfunctional β0 globin gene has a mutant splice donor site (IVS1.1 G to A) that produces several short-lived mRNAs from cryptic splice donor sites and no functional protein in mice.17 Heterozygous γHPFHδβ0 mice were generated from correctly targeted ES cells, and liver DNA was analyzed by Southern blot to confirm the targeted replacement of the mouse β globin genes (Figure 1B).
Figure 1.
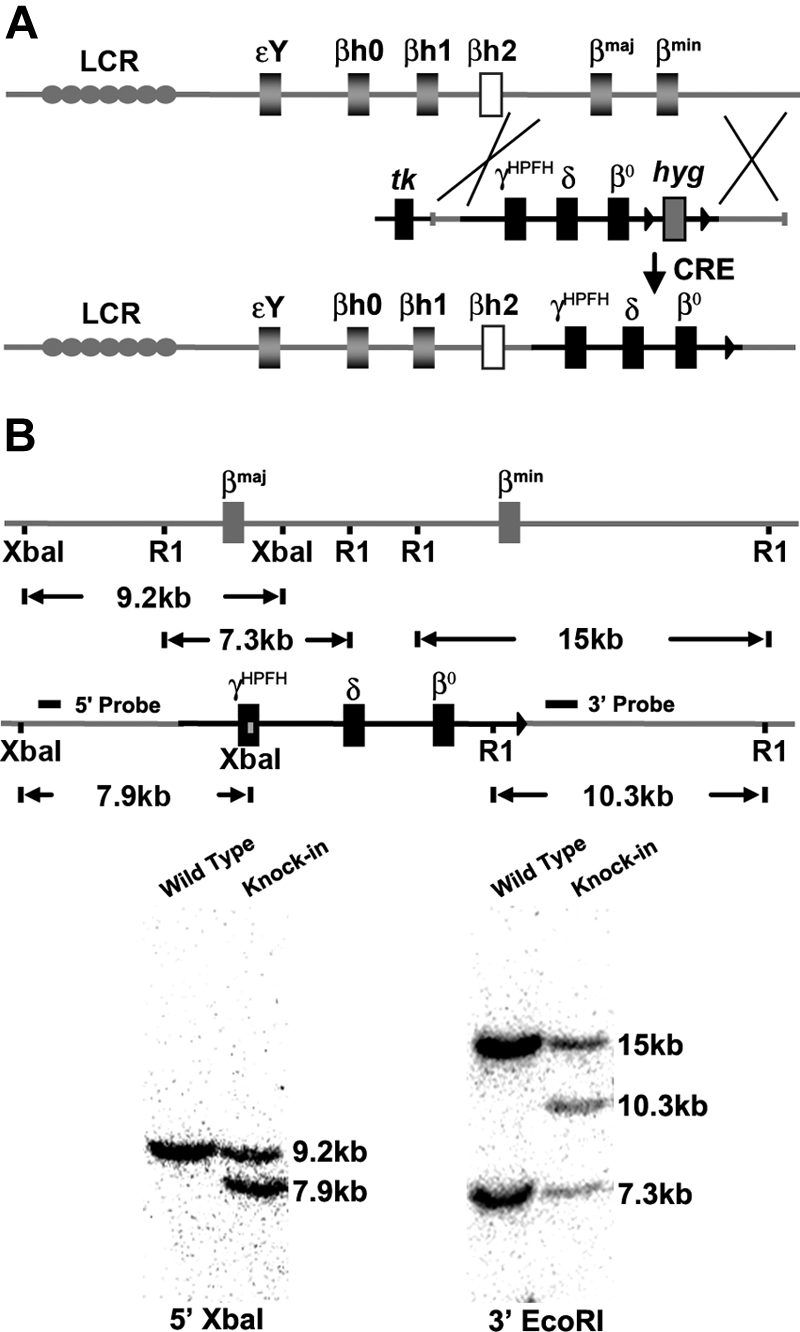
CA mice were generated by replacing the adult mouse β globin genes with a human γHPFHδβ0 globin gene cassette in mouse ES cells. (A) Scheme of targeted gene replacement of the adult mouse β globin genes by a human γHPFHδβ0 globin gene cassette in mouse ES cells. The hyg marker gene was deleted by breeding to CRE recombinase transgenic mice.20 (B) Southern blot confirmation of correct homologous recombination. The 5′ probe hybridizes with a 9.2-kb XbaI fragment from the wild-type allele and a 7.9-kb fragment from the human globin KI allele. The 3′ probe derived from part of the βmin globin gene anneals to a 14.8-kb EcoRI fragment from the βmin globin gene, a 7.3-kb fragment from the βmaj globin gene, and a 10.3-kb fragment from the human globin KI allele.
Heterozygous γHPFHδβ0 mice were bred with human adult α2α1 KI mice (generated by targeted gene replacement of both adult mouse α globin genes; T.M.R., unpublished data, December 2004) and human wild-type γβA KI mice29 to generate mice living on 100% human Hb. Fully humanized compound heterozygous γHPFHδβ0/γβA mice (β thalassemia minor) were bled weekly to analyze their fetal to adult Hb switch (Figure 2A). At birth compound heterozygous mice had 64% (± 1.7%; mean ± [SEM], n = 8) human γ globin chains, 36% (± 1.7%) human β globin chains, and no measureable human δ globin chains. Similar to newborn humans, the high fetal globin chain levels present at birth were gradually replaced by increasing levels of adult globins. The β-like globin chain composition stabilizes by 8 weeks of age with fetal γ globin chains reduced to 4.9% (± 0.8%) and the adult β and δ globin chains levels increased to 90.5% (± 0.7%) and 4.6%(± 0.2%), respectively.
Figure 2.
