

Abstract
As an alternative approach to blocking estrogen action, we have developed small molecules that directly disrupt the key estrogen receptor (ER)/coactivator interaction necessary for gene activation. The more direct, protein-protein nature of this disruption might be effective even in hormone-refractory breast cancer. We have synthesized a pyrimidine-core library of moderate size, members of which act as α-helix mimics to block ERα/coactivator interaction. Structure- activity relationships have been explored with various C, N, O and S-substituents on the pyrimidine core. Time-resolved fluorescence resonance energy transfer and cell-based reporter gene assays show that the most active members inhibit the ERα/steroid receptor coactivator interaction with Ki’s in the low micromolar range. Through these studies, we have obtained a refined pharmacophore model for activity in this pyrimidine series. Furthermore, the favorable activities of several of these compounds support the feasibility that this coactivator binding inhibition mechanism for blocking estrogen action might provide a potential alternative approach to endocrine therapy.
INTRODUCTION
Although historically considered female reproductive hormones, estrogens are now recognized to be major regulators of physiological functions in both reproductive and non-reproductive tissues in both men and women.1, 2 While estrogen activity is required or is beneficial in many cases, such as for fertility,3, 4 during pregnancy, and for bone health and metabolic function,5–9 the pro-proliferative effect of estrogens in many target tissues can be pathological.10, 11 Such is the case with breast cancer, where tumor growth is driven by estrogen in ca. one-third of tumors.12 In such patients, good therapeutic responses can often be achieved by blocking estrogen action with antiestrogens (estrogen antagonists).13
Both estrogens and antiestrogens work through the estrogen receptors, of which there are two subtypes, ERα and ERβ. 2, 14 The ERs are ligand-modulated transcription factors that act through cis-regulatory gene sequences in chromatin to regulate patterns of gene expression in target cells. Estrogen agonists bind to the ERs and stabilize conformations that recruit key coregulatory proteins, in particular, members of the p160 steroid receptor coactivator family (SRCs) (Figure 1A).15 The ER-SRC interaction involves protein-protein contact between signature LXXLL sequences in the SRC and a hydrophobic groove on the surface of the ligand-binding domain of the ER-agonist complex with which the three leucine residues of the helix interact. The coactivators that are recruited by the ER-agonist complex are responsible for altering chromatin architecture, loosening nucleosome structure, and activating RNA polymerase II (polII), as required to up-regulate gene transcription.16 Estrogen antagonists occupy the same ligand binding site as agonists, but they stabilize different ER conformations, ones that preclude or reduce the binding of the SRC coactivators, thereby indirectly interrupting estrogen signaling (Figure 1B).17
Figure 1.

Cartoon representation of classical vs. CBI antagonism of ER. A. Conformation of agonist-bound ER with helix 12 forming part of the steroid receptor coactivator (SRC) binding site; B. Conformation of antagonist-bound ER in which helix 12 occupies the SRC binding site, disrupting the ER/SRC interaction indirectly; C, Conformation of agonist-bound ER in which a CBI occupies the SRC binding site, disrupting ER/SRC interaction directly.
While in many cases antiestrogen therapy in hormone-responsive breast cancer can stop or reverse tumor progression, resistance develops with time, so that typically after ca. 5 years, tumor growth resumes despite continued treatment with antiestrogens.18, 19 In fact, in laboratory models of breast cancer and even in the clinic, it has been shown that antiestrogens can even become stimulatory in hormone resistant breast cancer.20 While a molecular-level understanding of the mechanism of resistance to endocrine therapy is still incomplete, it is clear that breast cancer cells can undergo adaptations such that antiestrogens can no longer block estrogen signaling. Nevertheless, tumor growth in this hormone-refractory state still appears to rely on the ER-SRC interaction.21
Such considerations motivated us to search for molecules that might inhibit this key ER-SRC interaction directly. Such small molecules, termed coactivator binding inhibitors (CBIs), would be designed to bind in the hydrophobic groove of the ER-agonist complex, thereby blocking the interaction between ER and SRC (Figure 1C).22 Because the CBIs would be blocking this key protein-protein interaction in a direct manner, rather than indirectly as do conventional estrogen antagonists, they might not be subject to the resistance mechanisms that compromise endocrine therapy with antiestrogens. The distinction between inhibition of estrogen signaling through ER by antiestrogens, which act in the ligand binding pocket, and CBIs, which act in the coactivator groove, is illustrated in Figure 1.
In designing CBIs, we have used a structure-guided approach, seeking to find simple cyclic systems that could be assembled in a modular fashion and would display three hydrophobic substituents that mimic the three key leucine residues in the LXXLL interaction sequence. In our earlier work, in which we explored a number of different approaches and core heterocyclic systems, we found that pyrimidine-core molecules appeared to be promising structures as based on their CBI activity and ease of preparation.22 Others have also reported on compounds with ER-CBI activity. Our initial publication was closely followed by a study from Shao et al. which identified ‘guanylhydrazone’ ER CBIs,23 and compounds based on bicyclo[2.2.2]octane24 and pyridylpyridone25 scaffolds have also been reported to have similar activity. Most recently, our laboratory has also published promising results utilizing the amphipathic nature of alternately subsititued trialkyl/triamino benzenes as disruptors of the ER/SRC interaction.26
In this study, we have explored a wide variety of trisubstituted pyrimidine-core systems as potential CBIs, and we have characterized their potency as inhibitors of the interaction of ERα and ERβ with SRC-3 using a time-resolved fluorescence resonance energy transfer assay (TR-FRET), as well as in cell-based assays of estrogen transcriptional activity. Through this study, we have developed a refined pharmacophore model for CBIs of this pyrimidine class, and we have obtained a number of compounds that have low micromolar potency as CBIs in blocking the ER-coactivator interaction in vitro. These compounds are also effective in blocking estrogen-stimulated transcriptional activity in breast cancer cells in a manner that is insurmountable by increased estrogen, a hallmark that critically distinguishes CBIs from conventional estrogen antagonists. Nearly all of these CBIs also have high selectivity for ERα over ERβ. The results of this study suggest that blocking estrogen action using CBIs might provide a viable alternative approach to endocrine therapies.
RESULTS AND DISCUSSION
Based on our initial success in developing ER-CBIs using 2,4-diamino-6-alkyl pyrimidines,22 we wanted to investigate this pharmacophore type further in hopes of increasing the affinity of the CBI. As shown in Figure 2, the design of the initial pyrimidine library was based on the roughly triangular orientation of the L690, L693, and L694 residues of an ER-bound LXXLL-containing peptide from SRC-2 (also known as glucocorticoid receptor interacting protein 1, GRIP-1).27 Although we first reported that the 6-alkyl-2,4-diaminopyrimidines bound with Ki’s in the mid micromolar range (29–49 μM as measured by a fluorescence polarization assay), when they were tested in a more sensitive TR-FRET assay, the best of these, 2,4-diisobutylamino-6-isoamylpyrimidine (Figure 2b), was found to bind with a Ki approaching submicromolar concentrations.24 The high affinity of these compounds, as well as the relative ease with which substituted pyrimidine heterocycles can be synthesized, provided a fruitful starting point for the preparation of an expanded ER-CBI library.
Figure 2.

Structure-based design of pyrimidine core molecules based on the ER/SRC-2 interaction (3erd). A, Rendering of SRC-2 peptide from 3erd crystal structure (the internal H691 and R692 residues are deleted for clarity); B, Minimized structure of CBI 2,4-diisobutylamino-6-isopentylpyrimidine; C, Overlay of the SRC-2 peptide and CBI; D, Side-view of SRC-2 peptide and CBI overlay in coactivator groove.
Library design and synthesis
In our initial attempts at expanding the pyrimidine library, we followed the synthetic route previously described.22 Although this path can be used to produce the desired substituted pyrimidines, it is laborious (due to sparsely soluble intermediates) and prohibitively low-yielding. Consequently, we quickly turned our attention to synthetic routes involving the preformed heterocycle, finally settling upon 2,4,6-trichloropyrimidine, which is a cheap, easily-modified starting material. As detailed below, we eventually applied a wide range of reactions, including aminations, alkylations, alkoxylations, and sulfide formation, on a variety of tri-, di-, and monochloropyrimidines, all of which proceeded in moderate to good yields. Additionally, alkylations and aminations could be applied to this precursor in a site-selective manner.28–31
We designed our pyrimidine-based library to incorporate the leucine and phenylalanine-mimicking substituents of the previously synthesized compounds, as well as tryptophan-mimicking naphthyl groups. In addition to the N- and C-side-arms previously described, we also incorporated other heteroatom-containing substituents (O, S, and SO2) into the pyrimidine core to probe, more deeply, the nature of the binding mode of the CBI in the coactivator groove. Triamino, trimercapto, and trialkoxy pyrimidines were also synthesized. The overall goal of this approach was to make a thorough exploration of the structure-activity relationships of the 2-, 4-and 6-positions of the pyrimidine ring with respect to substituent size, polarity, and hydrogen-bond donor/acceptor capability. In practice, the library design was an iterative activity, evolving with the project as progressive binding results were obtained.
Phenethyl and styryl pyrimidines
The first step toward formation of the CBI library involves the site-selective Suzuki-Miyaura cross-coupling reaction of trans-2-phenylvinylboronic acid with 2,4,6-trichloropyrimidine, which gives the 2,4-dichloro-6-styryl-pyrimidine 1 as the major isomer (73% yield), together with small amounts of the symmetric di-coupled 2-chloro-4,6-distyryl-pyrimidine as the only significant byproduct.29 From this point, the synthesis splits into three routes. The first route (top pathway in Scheme 1) involves the double addition of an amine or alkoxy nucleophile to 1 to produce ‘symmetrically’ substituted pyrimidines 2a d, followed by hydrogenation of the styryl double bond over 10% Pd on carbon at atmospheric pressure of H2 to give the final pyrimidine products 3a c. The second route (middle pathway in Scheme 1) involves the formation of intermediates that are labile under hydrogenation conditions (most notably thioethers). As such, it is necessary to first reduce the styryl double bond of 1 before addition of any nucleophile. The reduced 2,4-dichloro-6-phenethylpyrimidine (4) is subsequently reacted under SNAr conditions with either an excess of alkoxide or thiolate to form the ‘symmetrically’ substituted pyrimidines 5a c. The disulfanyl compounds 5b and 5c are then oxidized with mCPBA to form the disulfonyl pyrimidines 6a and 6b (for complete structural information see Tables 1 and 2 and Supporting Information).32, 33
Scheme 1.

Synthesis of ‘symmetric’ and unsymmetric 2,4-diamino-6-phenethylpyrimidines
Table 1.
Inhibitory activity of 6-alkyl-2,6-diaminopyrimidines on the ERα and ERβ/coactivator interaction as measured in TR-FRET and luciferase reporter gene assays.
 |
TR-FRETKi(μM) | Reporter Gene IC50(μM) | ||||
|---|---|---|---|---|---|---|
| Cmpd # | R1 | R2 | R3 | ERα | ERβ | ERα |
| control | SRC-1 BoxII | 0.065 | 0.066 | |||
| 2a | -styryl | -NHCH2CH(CH3)2 | -NHCH2CH(CH3)2 | 7.5 | 242 | 6.9 |
| 2b | -styryl | -NHCH2Ph | -NHCH2Ph | >1000 | >1000 | >1000 |
| 3a | -CH2CH2Ph | -NHCH2CH(CH3)2 | -NHCH2CH(CH3)2 | 5.8 | >1000 | 2.1 |
| 3b | -CH2CH2Ph | -NHCH2Ph | -NHCH2Ph | >1000 | >1000 | >1000 |
| 8b | -styryl | -N(Boc)CH2Ph | -NHCH2CH(CH3)2 | >1000 | 132 | |
| 9a | -CH2CH2Ph | -N(Boc)CH2CH(CH3)2 | -NHCH2Ph | >1000 | >1000 | |
| 9b | -CH2CH2Ph | -N(Boc)CH2Ph | -NHCH2CH(CH3)2 | >1000 | >1000 | |
| 10a | -styryl | -NHCH2CH(CH3)2 | -NHCH2Ph | >1000 | >1000 | 12 |
| 10b | -styryl | -NHCH2CH(CH3)2 | -NHCH2-1-Nap | >1000 | >1000 | >1000 |
| 10c | -styryl | -NHCH2Ph | -NHCH2CH(CH3)2 | 16 | >1000 | 9.8 |
| 10d | -styryl | -NHCH2Ph | -NHCH2-1-Nap | >1000 | >1000 | |
| 10g | -styryl | -NHCH2-1-Nap | -NHCH2-1-Nap | >1000 | >1000 | |
| 11a | -CH2CH2Ph | -NHCH2CH(CH3)2 | -NHCH2Ph | 7.6 | >1000 | 4.5 |
| 11b | -CH2CH2Ph | -NHCH2CH(CH3)2 | -NHCH2-1-Nap | >1000 | >1000 | >1000 |
| 11c | -CH2CH2Ph | -NHCH2Ph | -NHCH2CH(CH3)2 | 1.7 | >1000 | 1.9 |
| 11d | -CH2CH2Ph | -NHCH2Ph | -NHCH2-1-Nap | >1000 | >1000 | |
| 11f | -CH2CH2Ph | -NHCH2-1-Nap | -NHCH2Ph | >1000 | >1000 | >1000 |
| 11g | -CH2CH2Ph | -NHCH2-1-Nap | -NHCH2-1-Nap | >1000 | >1000 | >1000 |
| 13a | -CH2(CH2)2CH3 | -NH(CH2)2CH3 | -NH(CH2)2CH3 | 16 | >1000 | 13 |
| 13b | -CH2CH2CH(CH3)2 | -NHCH2CH(CH3)2 | -NHCH2CH(CH3)2 | 2.8 | >1000 | 3.1 |
| 13c | -CH2CH2CH(CH3)2 | -NHCH2Ph | -NHCH2Ph | 4.1 | >1000 | 8.3 |
| 13d | -CH2CH2CH(CH3)2 | -NHCH2-1-Nap | -NHCH2-1-Nap | >1000 | >1000 | >1000 |
| 13i | -CH2CH2-1-Nap | -NHCH2CH(CH3)2 | -NHCH2CH(CH3)2 | 2.5 | >1000 | 2.1 |
| 13j | -CH2CH2-1-Nap | -NHCH2Ph | -NHCH2Ph | >1000 | >1000 | |
| 13k | -CH2CH2-1-Nap | -NHCH2-1-Nap | -NHCH2-1-Nap | >1000 | >1000 | >1000 |
| 18a | -CH2CH3 | -NHCH2CH(CH3)2 | -NHCH2CH(CH3)2 | 10 | >1000 | 7.3 |
| 18b | -CH2CH2CH(CH3)2 | -NHCH2CH(CH3)2 | -NHCH2Ph | 7.9 | >1000 | 1.8 |
| 18c | -CH2CH2CH(CH3)2 | -NHCH2CH(CH3)2 | -NHCH2-1-Nap | 7.2 | >1000 | 4.1 |
| 18d | -CH2CH2CH(CH3)2 | -NHCH2Ph | -NHCH2CH(CH3)2 | 5.8 | >1000 | 5.8 |
| 18e | -CH2CH2CH(CH3)2 | -NHCH2Ph | -NHCH2-1-Nap | >1000 | >1000 | >1000 |
| 18f | -CH2CH2CH(CH3)2 | -NHCH2-1-Nap | -NHCH2CH(CH3)2 | 2.4 | >1000 | 6.7 |
| 18g | -CH2CH2CH(CH3)2 | -NHCH2-1-Nap | -NHCH2Ph | >1000 | >1000 | >1000 |
| 18h | -CH2CH2-1-Nap | -NHCH2CH(CH3)2 | -NHCH2Ph | >1000 | >1000 | |
| 18j | -CH2CH2-1-Nap | -NHCH2Ph | -NHCH2CH(CH3)2 | >1000 | >1000 | |
| 18l | -CH2CH2-1-Nap | -NHCH2-1-Nap | -NHCH2CH(CH3)2 | >1000 | >1000 | |
| 26 | -NHCH2CH(CH3)2 | -Cl | -NHCH2CH(CH3)2 | >1000 | >1000 | |
| 27d | -NHCH2CH(CH3)2 | -H | -NHCH2CH(CH3)2 | >1000 | >1000 | |
Table 2.
Inhibitory activity of various pyrimidines on the ERα and ERβ/coactivator interaction as measured in TR-FRET and luciferase reporter gene assays.
 |
TR-FRET Ki(μM) | Reporter Gene IC50(μM) | ||||
|---|---|---|---|---|---|---|
| Cmpd # | R1 | R2 | R3 | ERα | ERβ | ERα |
| control | SRC-1 BoxII | 0.065 | 0.066 | |||
| 3c | -CH2CH2Ph | -OCH2CH(CH3)2 | -OCH2CH(CH3)2 | >1000 | >1000 | >1000 |
| 5b | -CH2CH2Ph | -SCH2CH(CH3)2 | -SCH2CH(CH3)2 | >1000 | >1000 | |
| 6a | -CH2CH2Ph | -SO2CH2CH(CH3)2 | -SO2CH2CH(CH3)2 | >1000 | >1000 | |
| 13e | -CH2CH2CH(CH3)2 | -OCH2CH(CH3)2 | -OCH2CH(CH3)2 | >1000 | >1000 | |
| 13g | -CH2CH2CH(CH3)2 | -SCH2CH(CH3)2 | -SCH2CH(CH3)2 | >1000 | >1000 | |
| 14a | -CH2CH2CH(CH3)2 | -SO2CH2CH(CH3)2 | -SO2CH2CH(CH3)2 | >1000 | >1000 | |
| 21a | -NHCH2CH(CH3)2 | -NHCH2CH(CH3) 2 | -CH2CH2CH(CH3)2 | >1000 | >1000 | |
| 21d | -NHCH2CH(CH3)2 | -NHCH2-1-Nap | -CH2CH2CH(CH3)2 | >1000 | >1000 | >1000 |
| 23a | -CH2CH2CH(CH3)2 | -CH2CH2CH(CH3 )2 | -NHCH2CH(CH3)2 | >1000 | >1000 | |
| 23b | -CH2CH2CH(CH3)2 | -CH2CH2CH(CH3 )2 | -NHCH2Ph | >1000 | >1000 | >1000 |
| 23c | -CH2CH2CH(CH3)2 | -CH2CH2CH(CH3 )2 | -NHCH2-1-Nap | >1000 | >1000 | |
| 24a | -CH2CH2Ph | -CH2CH2Ph | -NHCH2CH(CH3)2 | >1000 | >1000 | >1000 |
| 25a | -NHCH2CH(CH3)2 | -NHCH2CH(CH3)2 | -NHCH2CH(CH3)2 | 5.7 | 299 | 2.4 |
| 25c | -OCH2CH(CH3)2 | -OCH2CH(CH3)2 | -OCH2CH(CH3)2 | >1000 | >1000 | >1000 |
| 25e | -SCH2CH(CH3)2 | -SCH2CH(CH3)2 | -SCH2CH(CH3)2 | 120 | >1000 | >1000 |
| 27a | -NHCH2CH(CH3)2 | -NHCH2Ph | -NHCH2CH(CH3)2 | 7.3 | >1000 | 9.5 |
| 27b | -NHCH2CH(CH3)2 | -OCH2CH(CH3)2 | -NHCH2CH(CH3)2 | >1000 | >1000 | |
| 27c | -NHCH2CH(CH3)2 | -SCH2CH(CH3)2 | -NHCH2CH(CH3)2 | >1000 | >1000 | |
| 28 | -NHCH2CH(CH3)2 | -SO2CH2CH(CH3)2 | -NHCH2CH(CH3)2 | >1000 | >1000 | |
| 29a | -CH2CH2CH(CH3)2 | -N(CH3)CH2CH(CH3)2 | -N(CH3)CH2CH(CH3)2 | >1000 | >1000 | |
| 29c | -CH2CH2-1-Nap | -N(CH3)CH2CH(CH3)2 | -N(CH3)CH2CH(CH3)2 | >1000 | >1000 | |
| 30 | -CH2CH2Ph | -N(CH3)CH2CH(CH3)2 | -N(CH3)CH2CH(CH3)2 | >1000 | >1000 | |
| 31a | -CH2CH2CH(CH3)2 | -NHCH2CH(CH3)2 | -N(CH3)CH2CH(CH3)2 | 7.2 | >1000 | 9.7 |
| 31c | -CH2CH2-1-Nap | -NHCH2CH(CH3)2 | -N(CH3)CH2CH(CH3)2 | >1000 | >1000 | |
| 32 | -CH2CH2Ph | -NHCH2CH(CH3)2 | -N(CH3)CH2CH(CH3)2 | >1000 | >1000 | |
| 34a | -CH2CH2CH(CH3)2 | -N(CH3)CH2CH(CH3)2 | -NHCH2CH(CH3)2 | >1000 | >1000 | |
| 34c | -CH2CH2-1-Nap | -N(CH3)CH2CH(CH3)2 | -NHCH2CH(CH3)2 | 2.4 | >1000 | 4.4 |
| 35 | -CH2CH2Ph | -N(CH3)CH2CH(CH3)2 | -NHCH2CH(CH3)2 | 8.4 | >1000 | 5.5 |
| 37a | -CH2CH2CH(CH3)2 | -NHCH2CH(CH3)2 | -OCH2CH(CH3)2 | >1000 | >1000 | 6.9 |
| 37b | -CH2CH2CH(CH3)2 | -NHCH2CH(CH3)2 | -SCH2CH(CH3)2 | >1000 | >1000 | |
| 39 | -CH2CH2CH(CH3)2 | -NHCH2CH(CH3)2 | -SO2CH2CH(CH3)2 | >1000 | >1000 | |
The third pathway of phenethylpyrimidine synthesis involves the formation of unsymmetrically substituted 2,4-diaminopyrimidines, as shown in the bottom pathway of Scheme 1. Starting from 1, the chloride at the 4-pyrimidinyl position is selectively displaced using an NaH-deprotonated tert-butyl carbamate to form the 2-chloro-6-styryl-pyrimidin-4-yl carbamates 7a c.30 The pyrimidinyl carbamates then undergo addition of a second amine to form the SNAr products 8a f. At this point, either the double bond of the styryl group is reduced under mild hydrogenation conditions (10% Pd/C, 1 atm H2) to give phenethyl derivatives 9a d, or the Boc group is removed in TFA/CH2Cl2, followed by hydrogenation of the alkene, producing the unsymmetrical 2,4-diamino-6-phenethylpyrimidines 11a f. This synthetic approach allowed for all four of the final unsymmetrical product types, stemming from and including the aminopyrimidinyl carbamates 8a f, to be prepared and submitted for CBI assays.
Isoamyl and naphthethylpyrimidines
Because of the limited accessibility of the associated boronic acids or esters, we turned to the direct coupling of sp3-hybridized alkanes with the pyrimidinyl trichloride to form the 6-isoamyl and 6-naphthethylpyrimidine series. This was effectively accomplished through the use of a site-selective Fe(acac)3-catalyzed Kumada-type coupling involving alkyl Grignard reagents, as developed by the Furstner laboratory.28, 34, 35 Following their procedure, we obtained the desired 6-alkyl pyrimidines 12a c in moderate yields. The major byproduct is the ‘symmetric’, disubstituted 4,6-dialkylpyrimidine, with both the 2-naphthethylpyrimidine and the asymmetric, 2,6-dinaphthethylpyrimidine also isolated as minor products. The 2,4-dichloro-6-alkylpyrimidines 12a c were then reacted with O-, N-, and S- nucleophiles to produce the ‘symmetrically’ substituted pyrimidines 13a k. Additionally, the dithioethers 13g and 13h were oxidized with mCBPA in CHCl3 to give the respective disulfonylpyrimidines 14a and 14b (top pathway in Scheme 2).
Scheme 2.

Synthesis of alkylpyrimidines: A, synthesis of 6-isoamyl and 6-naphthethyl pyrimidines; B, synthesis of 2-isoamyl and 2-naphthethyl pyrimidines.
Unfortunately, it proved more difficult to create unsymmetrically-substituted diamines from the alkylpyrimidines 12b and 12c than from the styryl-substituted pyrimidine 1. When 12b was submitted to the same SNAr conditions used to form 7a, an unsymmetric dimer of the starting material was isolated as the major product instead of the desired carbamate. This reaction presumably proceeds through deprotonation of the isoamyl α-carbon of one pyrimidine, followed by displacement of the aromatic chloride on a second molecule. This problem is easily avoided by first introducing the carbamate site-selectively into 2,4,6-trichloropyrimidine at the 4-position (compounds 15a c). This is followed by the Fe(acac)3-catalyzed coupling of the alkyl Grignard to produce the 6-alkyl-pyrimidin-4-yl carbamates 16a g. Amination under the same conditions used for the chlorostyrylpyrimidines leads to the aminoalkylpyrimidinyl carbamates 17a m. The Boc group was subsequently removed with TFA in CH2Cl2 to produce the final 2,4-diamino-6-alkylpyrimidines 18a m in modest overall yield (bottom pathway in Scheme 2a).
It should be noted that, for both of the site-selective steps described in the paragraph above, a small amount of the undesired isomer is also formed. To explore the effect of the exchange of the 6-alkyl and the 4-amino group on the pyrimidine CBIs, four of these minor isomers, 19a d, were subjected to SNAr conditions with isobutylamine, with subsequent removal of the Boc group to produce the 4,6-diamino-2-isoamylpyrimidines 21a d, as shown in Scheme 2b.
To explore the SAR space of the pyrimidine library further, 4,6-dialkyl-2-aminopyrimidines were prepared from the 4,6-dialkyl-2-chloropyrimidines 22a and 22b, which were obtained by either Suzuki-Miyaura coupling of two equivalents of trans-2-phenylvinylboronic acid to 2,4,6-trichloropyrimidine or as the major byproduct from the reaction forming 12a. The final monoamine products were obtained by amination of 22a and 22b, followed by reduction of the styryl groups of compounds 23d f to provide the 2-amino-4,6-diisopropylpyrimidines 23a c and 2-amino-4,6-diphenethylpyrimidines 24a c (Scheme 3).
Scheme 3.

Synthesis of 4,6-dialkyl-2-aminopyrimidines
In addition to the alkyldiaminopyrimidines and dialkylaminopyrimidines, we also wanted to probe the effect of three electronegative substituents on the binding of the pyrimidine core molecules. Thus, we formed the ‘symmetric’ trisubstituted pyrimidines 25a f with three heteroatom-containing substituents by the reaction of 2,4,6-trichloropyrimidine with O, N, and S nucleophiles (Scheme 4). In addition to the ‘symmetrically’ substituted triisobutylaminopyrimidine 25a, at lower temperature and shorter reaction time, the unsymmetrical diamination byproduct, 2,4-diisobutylamino-6-chloropyrimidine, 26, was also produced in modest yield from the reaction of isobutylamine with 2,4,6-trichloropyrimidine. Compound 26 was subsequently substituted with benzyl amine, isobutylalcohol, or isobutylmercaptan to form the unsymmetric tri-heteroatom substitutued pyrimidines 27a c. Reductive dechlorination of 26 gave the 6-proteopyrimidine 27d, and the isobutylsulfanyl pyrimidine 27c was oxidized with mCPBA in CHCl3 to form the diaminosulfone 28.
Scheme 4.

Synthesis of tri-heteroatom-substituted pyrimidines
Methylated aminopyrimidines
During the course of this study, it became clear that there was an important relationship between the activity of the CBI and the nature of the heteroatom substituents, suggesting that simple insertion of an electronegative atom was not sufficient to produce high-affinity compounds (see Table 2). The apparent requirement for a nitrogen-containing substituent at the 2- and/or 4-position of the pyrimidine suggested that H-bond donation might be playing a role in supporting the binding of the diaminopyrimidines. To probe this hypothesis, we synthesized N2-methyl, N4-methyl, and N2,N4-dimethyl diaminopyrimidines, as shown in Scheme 5. Methylation at these sites should obliterate the H-bond donor capability of the secondary amine, while retaining the polarity and planarity induced by the aryl nitrogen.
Scheme 5.

Synthesis of N-methylated pyrimidines
Both the N2-methylated and the ‘symmetrically’ substituted N2,N4-dimethylated pyrimidines were easily synthesized by the non-selective amination with N-methylisobutylamine and subsequent reduction of compounds 1 and 12b,c; and 7a and 16b,e (see the middle and top pathways of Scheme 5, respectively). The N4-methylated compounds could be accessed through a K2CO3-catalyzed site-selective addition of N-methylisobutylamine to 12b,c or as a byproduct from the formation of 29a, followed by amination with isobutylamine and reduction of the styryl moieties when present (bottom pathway of Scheme 5).
The binding results for the N-methylated 6-isoamylpyrimidines 29a, 31a, and 34a show that there is a strong preference for a free N H at the 4-position of the isoamyl-substituted pyrimidine ring (see Table 2). As H-bonding at the 2-position seemed less important to CBI binding, the O, S, and SO2-containing 4-isobutylamino-6-isoamylpyrimidines 37a,b and 39 were also synthesized according to the methods described above and as shown in Scheme 6. Based purely on polarity, these compounds should show CBI activity similar to the diaminopyrimidines 13b and 31a. On the other hand, if H-bond donation or overall planarity of the CBI is important, decreased affinity would be expected.
Scheme 6.

Synthesis 2-alkoxy, 2-sulfanyl, and 2-sulfonylpyrimidines.
In vitro time-resolved FRET assay of the inhibition of coactivator binding to ERα and ERβ by pyrimidine-core CBIs
The inhibitory activity of members of our pyrimidine CBI library for coactivator binding to both ERα and ERβ systems was measured using a TR-FRET assay. This assay employs a site-specifically labeled terbium/streptavidin-biotin/ER-LBD construct and a fluorescein-labeled nuclear receptor domain of the steroid receptor coactivator 3 (SRC-3-NRD). In the presence of agonist (17β-estradiol) and the absence of a CBI, the SRC-3-NRD binds to the ER-LBD, allowing transfer of fluorescence resonance energy from the Tb donor (D) to the fluorescein acceptor (A). With increasing concentrations of CBI, the ER/SRC-3 complex is disrupted, and fluorescence resonance energy transfer decreases. This provides a dose-dependent inhibition curve, typically plotted on an A/D*1000 scale, from which Ki values for the various compounds can be calculated. An unlabeled peptide containing the NR Box II (LXXLL motif) of SRC-1, a natural coactivator of ER, is used as a positive control. The results from these binding studies are summarized in Tables 1 and 2 (additional binding data can be found in Supporting Information).
Initially, what is most striking about the data is the almost universal selectivity of the pyrimidine core CBIs for ERα over ERβ coactivator binding inhibition. With the exception of three compounds that show only very modest affinity for ERβ (2a, 8b, and 25a), all compounds assayed show binding only to ERα. (These results have been echoed in preliminary studies in cell-based reporter gene systems, which have also confirmed the ERα selectivity of pyrimidine compounds 3a, 13b, and 27a, which show no mechanism-based inhibition of ERβ.) Of these three, 2a and 25a are still over 30-fold ERα selective, while only the Boc-protected compound, 8b, shows complete selectivity for ERβ with no activity for ERα. Loss of ERβ activity occurs readily, as observed in the relationship of compound 2a to 3a (converted by hydrogenation of the styryl double bond) and compound 8b to 10c (involving simple removal/addition of a Boc group from the 4-aminopyrimidinyl position). These two factors seem to be important in increasing ERβ affinity, while also decreasing affinity for ERα (compare 3a to 2a, 11a to 10a, and 11c to 10c). It is likely that the combination of the styryl functionality and the Boc-protecting group gives 8b its ERβ selectivity, and this suggests that the ERβ binding groove prefers compounds with greater rigidity coupled with increased hydrophobicity. Selectivity of small-molecules for ERα/ERβ CBI activity has been mentioned only briefly before,23 and seems surprising, since an examination of the coactivator groove of ERα and ERβ bound to LXXLL-containing peptides shows very little dissimilarity. The homology of the binding groove is again supported by assay of the control SRC-1 BoxII peptide (LXXLL motif), which has approximately equivalent affinity for both receptors. Nonetheless, the preference of other peptide-based CBIs for ERα over ERβ has been reported,36–38 and this selectivity is significant in that it may increase the ultimate therapeutic utility of these compounds.39–41
Table 1 contains the binding data for the ‘symmetrically’ and ‘unsymmetrically’ substituted 2,4-diamino-6-alkylpyrimidines and 2,4-diamino-6-styrylpyrimidines. Because these compounds were the initial synthetic targets of this study, as well as the compounds most closely related to those reported in our first ER-CBI paper,22 we were hopeful that these alternative and ‘unsymmetrical’ additions would provide increased CBI activity. While somewhat disappointed by the lack of significant increase in affinity, we were nonetheless pleased that this approach produced many compounds with CBI activity having comparable or slightly higher affinity for ERα.
Included in Table 1 are compounds 3b, 13a, and 13b, which represent the high affinity pyrimidine CBIs reported previously.22 Interestingly, while 13a and 13b show lower Ki values in our more sensitive TR-FRET assay than those we reported using our earlier fluorescence polarization assay, compound 3b shows no measurable binding to either ERα or ERβ by TR-FRET (Ki = 49, 32, and 29 μM by FP vs. >1000, 16, and 2.8 μM by TR-FRET, respectively, for 3b, 13a, and 13b). We believe that the lower affinity value that we obtain for 3b by the TR-FRET assay is the correct one, because it is consistent with a trend we have observed throughout this series of compounds, namely, the lack of affinity of the ERα coactivator groove for extremely bulky ligands. This is apparent in the general exclusion of pyrimidines that contain a naphthyl group, exceptions being those pyrimidines (13i, 18c, and 18f) that contain only two small isobutyl/isoamyl side-arms in addition to the larger aromatic group. Additionally, while the coactivator groove readily tolerates CBIs with two phenyl groups and a single isoamyl/isobuyl group (11a, 11c, and 13c), addition of a third aromatic substituent (3b) results in complete loss of binding. In general, it appears that those compounds that bind best to the coactivator groove are those that fill it snugly, without exceeding certain steric limitations, as in compounds 11c, 13i, and 18f.