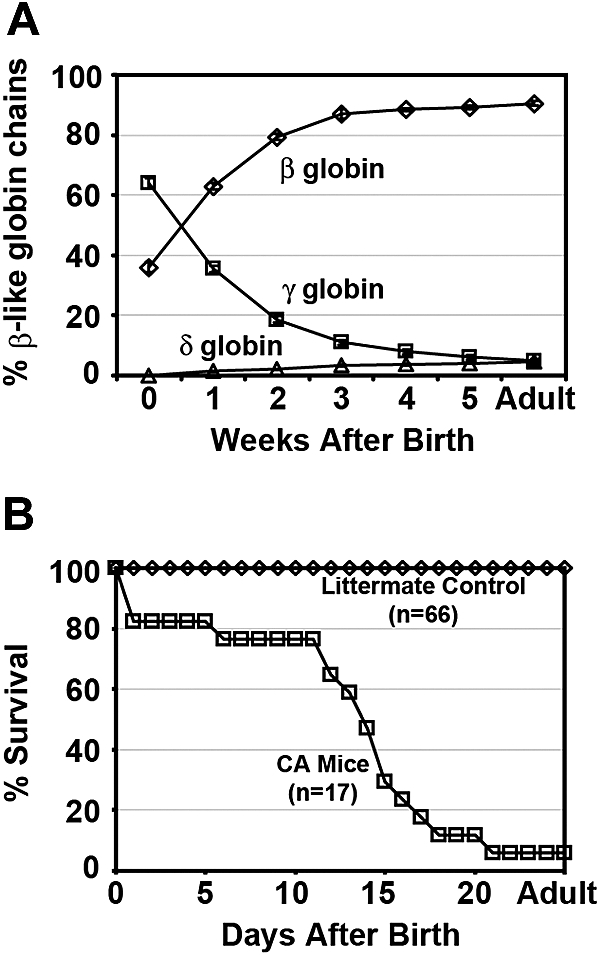
Hb switching and survival curves of humanized γHPFH δβ0 CA mice. (A) Humanized compound heterozygous γHPFHδβ0/γβA mice complete the switch from high levels of fetal γ globin to adult δ and β globins after birth. Weekly hemolysates from peripheral blood were analyzed by HPLC to quantify the β-like globin chains. The fractional percentage of γ (□), β (◇), and δ (▵) globin chains relative to total β-like chains is plotted over time. Values represent mean ± SEM, n = 8. (B) Survival curves of humanized homozygous γHPFHδβ0 CA (□) and littermate control (◇) mice. The majority of fully humanized γHPFHδβ0 CA mice expire within 3 weeks of age as the human γ globin switches to the human δ and nonfunctional human β0 globin genes. Moribund homozygous CA mice were humanely euthanized before death, and their genotype was confirmed by PCR of tail tip DNA. All humanized littermate control mice (γHPFHδβ0/γβA and γβA/γβA) survived beyond 5 weeks of age.
The lifespan of humanized homozygous γHPFHδβ0 CA mice was determined to assess whether the elevated levels of HbF at birth and the presence of increasing levels of minor HbA2 after birth would increase their postnatal survival in the absence of any functional HbA. Compound heterozygous γHPFHδβ0/γβA mice were bred together to produce homozygous γHPFHδβ0 CA mice. Fully humanized CA mice had a median survival of 14 days after birth (Figure 2B). Sixty-five percent of CA mice died within a 1-week period between 11 and 18 days of age. Whereas some CA mice expired as early as 1 day after birth, 1 CA mouse survived into adulthood solely upon human HbF (88%) and HbA2 (12%). By contrast, all of the littermate controls, which include γHPFHδβ0/γβA KI mice and γβA/γβA KI mice, survived into adulthood. Thus, incorporation of the HPFH and minor adult globin alleles in the humanized γHPFHδβ0 CA mice increased their survival beyond birth and generated a Hb-switching pattern that more closely mimics that seen in β thalassemia patients.30
Humanized γHPFHδβ0 CA mice have a β thalassemia major phenotype
Concomitant with the decrease in fetal Hb during the postnatal period of development, the humanized homozygous γHPFHδβ0 CA mice (β thalassemia major) became severely anemic. At the time of their euthanasia, CA mice had a marked reduction in their RBC counts, PCVs, and Hb levels compared with littermate HbA controls (Table 1). This anemia induced a massive erythroid hyperplasia in the humanized CA mice. The spleen, a normal site of stress erythropoiesis in mice, becomes greatly expanded in models of hemolytic anemia. The percentage of cells in the bone marrow and spleen that express the erythroid-specific cell-surface antigen Ter119 was 60% and 83%, respectively, in CA mice compared with 40% and 50% in control animals (Figure 3A and Table 2). In addition to the fractional increase in erythroid cells in the spleen, there was also an average 14-fold increase in the size of the spleen (Table 1), resulting in an overall 23-fold increase in splenic erythroid cells in CA mice. This splenomegaly was characterized by a loss of the normal architecture of red (erythroid) and white (lymphoid) pulp regions that were replaced with mostly early stage erythroblasts (Figures 3A, 4, and Table 2). Extramedullary hematopoiesis was observed as clusters of erythroblasts throughout the liver (Figure 4). The RBC morphology in peripheral blood smears was typical of severe β thalassemia. Marked anisopoikilocytosis, polychromatophilia, spherocytosis, and numerous nucleated erythroblasts were present (Figure 4). There was a doubling of the RBC distribution width, RDW, due to the mixture of large nascent reticulocytes with small mature spherocytes (Table 1). Excess precipitated α globin chains were observed after vital staining with crystal violet (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). In the absence of any therapeutic intervention, CA mice died due to their lethal anemia before weaning age (Figure 2B).
Table 1.
Hematology of humanized HbA control, β thalassemia minor, and β thalassemia major mice before and after transfusion
| Human Hb mice | n | RBC, 106/μL | Hb, g/dL | PCV, % | Retic, % | RDW, % | Spleen, % of BW |
|---|---|---|---|---|---|---|---|
| HbA control | 9 | 10.9 (± 0.3) | 13.6 (± 0.1) | 44.8 (± 0.4) | 2.8 (± 0.2) | 19.6 (± 0.2) | 0.35 (± 0.02) |
| β thalassemia minor | 10 | 11.8 (± 0.2)* | 12.7 (± 0.3)* | 42.0 (± 0.7)* | 4.1 (± 0.3)* | 20.2 (± 0.3)n.s. | 0.53 (± 0.04)* |
| β thalassemia major untransfused CA | 9 | 2.5 (± 0.3)** | 3.0 (± 0.4)** | 15.0 (± 1.9)** | 72.1 (± 2.2)** | 38.5 (± 2.4)** | 5.1 (± 1.5)* |
| β thalassemia major hypertransfused CA | 5 | 9.8 (± 0.5)n.s. | 15.1 (± 0.6)* | 42.2 (± 1.8)n.s. | ND | 16.0 (± 0.2)** | 0.79 (± 0.07)** |
| β thalassemia major hypotransfused CA | 5 | 5.9 (± 0.1)** | 7.4 (± 0.4)** | 25.8 (± 0.7)** | ND | 18.4 (± 0.5)* | 4.1 (± 0.4)** |
Genotypes of human Hb mice are as follows: HbA (α2α1/α2α1 γβA/γβA), β thalassemia minor (α2α1/α2α1 γHPFHδβ0/γβA), and β thalassemia major (α2α1/α2α1 γHPFHδβ0/γHPFHδβ0). Mice are analyzed 8 weeks after birth, except the untransfused CA mice, which are analyzed at the time of their euthanasia (age of 19 ± 6 days). Values represent mean (± SEM). Statistical significances are determined for the β thalassemic major and minor mice compared with the HbA control mice. P values were calculated by 2-tailed unpaired Student t test.
RBC indicates red blood cell; Hb, hemoglobin; PCV, packed cell volume; Retic, reticulocyte; RDW, red cell distribution width; BW, body weight; ND, not determined; and n.s., not significant.
P < .05.
P < .0001.
Figure 3.

Erythroid hyperplasia, ineffective erythropoiesis, and hemolysis in humanized γHPFHδβ0 CA mice. (A) Erythroid hyperplasia in the bone marrow and spleen of CA mice was measured by flow cytometry. Bone marrow and spleen cells from age- and sex-matched CA and wild-type mice were stained with fluorescently labeled antibodies to the erythroid antigen Ter119 and transferrin receptor CD71 and fluorescent-labeled annexin V. Dead cells were excluded by 7-aminoactinomycin D (7AAD) staining. The percentages of proerythroblasts (region I: CD71HIGH, Ter119LOW), early erythroblasts (region II: CD71+, Ter119+), late erythroblasts (region III: CD71−, Ter119+), and total Ter119+ erythroid cells (total Ter119+) in the bone marrow and spleen are shown in Table 2. Erythroid populations of humanized HbA mice (α2α1/α2α1 γβA/γβA) and C57BL/6J wild-type mice did not differ significantly (data not shown). (B) Demonstration of ineffective erythropoiesis in CA mice by apoptosis of erythroid progenitors by annexin V–binding assay.32 Representative histograms are shown of annexin V staining of early (region II) and late (region III) erythroblasts in CA and control mouse bone marrow and spleen cells from panel A. No antibody control samples were stained with all the antibodies in panel A, except annexin V. Annexin V+ cell ratios are quantified in Table 3. Annexin V+ erythroid populations of humanized HbA mice (α2α1/α2α1 γβA/γβA) and C57BL/6J wild-type mice did not differ significantly (data not shown). (C) Bilirubin levels increased significantly in untransfused CA mice compared with age- and sex-matched wild-type control mice, indicating increased hemolysis in CA mice. Bilirubin levels returned to control levels in hypertransfused, but not hypotransfused CA mice. *P < .05; **P < .0001; n = 3 in each group.
Table 2.
Percentages of erythroid progenitor populations in the bone marrow and spleen of humanized CA and control mice by flow cytometry
| Tissue | Cells | Region | Percentage of total cells (%) |
P | |
|---|---|---|---|---|---|
| Control mice | CA mice | ||||
| Bone marrow | Proerythroblasts | I | 0.13 (± 0.03) | 0.82 (± 0.21) | .0188 |
| Early erythroblasts | II | 27.9 (± 5.6) | 56.0 (± 3.1) | .0046 | |
| Late erythroblasts | III | 11.9 (± 2.9) | 4.0 (± 0.8) | .04 | |
| Total Ter119+ | II + III | 39.7 (± 4.2) | 60.1 (± 2.6) | .0059 | |
| Spleen | Proerythroblasts | I | 0.10 (± 0.04) | 0.70 (± 0.16) | .0126 |
| Early erythroblasts | II | 15.6 (± 6.7) | 74.9 (± 5.6) | .0005 | |
| Late erythroblasts | III | 34.1 (± 3.6) | 8.3 (± 1.9) | .0007 | |
| Total Ter119+ | II + III | 49.7 (± 3.4) | 83.2 (± 6.9) | .0048 | |
Statistical significances were determined for the CA mice compared with control mice. Erythroid populations of humanized HbA mice (α2α1/α2α1 γβA/γβA) and C57BL/6J control mice do not differ significantly (data not shown). Percentages are mean (± SEM). n = 4 in each group. P values were calculated by 2-tailed unpaired Student t test.
Figure 4.