Comparison of the pairs of compounds 11a and 11c, 18b and 18d, and 18c and 18f in Table 1 yields another general trend in CBI affinity. For each of these pairs, affinity is increased 1.5 to 3-fold when the amino substituents at the 2 and 4-positions are situated so that the larger moiety is in the 4-position. This effect becomes more dramatic as the size difference in the groups increases (18c and 18f vs. 18b and 18d). Conversely, it appears that the size of the substituent on the alkyl group at the 6-position has little effect on binding, as both the 2,4-diisobutylaminopyrimidines with the large naphthethyl group (13i) and the isoamyl group (13b) bind with comparable affinity. Nevertheless, if the size of the group at the 6-position is reduced drastically, binding is negatively affected, as in the 6-ethylpyrimidine 18a (Ki =10 μM), and substitution with a chlorine or hydrogen atom at the same position results in complete loss of activity (26 and 27d). As mentioned above, introduction of a double bond at the 6-position decreases or, in extreme cases, obliterates ERα affinity, and, in all cases, pyrimidines with Boc-protection at the N4-amine fail to bind ERα (see Supporting Information for further examples of these compounds).
To better understand the effect of substituent type and placement on the pyrimidine ring, we next investigated the binding of 4,6-dialkyl-2-amino- and 2-alkyl-4,6-diaminopyrimidines, as shown in Table 2. Despite the overall hydrophobic nature of the coactivator groove and the distinct structural homology of the 4,6-dialkylpyrimidines 23a 24c to the high affinity 2,4-diamino compounds, we failed to observe binding with any members of this pyrimidine derivative class. Possibly more surprising is the complete lack of binding exhibited by the 2-alkyl-4,6-diaminopyrimidines 21a 21d. Although they are simple isomers of compounds 13b, 13i, 18b, 18c, 18d, and 18f, which all have Ki values below 10 μM, the compounds with a methylene group replacing the amino group at the 2-position suffer complete loss of CBI binding activity. Together, these results suggest that at least two electronegative substituents on the pyrimidine ring are required for CBI activity. Moreover, it is not sufficient for these substituents to have a general 1,3-relationship; rather, binding requires specific substitution at the 2 and 4 positions. Along with the two nitrogens of the pyrimidine core, these two substituent nitrogen atoms form a tetrad of electronegative atoms which appears to provide a unique and essential binding motif.
In contrast to the importance of the presence of the amino substituents at the 2 and 4 positions of the pyrimidine ring, the nature of the C-6 substituent seems to be of minimal importance, as the alkyl group can be replaced with a third amino substituent with minimal loss in binding affinity. Examples of this are the triamino CBIs 25a and 27a (Table 2), which have potencies similar to that of their alkyl-diamino analogs 13b and 18d (Table 1), with Ki values of 5.7 and 7.3, and 2.8 and 5.8, respectively. Nonetheless, there is some selectivity for atom type at the 6-position as is evidenced by the lack of activity of the 2,4-diisobutylaminopyrimidines 27b,c, and 28 which contain O, S, and SO2-linked substituents at that site (Table 2).
In addition to the amino pyrimidines already described, a number of other heteroatom-containing pyrimidines were also analyzed for CBI activity. Incorporation of the alkoxy, sulfanyl, and sulfonyl pyrimidines into the library (Table 2 and Supporting Information) allowed for further evaluation of the role of the amino substituents in CBI binding. Although we had anticipated that any electronegative atom or group would provide affinity similar to the previously assayed aminopyrimidines, we found that none of the pyrimidines incorporating two or three O or S substituents exhibited any inhibition of coactivator binding. (The lone exception to this is the triisobutylsulfanylpyrimidine 25e, which bound ERα with the relatively high Ki of 120 μM.) Even oxidation of the thioether function to the highly polar sulfone failed to elicit activity.
At this point, it became evident that there is a necessary relationship between the presence of nitrogen-bearing substituents at a specific position on the pyrimidine and the potency of the CBI. We rationalized that the importance of the N heteroatom was most likely based on either its ability to act as a hydrogen-bond donor or its planarity due to resonance interaction between the nitrogen lone pair and the aryl π-system. If H-bond donation was required for CBI binding, it was likely that it would be position specific. To probe these possibilities, we synthesized and assayed the N2-methyl, N4-methyl, and N2, N4-dimethyl diaminopyrimidine compounds, 29a 35.
As shown in Table 2, when both the N2- and N4-nitrogens are methylated, affinity for ERα is completely lost (29a d). Interestingly, some activity is regained in the selectively monomethylated compounds: When the N4 of the 6-isoamylpyrimidine is methylated, no binding is observed (34a), but activity is nearly fully restored when the methyl group is switched to N2 (31a). In the phenethyl and naphthethyl compounds, the opposite is observed, and the compounds with a free N H at the N2-position bind (34c and 35), whereas the N4 isomers are inactive (31c and 32).
This restoration of activity associated with monomethylation suggests that N H H-bond donation is indeed important to binding. That this binding is specific to either the 2- or 4-position of the aromatic ring, depending on the nature and/or size of the C-6 substituent, implies that more subtle electronic effects are also playing a role in orientation of the CBI in the coactivator groove: sterically, the 2- and 4-positions of the pyrimidine should be practically equivalent, but the dipole moment of the heterocycle relative to the methyl-substituted amine is position-dependent, and the differential ability of the aromatic nitrogen atoms to act as hydrogen bond acceptors must also be considered. The preference for the free N H to switch from the 4- to 2-position as the steric bulk of the alkyl group increases from isoamyl to phenethyl may indicate that a different binding mode (i.e., 120° rotation) is involved in inhibition with these larger molecules. The manner in which these two somewhat contradictory observations might be understood has been analyzed using computation docking experiments and is discussed below.
After showing the selective importance of at least one free amine N H at either the 2- or 4-position of the pyrimidine, we wanted to probe the ability of other electronegative atoms to offer the same binding properties as the non-H-bonding N-methyl amine. To accomplish this, we prepared the 6-isoamyl-4-isobutylpyrimidines 37a,b and 39. None of these O- or S-containing pyrimidines showed any inhibition of the receptor/coactivator complex. Although not conclusive, this gives strong support for the need of two amino substituents at both the N2- and N4-positions of the pyrimidine ring. Although it is possible that subtle electronic changes are the basis of this marked affinity differential, it is more likely that the planarity and associated rigidity of the N aryl bond creates an overall CBI core structure that is more conducive to binding. A summary of the structure-activity relationships of the pyrimidine-core CBIs is shown in Figure 3.
Figure 3. In vitro.
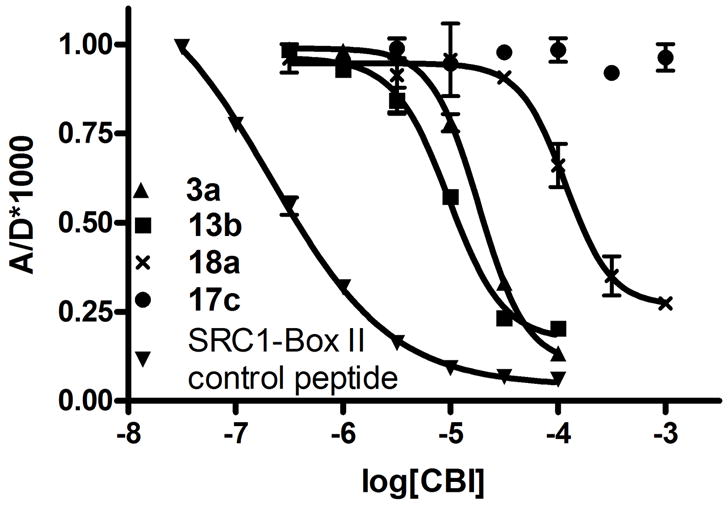
coactivator binding inhibition assay. TR-FRET assay shows displacement of SRC-3-NRD-Fl by control peptide and pyrimidine CBIs.
In vitro radiometric assay of CBI binding affinity for the ligand binding pocket of ERα and ERβ
To further ensure that the inhibition we observe with these compounds in the TR-FRET CBI assay is not due to the compound binding as an antagonist at the ligand binding pocket, we conducted a competitive radiometric ligand binding assay using tritium-labeled estradiol as a tracer and full-length purified human ERα and ERβ. The binding affinities of the CBIs relative to the tracer and standard, estradiol, are expressed as relative binding affinity (RBA) values. The pyrimidine CBIs generally have RBAs for ERα of less than 0.005% (estradiol = 100%), and while there are a few significant exceptions, even the CBI with the highest affinity for the ligand binding pocket (11a) binds with only 0.051% the affinity of estradiol.
To properly interpret these results, it must be remembered that estradiol has a Kd for ERα of 0.2 nM; therefore, at the concentration of estradiol used for the TR-FRET CBI assay (1μM), a compound with an RBA of even 0.1% would show an IC50 value of minimally 1 mM in the assay if it were acting as a conventional antagonist (i.e., competing with estradiol), rather than as a CBI (i.e., competing with SRC). Thus, we conclude that the inhibitions we are observing in our TR-FRET assay could not be due to the competition of these pyrimidines for the natural ligand estradiol with concomitant induction of an antagonist, non-coactivator binding conformation of the ER. Rather, the active pyrimidine CBIs reported in this library are disrupting the ER/SRC interaction by direct binding to the protein surface at the coactivator binding groove, not to the traditional, internal ligand binding site. (See Supporting Information for specific RBAs.)
Cell-based assay of the inhibition of estrogen-induced reporter gene transcriptional activity by the CBIs
To investigate the cellular activity of our pyrimidine core CBIs, we utilized a estrogen-responsive luciferase reporter gene assay performed in human endometrial cancer (HEC-1) cells lacking endogenous estrogen receptor, but transfected with a full-length ERα expression vector, an estrogen-responsive luciferase reporter gene plasmid (2ERE Luc), and pCMV β-galactosidase (β-gal; internal control). The cells were incubated with two concentrations of estradiol (1 nM and 100 nM) and titrated concentrations of CBI. After incubation for 24 hours, cells were lysed and assayed for luciferase reporter gene and β-galactosidase activity.
Those CBIs that were active showed not only a concentration-dependent reduction in reporter gene activity, but they also gave IC50 values that were unaffected by changing the estradiol concentration. This inhibition, insurmountable by 100-fold excess estradiol, indicates that the CBIs are not competing against the ligand estradiol, as a conventional antagonist would; rather, they are acting through the CBI mechanism to block coactivator binding. Examples of the estrogen insurmountable reporter gene inhibitory activity of two representative CBIs (3a and 13b) are shown in Figure 5a (TR-FRET curves for the same compounds are shown in Figure 3).
Figure 5.

Coactivator binding inhibition assays. A, Dose-response of CBI inhibition of ER-mediated transcription in cell-based reporter gene assay run at two concentrations of estradiol; B, Plot of CBI Ki (μM) as calculated from in vitro TR-FRET assay vs. CBI IC50 as determined by cell-based reporter gene assay.
ERI-5, a previously published CBI inhibitor of the ERα-SRC interaction reported to have an IC50 of 5.5 μM in a COS-7 cell mammalian 2 hybrid assay,23 was used as a positive control in these reporter gene assays; in our hands, it showed an IC50 value of 1.4 μM. Because of its cellular impermeability, the SRC-1 Box II control peptide used for the in vitro TR-FRET work could not be used in the cell-based assay. Measurement of β-galactosidase activity simultaneously with CBI testing serves as an internal control to confirm that decreased luciferase activity corresponds to a CBI blockade of ERα-coactivator binding, rather than to a generalized mechanism of cellular toxicity. The activity of the internal β-galactosidase control was affected by only the highest concentration of CBI used in the assay, 20 μM, which decreased the activity of the control to approximately 70% of the expected maximal value for only the most potent pyrimidine compounds. A functionally different type of viability assay, probing cellular mitochondrial function, was also employed to confirm that the decrease in luciferase activity we observed was truly due to compound activity. This assay (CellTiter 96R AQueous One Solution Cell Proliferation Assay, Promega) was run using compounds that showed the greatest discrepancy in IC50 values between in vitro and cellular assays (i.e. 3a, 10a, 18b, 25a, and 37a) and provided results in agreement with the β-galactosidase reporter gene assay controls, also showing that viable cell counts decreased significantly only at the highest doses of compound (20 μM). Because the IC50 values we see for these compounds are significantly lower than 20 μM where cell toxicity first becomes apparent, we have concluded that the inhibition of estrogen-stimulated reporter gene activity by the CBIs was a transcription-based response rather than the result of general cell death.
In all instances where activity was observed in the TR-FRET protein-binding assay, comparable potencies or, in a number of cases, increased potencies were observed in the luciferase reporter gene assay (Tables 1 and 2). A comparison of potencies in the cell-based activity assay vs. the in vitro binding assay is illustrated in Figure 5b. The points below the dotted line represent cases where higher potency was evident in cell-based assay. While we can only speculate at this point, enhanced potency in the cell-based assays could be the result of decreased available concentrations of CBI compound in vitro because of sequestration by the small amount of detergent that needs to be present in the buffer of the TR-FRET assay. It could also be due to cell-based mechanisms that increase the local concentrations of the CBI by increased cellular uptake. In either case, we are encouraged by the increased potencies of these compounds when tested in a more physiologically relevant cell-based assay: not only do these results confirm the CBI activity of the library compounds observed in the TR-FRET assays, but they also provide evidence that these compounds are soluble and cell-permeable at concentrations necessary for inhibition. In a very low percentage of compounds, we have seen activity in the cell-based assays that was not discovered using the TR-FRET assay (10a and 37a). It is possible that these results are due to an alternative mechanism of inhibition involving the AF-1 and/or DNA-binding domains of ER, which are present in the full-length protein used in the reporter gene assays but not in the ligand-binding domain of ER employed in the TR-FRET assay. Together, these data provide support that these pyrimidine CBI compounds could be effective as novel inhibitors of ERα-SRC interactions and, thus, ER-mediated transcription.
Modeling of CBI docking in ERα
To better understand the binding mode(s) involved with the pyrimidine-core CBIs, we have performed extensive docking experiments using, as a starting point, the structure of ER-LBD co-crystallized with the agonist diethylstilbestrol and a SRC-2 peptide (see http://www.rcsb.org entry 3erd). After work-up of the crystal structure, the coactivator peptide was removed, and the CBIs were docked into the resulting empty binding groove using the FlexiDock module of SYBYL® 7.3 from Tripos™.
As has been noted previously,22 these experiments confirm that the coactivator binding groove of ER is flexible in nature, and modeling that is representative of reality is often complicated by the apparent accommodation of a variety of orientations of a single ligand. This is especially problematic when docking quasi-symmetrical compounds such as 13b. Fortunately, while analyzing the various docking results, we have been able to apply the empirical binding and activity data described above to produce ER/CBI-binding models that are satisfying chemically, computationally, and experimentally (see Figure 6).
Figure 6.

Docking of pyrimidine-core CBIs into the coactivator groove of ERα-LBD. A, electrostatic rendering of 13b bound to ERα-LBD; B, cut-away showing the interaction of Glu542 and Lys362 with 13b; C, electrostatic rendering of 13i bound to ERα-LBD; D, cut-away showing the interaction of Glu542 and Lys362 with 13i.
We have been especially interested in comparing the binding orientation of the isopentyl pyrimidine 13b and the analogous naphthylethyl pyrimidine 13i, as these compounds show essentially equivalent binding affinity in the TR-FRET assay, yet exhibit opposite selectivity for their corresponding monomethylated aromatic amines. Additionally, the pre-existing knowledge of which N-methylated CBI (N2 vs. N4) binds to the surface of the LBD, greatly aids in determination of the binding mode of the unmethylated compound.
In general, the coactivator groove consists of three major binding sites: a small pocket (shown on the right of the binding site in Figure 6), a medium-size pocket (on the left) and a large shelf (bottom center). Another important feature of the binding site is the ‘charge clamp’ composed of Glu542 and Lys362 of the ERα-LBD, which, in the presence of a coactivator, interact in a productive manner with the inherent dipole associated with α helical peptide backbone of the LXXLL sequence. As shown in Figures 6a and b, 13b binds to the ER coactivator groove in such a manner as to optimize its interaction with, not only the three hydrophobic pockets, but also both the carboxyl and amine groups of the charge clamp. The isopentyl alkyl group fills the small pocket on the right, pushing the amine in the 4-position into the medium sized pocket on the left and towards Glu 542, allowing for a positive interaction between partially positively charged N-H of the amine and the negatively charged carboxyl group. The second isobutylamine substituent occupies the coactivator shelf, so that there is minimal steric interaction between the alkyl group and the charged amine of Lys362. This allows for positive electrostatic interaction between Lys362 and the lone pair of the pyrimidinyl ring nitrogen. Additionally, docking of the associated monomethylated products 31a and 34a in this orientation allows for rationalization of the binding selectivity observed: When the amine at the 2-position is methylated (31a), there is very little rearrangement of either the ligand or the protein, and the interaction between the Lys362 and the pyrimidine nitrogen is maintained. Methylation at the N4-position of the pyrimidine (34a) completely disrupts the interaction between the N-H and the Glu542, and, consequently, affinity for the receptor is lost.