Histopathology of blood, spleen, and liver of humanized HbA control and CA mice before and after transfusion. Peripheral blood smears of untransfused CA mice exhibit severe anemia compared with HbA littermate control mice. There are significant numbers of reticulocytes, circulating erythroblasts, and microcytic, hypochromic, and fragmented RBCs in untransfused CA mouse blood. In addition, the normal structure of red pulp and white pulp in the spleen is absent. In the liver of untransfused CA mice, there are extensive clusters of extramedullary hematopoiesis and increased iron staining. In contrast, the hypertransfused CA mouse has a normal peripheral blood smear and greatly improved histology similar to the HbA control, except excess iron is present in the liver. Hypotransfused CA mice are still anemic, have significant numbers of erythroblasts and thalassemic RBCs in the blood, and have a histopathology similar to the untransfused CA mouse. All mice are 8 weeks of age, except for the untransfused CA mouse, which is only 2 weeks old. Scale bars: blood smear, 10 μm; spleen, 100 μm; liver (low power), 50 μm; liver (high power), 10 μm; liver iron, 50 μm.
Marked ineffective erythropoiesis and hemolysis in humanized γHPFHδβ0 CA mice
Because the premature destruction of erythroid progenitors in the bone marrow (ineffective erythropoiesis) and erythrocytes from the circulation (hemolysis) are hallmarks of β thalassemia, their roles in the progression of anemia in the humanized CA mice were analyzed. Erythroid progenitors in the bone marrow and spleen of wild-type and homozygous γHPFHδβ0 CA mice were stained with fluorescently labeled antibodies to the erythroid-specific antigen, Ter119, and the transferrin receptor, CD71, to distinguish pro-, early, and late erythroblasts, as previously described.31 Ter119+ erythroid populations may also include enucleated erythroid cells circulating through the bone marrow and spleen that are not erythroblasts. Figure 3A and Table 2 demonstrate that both proerythroblasts (region I: CD71HIGH, Ter119LOW) and early erythroblasts (region II: CD71+, Ter119+) in the bone marrow and spleen of CA mice were increased compared with control. Furthermore, the ratio of early (region II) to late erythroblasts (region III: CD71−, Ter119+) was markedly increased in both tissues of the CA mice relative to controls. Thus, the erythroid tissues of CA mice demonstrated a marked expansion of progenitors that was skewed toward earlier erythroblasts.
In humans, free α globin chains in differentiating thalassemic erythroblasts can cause premature cell death by apoptosis.1–3 Apoptosis of early and late erythroblasts was analyzed by flow cytometry after binding to fluorescent-labeled annexin V (Figure 3B and Table 3).32 There was a significant increase in annexin V+ early and late erythroblasts (regions II and III) in the bone marrow of CA mice. However, in the CA spleen, only the late erythroblasts (region III) had significantly increased levels of annexin V+ cells compared with controls. These data demonstrate increased levels of ineffective erythropoiesis caused by apoptosis of early and late erythroblasts in the bone marrow and late erythroblasts in the spleen of CA mice.
Table 3.
Percentages of annexin V+ staining erythroid progenitors in the bone marrow and spleen of humanized CA versus control mice
| Tissue | Cells (region) | Annexin V+ (%) |
P | |
|---|---|---|---|---|
| Control mice | CA mice | |||
| Bone marrow | Early erythroblasts (II) | 13.9 (± 3.3) | 33.7 (± 3.5) | .0059 |
| Late erythroblasts (III) | 1.8 (± 0.3) | 50.0 (± 2.5) | < .0001 | |
| Spleen | Early erythroblasts (II) | 13.6 (± 4.5) | 20.9 (± 4.8) | .3099 |
| Late erythroblasts (III) | 0.9 (± 0.3) | 53.2 (± 1.6) | < .0001 | |
Statistical significances were determined for the CA mice compared with control mice. Annexin V+ erythroid populations of humanized HbA mice (α2α1/α2α1 γβA/γβA) and C57BL/6J control mice do not differ significantly (data not shown). Percentages are mean (± SEM). n = 4 in each group. P values were calculated by 2-tailed unpaired Student t test.
Hemolysis of RBCs was another cause of anemia in humanized CA mice. Peripheral blood smears showed numerous damaged RBCs, intracellular inclusions, and reticulocytes (Figures 4, S1). Elevated reticulocytosis with decreased RBC count, a direct indicator of hemolysis in β thalassemia, was confirmed by flow cytometry of thiazole orange-stained blood.21 In CA mice, the reticulocyte count was 72.1% (± 2.2%) compared with 2.8% (± 0.2%) in γβA/γβA littermate control mice (Table 1). Another indicator of hemolysis is increased plasma bilirubin, a product of Hb degradation. The total plasma bilirubin levels in CA mice compared with control mice increased from 0.2 mg/dL to 18.4 (± 0.8) mg/dL. Direct bilirubin levels increased from 0.1 mg/dL in wild-type mice to 2.8 (± 0.8) mg/dL in CA mice. Hence, indirect bilirubin increased from 0.1 mg/dL to 15.6 gm/dL approximately 150-fold in CA mice (Figure 3C). These data demonstrate that hemolysis contributes to the severe anemia in humanized CA mice.
Rescue of humanized γHPFHδβ0 CA mice from lethal anemia by transfusion therapy
Regular lifelong blood transfusion therapy is essential for the survival and improved quality of life of β thalassemia major patients. An important goal of transfusion therapy is to maintain Hb levels high enough to alleviate anemia, suppress ineffective erythropoiesis, and allow for normal growth and development, while simultaneously limiting adverse transfusion reactions and toxicity from iron overload.
Humanized γHPFHδβ0 CA mice were given blood transfusions to rescue them from lethal anemia. Two groups of CA mice were transfused weekly for a total of 8 weeks with fresh, leukocyte reduced, packed RBCs from GFP transgenic mouse blood donors.33 Hypertransfused mice received enough donor blood to maintain Hb levels at approximately 14 g/dL. Hypotransfused mice were transfused to raise Hb levels only to approximately 7 g/dL (Figure 5A). Both groups of CA mice receiving transfusions had stable Hb and GFP+ RBC chimerism levels by 1 month of age (Figure 5A,B). Transfused CA mice survived well beyond the lethal end point of untransfused CA mice (compare age in Figure 2B with Figure 5).
Figure 5.

Hb levels and GFP+ RBC chimerism in peripheral blood of hyper- and hypotransfused CA mice. Humanized CA mice were transfused weekly with packed donor RBCs from GFP+ transgenic mice. Two days after transfusion, mice were bled for determination of Hb level by spectrophotometry (A) and percentage of donor GFP+ RBCs by flow cytometry (B). Values represent mean (± SEM), n = 5 in each group.
After 8 weeks of therapy, the anemia in both groups of transfused CA mice was markedly improved compared with untransfused CA mice. RBC indices in hypertransfused mice were similar to wild-type controls (Table 1). Less than 1% of the circulating RBCs in hypertransfused CA mice was derived from endogenous erythropoiesis, suggesting that the erythropoietic activity was effectively suppressed (Figure 5B). Suppression of endogenous ineffective erythropoiesis was confirmed in the bone marrow, where the percentage of Ter119+ cells was decreased 63% from untransfused CA mice (60%-22%) and 33% from wild-type controls (33% to 22%, Figure 6A). No extramedullary hematopoiesis was found in the livers of hypertransfused CA mice, splenomegaly was markedly reduced with restoration of normal red and white pulp, and plasma bilirubin levels returned to normal (Figures 3C, 4).
Figure 6.

Erythropoietic activity and storage iron levels in hypertransfused and hypotransfused CA mice. (A) Erythroid progenitors in the bone marrow of untransfused CA mice, wild-type controls, and transfused CA mice were determined by flow cytometry analysis of Ter119 and CD71 stained bone marrow cells. Dead cells and transfused GFP+ RBCs were excluded from analysis by using the 7AAD− and GFP− populations, respectively. Percentages of Ter119+ erythroid cells are shown. Untransfused and hypotransfused CA mice had increased erythropoietic activity compared with control, whereas hypertransfused CA mice had reduced erythropoietic activity. All mice were 8 weeks of age, except the untransfused CA mice, which were only 2 weeks old. (B) Storage iron concentrations in livers and hearts were quantified in male and female CA mice after 8 weeks of transfusion. Compared with littermate control γβA/γβA mice, the storage iron in the livers of both hyper- and hypotransfused CA mice increased significantly (P ≤ .0001). There was no significant increase in the hearts of either transfused group. P values were calculated by 2-tailed unpaired Student t test, n ≥ 5 in each group.
In contrast, hypotransfused CA mice were still anemic with low RBC counts, Hb levels, and PCVs compared with wild-type control animals (Table 1). Hypotransfused CA mice still had approximately 20% endogenous thalassemic RBCs in the peripheral blood (GFP−, Figure 5B) with numerous circulating erythroblasts (Figure 4) and elevated plasma bilirubin (Figure 3C), suggesting continued robust endogenous erythropoietic activity and hemolysis. Expanded ineffective erythropoiesis was still evident in the bone marrow, where the percentage of erythroid cells was 70% above wild-type mice and only slightly decreased from untransfused CA mice (Figure 6A). The histopathologic phenotype of hypotransfused mice was similar to 2-week-old untransfused CA mice. There were continued erythroid hyperplasia, ineffective erythropoiesis, and extramedullary hematopoiesis (Figure 4).
Because mammals have no active mechanism for removal of excess iron, regular transfusion therapy can lead to increased iron stores in the tissues. Both transfusion regimens led to increased Prussian blue iron staining in the liver similar to untransfused CA mice (Figure 4). Mouse liver and heart storage iron from both transfused groups was quantified. Compared with humanized γβA/γβA littermate control mice, the liver storage iron concentration increased significantly under both transfusion regimens. However, there was no increase in the heart storage iron concentration after 8 weeks of transfusion therapy (Figure 6B).
Discussion
This study describes a mouse model of β thalassemia major that has numerous improvements over previous models. This novel CA model is heritable; survives postnatally solely on human HbF and HbA2; has marked erythroid hyperplasia, ineffective erythropoiesis, reticulocytosis, and hemolysis; and rescue from death by regular transfusion results in iron overload. Because all these characteristics are shared with β thalassemia major patients, this humanized CA model is suitable for both basic and preclinical studies.
The major weakness of earlier models of CA was their early prenatal death due to differences in the developmental timing of Hb switching in humans and mice. Mice switch from primitive embryonic globins directly to definitive adult globins early in fetal life.7,8 Therefore, deletions of the adult mouse β globin genes lead to severe β thalassemia and in utero death.9,11,12,14 Similarly, transgenic mice that contain large portions of the human β globin locus also switch fetal to adult Hb too early in development.34–38 However, linked human γ and β globin transgenes with the intergenic region deleted exhibit a delay in the fetal to adult Hb switch.39 Humanized mice containing such transgenes have high levels of human HbF at birth.39 Recently, we incorporated such a delayed γ to β Hb switching cassette (γβ) directly into the mouse β globin locus replacing the murine adult β globin genes.17,29 Humanized mice with a delayed Hb-switching γβ0 cassette are born solely with human HbF in their RBCs, but expired within hours due to anemia.17 By incorporating an HPFH mutation into the γ globin gene promoter and the minor adult δ globin gene in the γHPFHδβ0 KI cassette, the humanized CA mice reported in this study have an extended postnatal lifespan compared with the previous γβ0 CA mouse model.17
A simple lifespan comparison of mice (2 years) with humans (70 years) suggests each day of life in the mouse would represent approximately 35 days in humans. Using this simple age-scaling model, a median lifespan of 14 days in the humanized CA mice represents 490 days in humans or approximately 16 months. This is approximately the same timeframe that infants with CA become severely anemic and require blood transfusions for survival. The similarity in timing of disease onset between humans and CA mice allows the study of the effects of anemia on disease progression during development and the testing of therapeutic strategies designed to cure β thalassemia in humans before onset of severe disease.