In contrast to the quasi-symmetric 13b, the large naphthyl group of 13i constrains the CBI to only two possible orientations, both of which place the bicyclic aromatic on the large non-polar shelf. Of these two orientations, only that shown in Figures 6c and d is situated so that monomethylation at the 4-position is tolerated while methylation at the 2-position causes disruption of the ER/CBI complex. As seen in the electrostatic rendering of the complex (Figure 6c), this orientation also correctly matches the negatively and positively-charged residues of the ‘charge clamp’ with the electron-deficient N-H and the electron-rich pyrimidine N. The cut-away rendering in Figure 6d more explicitly shows this orientation. Although not shown, Figure 6d also provides an illustration of the effects of mono-N-methylation of 13i: While there is ample room for methylation at N4 (this in effect pushes the CBI to the right, deeper into the small pocket), methylation at N2 disrupts the pyrimidine nitrogen/Lys362 interaction, causing the CBI to be bumped out of the binding pocket and the lysine residue to be pushed back and to the right.
CONCLUSIONS
In this report, we describe the progressive optimization of the structure of 2,4,6-trisubstituted pyrimidines as inhibitors of the interaction between agonist-liganded estrogen receptor (ER) and a key coactivator protein, steroid receptor coactivator 3 (SRC-3). By inhibiting the interaction of ER with this important mediator of ER transcriptional activity, these coactivator binding inhibitors (CBIs) are able to block ER activity at a point after ligand binding, and we have demonstrated in cell-based reporter gene assays that this inhibition is insurmountable by increasing estrogen concentration. In this sense, inhibition of ER activity by CBIs is direct and distinct from that of conventional estrogen antagonists, which compete for agonist binding and interfere with SRC binding indirectly, by altering the topology of the ER from an agonist conformation that recruits SRCs to an antagonist conformation that does not.
We have achieved this optimization by developing an expedited synthesis of 2,4,6-trisubstituted pyrimidines, which has enabled us to systematically generate a series of focused pyrimidine libraries having alkyl and aralkyl substituents of various size and positioning, linked to the heterocyclic core through carbon and heteroatoms. Additionally, we have probed for the role of potential hydrogen bonding involvement of NH-linking atoms.
Through these studies, we have developed a rather complete structure-activity profile outlining a pharmacophore for the CBI activity of these trisubstituted pyrimidines. This pharmacophore specifies the following:
The need for N-linked side chains at the 2 and 4-positions of the pyrimidine ring, with a preference for a smaller group (isobutyl) at the 2 position.
Ready accommodation of either a C or N-bound moiety at the 6-position of the pyrimidine.
A strict size limit to the CBI binding pocket, with no more than two medium (phenyl) groups or one large (naphthyl) group tolerated at one time on the tridentate ligand.
Electrostatic effects and/or hydrogen bonding that require at least one free aryl N-H, the preferred position of which being dependant on the size of the alkyl group at the 6-position of the pyrimidine ring.
The best compounds we have obtained have Ki values (as inhibitors of coactivator binding in vitro) and IC50 values (for inhibition of reporter gene activity in cells) in the low micromolar region, and the vast majority of these compounds show strong selectivity for ERα over ERβ. The most promising of these compounds are being evaluated in further cell-based and in other more biologically advanced activity assays. The results of this study suggest that blocking estrogen action using CBIs might provide a viable alternative approach to endocrine therapies.
EXPERIMENTAL SECTION
General Synthetic Methods
All reagents were used as purchased except where noted. THF, Ether, CH2Cl2, and DMF used in reactions were dried using a solvent delivery system (neutral alumina column). Solvents used for extraction and flash chromatography were reagent or ultima grade purchased from either Aldrich or Fisher Scientific. All reactions were run under dry N2 atmosphere except where noted. Flash column chromatography was performed on Silica P Flash Silica Gel (40–64 μM, 60A) from SiliCycle®. 1HNMR and 13CNMR spectra were obtained on 500 MHz Varian® FT-NMR spectrometers. Except where noted, both low and high resolution mass spectra were obtained using electrospray ionization on either a Micromass Q-Tof Ultima or Waters Quattro instrument. HPLC analysis was performed using a Waters 1525 binary HPLC pump equipped with a Waters in-line degasser AF, Waters 2487 Dual γ absorbance detector, and a Waters Symmetry® C18 5μM, 4.6 X 150 mm column. Microanalysis was performed with a CE 440 CHN analyzer.
Representative Syntheses (For complete synthetic details and HPLC analysis of active compounds please see Supporting Information). 2,4-Dichloro-6-styryl-pyrimidine (1)
Based on the coupling described by Tan et al.29 Trans-2-Phenylvinylboronic acid (2.546 g, 17.2 mmol), K3PO4 (7.307 g, 34.4 mmol), and PdCl2(PPh3)2 (0.362 g, 0.52 mmol) were dissolved in 100 mL THF. To this mixture, 2,4,6-trichloropyrimidine (3.156 g, 17.2 mmol) dissolved in 20 mL THF was added producing a cloudy yellow suspension. H2O (15 mL) were added and the now clear solution was heated at reflux for 7 h. Approximately 100 mL of H2O were added, and the biphasic mixture was extracted three times with ether. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent removed with a rotary evaporator. The product purified by column chromatography (gradient elution 5 10% EtOAc in hexanes) to provide 1 (3.1613 g, 73%). 1H NMR (500 MHz, CDCl3) δ ppm 6.95 (d, J=15.87 Hz, 1 H), 7.22 (s, 1 H), 7.41 (m, 3 H), 7.59 (dd, J=7.45, 2.08 Hz, 2 H), 7.96 (d, J=15.87 Hz, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 117.1, 123.0, 128.2, 129.2, 130.6, 134.8, 140.9, 160.7, 162.8, 166.6. HRMS (ESI+) m/z calcd for C12H9N2Cl2+ 251.0143, found 251.0101.
N,N'-diisobutyl-(6-styryl-pyrimidin-2,4-yl)-diamine (2a)
Compound 1 (0.111 g, 0.44 mmol) was dissolved in 4 mL of isobutylamine and heated with stirring in a sealed high-pressure tube at 115 °C for 43 h. The reaction was allowed to cool to room temperature and the excess amine removed in vacuo. The residue was extracted from H2O three times with ether. The combined organic layers were washed twice with brine, dried over MgSO4, and the solvent removed by rotary evaporator. Purification was accomplished with column chromatography (50% EtOAc in hexanes) to give 2a (67 mg, 47%). 1H NMR (500 MHz, CDCl3) δ ppm 0.97 (d, J=6.65 Hz, 6 H), 0.98 (d, J=6.65 Hz, 6 H), 1.88 (sept, J=6.65 Hz, 1 H), 1.89 (sept, J=6.65 Hz, 1 H), 3.12 (bs, 2 H), 3.25 (t, J=6.32 Hz, 2 H), 4.78 (bs, 1 H), 4.90 (bs, 1 H), 5.74 (s, 1 H), 6.83 (d, J=15.87 Hz, 1 H), 7.28 (t, J=7.29 Hz, 1 H), 7.35 (t, J=7.50 Hz, 2 H), 7.55 (d, J=7.50 Hz, 2 H), 7.66 (d, J=15.86 Hz, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.5, 20.6, 28.7, 28.8, 49.3, 127.5, 127.7, 128.6, 128.9, 134.0, 136.8, 162.8, 164.4. HRMS (ESI+) m/z calcd for C20H29N4+ 325.2392, found 325.2393. Anal. (C20H28N4) H. C: calcd, 74.03; found, 73.44. N: calcd, 17.27; found, 16.82.
2,4-Diisobutoxy-6-styryl-pyrimidine (2c)
Clean Na0 (82 mg, 1.4 mmol) was dissolved in 2 mL of isobutylalcohol while heating at 70 °C with stirring (~2 h). To the resulting slightly cloudy solution, 150 mg (0.60 mmol) of 1 were added with stirring forming an opaque pale orange mixture. One more mL of isobutylalcohol was added, and the reaction was stirred at 70°C for 4 h. The reaction was allowed to cool to room temperature, and extracted from H2O three times with ether. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent and excess alcohol removed with a rotary evaporator. Purification was accomplished with column chromatography (10% EtOAc in hexanes) to give 2c (0.172 g, 88%). 1H NMR (500 MHz, CDCl3) δ ppm 1.01 (d, J=6.59 Hz, 6 H), 1.06 (d, J=6.59 Hz, 6 H), 2.08 (sept, J=6.84 Hz, 1 H), 2.19 (sept, J=6.84 Hz, 1 H), 4.14 (d, J=6.59 Hz, 2 H), 4.18 (d, J=6.84 Hz, 2 H), 6.33 (s, 1 H), 6.93 (d, J=15.87 Hz, 1 H), 7.32 (t, J=7.20 Hz, 1 H), 7.37 (t, J=7.32 Hz, 2 H), 7.57 (d, J=7.57 Hz, 2 H), 7.82 (d, J=15.87 Hz, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 19.4, 19.6, 28.0, 28.1, 72.9, 73.9, 99.6, 126.0, 127.7, 128.9, 129.1, 136.0, 136.1, 164.3, 165.3, 172.4. HRMS (ESI+) m/z calcd for C20H27N2O2+ 327.2073, found 327.2085. Anal. (C20H26N2O2) C, H, N.
N,N'-dibenzyl-(6-phenethyl-pyrimidin-2,4-yl)-diamine (3b)
Compound 2b (80 mg, 0.20 mmol) and 10% Pd/C (22 mg, 0.020 mmol) were dissolved in 16 mL of MeOH. The resulting suspension was stirred under 1 atm H2 for 1h. The solvent was removed in vacuo and the residue purified with column chromatography (25% EtOAc in hexanes) to give 3b (42 mg, 52%). 1H NMR (500 MHz, CDCl3) δ ppm 2.72 (distorted dd, J=10.51, 8.36 Hz, 2 H), 2.95 (distorted dd, J=10.51, 8.58 Hz, 2 H), 4.48 (d, J=4.07 Hz, 2 H), 4.61 (d, J=6.00 Hz, 2 H), 4.90 (bs, 1 H), 5.24 (bs, 1 H), 5.56 (s, 1 H), 7.16 7.37 (m, 15 H). 13C NMR (500 MHz, CDCl3) δ ppm 34.9, 39.7, 45.6, 50.4, 126.1, 127.1, 127.6, 127.7, 127.8, 128.5, 128.6, 128.7, 128.9, 140.3, 141.9, 162.4. HRMS (ESI+) m/z calcd for C26H27N4+ 395.2236, found 395.2245.
2,4-Dichloro-6-phenethyl-pyrimidine (4)
Compound 1 (1.00 g, 4.0 mmol) and 10% Pd/C (118 mg, 0.11 mmol) were dissolved in 40 mL of MeOH. The resulting suspension was stirred under 1 atm H2 for 1 h. The solvent was removed in vacuo and the residue purified with column chromatography (10% EtOAc in hexanes) to give 4 (0.521 mg, 52%). 1H NMR (500 MHz, CDCl3) δ ppm 3.06 (s, 4 H), 7.05 (s, 1 H), 7.17 (d, J=6.86 Hz, 2 H), 7.22 (t, J=7.29 Hz, 1 H), 7.29 (t, J=7.40 Hz, 2 H). 1H NMR (500 MHz, acetone-D6) δ ppm 3.08 (AA’BB’, 2 H) 3.13 (AA’BB’, 2 H), 7.20 (t, J=6.96 Hz, 1 H), 7.28 (m, 4 H), 7.53 (s, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 34.4, 39.2, 119.4, 126.7, 128.5, 128.8, 139.9, 160.7, 162.5, 174.6. HRMS (ESI+) m/z calcd for C12H11N2Cl2+ 253.0299, found 253.0301.
2,4-Bis-benzyloxy-6-phenethyl-pyrimidine (5a)
Clean Na0 (54 mg, 2.3 mmol) was dissolved in 2 mL of benzylalcohol while heating at 70 °C with stirring (~2 h). To the resulting pale yellow solution, 100 mg (0.40 mmol) of 4 were added, and the reaction was stirred at 70 °C for 16 h. The reaction was allowed to cool to room temperature, and extracted from H2O three times with ether. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent and excess alcohol removed in vacuo (removal of the alcohol required heating to 80 °C). Purification was accomplished with column chromatography (10% EtOAc in hexanes) to give 5a (0.130 g, 83%). 1H NMR (500 MHz, CDCl3) δ ppm 2.93 (distorted dd, J=9.77, 9.03 Hz, 2 H), 3.03 (distorted dd, J=10.25, 8.79 Hz, 2 H), 5.40 (s, 2 H), 5.45 (s, 2 H), 6.25 (s, 1 H), 7.20 (m, 3 H), 7.26 7.43 (m, 10 H), 7.51 (d, J=7.08 Hz, 2 H). 13C NMR (500 MHz, CDCl3) δ ppm 34.6, 39.3, 68.3, 69.2, 100.6, 126.2, 128.1, 128.2, 128.4, 128.56, 128.6, 128.7, 136.4, 137.0, 141.3, 164.8, 171.5, 172.3. HRMS (ESI+) m/z calcd for C26H25N2O2+ 397.1916, found 397.1916. Anal. (C26H24N2O2) H, N. C: calcd, 78.76; found, 78.22.
2,4-Bis-isobutylsulfanyl-6-phenethyl-pyrimidine (5b)
To a suspension of 48 mg (1.2 mmol) NaH (60% dispersion in mineral oil) in 5 mL of THF, 2-methyl-1-propanethiol (127 μL, 1.2 mmol) was added and the resulting mixture was stirred for 30 min. To this compound, 4 (0.100 g, 0.40 mmol) in 1 mL THF was added and suspension became a cloudy pink. The reaction was stirred 16 h at room temperature and the extracted from H2O three times with CH2Cl2. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent removed with a rotary evaporator. Purification was accomplished with column chromatography (50% to 75% CH2Cl2 in hexanes) to give 5b (0.131 g, 92%). 1H NMR (500 MHz, CDCl3) δ ppm 1.04 (d, J=6.65 Hz, 6 H), 1.07 (d, J=6.65 Hz, 6 H), 1.95 (sept, J=6.65 Hz, 1 H), 2.01 (sept, J=6.65 Hz, 1 H), 2.86 (distorted dd, J=10.29, 9.22 Hz, 2 H), 3.00 (distorted dd, J=10.51, 8.79 Hz, 2 H), 3.07 (d, J=6.65 Hz, 4 H), 6.60 (s, 1 H), 7.20 (m, 3 H), 7.28 (t, J=7.40 Hz, 2 H). 13C NMR (500 MHz, CDCl3) δ ppm 22.1, 22.2, 28.6, 28.7, 34.7, 37.6, 39.2, 39.5, 113.2, 126.3, 128.59, 128.64, 141.2, 167.3, 170.1, 171.5. HRMS (ESI+) m/z calcd for C20H29N2S2+ 361.1772, found 361.1781.
2,4-Bis-(2-methyl-propane-1-sulfonyl)-6-phenethyl-pyrimidine (6a)
From the procedure described by Hurst.32, 33 mCPBA (77%, 53 mg, 0.24 mmol) and 5b (18 mg, 0.050 mmol) were dissolved in 1.1 mL CHCl3 with stirring. The solution was then allowed to stand at room temperature for 36 hours. CHCl3 (5 mL) was added and the solution was washed 2 times with sat. NaHCO3 sol. The aqueous layers were extracted once with CHCl3 and the combined organic layers were washed with sat. NaCl sol. and dried over MgSO4. The solvent was removed in vacuo, and chromatography of the residue (30% EtOAc in hexanes) gave 6a in quantitative yield. 1H NMR (500 MHz, CDCl3) δ ppm 1.10 (d, J=6.86 Hz, 6 H), 1.13 (d, J=6.86 Hz, 6 H), 2.29 (sept, J=6.65 Hz, 1 H), 2.40 (sept, J=6.65 Hz, 1 H), 3.16 (t, J=7.72 Hz, 2 H), 3.37 (m, 4 H), 3.42 (d, J=6.43 Hz, 2 H), 7.16 (d, J=7.08 Hz, 2 H), 7.22 (t, J=7.40 Hz, 1 H), 7.29 (t, J=7.40 Hz, 2 H), 7.91 (s, 1 H). 13C NMR (500 MHz, CDCl3)δ ppm 22.8, 22.9, 24.0, 24.1, 34.4, 40.0, 58.6, 58.7, 118.8, 127.0, 128.6, 129.0, 139.2, 166.4, 167.2, 177.2. HRMS (ESI+) m/z calcd for C20H29N2O4S2+ 425.1569, found 425.1588.
(2-Chloro-6-styryl-pyrimidin-4-yl)-naphthalen-1-ylmethyl-carbamic acid tert-butyl ester (7c)
From the procedure described by Zanda, et al.30 Compound 1 (0.700 g, 2.8 mmol) and Naphthalen-1-ylmethyl-carbamic acid tert-butyl ester (0.745 g, 3.06 mmol) were dissolved in 20 mL DMF. To this solution, NaH (60% dispersion in mineral oil, 0.134 g, 3.3 mmol) was added under nitrogen causing the solution to become a dark turquoise. The reaction was stirred at room temperature for 1 h. The DMF was removed in vacuo with heating and the residue extracted from brine twice with ether and once with CH2Cl2. The combined organic layers were washed with sat. NaCl sol. and dried over MgSO4. The solvent was removed in vacuo, and chromatography of the residue (1:1 CH2Cl2:hexanes to CH2Cl2) gave 7c (573 mg, 44%). 1H NMR (500 MHz, CDCl3) δ ppm 1.33 (s, 9 H), 5.79 (s, 2 H), 7.05 (d, J=15.87 Hz, 1 H), 7.21 (d, J=7.32 Hz, 1 H), 7.38 (m, 4 H), 7.50 7.62 (m, 4 H), 7.76 (d, J=8.06 Hz, 1 H), 7.91 (m, 2 H), 8.09 (d, J=8.30 Hz, 1 H), 8.10 (s, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 28.0, 46.3, 83.5, 109.0, 122.99, 123.03, 125.2, 125.5, 125.8, 126.3, 127.6, 128.0, 128.99, 129.05, 129.7, 131.0, 133.6, 133.8, 135.7, 138.1, 153.4, 160.0, 162.7, 165.5. HRMS (ESI+) m/z calcd for C28H27ClN3O2+ 472.1792, found 472.1790.
(2-Benzylamino-6-styryl-pyrimidin-4-yl)-isobutyl-carbamic acid tert-butyl ester (8a)
Compound 7a (0.059 g, 0.15 mmol) and benzylamine (50 μL, 0.45 mmol) were dissolved in 3 mL DMSO and heated with stirring in a sealed high pressure tube at 90 °C for 16 h. As the reaction had not yet progressed to completion (as shown by TLC), at this point the temperature was raised to 110 °C and the solution was heated an additional 22 h. The DMSO was removed in vacuo with heating, and the resulting residue was extracted from brine three times with ether. The combined organic layers were washed with brine and dried over MgSO4. The solvent was removed in vacuo, and chromatography of the residue (5% EtOAc in hexanes) produced 8a (49 mg, 70%). 1H NMR (500 MHz, CDCl3) δ ppm 0.80 (d, J=6.59 Hz, 6 H), 1.55 (s, 9 H), 1.98 (sept, J=6.84 Hz, 1 H), 3.79 (d, J=7.32 Hz, 2 H), 4.65 (d, J=5.86 Hz, 2 H), 5.35 (bs, 1 H), 6.94 (d, J=16.11 Hz, 1 H), 7.25 (t, J=7.20 Hz, 1 H), 7.28 7.39 (m, 8 H), 7.55 (d, J=7.57 Hz, 2 H), 7.72 (d, J=15.87 Hz, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.4, 28.2, 28.4, 45.8, 52.2, 81.9, 127.2, 127.49, 127.51, 127.6, 128.7, 128.86, 128.91, 135.0, 136.5, 140.0, 154.3, 161.8, 162.4, 163.6. HRMS (ESI+) m/z calcd for C28H35N4O2+ 459.2760, found 459.2760.
(2-Benzylamino-6-phenethyl-pyrimidin-4-yl)-isobutyl-carbamic acid tert-butyl ester (9a)
Compound 8a (6 mg, 0.013 mmol) and 10% Pd/C (2 mg, 0.002 mmol) were dissolved in 2 mL of MeOH and 2 mL CH2Cl2. The resulting suspension was stirred under 1 atm H2 for 1 h. The solvent was removed in vacuo, and the residue purified with column chromatography (25% EtOAc in hexanes) to give 9a in quantitative yield. 1H NMR (500 MHz, CDCl3) δ ppm 0.78 (d, J=6.84, 6 H), 1.51 (s, 9 H), 1.95 (sept, J=6.84 Hz, 1 H), 2.84 (m, 2 H), 2.99 (distorted dd, J=11.23, 8.79 Hz, 2 H), 3.76 (d, J=7.32 Hz, 2 H), 4.60 (d, J=5.86 Hz, 2 H), 5.32 (bs, 1 H), 7.08 (s, 1 H), 7.16 7.35 (m, 10 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.4, 28.2, 28.4, 35.2, 40.2, 45.7, 52.2, 81.8, 102.6, 126.1, 127.2, 127.4, 128.57, 128.65, 128.7, 133.6, 135.0, 140.0, 154.2. HRMS (ESI+) m/z calcd for C28H37N4O2+ 461.2917, found 461.2920.
N2-Benzyl-N4-isobutyl-6-styryl-pyrimidine-2,4-diamine (10a)
Compound 8a (16 mg, 0.035 mmol) was dissolved in 1 mL CH2Cl2, and, to this solution, 1 mL TFA was added with stirring, causing the solution to become bright yellow. The reaction was stirred at room temperature for 30 min, and the solvent was removed in vacuo. The resulting residue was extracted from H2O three times with ether. The combined organic layers were washed with brine, dried over MgSO4, and the solvent removed by rotary evaporator. Column chromatography of the residue (40% EtOAc in hexanes) gave 10a (12 mg, 98%). 1H NMR (500 MHz, CDCl3) δ ppm 0.96 (d, J=6.59 Hz, 6 H), 1.86 (sept, J=6.59 Hz, 1 H), 3.12 (bs, 2 H), 4.66 (d, J=5.86 Hz, 2 H), 4.74 (bs, 1 H), 5.12 (bs, 1 H), 5.78 (s, 1 H), 6.83 (d, J=15.87 Hz, 1 H), 7.23 7.42 (m, 8 H), 7.54 (d, J=7.57 Hz, 2 H), 7.67 (d, J=15.87 Hz, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.5, 28.7, 45.8, 127.1, 127.5, 127.6, 127.8, 128.6, 128.9, 134.1, 136.8, 140.4, 162.5, 164.5. HRMS (ESI+) m/z calcd for C23H27N4+ 359.2236, found 359.2254.
N2-Benzyl-N4-isobutyl-6-phenethyl-pyrimidine-2,4-diamine (11a)
Compound 10a (9 mg, 0.025 mmol) and 10% Pd/C (12 mg, 0.011 mmol) were dissolved in 4 mL MeOH. The resulting suspension was stirred under 1 atm H2 for 1 h. The solvent was removed in vacuo, and the residue purified with column chromatography (50% EtOAc in hexanes) to give 11a in quantitative yield. 1H NMR (500 MHz, CDCl3) δ ppm 0.92 (d, J=6.59 Hz, 6 H), 1.80 (sept, J=6.84 Hz, 1 H), 2.73 (distorted dd, J=10.25, 8.30 Hz, 2 H), 2.97 (distorted dd, J=10.50, 8.55 Hz, 2 H), 3.03 (bs, 2 H), 4.61 (d, J=5.86 Hz, 2 H), 4.63 (bs, 1 H), 5.08 (bs, 1 H), 5.52 (s, 1 H), 7.16 7.34 (m, 8 H), 7.37 (d, J=6.84 Hz, 2 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.5, 28.6, 35.0, 39.8, 45.7, 126.0, 127.1, 127.8, 128.5, 128.6, 128.7, 142.0, 162.5, 164.1. HRMS (ESI+) m/z calcd for C23H29N4+ 361.2392, found 361.2398.
2,4-Dichloro-6-(3-methyl-butyl)-pyrimidine (12b)
Based on methodology described by Furstner et al.28, 34, 35 Isopentylmagnesium bromide was formed by slow addition without stirring of isoamylbromide (5.0 mL, 41.7 mmol) to the bottom of a 3-neck flask fitted with a condenser that contained magnesium metal (1.116 g, 45.9 mmol) and 1,2-dibromoethane (500 μL, 5.8 μmol) in ~100 mL of diethyl ether. Once bubbles began to form, the solution was stirred until evolution ceased (1.5 h). Before addition to the following reaction mixture the Grignard was titrated using menthol and 1,10-phenanthroline dissolved in THF. In a separate flask, 2,4,6- trichloropyrimidine (2.20 g,. 11.99 mmol), Fe(acac)3 (0.217 g, 0.614 mmol), and N-methyl-2- pyrolidinone (6.5 ml) were combined in dry THF (85 mL) producing an orange solution. To this solution, the isopentylmagnesium bromide (66mL, 0.181 M in ether, 11.05 mmol) was added dropwise over the course of 20 minutes, immediately producing a cloudy dark orange precipitate. The reaction was stirred for 1 h at room temperature and then quenched with 1 M aqueous HCl. The mixture was extracted three times with ether, and the combined organic layers were washed with sat. NaHCO3 sol. and brine, dried over MgSO4, and the solvent removed in vacuo. Purification was accomplished by column chromatography (CH2Cl2) to give 12b as the major product (1.377 g, 52%). 1H NMR (500 MHz, CDCl3) δ ppm 0.91 (d, J=6.43 Hz, 6 H), 1.58 (m, 3 H), 2.71 (AA’XX’, 2 H), 7.14 (s, 1 H). 13C NMR (500 MHz, CDCl3)δ ppm 22.4, 28.0, 35.7, 37.69, 118.9, 160.5, 162.4, 176.4. HRMS (ESI+) m/z calcd for C9H13N2Cl2+ 219.0456, found 219.0462.
2,4-dibenzylamino-6-isopentylpyrimidine (13c)
Compound 12b (0.071 g, 0.32 mmol) was dissolved in benzylamine (1.5 mL) and heated sequentially with stirring in a sealed high-pressure tube at 110 °C for 5 h, 140 °C for 6h, and finally 155 °C for 13h. The solution was cooled and extracted from sat. NaHCO3 solution once with CHCl3 and twice with CH2Cl2. The combined organic layers were washed with sat. NaHCO3 solution, H2O, and brine, dried over anhydrous MgSO4 and the solvent was removed with a rotary evaporator. Purification was accomplished with column chromatography (50% EtOAc in hexanes) to give 13c in quantitative yield. 1H NMR (500 MHz, CDCl3) δ ppm 0.91 (d, J=6.65 Hz, 6 H), 1.51 (m, 2 H), 1.59 (sept, J=6.65 Hz, 1 H), 2.42 (t, J=8.15 Hz, 2 H), 4.49 (bs.d, J=5.15 Hz, 2 H), 4.59 (d, J=6.00 Hz, 2 H), 4.98 (bs, 1 H), 5.20 (bs, 1 H), 5.61 (s, 1 H), 7.21 7.38 (m, 10 H). 13C NMR (500 MHz, CDCl3) δ ppm 22.7, 28.1, 36.0, 37.9, 45.6, 53.3, 105.5, 127.0, 127.1, 127.5, 127.71, 127.74, 128.4, 128.57, 128.6, 128.8, 140.3, 162.4, 163.8, 170.4. HRMS (ESI+) m/z calcd for C23H29N4+ 361.2392, found 361.2386. Anal. (C23H28N4) H, N. C, calcd, 76.63; found, 77.36.
2,4-Diisobutoxy-6-(3-methyl-butyl)-pyrimidine (13e)
Clean Na0 (63 mg, 2.7 mmol) was dissolved in 2mL of isobutyl alcohol while heating at 70 °C. To this solution, 12b (0.100 g, 0.46 mmol) dissolved in 0.5 mL of isobutyl alcohol was added and solution became a cloudy pale orange. The reaction was stirred at 70 °C for 12 h and allowed to cool to room temperature. The solution was extracted from H2O three times with ether. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent removed with a rotary evaporator. Purification was accomplished with column chromatography (10% EtOAc in hexanes) to give 13e (0.102 g, 76%). 1H NMR (500 MHz, CDCl3) δ ppm 0.90 (d, J=6.22 Hz, 6 H), 0.97 (d, J=6.65 Hz, 6 H), 1.00 (d, J=6.65 Hz, 6 H), 1.49 1.64 (m, 3 H), 2.03 (sept, J=6.86 Hz, 1 H), 2.10 (sept, J=6.65 Hz, 1 H), 2.55 (t, J=7.72 Hz, 2 H), 4.06 (d, J=6.65 Hz, 2 H), 4.07 (d, J=6.65 Hz, 2 H), 6.16 (s, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 19.4, 19.6, 22.6, 27.97, 28.04, 28.06, 35.70, 35.75, 72.6, 73.7, 99.4, 165.3, 171.9, 173.6. HRMS (ESI+) m/z calcd for C17H31N2O2+ 295.2386, found 295.2392. Anal. (C17H30N2O2) H, N. C: calcd, 69.35; found, 69.86.
2,4-Bis-isobutylsulfanyl-6-(3-methyl-butyl)-pyrimidine (13g)
To a suspension of 41 mg (1.03 mmol) NaH in 4 mL of THF, 2-methyl-1-propanethiol (93 mg, 1.03 mmol) was added and the resulting mixture was stirred for 20 min. To this, 12b (0.078 g, 0.35 mmol) in 3 mL THF was added and the suspension became a cloudy yellow. The reaction was stirred 16 h at room temperature and the extracted from H2O three times with ether. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent removed with a rotary evaporator. Purification was accomplished with column chromatography (50% CH2Cl2 in hexanes) to give 13g (0.114 g, 98%). 1H NMR (500 MHz, CDCl3) δ ppm 0.91 (d, J=6.35 Hz, 6 H), 1.03 (d, J=6.59 Hz, 6 H), 1.04 (d, J=6.59 Hz, 6 H), 1.49 1.63 (m, 3 H), 1.89 2.03 (m, 2 H), 2.54 (t, J=7.81 Hz, 2 H), 3.05 (d, J=6.84 Hz, 2 H), 3.08 (d, J=6.84 Hz, 2 H), 6.64 (s, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 22.15, 22.22, 22.6, 28.0, 28.7, 35.6, 37.6, 37.8, 39.5, 112.9, 168.9, 169.8, 171.3. HRMS (ESI+) m/z calcd for C17H31N2S2+ 327.1929, found 327.1941.
4-(3-Methyl-butyl)-2,6-bis-(2-methyl-propane-1-sulfonyl)-pyrimidine (14a)
From the procedure described by Hurst.32, 33 mCPBA (77%, 142 mg, 0.63 mmol) in 1.5 mL of CHCl3 was added to a solution of 13g (43 mg, 0.13 mmol) in 1 mL CHCl3 with stirring. The solution was then allowed to stand at room temperature for 36 hours. CHCl3 (10 mL) was added and the solution was washed 2 times with sat. NaHCO3 sol. The aqueous layers were extracted once with CHCl3 and the combined organic layers were washed with sat. NaCl sol. and dried over MgSO4. The solvent was removed in vacuo, and chromatography of the residue (25% EtOAc in hexanes) gave 14a (0.026 g, 50%). 1H NMR (500 MHz, CDCl3) δ ppm 0.97 (d, J=6.43 Hz, 6 H), 1.13 (d, J=6.86 Hz, 6 H), 1.14 (d, J=6.86 Hz, 6 H), 1.62 1.72 (m, 3 H), 2.36 (sept, J=6.86 Hz, 1 H), 2.41 (sept, J=6.86 Hz, 1 H), 3.03 (m, 2 H), 3.39 (d, J=6.65 Hz, 2 H), 3.46 (d, J=6.65 Hz, 2 H), 8.02 (s, 1 H). 13C NMR (500 MHz, CDCl3)δ ppm 22.5, 22.87, 22.92, 24.0, 24.2, 28.2, 36.7, 37.7, 58.5, 58.7, 118.4, 166.3, 167.2, 179.0. HRMS (ESI+) m/z calcd for C17H31N2O4S2+ 391.1725, found 391.1732.
(2,6-Dichloro-pyrimidin-4-yl)-isobutyl-carbamic acid tert-butyl ester (15a)
Based on the method described by Zanda et al.30 2,4,6-Trichloropyrimidine (0.500 g, 2.73 mmol) and N-isobutyl-tert-butyl carbamate (0.473 g, 2.73 mmol) were dissolved in 15 mL of DMF. NaH (60% dispersion in mineral oil, 0.163 g, 4.07 mmol) was added and the solution turned yellow. The reaction was stirred at room temperature overnight, quenched with sat. NH4Cl sol, and then extracted three times with ether. The combined organic phases were washed with sat. NaCl sol., and dried over MgSO4. The solvent was removed in vacuo, and column chromatography (1:1 CH2Cl2:hexanes) of the residue produced 15a (0.642 g, 74%). 1H NMR (500 MHz, CDCl3)δ ppm 0.89 (d, J=6.86 Hz, 6 H), 1.54 (s, 9 H), 2.03 (sept, J=6.86 Hz, 1 H), 3.87 (d, J=7.29 Hz, 2 H), 8.08 (s, 1 H). 13C NMR (500 MHz, CDCl3)δ ppm 20.3, 28.0, 28.2, 52.7, 83.8, 110.4, 153.3, 158.9, 162.0, 163.0. HRMS (ESI+) m/z calcd for C13H20N3O2Cl2+ 320.0933, found 320.0939.