Whereas there are numerous nondeletional HPFH mutations in humans that cause asymptomatic elevation of fetal Hb through adulthood,25,40–43 the Greek nondeletional HPFH −117 mutation was used to generate this humanized model of CA.25 It is possible that other HPFH mutations in similar context would also generate viable humanized CA mice whose postnatal survival correlates with γ globin expression level. Some may even live into adulthood.
The level of minor adult HbA2 is usually elevated in heterozygous β thalassemia, and its measurement is an important diagnostic tool for identifying carriers of β thalassemia.30,44–46 There is a clear postnatal Hb switch from γ to δ in the compound heterozygous γHPFHδβ0/γβA mice (Figure 2A), and the final adult HbA2 level of 4.6% (± 0.2%) is comparable with that measured in human β thalassemia trait patients.30,44–46 The HbA2 levels could potentially be greater at birth if a wild-type rather than an HPFH γ globin promoter had been used in the KI cassette because the HPFH promoter is most likely delaying the γ to δ Hb switch. Regardless, the presence of even small amounts of δ globin chains would beneficially reduce excess α globin chains in the thalassemic erythrocytes in this humanized CA model.
Homeostasis of the erythron requires a balance between erythroid production and destruction. Excess free α globin chains in CA cause premature destruction of erythroid progenitors in the marrow and erythrocytes in the circulation, resulting in anemia even though there is massive erythroid hyperplasia. In this study, the reticulocytosis, hemolysis, and annexin V staining of erythroblasts in the bone marrow and spleen of humanized CA mice differ from previously published results in an adult onset transplantation mouse model of CA.15,16 The difference is probably due to the lack of any β-like globin chains in the adult transplant CA model, whereas human CA patients and the humanized CA mice in this report have γ and δ globin chains available for Hb formation with excess α globin chains. Interestingly, high levels of annexin V+ early and late erythroblasts were measured in vivo in the bone marrow of humanized CA mice, whereas in in vitro expanded human CD34+ erythroid cultures from CA patients the majority of annexin V staining was observed in the early polychromatophilic erythroblasts.1 Recent studies have shown that death receptor and ligand-mediated apoptosis play a critical role in both normal and stress erythropoiesis.47 Humanized CA mice will be an interesting model system to study mechanisms of erythroid homeostasis during development and disease.
The lifespan of humanized CA mice was extended by regular blood transfusion. Whereas hypertransfusion effectively suppressed erythropoiesis in the marrow, both hyper- and hypotransfused mice had increased stores of iron in the liver. A more moderate transfusion regimen that maintains Hb between 10 and 11 g/dL may be more effective at reducing both endogenous erythropoiesis and iron overload.48 Heart storage iron yielded no significant difference between transfused CA mice and control mice, indicating that longer term transfusion may be required to observe increased iron storage in the heart.
Reactivation of the human γ globin gene could ameliorate the anemia in β thalassemia. The human γHPFH globin KI allele was regulated in a manner similar to humans in terms of the timing of the Hb switch. This suggests that study of the epigenetic and transcriptional regulation of the human γHPFH KI allele may provide insight into the mechanism of HPFH in humans. Mechanistic studies of Hb switching in this humanized model of CA could prove useful in the design of new therapeutic strategies for patients with β thalassemia major.
Acknowledgments
We thank Jin-Xiang Ren for her expert technical assistance. We thank Mary Hamilton, Maya Spivey, and Albert Tousson from the UAB CPL and High-Resolution Imaging Facility cores for their technical assistance. We thank the Cooley's Anemia Foundation's Request for Application for Translational Research in Adult Thalassemia for support. We thank Joseph Ruisi, UNICO, and the Thalassemia-Cooley's Anemia Group at UAB.
This work was supported by National Institutes of Health grants R01 HL072351 and R01 HL073440 (T.M.R.), and T32 GM008111 (S.C.M.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: Y.H. designed and performed experiments, analyzed research, made figures, and wrote the paper; S.C.M. performed experiments and analyzed research; and T.M.R. designed and performed experiments, analyzed research, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Dr Thomas M. Ryan, Department of Biochemistry and Molecular Genetics, University of Alabama at Birmingham, 1918 University Blvd, MCLM 566A, Birmingham, AL 35294; e-mail: tryan@uab.edu.
References
- 1.Mathias LA, Fisher TC, Zeng L, et al. Ineffective erythropoiesis in β-thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp Hematol. 2000;28:1343–1353. doi: 10.1016/s0301-472x(00)00555-5. [DOI] [PubMed] [Google Scholar]
- 2.Centis F, Tabellini L, Lucarelli G, et al. The importance of erythroid expansion in determining the extent of apoptosis in erythroid precursors in patients with β-thalassemia major. Blood. 2000;96:3624–3629. [PubMed] [Google Scholar]
- 3.Yuan J, Angelucci E, Lucarelli G, et al. Accelerated programmed cell death (apoptosis) in erythroid precursors of patients with severe β-thalassemia (Cooley's anemia). Blood. 1993;82:374–377. [PubMed] [Google Scholar]
- 4.Cazzola M, Alessandrino P, Barosi G, Morandi S, Stefanelli M. Quantitative evaluation of the mechanisms of the anaemia in heterozygous β-thalassaemia. Scand J Haematol. 1979;23:107–114. doi: 10.1111/j.1600-0609.1979.tb02680.x. [DOI] [PubMed] [Google Scholar]
- 5.Kean LS, Brown LE, Nichols JW, Mohandas N, Archer DR, Hsu LL. Comparison of mechanisms of anemia in mice with sickle cell disease and β-thalassemia: peripheral destruction, ineffective erythropoiesis, and phospholipid scramblase-mediated phosphatidylserine exposure. Exp Hematol. 2002;30:394–402. doi: 10.1016/s0301-472x(02)00780-4. [DOI] [PubMed] [Google Scholar]
- 6.Olivieri NF. The β-thalassemias. N Engl J Med. 1999;341:99–109. doi: 10.1056/NEJM199907083410207. [DOI] [PubMed] [Google Scholar]
- 7.Kingsley PD, Malik J, Emerson RL, et al. “Maturational” globin switching in primary primitive erythroid cells. Blood. 2006;107:1665–1672. doi: 10.1182/blood-2005-08-3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitelaw E, Tsai SF, Hogben P, Orkin SH. Regulated expression of globin chains and the erythroid transcription factor GATA-1 during erythropoiesis in the developing mouse. Mol Cell Biol. 1990;10:6596–6606. doi: 10.1128/mcb.10.12.6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ciavatta DJ, Ryan TM, Farmer SC, Townes TM. Mouse model of human β zero thalassemia: targeted deletion of the mouse β maj- and β min-globin genes in embryonic stem cells. Proc Natl Acad Sci U S A. 1995;92:9259–9263. doi: 10.1073/pnas.92.20.9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jamsai D, Zaibak F, Khongnium W, et al. A humanized mouse model for a common β0-thalassemia mutation. Genomics. 2005;85:453–461. doi: 10.1016/j.ygeno.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 11.Lewis J, Yang B, Kim R, et al. A common human β globin splicing mutation modeled in mice. Blood. 1998;91:2152–2156. [PubMed] [Google Scholar]
- 12.Shehee WR, Oliver P, Smithies O. Lethal thalassemia after insertional disruption of the mouse major adult β-globin gene. Proc Natl Acad Sci U S A. 1993;90:3177–3181. doi: 10.1073/pnas.90.8.3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vadolas J, Nefedov M, Wardan H, et al. Humanized β-thalassemia mouse model containing the common IVSI-110 splicing mutation. J Biol Chem. 2006;281:7399–7405. doi: 10.1074/jbc.M512931200. [DOI] [PubMed] [Google Scholar]
- 14.Yang B, Kirby S, Lewis J, Detloff PJ, Maeda N, Smithies O. A mouse model for β 0-thalassemia. Proc Natl Acad Sci U S A. 1995;92:11608–11612. doi: 10.1073/pnas.92.25.11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rivella S, May C, Chadburn A, Riviere I, Sadelain M. A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human β-globin gene transfer. Blood. 2003;101:2932–2939. doi: 10.1182/blood-2002-10-3305. [DOI] [PubMed] [Google Scholar]
- 16.Libani IV, Guy EC, Melchiori L, et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in β-thalassemia. Blood. 2008;112:875–885. doi: 10.1182/blood-2007-12-126938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huo Y, McConnell SM, Liu S-R, et al. Humanized mouse model of Cooley's anemia. J Biol Chem. 2009;284:4889–4896. doi: 10.1074/jbc.M805681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang J, Deng K, Wu H, et al. Efficient production of mice from embryonic stem cells injected into four- or eight-cell embryos by piezo micromanipulation. Stem Cells. 2008;26:1883–1890. doi: 10.1634/stemcells.2008-0164. [DOI] [PubMed] [Google Scholar]
- 19.Poueymirou WT, Auerbach W, Frendewey D, et al. F0 generation mice fully derived from gene-targeted embryonic stem cells allowing immediate phenotypic analyses. Nat Biotechnol. 2007;25:91–99. doi: 10.1038/nbt1263. [DOI] [PubMed] [Google Scholar]
- 20.Nozaki M, Ohishi K, Yamada N, Kinoshita T, Nagy A, Takeda J. Developmental abnormalities of glycosylphosphatidylinositol-anchor-deficient embryos revealed by Cre/loxP system. Lab Invest. 1999;79:293–299. [PubMed] [Google Scholar]
- 21.Ferguson DJ, Lee SF, Gordon PA. Evaluation of reticulocyte counts by flow cytometry in a routine laboratory. Am J Hematol. 1990;33:13–17. doi: 10.1002/ajh.2830330104. [DOI] [PubMed] [Google Scholar]
- 22.Torrance JD, Bothwell TH. A simple technique for measuring storage iron concentrations in formalinised liver samples. S Afr J Med Sci. 1968;33:9–11. [PubMed] [Google Scholar]
- 23.Gelinas R, Endlich B, Pfeiffer C, Yagi M, Stamatoyannopoulos G. G to A substitution in the distal CCAAT box of the Aγ-globin gene in Greek hereditary persistence of fetal haemoglobin. Nature. 1985;313:323–325. doi: 10.1038/313323a0. [DOI] [PubMed] [Google Scholar]
- 24.Berry M, Grosveld F, Dillon N. A single point mutation is the cause of the Greek form of hereditary persistence of fetal haemoglobin. Nature. 1992;358:499–502. doi: 10.1038/358499a0. [DOI] [PubMed] [Google Scholar]
- 25.Collins FS, Metherall JE, Yamakawa M, Pan J, Weissman SM, Forget BG. A point mutation in the Aγ-globin gene promoter in Greek hereditary persistence of fetal haemoglobin. Nature. 1985;313:325–326. doi: 10.1038/313325a0. [DOI] [PubMed] [Google Scholar]