[2-Chloro-6-(3-methyl-butyl)-pyrimidin-4-yl]-isobutyl-carbamic acid tert-butyl ester (16b)
Based on methodology described by Furstner et al.28, 34, 35 Isopentylmagnesium bromide was formed by slow addition without stirring of isoamylbromide (0.700 mL, 5.84 mmol) to the bottom of a 3-neck flask fitted with a condenser that contained magnesium metal (0.50 g, 20.6 mmol) and 1,2-dibromoethane (75 μL, 0.87 μmol) in ~20 mL of diethyl ether. Once bubbles began to form, the solution was stirred until evolution ceased (1.5 h). Before addition to the following reaction mixture, the Grignard was titrated using menthol and 1,10-phenanthroline dissolved in THF. Compound 15a (0.220 g,. 0.69 mmol), Fe(acac)3 (0.017 g, 0.048 mmol), and N-methyl-2-pyrolidinone (0.4 mL) were combined in dry THF (5.0 mL). To this solution, isopentylmagnesium bromide (0.29 M in ether, 3.55 mL, 1.03 mmol) was added dropwise, causing a progression from the starting brown solution to a cloudy orange followed by a dark brown and finally a clear solution containing solid salts. The reaction was stirred for 4 h at room temperature and then quenched with 1 M aqueous HCl. The mixture was extracted three times with ether, and the combined organic layers were washed with brine, dried over MgSO4, and the solvent removed in vacuo. Purification was accomplished by column chromatography (CH2Cl2 followed by 3% EtOAc in CH2Cl2) to give 16b (128 mg, 52%). 1H NMR (500 MHz, CDCl3)δ ppm 0.88 (d, J=6.59 Hz, 6 H), 0.92 (d, J=6.35 Hz, 6 H), 1.52 1.67 (m, 12 H), 2.03 (sept, J=6.84 Hz, 1 H), 2.66 (m, 2 H), 3.87 (d, J=7.08 Hz, 2 H), 7.78 (s, 1 H). 13C NMR (500 MHz, CDCl3)δ ppm 20.3, 22.6, 28.2, 28.3, 36.3, 37.2, 38.2, 52.5, 82.9, 109.8, 153.7, 159.3, 162.6, 174.4. HRMS (ESI+) m/z calcd for C18H31N3O2Cl+ 356.2105, found 356.2114.
[2-Benzylamino-6-(3-methyl-butyl)-pyrimidin-4-yl]-isobutyl-carbamic acid tert-butyl ester (17a)
Compound 16b (0.020 g, 0.056 mmol) was dissolved in benzylamine (1 mL) and heated with stirring in a sealed high-pressure tube at 100 °C for 24 h. The solution was cooled and the excess amine removed in vacuo with heating. The residue was extracted from sat. NaHCO3 solution three times with CH2Cl2. The combined organic layers were washed with H2O and brine, dried over anhydrous MgSO4 and the solvent removed with a rotary evaporator. Purification was accomplished with column chromatography (15% EtOAc in hexanes) to give 17a in quantitative yield. 1H NMR (500 MHz, CDCl3) δ ppm 0.78 (d, J=6.86 Hz, 6 H), 0.92 (d, J=6.43 Hz, 6 H), 1.50 1.66 (m, 12 H), 1.95 (sept, J=6.86 Hz, 1 H), 2.53 (m, 2 H), 3.75 (d, J=7.29 Hz, 2 H), 4.58 (d, J=6.00 Hz, 2 H), 5.31 (bs, 1 H), 7.06 (s, 1 H), 7.21 7.34 (m, 5 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.3, 22.7, 28.2, 28.3, 28.4, 36.5, 38.3, 45.6, 52.2, 81.7, 127.1, 127.4, 128.7, 140.0, 145.3, 154.3, 161.7, 161.8, 172.6. HRMS (ESI+) m/z calcd for C25H39N4O2+ 427.3073, found 427.3068.
N4-Benzyl-N2-isobutyl-6-(3-methyl-butyl)-pyrimidine-2,4-diamine (18d)
Compound 17c (0.030 g, 0.071 mmol) was dissolved in 1.5 mL CH2Cl2 followed by addition of 1.5 mL TFA with stirring. The reaction was stirred at room temperature for 1.5 h, and the solvent and excess TFA were removed by rotary evaporator. Direct purification of the residue was accomplished with column chromatography (20% EtOAc and 2% TEA in hexanes) to give 18d in quantitative yield. 1H NMR (500 MHz, CDCl3) δ ppm 0.90 (d, J=6.65 Hz, 6 H), 0.93 (d, J=6.86 Hz, 6 H), 1.49 (m, 2 H), 1.58 (sept, J=6.65 Hz, 1 H), 1.82 (sept, J=6.65 Hz, 1 H), 2.39 (m, 2 H), 3.18 (t, J=6.32 Hz, 2 H), 4.50 (d, J=5.15 Hz, 2 H), 4.84 (bs, 1 H), 4.88 (bs, 1 H), 5.56 (s, 1 H), 7.30 (m, 5 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.5, 22.7, 28.2, 28.8, 36.1, 38.0, 45.5, 49.2, 91.9, 127.5, 127.7, 128.8, 162.8, 163.8. HRMS (ESI+) m/z calcd for C20H31N4+ 327.2549, found 327.2553.
[6-Chloro-2-(3-methyl-butyl)-pyrimidin-4-yl]-isobutyl-carbamic acid tert-butyl ester (19a)
Formed as a byproduct from the synthesis of 16b (30 mg, 12%). 1H NMR (500 MHz, CDCl3) δ ppm 0.89 (d, J=6.84 Hz, 6 H), 0.93 (d, J=6.35 Hz, 6 H), 1.55 (s, 9 H), 1.60 (sept, J=6.59 Hz, 1 H), 1.66 (m, 2 H), 2.02 (sept, J=6.84 Hz, 1 H), 2.82 (distorted t, J=8.06 Hz, 2 H), 3.92 (d, J=7.08 Hz, 2 H), 7.88 (s, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.4, 22.7, 27.9, 28.2, 28.3, 37.26, 37.32, 52.5, 82.9, 109.7, 153.8, 160.9, 161.6, 171.1. HRMS (ESI+) m/z calcd for C18H31N3O2Cl+ 356.2105, found 356.2099.
Isobutyl-[6-isobutylamino-2-(3-methyl-butyl)-pyrimidin-4-yl]-carbamic acid tert-butyl ester (20a)
Compound 19a (0.019 g, 0.053 mmol) was dissolved in isobutylamine (2 mL) and heated with stirring in a sealed high-pressure tube at 100 °C for 16 h. The solution was cooled and the excess amine removed in vacuo. Direct purification of the residue was accomplished with column chromatography (15% EtOAc in hexanes) to give 20a (18 mg, 88%). 1H NMR (500 MHz, CDCl3) δ ppm 0.86 (d, J=6.84 Hz, 6 H), 0.92 (d, J=6.35 Hz, 6 H), 0.98 (d, J=6.84 Hz, 6 H), 1.52 (s, 9 H), 1.62 (m, 3 H), 1.86 (sept, J=6.59 Hz, 1 H), 1.95 (m, J=6.84 Hz, 1 H), 2.63 (distorted t, J=8.06 Hz, 2 H), 3.06 (t, J=5.74 Hz, 2 H), 3.87 (d, J=7.32 Hz, 2 H), 4.89 (bs, 1 H), 6.68 (s, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.4, 20.6, 22.8, 28.0, 28.4, 28.51, 28.54, 37.4, 49.5, 52.4, 81.4, 110.0, 157.3, 164.1.
N,N'-Diisobutyl-2-(3-methyl-butyl)-pyrimidine-4,6-diamine (21a)
Compound 20a (0.018 g, 0.047 mmol) was dissolved in 2 mL CH2Cl2 followed by addition of 2 mL TFA with stirring. The reaction was stirred at room temperature for 1 h, and the solvent and excess TFA were removed by rotary evaporator. Direct purification of the residue was accomplished with column chromatography (25% EtOAc and 2% TEA in hexanes) to give 21a (6 mg, 47%). 1H NMR (500 MHz, CDCl3) δ ppm 0.92 (d, J=6.35 Hz, 6 H), 0.99 (d, J=6.84 Hz, 12 H), 1.61 (m, 3 H), 1.87 (sept, J=6.84 Hz, 2 H), 2.51 (m, 2 H), 2.98 (t, J=6.10 Hz, 4 H), 4.83 (bs, 2 H), 5.05 (s, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.6, 22.8, 28.48, 28.54, 37.7, 37.9, 49.8, 110.0, 163.6. HRMS (ESI+) m/z calcd for C17H33N4+ 293.2705, found 293.2696.
2-Chloro-4,6-distyryl-pyrimidine (22a)
Based on the coupling described by Tan et al.29 Trans-2-Phenylvinylboronic acid (0.148 g, 1.00 mmol), K3PO4 (0.430 g, 2.03 mmol), and PdCl2(PPh3)2 (0.010 g, 0.015 mmol) were combined in a 25 ml round-bottom flask. To this mixture, 2,4,6-trichloropyrimidine (0.093 g, 0.50 mmol) dissolved in THF (1.5 mL) was added producing a cloudy suspension. More THF (4.5 ml) and H2O (0.11 mL) was added, producing a white cloudy suspension, which was heated at reflux for 19 h. 0.5 mL more of H2O were added, eventually causing the precipitate to dissolve and the solution to turn dark orange. After 5 more hours at reflux, the solution was allowed to cool to room temperature. 35 mL of H2O were added and the biphasic mixture was extracted three times with ether. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent removed with a rotary evaporator. The product was recrystallized from 5% EtOAc/hexanes and the filtrate purified with by clumn chromatography (5 % EtOAc/hexanes) to provide 22a (0.084 g, 52%). 1H NMR (500 MHz, CDCl3) δ ppm 7.04 (d, J=15.87 Hz, 2 H), 7.20 (s, 1 H), 7.36 7.45 (m, 6 H), 7.62 (d, J=6.84 Hz, 4 H), 7.95 (d, J=15.87 Hz, 2 H). 13C NMR (500 MHz, CDCl3) δ ppm 114.9, 124.6, 128.1, 129.2, 130.0, 135.5, 139.0, 161.7, 165.9. HRMS (ESI+) m/z calcd for C20H16N2Cl+ 319.1002, found 319.1005.
2-Chloro-4,6-bis-(3-methyl-butyl)-pyrimidine (22b)
Formed as a byproduct from the synthesis of 12b (279 mg, 9%). 1H NMR (500 MHz, CDCl3) δ ppm 0.91 (d, J=6.22 Hz, 12 H), 1.53 1.63 (m, 6 H), 2.67 (distorted t, J=8.36 Hz, 4 H), 6.93 (s, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 22.5, 28.2, 35.8, 38.0, 117.5, 161.0, 174.6. HRMS (ESI+) m/z calcd for C14H24N2Cl+ 255.1628, found 255.1620.
[4,6-Bis-(3-methyl-butyl)-pyrimidin-2-yl]-isobutylamine (23a)
Compound 22b (0.050 g, 0.20 mmol) was added to 1.5 mL of isobutylamine immediately forming a bright yellow solution. With stirring at room temperature (~5 min), the solution became a light brown. The reaction mixture was heated in a sealed high-pressure tube at 95 °C for 16 h. The reaction was cooled and the excess isobutylamine was removed in vacuo. The resulting residue was extracted from H2O three times with ether. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent removed with a rotary evaporator. Purification was accomplished with column chromatography (10% EtOAc in hexanes) to give 23a in quantitative yield. 1H NMR (500 MHz, CDCl3) δ ppm 0.92 (d, J=6.35 Hz, 12 H), 0.95 (d, J=6.84 Hz, 6 H), 1.50 1.64 (m, 6 H), 1.85 (sept, J=6.59 Hz, 1 H), 2.50 (m, 4 H), 3.24 (dd, J=6.71, 5.98 Hz, 2 H), 4.99 (bt, J=5.37 Hz, 1 H), 6.24 (s, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.5, 22.7, 28.1, 28.8, 36.0, 38.0, 49.2, 108.1, 162.8, 171.7. HRMS (ESI+) m/z calcd for C18H34N3+ 292.2753, found 292.2755.
(4,6-Diphenethyl-pyrimidin-2-yl)-isobutylamine (24a)
Compound 23d (0.089 g, 0.25 mmol) was dissolved in suspension of 13 mg (0.012 mmol) 10% Pd/C in 20 mL of MeOH, forming a bright yellow mixture. The suspension was stirred under 1 atm of H2 for 45 min. producing a clear mixture. The MeOH was removed in vacuo and the residue purified with column chromatography (25% EtOAc in hexanes) to give 24a (57 mg, 64%). 1H NMR (500 MHz, CDCl3) δ ppm 1.00 (d, J=6.65 Hz, 6 H), 1.90 (sept, J=6.65 Hz, 1 H), 2.82 (distorted dd, J=10.29, 8.79 Hz, 4 H), 2.98 (distorted dd, J=10.29, 8.79 Hz, 4 H), 3.30 (t, J=6.22 Hz, 2 H), 5.10 (t, J=5.15 Hz, 1 H), 6.19 (s, 1 H), 7.20 (m, 6 H), 7.29 (m, 4 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.5, 28.8, 34.9, 39.6, 49.2, 108.7, 126.2, 128.56, 128.62, 141.7, 162.9, 170.3. HRMS (ESI+) m/z calcd for C24H30N3+ 360.2440, found 360.2435. Anal (C24H29N3) C, H, N.
N,N',N''-Triisobutyl-pyrimidine-2,4,6-triamine (25a)
2,4,6-trichloropyrimidine (0.182 g, 0.11 mmol) was added to 4 mL of isobutylamine, producing a violent reaction accompanied by the evolution of light smoke. The reaction mixture was heated with stirring in a sealed high-pressure tube at 115 °C for 43 h. The reaction was removed from the heat, allowed to cool to room temperature, and the excess amine was removed in vacuo. The resulting residue was extracted from H2O three times with CH2Cl2. The combined organic layers were washed with sat. NaHCO3 and brine and dried over anhydrous MgSO4. The solvent was removed with a rotary evaporator and purification of the residue was accomplished with column chromatography (100% EtOAc) to give 25a (175 mg, 60%). 1H NMR (500 MHz, CDCl3) δ ppm 0.91 (d, J=6.84 Hz, 6 H), 0.94 (d, J=6.59 Hz, 12 H), 1.81 (m, 3 H), 2.96 (t, J=6.35 Hz, 4 H), 3.12 (t, J=6.59 Hz, 2 H), 4.52 (m, 3 H), 4.74 (s, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.5, 20.6, 28.6, 28.8, 49.1, 49.6, 71.6, 162.4, 164.5. HRMS (ESI+) m/z calcd for C16H32N5+ 294.2658, found 294.2656. Anal. (C16H31N5) C, H, N.
2,4,6-Triisobutoxy-pyrimidine (25c)
Clean Na0 (83 mg, 3.6 mmol) was dissolved in 4 mL of isobutylalcohol while heating at 70 °C. The resulting solution was cooled to room temperature and 104 mg (0.57 mmol) 2,4,6-trichloropyrimidine dissolved in 0.5 mL of isobutyl alcohol were added with stirring, causing the solution to immediately become cloudy. The reaction was stirred at room temperature for 1 h and then at 60 °C for 24 h. The reaction was allowed to cool to room temperature, extracted from H2O twice with ether and once with CH2Cl2. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent and excess alcohol removed with a rotary evaporator. Purification was accomplished with column chromatography (5% EtOAc in hexanes) to give 25c (0.151 g, 90%). 1H NMR (500 MHz, CDCl3) δ ppm 0.96 (d, J=6.65 Hz, 12 H), 0.99 (d, J=6.65 Hz, 6 H), 2.03 (sept, J=6.65 Hz, 2 H), 2.11 (sept, J=6.65 Hz, 1 H), 4.02 (d, J=6.86 Hz, 4 H), 4.05 (d, J=6.86 Hz, 2 H), 5.66 (s, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 19.4, 19.5, 28.0, 28.1, 73.1, 73.9, 83.4, 164.9, 172.8. HRMS (ESI+) m/z calcd for C16H29N2O3+ 297.2178, found 297.2179. Anal. (C16H28N2O3) H, N. C: calcd, 64.83; found, 65.26.
2,4,6-Tris-isobutylsulfanyl-pyrimidine (25e)
To a suspension of 147 mg NaH (3.67 mmol, 60% dispersion in mineral oil) in 15 mL of THF, 2-methyl-propane-1-thiol (0.35 mL, 3.3 mmol) was added dropwise and the resulting mixture was stirred for 20 min. To this, 2,4,6-trichloropyrimidine (0.150 g, 0.82 mmol) was added. The reaction was stirred 16 h at room temperature, quenched with H2O, and extracted three times with ether. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent removed with a rotary evaporator. Purification was accomplished with column chromatography (50% CH2Cl2 in hexanes) to give 25e (0.226 g, 94%). 1H NMR (500 MHz, CDCl3) δ ppm 1.01 (d, J=6.65 Hz, 12 H), 1.04 (d, J=6.65 Hz, 6 H), 1.92 (sept, J=6.65 Hz, 2 H), 1.98 (sept, J=6.65 Hz, 1 H), 3.02 (d, J=6.86 Hz, 2 H), 3.03 (d, J=6.86 Hz, 4 H), 6.64 (s, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 22.1, 22.2, 28.6, 28.7, 37.8, 39.5, 110.8, 168.0, 170.9. HRMS (ESI+) m/z calcd for C16H29N2S3+ 345.1493, found 345.1479. Anal. (C16H28N2S3) C, H, N.
2,4-diisobutylamino-6-chloropyrimidine (26)
Isobutylamine (1 mL) was added dropwise to 0.337 g (1.8 mmol) of 2,4,6-trichloropyrimidine producing a violent reaction accompanied by the evolution of white smoke. The resulting mixture was heated in a sealed pressure tube at 75 °C for 1 h. The reaction was cooled to room temperature and extracted three times from sat. NaHCO3 solution with CH2Cl2. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent removed with a rotary evaporator. Purification was accomplished with column chromatography (5% EtOAc in hexanes) to give 26 (0.216 g, 45%). 1H NMR (500 MHz, CDCl3) δ ppm 0.90 (d, J=6.59 Hz, 6 H), 0.92 (d, J=6.59 Hz, 6 H), 1.76 1.88 (m, 2 H), 3.03 (bs, 2 H), 3.15 (t, J=6.35 Hz, 2 H), 4.98 (bs, 1 H), 5.16 (bs, 1 H), 5.66 (s, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.4, 28.5, 28.6, 49.0, 162.1, 164.4. HRMS (ESI+) m/z calcd for C12H22N4Cl+ 257.1533, found 257.1522.
2,4-diisobutylamino-pyrimidine (27d)
Compound 26 (0.035 g, 0.14 mmol) was dissolved in a suspension of 3 mL MeOH, 23 μL Et3N, and 24 mg (0.022 mmol) 10% Pd/C. The suspension was stirred under 1 atm of H2 for 24 h. The reaction was filtered using a 2μM syringe filter and the solvent removed using a rotary evaporator. The residue was extracted three times from H2O with CH2Cl2. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent removed in vacuo. Purification was accomplished with column chromatography (5% EtOAc and 1% TEA in hexanes) to give 27d (19 mg, 63%). 1H NMR (500 MHz, CDCl3) δ ppm 0.95 (d, J=6.84 Hz, 6 H), 0.95 (d, J=6.59 Hz, 6 H), 1.80 1.90 (m, 2 H), 3.07 (bs, 2 H), 3.17 (dd, J=6.71, 5.98 Hz, 2 H), 4.73 (bs, 1 H), 4.86 (s, 1 H), 5.66 (d, J=5.86 Hz, 1 H), 7.81 (bs, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.47, 20.51, 28.6, 28.7, 49.1, 156.3, 162.6, 163.4. HRMS (ESI+) m/z calcd for C12H23N4+ 223.1923, found 223.1915.
N2,N4-dimethyl-N2,N4-diisobutyl-(6-isopentyl-pyrimidin-2,4-yl)-diamine (29a)
N-methylisobutylamine (1 mL) was added dropwise to 12b (0.055 g, 0.22 mmol), causing the immediate formation of a precipitate. The reaction mixture was stirred in a sealed high-pressure tube at 80 °C for 16h, allowed to cool to room temperature, and extracted three times from aq. NaHCO3 with CH2Cl2. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent removed in vacuo. Purification was accomplished with column chromatography (10% EtOAc in hexanes) to give 29a (25 mg, 31%). 1H NMR (500 MHz, CDCl3) δ ppm 0.89 (d, J=6.65 Hz, 12 H), 0.92 (d, J=6.43 Hz, 6 H), 1.52 1.66 (m, 3 H), 2.06 (nonet, J=6.86 Hz, 2 H), 2.45 (t, J=7.93 Hz, 2 H), 2.99 (s, 3 H), 3.12 (s, 3 H), 3.28 (d, J=6.22 Hz, 2 H), 3.39 (d, J=7.29 Hz, 2 H), 5.57 (s, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.58, 20.63, 22.8, 27.7, 27.8, 28.0, 36.2, 36.5, 36.7, 38.1, 57.0, 57.2, 89.6, 162.2, 163.2, 169.7. HRMS (ESI+) m/z calcd for C19H37N4+ 321.3018, found 321.3014.
Isobutyl-[2-(isobutyl-methyl-amino)-6-styryl-pyrimidin-4-yl]-carbamic acid tert-butyl ester (31b0)
Compound 7a (0.063 g, 0.16 mmol) was dissolved in 1.5 mL of N-methylisobutylamine and heated with stirring in a sealed high-pressure tube at 100 °C for 16 h. The reaction was allowed to cool to room temperature and the excess amine removed in vacuo. The resulting residue was extracted three times from H2O with ether. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent removed by rotary evaporator. Purification was accomplished with column chromatography (CH2Cl2 followed by 10% EtOAc in hexanes) to give 31b0 (32 mg, 45%). 1H NMR (500 MHz, CDCl3) δ ppm 0.90 (d, J=6.84 Hz, 6 H), 0.93 (d, J=6.84 Hz, 6 H), 1.56 (s, 9 H), 2.11 (m, 2 H), 3.21 (s, 3 H), 3.45 (d, J=7.08 Hz, 2 H), 3.87 (d, J=7.32 Hz, 2 H), 6.95 (d, J=15.63 Hz, 1 H), 7.17 (s, 1 H), 7.29 (t, J=7.32 Hz, 1 H), 7.37 (t, J=7.45 Hz, 2 H), 7.56 (d, J=7.57 Hz, 2 H), 7.76 (d, J=15.87 Hz, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.5, 20.6, 27.6, 28.2, 28.5, 36.4, 52.3, 57.2, 81.7, 100.9, 127.6, 128.3, 128.6, 128.9, 134.4, 136.8, 154.4, 161.7, 161.8, 162.9. HRMS (ESI+) m/z calcd for C26H39N4O2+ 439.3073, found 439.3076.
N2-methyl-N2,N4-diisobutyl-(6-styryl-pyrimidin-2,4-yl)-diamine (31b)
Compound 31b0 (0.032 g, 0.073 mmol) was dissolved in 1.5 mL CH2Cl2 and to this solution 1.5 mL of TFA was added dropwise with stirring. The reaction was allowed to stir at room temperature for 2.5 h. The solvent was removed in vacuo, and the resulting residue was directly purified by column chromatography (5% EtOAc and 1% TEA in hexanes) to give 31b in quantitative yield. 1H NMR (500 MHz, CDCl3) δ ppm 0.93 (d, J=6.59 Hz, 6 H), 0.97 (d, J=6.84 Hz, 6 H), 1.90 (sept, J=6.84 Hz, 1 H), 2.12 (sept, J=6.59 Hz, 1 H), 3.15 (bs, 2 H), 3.19 (s, 3 H), 3.45 (d, J=7.32 Hz, 2 H), 4.64 (bs, 1 H), 5.67 (s, 1 H), 6.83 (d, J=15.87 Hz, 1 H), 7.28 (t, J=7.32 Hz, 1 H), 7.35 (t, J=7.57 Hz, 2 H), 7.55 (d, J=7.32 Hz, 2 H), 7.71 (d, J=15.87 Hz, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.57, 20.64, 27.7, 28.8, 36.3, 49.2, 57.1, 127.5, 128.37, 128.43, 128.8, 133.5, 162.4. HRMS (ESI+) m/z calcd for C21H31N4+ 339.2549, found 339.2542.
N2-methyl-N2,N4-diisobutyl-(6-phenethyl-pyrimidin-2,4-yl)-diamine (32)
Compound 31b (0.022 g, 0.064 mmol) was dissolved in a suspension of 16 mg (0.015 mmol) 10% Pd/C in 4 mL MeOH. The reaction was stirred under 1 atm of H2 for 45 min. The solvent was removed in vacuo and residue was purified directly by column chromatography (10% EtOAc in hexanes) to give 32 (16 mg, 73%). 1H NMR (500 MHz, CDCl3) δ ppm 0.90 (d, J=6.59 Hz, 6 H), 0.94 (d, J=6.59 Hz, 6 H), 1.85 (sept, J=6.59 Hz, 1 H), 2.07 (sept, J=6.59 Hz, 1 H), 2.74 (bt, J=6.84 Hz, 2 H), 3.00 (dd, J=9.28, 6.84 Hz, 2 H), 3.06 (bs, 2 H), 3.12 (s, 3 H), 3.42 (d, J=7.32 Hz, 2 H), 4.52 (bs, 1 H), 5.44 (s, 1 H), 7.17 (t, J=7.20 Hz, 1 H), 7.22 (distorted d, J=6.84 Hz, 2 H), 7.27 (t, J=7.57 Hz, 2 H). HRMS (ESI+) m/z calcd for C21H33N4+ 341.2705, found 341.2705.
(2-Chloro-6-styryl-pyrimidin-4-yl)-isobutyl-methyl-amine (33b)
Compound 1 (50 mg, 0.20 mmol) and K2CO3 (82 mg, 0.60 mmol) were mixed in 2.5 mL of DMF. To this suspsension, 24 μL (0.20 mmol) of N-methylisobutylamine were added with stirring at room temperature. Consumption of starting material was observed by TLC after 3 h. The reaction mixture was diluted with H2O and extracted three times with ether. The combined organic layers were washed with brine, dried over MgSO4, and the solvent removed by rotary evaporator. Purification of the residue by column chromatography (CH2Cl2 followed by 10% EtOAc in hexanes) afforded 33b (50 mg, 84%). 1H NMR (500 MHz, CDCl3) δ ppm 0.94 (d, J=6.35 Hz, 6 H), 2.08 (sept, J=6.84 Hz, 1 H), 3.11 (bs, 3 H), 3.42 (bs, 2 H), 6.25 (s, 1 H), 6.88 (d, J=15.87 Hz, 1 H), 7.32 (t, J=7.20 Hz, 1 H), 7.37 (t, J=7.32 Hz, 2 H), 7.56 (d, J=7.32 Hz, 2 H), 7.80 (d, J=15.87 Hz, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.3, 27.5, 37.1, 125.6, 127.7, 129.0, 129.2, 136.1, 136.3 164.1. HRMS (ESI+) m/z calcd for C17H21N3Cl+ 302.1424, found 302.1421.
N4-methyl-N2,N4-diisobutyl-(6-styryl-pyrimidin-2,4-yl)-diamine (34b)
Compound 33b (0.050 g, 0.17 mmol) was dissolved in 2.5 mL of isobutylamine and heated with stirring in a sealed high-pressure tube at 100 °C for 24 h. The reaction was allowed to cool to room temperature and the excess amine removed in vacuo. Purification was accomplished with column chromatography (5% EtOAc and 1% TEA in hexanes) to give 34b (27 mg, 47%). 1H NMR (500 MHz, CDCl3) δ ppm 1H 0.92 (d, J=6.59 Hz, 6 H), 0.98 (d, J=6.59 Hz, 6 H), 1.91 (sept, J=6.59 Hz, 1 H), 2.08 (sept, J=6.84 Hz, 1 H), 3.06 (s, 3 H), 3.25 (t, J=6.35 Hz, 2 H), 3.33 (bs, 2 H), 4.93 (bs, 1 H), 5.83 (s, 1 H), 6.85 (d, J=15.87 Hz, 1 H), 7.28 (t, J=7.32 Hz, 1 H), 7.35 (t, J=7.57 Hz, 2 H), 7.55 (d, J=7.32 Hz, 2 H), 7.66 (d, J=15.87 Hz, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.5, 20.6, 27.8, 28.9, 36.8, 49.4, 57.3, 92.2, 127.4, 128.1, 128.4, 128.8, 133.5, 136.9, 162.5, 163.8. HRMS (ESI+) m/z calcd for C21H31N4+ 339.2549, found 339.2562.
N4-methyl-N2,N4-diisobutyl-6-[(2-naphthalen-1-yl-ethyl)-pyrimidin-2,4-yl]-diamine (34c)
Compound 33c (0.012 g, 0.03 mmol) was dissolved in 4 mL of isobutylamine and heated with stirring in a sealed high-pressure tube at 95 °C for 20 h. The reaction was allowed to cool to room temperature and the excess amine removed in vacuo. The resulting residue was extracted three times from H2O with CH2Cl2. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent removed by rotary evaporator. Purification was accomplished with column chromatography (10% EtOAc and 1% TEA in hexanes) to give 34c (3 mg, 21%). 1H NMR (500 MHz, CDCl3) δ ppm 0.86 (d, J=6.59 Hz, 6 H), 0.98 (d, J=6.84 Hz, 6 H), 1.87 2.03 (m, 2 H), 2.86 (m, 2 H), 2.97 (s, 3 H), 3.24 (t, J=6.35 Hz, 4 H), 3.44 (distorted t, J=8.30 Hz, 2 H), 5.03 (bs, 1 H), 5.52 (s, 1 H), 7.37 (m, 2 H), 7.45 7.54 (m, 2 H), 7.71 (d, J=7.57 Hz, 1 H), 7.85 (dd, J=7.93, 1.34 Hz, 1 H), 8.15 (d, J=8.30 Hz, 1 H). HRMS (ESI+) m/z calcd for C25H35N4+ 391.2862, found 391.2863.
N4-dimethyl-N2,N4-diisobutyl-(6-phenethyl-pyrimidin-2,4-yl)-diamine (35)
Compound 34b (0.016 g, 0.05 mmol) was dissolved in a suspension of 10 mg (0.009 mmol) 10% Pd/C in 2 mL MeOH. The reaction was stirred under 1 atm of H2 for 45 min. The solvent was removed in vacuo and residue was purified directly by column chromatography (10% EtOAc and 1% TEA in hexanes) to give 35 (14 mg, 85%). 1H NMR (500 MHz, CDCl3) δ ppm 0.88 (d, J=6.59 Hz, 6 H), 0.96 (d, J=6.59 Hz, 6 H), 1.88 (sept, J=6.84 Hz, 1 H), 2.00 (sept, J=6.59 Hz, 1 H), 2.72 (m, 2 H), 2.97 (m, 5 H), 3.21 (t, J=6.47 Hz, 2 H), 3.26 (bs, 2 H), 4.92 (bs, 1 H), 5.55 (s, 1 H), 7.18 (t, J=7.20 Hz, 1 H), 7.22 (d, J=6.84 Hz, 2 H), 7.27 (t, J=7.57 Hz, 2 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.5, 20.6, 27.7, 28.8, 35.4, 36.7, 40.0, 49.4, 57.2, 91.3, 126.0, 128.5, 128.7, 142.1, 162.3, 163.4. HRMS (ESI+) m/z calcd for C21H33N4+ 341.2705, found 341.2713.
Isobutyl-[2-isobutylsulfanyl-6-(3-methyl-butyl)-pyrimidin-4-yl]-carbamic acid tert-butyl ester (36b)
To a suspension of 45 mg (1.1 mmol,) NaH (60% dispersion in mineral oil) in 3 mL of THF, 2-methyl-propane-1-thiol (122 μL, 1.1 mmol) was added dropwise and the resulting mixture was stirred for 15 min. To this mixture, 16b (0.040 g, 0.11 mmol) was added dissolved in 1 mL THF. The reaction was stirred 16 h at room temperature, quenched with H2O, and extracted three times with ether. The combined organic layers were washed with brine, dried over anhydrous MgSO4, and the solvent removed with a rotary evaporator. Purification was accomplished with column chromatography (10% EtOAc in hexanes) to give 36b (0.036 g, 78%). 1H NMR (500 MHz, CDCl3) δ ppm 0.88 (d, J=6.84 Hz, 6 H), 0.92 (d, J=5.86 Hz, 6 H), 1.04 (d, J=6.59 Hz, 6 H), 1.54 (s, 9 H), 1.58 (bs, 3 H), 1.95 (sept, J=6.84 Hz, 1 H), 2.04 (sept, J=6.84 Hz, 1 H), 2.62 (bt, J=7.32 Hz, 2 H), 3.00 (d, J=6.35 Hz, 2 H), 3.89 (d, J=7.57 Hz, 2 H), 7.44 (s, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 20.3, 22.3, 22.7, 28.1, 28.3, 28.4, 28.7, 36.3, 38.1, 39.5, 52.2, 82.3, 107.4, 154.0, 160.7, 170.3, 171.8. HRMS (ESI+) m/z calcd for C16H29N2S3+ 345.1493, found 345.1479.