- 26.Peterson KR, Li QL, Clegg CH, et al. Use of yeast artificial chromosomes (YACs) in studies of mammalian development: production of β-globin locus YAC mice carrying human globin developmental mutants. Proc Natl Acad Sci U S A. 1995;92:5655–5659. doi: 10.1073/pnas.92.12.5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kollia P, Karababa PH, Sinopoulou K, et al. Beta-Thalassaemia mutations and the underlying β gene cluster haplotypes in the Greek population. Gene Geogr. 1992;6:59–70. [PubMed] [Google Scholar]
- 28.Treisman R, Orkin SH, Maniatis T. Specific transcription and RNA splicing defects in five cloned β-thalassaemia genes. Nature. 1983;302:591–596. doi: 10.1038/302591a0. [DOI] [PubMed] [Google Scholar]
- 29.Wu LC, Sun CW, Ryan TM, Pawlik KM, Ren J, Townes TM. Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood. 2006;108:1183–1188. doi: 10.1182/blood-2006-02-004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huisman TH. Levels of Hb A2 in heterozygotes and homozygotes for β-thalassemia mutations: influence of mutations in the CACCC and ATAAA motifs of the β-globin gene promoter. Acta Haematol. 1997;98:187–194. doi: 10.1159/000203622. [DOI] [PubMed] [Google Scholar]
- 31.Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a−/−5b−/− mice due to decreased survival of early erythroblasts. Blood. 2001;98:3261–3273. doi: 10.1182/blood.v98.12.3261. [DOI] [PubMed] [Google Scholar]
- 32.Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84:1415–1420. [PubMed] [Google Scholar]
- 33.Schaefer BC, Schaefer ML, Kappler JW, Marrack P, Kedl RM. Observation of antigen-dependent CD8+ T-cell/dendritic cell interactions in vivo. Cell Immunol. 2001;214:110–122. doi: 10.1006/cimm.2001.1895. [DOI] [PubMed] [Google Scholar]
- 34.Behringer RR, Ryan TM, Palmiter RD, Brinster RL, Townes TM. Human γ- to β-globin gene switching in transgenic mice. Genes Dev. 1990;4:380–389. doi: 10.1101/gad.4.3.380. [DOI] [PubMed] [Google Scholar]
- 35.Gaensler KM, Kitamura M, Kan YW. Germ-line transmission and developmental regulation of a 150-kb yeast artificial chromosome containing the human β-globin locus in transgenic mice. Proc Natl Acad Sci U S A. 1993;90:11381–11385. doi: 10.1073/pnas.90.23.11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaufman RM, Pham CT, Ley TJ. Transgenic analysis of a 100-kb human β-globin cluster-containing DNA fragment propagated as a bacterial artificial chromosome. Blood. 1999;94:3178–3184. [PubMed] [Google Scholar]
- 37.Peterson KR, Clegg CH, Huxley C, et al. Transgenic mice containing a 248-kb yeast artificial chromosome carrying the human β-globin locus display proper developmental control of human globin genes. Proc Natl Acad Sci U S A. 1993;90:7593–7597. doi: 10.1073/pnas.90.16.7593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strouboulis J, Dillon N, Grosveld F. Developmental regulation of a complete 70-kb human β-globin locus in transgenic mice. Genes Dev. 1992;6:1857–1864. doi: 10.1101/gad.6.10.1857. [DOI] [PubMed] [Google Scholar]
- 39.Ryan TM, Ciavatta DJ, Townes TM. Knockout-transgenic mouse model of sickle cell disease. Science. 1997;278:873–876. doi: 10.1126/science.278.5339.873. [DOI] [PubMed] [Google Scholar]
- 40.Collins FS, Boehm CD, Waber PG, et al. Concordance of a point mutation 5′ to the Gγ globin gene with Gγβ+: hereditary persistence of fetal hemoglobin in the black population. Blood. 1984;64:1292–1296. [PubMed] [Google Scholar]
- 41.Surrey S, Delgrosso K, Malladi P, Schwartz E. A single-base change at position −175 in the 5′-flanking region of the Gγ-globin gene from a black with Gγ-β+ HPFH. Blood. 1988;71:807–810. [PubMed] [Google Scholar]
- 42.Tuan D, Feingold E, Newman M, Weissman SM, Forget BG. Different 3′ end points of deletions causing δβ-thalassemia and hereditary persistence of fetal hemoglobin: implications for the control of γ-globin gene expression in man. Proc Natl Acad Sci U S A. 1983;80:6937–6941. doi: 10.1073/pnas.80.22.6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang KG, Stoming TA, Fei YJ, et al. Identification of base substitutions in the promoter regions of the Aγ- and Gγ-globin genes in Aγ- (or Gγ)-β+-HPFH heterozygotes using the DNA-amplification-synthetic oligonucleotide procedure. Blood. 1988;71:1414–1417. [PubMed] [Google Scholar]
- 44.Craver RD, Abermanis JG, Warrier RP, Ode DL, Hempe JM. Hemoglobin A2 levels in healthy persons, sickle cell disease, sickle cell trait, and β-thalassemia by capillary isoelectric focusing. Am J Clin Pathol. 1997;107:88–91. doi: 10.1093/ajcp/107.1.88. [DOI] [PubMed] [Google Scholar]
- 45.Fortova H, Slavikova V, Musil F, Suttnar J, Brabec V. [Diagnosis of β-thalassemia on the basis of HbA2 determination]. Vnitr Lek. 1995;41:302–306. [PubMed] [Google Scholar]
- 46.Steinberg MH, Coleman MB, Adams JG., III β-Thalassemia with exceptionally high hemoglobin A2: differential expression of the δ-globin gene in the presence of β-thalassemia. J Lab Clin Med. 1982;100:548–557. [PubMed] [Google Scholar]
- 47.Liu Y, Pop R, Sadegh C, Brugnara C, Haase VH, Socolovsky M. Suppression of Fas-FasL coexpression by erythropoietin mediates erythroblast expansion during the erythropoietic stress response in vivo. Blood. 2006;108:123–133. doi: 10.1182/blood-2005-11-4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cazzola M, Borgna-Pignatti C, Locatelli F, Ponchio L, Beguin Y, De Stefano P. A moderate transfusion regimen may reduce iron loading in β-thalassemia major without producing excessive expansion of erythropoiesis. Transfusion. 1997;37:135–140. doi: 10.1046/j.1537-2995.1997.37297203514.x. [DOI] [PubMed] [Google Scholar]