[2-Isobutoxy-6-(3-methyl-butyl)-pyrimidin-4-yl]-isobutylamine (37a)
NaH (60% dispersion in mineral oil, 34 mg, 0.85 mmol) was added to 3 mL of isobutanol and stirred for 30 minutes at room temperature. To this solution, 28 mg (0.078 mmol) of 16b dissolved in 3 mL of isobutanol were added. The resulting solution was heated with stirring in a sealed high-pressure tube at 100 °C for 4 h. The solution was allowed to cool to room temperature and stirred an additional 16 h. Extraction of the reaction mixture was performed three times from H2O with ether, and the combined organic layers were washed with brine, dried over MgSO4, and the solvent removed by rotary evaporator. The resulting residue was dissolved in 2 mL CH2Cl2 and 2 mL of TFA were added with stirring. The reaction was stirred for 1.5 h at room temperature and the solvent was removed in vacuo. Purification of the residue was accomplished with column chromatography (10% EtOAc and 2% TEA in hexanes) to give 37a (22 mg, 98%). 1H NMR (500 MHz, CDCl3) δ ppm 0.92 (d, J=6.35 Hz, 6 H), 0.96 (d, J=6.84 Hz, 6 H), 1.00 (d, J=6.59 Hz, 6 H), 1.51 1.64 (m, 3 H), 1.86 (sept, J=6.84 Hz, 1 H), 2.08 (sept, J=6.59 Hz, 1 H), 2.50 (distorted t, J=8.06 Hz, 2 H), 3.09 (bs, 2 H), 4.02 (d, J=6.84 Hz, 2 H), 4.90 (bs, 1 H), 5.81 (s, 1 H). 13C NMR (500 MHz, CDCl3) δ ppm 19.6, 20.5, 22.7, 28.16, 28.18, 28.6, 37.9, 73.3, 165.0. HRMS (ESI+) m/z calcd for C17H32N3O+ 294.2545, found 294.2538.
Isobutyl-[2-isobutylsulfanyl-6-(3-methyl-butyl)-pyrimidin-4-yl]-amine (37b)
Compound 36b (18 mg, 0.043 mmol) was dissolved in 2 mL CH2Cl2, to which 2 mL of TFA were added with stirring. The reaction was stirred for 1.5 h at room temperature and the solvent was removed in vacuo. Purification of the residue was accomplished with column chromatography (10% EtOAc and 2% TEA in hexanes) to give 37b in quantitative yield. 1H NMR (500 MHz, CDCl3) δ ppm 0.89 (d, J=6.35 Hz, 6 H), 0.95 (d, J=6.59 Hz, 6 H), 1.02 (d, J=6.59 Hz, 6 H), 1.47 1.61 (m, 3 H), 1.86 2.01 (m, 2 H), 2.52 (distorted t, J=7.81 Hz, 2 H), 3.01 (d, J=6.59 Hz, 2 H), 3.22 (bs, 2 H), 6.09 (bs, 1 H). HRMS (ESI+) m/z calcd for C17H32N3S+ 310.2317, found 310.2309.
Isobutyl-[6-(3-methyl-butyl)-2-(2-methyl-propane-1-sulfonyl)-pyrimidin-4-yl]-carbamic acid tert-butyl ester (38)
From the procedure described by Hurst, et al.32, 33 mCPBA (77%, 27 mg, 0.12 mmol) and 20 mg of 36b were dissolved in 2 mL of CHCl3 and mixed well. The solution was then allowed to stand at room temperature for 16 hours. 20 mL of CHCl3 was added and the solution was washed 3 times with sat. NaHCO3 sol. The aqueous layers were extracted twice with CHCl3 and the combined organic layers were washed with sat. NaCl sol. and dried over MgSO4. The solvent was removed in vacuo, and chromatography of the residue (10% EtOAc in hexanes) gave 38 (9 mg, 40%). 1H NMR (500 MHz, CDCl3) δ ppm 0.91 (d, J=6.59 Hz, 6 H), 0.94 (d, J=6.59 Hz, 6 H), 1.10 (d, J=6.84 Hz, 6 H), 1.57 (s, 9 H), 1.59 1.67 (m, 3 H), 2.07 (sept, J=6.84 Hz, 1 H), 2.37 (sept, J=6.59 Hz, 1 H), 2.80 (m, 2 H), 3.39 (d, J=6.59 Hz, 2 H), 3.94 (d, J=7.32 Hz, 2 H), 8.05 (s, 1 H). 13C NMR (500 MHz, CDCl3)δ ppm 20.4, 22.6, 23.0, 24.1, 28.18, 28.24, 28.3, 36.3, 38.1, 52.8, 58.4, 83.4, 113.3, 153.7, 161.8, 164.5, 173.6.
Isobutyl-[6-(3-methyl-butyl)-2-(2-methyl-propane-1-sulfonyl)-pyrimidin-4-yl]-amine (39)
Compound 38 (9 mg, 0.019 mmol) was dissolved in 2 mL CH2Cl2, to which 2 mL of TFA were added with stirring. The reaction was stirred for 2.5 h at room temperature and the solvent was removed in vacuo. Purification of the residue was accomplished with column chromatography (25% EtOAc and 2% TEA in hexanes) to give 39 in quantitative yield. 1H NMR (500 MHz, CDCl3) δ ppm 0.93 (d, J=6.35 Hz, 6 H), 0.99 (d, J=6.59 Hz, 6 H), 1.10 (d, J=6.59 Hz, 6 H), 1.54 1.65 (m, 3 H), 1.89 (sept, J=6.59 Hz, 1 H), 2.37 (sept, J=6.59 Hz, 1 H), 2.66 (distorted t, J=8.06 Hz, 2 H), 3.06 (bs, 2 H), 3.37 (d, J=6.35 Hz, 2 H), 5.55 (bs, 1 H), 6.23 (s, 1 H). HRMS (ESI+) m/z calcd for C17H32N3O2S+ 342.2215, found 342.2209.
Biological Methods
TR-FRET CBI Assay
Purified ERα-417, labeled site-specifically with maleimide-biotin (Quanta BioDesign) and SRC-3-NRD, labeled non-specifically through cysteine residues with iodoacetamide-fluorescein (Invitrogen), were used in the time-resolved FRET assays.42 A 5-μL volume of a stock solution mix of ERα-417 (8 nM), estradiol (4 μM), and LanthaScreen Streptavidin-Terbium (Invitrogen) (2 nM) in TR-FRET buffer (20 mM Tris, pH 7.5, 0.01% NP40, 50 mM NaCl) were placed in separate wells of a black 96-well Molecular Devices HE high efficiency microplate (Molecular Devices, Inc., Sunnyvale, CA). In a second 96-well Nunc polypropylene plate (Nalge Nunc International, Rochester, NY), a 0.02 M solution of each coactivator binding inhibitor was serially diluted in a 1:10 fashion into DMF. Each concentration of coactivator binding inhibitor or vehicle was then diluted 1:10 into TR-FRET buffer, and 10 μL of this solution was added to the stock ERα solution in the 96-well plate. After a twenty- minute incubation, 5 μL of 200 nM fluorescein-SRC-3-NRD was added to each well. This mixture was allowed to incubate for 1 h at room temperature in the dark. TR-FRET was measured using an excitation filter at 340/10 nm, and emission filters for terbium and fluorescein at 495/20 and 520/25 nm, respectively. The final concentrations of the reagents were as follows: ERα-417 (2 nM), streptavidin-terbium (0.5 nM), estradiol (1 μM), coactivator binding inhibitor (0–1 mM), SRC-3-NRD (50 nM).
Luciferase Reporter Gene Assay
Human endometrial cancer (HEC-1) cells were maintained in culture and transfected in 24 well plates as previously described.43 HBSS (50 μL/well), Holo-transferrin (Sigma T1408) (20 μL/well), and lipofectin (Invitrogen #18292–011) (5 μL/well) were incubated together at room temperature for 5 minutes. A DNA mixture containing 200 ng of pCMVβ-galactosidase as an internal control, 500 ng of the estrogen responsive reporter gene plasmid 2ERE Luc, and 100 ng of full-length ER alpha expression vector with 75 μL HBSS per well was added to the first mixture and allowed to incubate for 20 minutes at room temperature. After changing the cell media to Opti-MEM (350 μL/well), 150 μL of the transfection mixture was added to each well. The cells were incubated at 37 °C in a 5% CO2 containing incubator for 6 h before the medium was replaced with fresh medium containing 5% charcoal-dextran-treated calf serum and the desired concentrations of ligands. Luciferase reporter gene activity was assayed 24 h after ligand addition as described.43
In the initial screen, compounds were assayed in a dose-response format at concentrations ranging from 20 μM to 0.6 μM; their inhibitory potential was determined by performing the assay in the presence of 10−9 M estradiol (E2). Upon validation of antagonistic activity, mechanism of action was examined by repeating the compound titration in the presence of both 10−7 and 10−9 M E2 with an expectation that changing the concentration of E2 100-fold would not change the IC50 of true coactivator binding inhibitors.
Estrogen Receptor-Relative Binding Affinity Assay
Relative binding affinities were determined using a modification of a competitive radiometric binding assay as previously described, using 2 nM [3H]estradiol as tracer ([2,4,6,7-3H]estra-1,3,5[10]-triene-3,17β-diol, 84– 85 Ci/mmol, Amersham/GE Health Care, Piscataway, NJ); purified full-length human ERα and ERβ were purchased from PanVera/Invitrogen (Carlsbad, CA). Incubations were conducted for 18–24 hr at 0 °C. Hydroxyapatite (BioRad, Hercules, CA) was used to absorb the receptor-ligand complexes, and free ligand was washed away. The binding affinities are expressed as relative binding affinity (RBA) values with the RBA of estradiol set to 100%. Estradiol binds to ERα with a Kd of 0.2 nM and to ERβ with a Kd of 0.5 nM.44,45
Molecular Modeling of the CBI/ER Interaction
All Modeling of the ER-LBD was performed using the Sybyl® 7.3 module from Tripos . The crystal structure of ER-LBD co-crystallized with diethylstilbestrol and an NR-box II (LXXLL motif) containing peptide from the steroid receptor coactivator 2 (SRC-2) was used as the basis for coactivator groove modeling (http://www.rcsb.org ID 3erd). The crystal structure was prepared for docking experiments by addition of the missing internal amino acids of the LBD using information contained in RCSB website and the loop search (PRODAT database) feature of Sybyl®. Hydrogens, missing sidechains, and charged end groups were added to the protein and peptide, and the ER/SRC-2 structure was minimized using MMFF94 calculated charges and an MMFF94s force field in three steps (inserted loops, water molecules, entire protein) with appropriate aggregates applied for each. CBI structures were minimized using the Tripos forcefield to termination gradient of 0.05 kcal/(mol*A). Initial placement of the CBI was performed using least squares multiple atom fitting of the alkyl groups on the CBI with the ER-interacting leucine residues of the SRC-2 peptide. Subsequent CBI orientations used for docking were manipulated into the coactivator groove by hand. Before docking, the SRC-2 peptide and all water molecules were removed from the ER-LBD structure. Docking experiments were performed using the Sybyl® FlexiDock module, with all bonds in the CBI and all sidechains within 10 A of the coactivator groove designated as rotatable. H-bond donor and acceptor sites were designated for the appropriate atoms in the CBI as well as for Glu542 and Lys362. Docking runs were begun from a random seed and allowed to proceed for 100,000 iterations. In most instances, duplicate runs were performed to ensure validity of the docking results. The lowest energy conformation of CBI/receptor thus produced was then further minimized with the MMFF94s force field to a termination gradient of 0.10 kcal/(mol*A), following a four step procedure to produce the final structures [(1) 10 A surrounding CBI with CBI and backbone static, (2) 10 A surrounding CBI with backbone static, (3) 10 A surrounding CBI with CBI static, and (4) 10 A surrounding CBI)].
Supplementary Material
Figure 4.
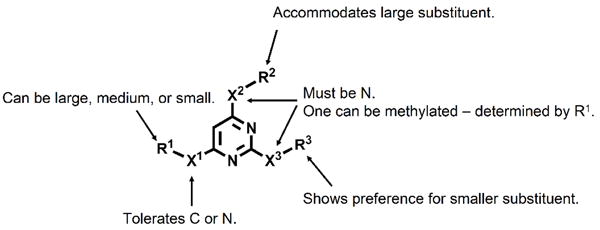
Summary of the structure-activity relationships of the pyrimidine-core CBIs.
Acknowledgments
We greatly acknowledge support of this research from the National Institutes of Health (PHS 5R37DK15556). JRG was supported by a David Robertson Fellowship and an NIH Fellowship NRSA 1 F30 ES016484-01 and Training Grant NRSA 5 T32 GM070421. AAP received support from an Illinois Distinguished Fellowship and a Robert D. Doolen Fellowship. We are grateful for the helpful suggestions of Dr. Sung Hoon Kim.
Abbreviations
- ER
estrogen receptor
- LBD
ligand binding domain
- SRC
steroid receptor coactivator
- CBI
coactivator binding inhibitor
- TR-FRET
time-resolved fluorescence resonance energy transfer
- RBA
relative binding affinity
Footnotes
Supporting Information Available: See for complete synthetic details, HPLC and CHN analyses, and more extensive biological data, including RBA values. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Stroem A, Treuter E, Warner M, Gustafsson JA. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 2.Gustafsson JA. What pharmacologists can learn from recent advances in estrogen signalling. Trends Pharmacol Sci. 2003;24:479–485. doi: 10.1016/S0165-6147(03)00229-3. [DOI] [PubMed] [Google Scholar]
- 3.Hess RA. Estrogen in the adult male reproductive tract: a review. Rep Biol Endocrinol. 2003;1(52):1–14. doi: 10.1186/1477-7827-1-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hewitt SC, Korach KS. Estrogen receptor knockout mice: Roles for estrogen receptors a and b in reproductive tissues. Reproduction (Cambridge, United Kingdom) 2003;125:143–149. doi: 10.1530/rep.0.1250143. [DOI] [PubMed] [Google Scholar]
- 5.Lakoski SG, Herrington DM. Effects of oestrogen receptor-active compounds on lipid metabolism. Diabetes, Obesity and Metabolism. 2005;7:471–477. doi: 10.1111/j.1463-1326.2004.00412.x. [DOI] [PubMed] [Google Scholar]
- 6.Schneider JG, Tompkins C, Blumenthal Roger S, Mora S. The metabolic syndrome in women. Cardiol Rev. 2006;14:286–291. doi: 10.1097/01.crd.0000233757.15181.67. [DOI] [PubMed] [Google Scholar]
- 7.Tikkanen MJ. Estrogens, progestins and lipid metabolism. Maturitas. 1996;23:S51–S55. doi: 10.1016/0378-5122(96)01012-2. [DOI] [PubMed] [Google Scholar]
- 8.Syed F, Khosla S. Mechanisms of sex steroid effects on bone. Biochem Biophys Res Commun. 2005;328:688–696. doi: 10.1016/j.bbrc.2004.11.097. [DOI] [PubMed] [Google Scholar]
- 9.Jaervinen TLN, Kannus P, Sievaenen H. Estrogen and bone-a reproductive and locomotive perspective. J Bone Miner Res. 2003;18:1921–1931. doi: 10.1359/jbmr.2003.18.11.1921. [DOI] [PubMed] [Google Scholar]
- 10.Carruba G. Estrogens and mechanisms of prostate cancer progression. Ann N Y Acad Sci. 2006;1089:201–217. doi: 10.1196/annals.1386.027. [DOI] [PubMed] [Google Scholar]
- 11.Vergote I, Neven P, van Dam P, Serreyn R, De Prins F, De Sutter P, Albertyn G. The estrogen receptor and its selective modulators in gynecological and breast cancer. Eur J Cancer. 2000;36:S1–S9. doi: 10.1016/s0959-8049(00)00203-3. [DOI] [PubMed] [Google Scholar]
- 12.Hanstein B, Djahansouzi S, Dall P, Beckmann MW, Bender HG. Insights into the molecular biology of the estrogen receptor define novel therapeutic targets for breast cancer. Eur J Endocrinol. 2004;150:243–255. doi: 10.1530/eje.0.1500243. [DOI] [PubMed] [Google Scholar]
- 13.Nicholson RI, Johnston SR. Endocrine therapy - current benefits and limitations. Breast Cancer Res Treat. 2005;93:S3–S10. doi: 10.1007/s10549-005-9036-4. [DOI] [PubMed] [Google Scholar]
- 14.Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor transcription and transactivation. Estrogen receptor alpha and estrogen receptor beta. Regulation by selective estrogen receptor modulators and importance in breast cancer. Breast Cancer Res. 2000;2:335–344. doi: 10.1186/bcr78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKenna NJ, O'Malley BW. Minireview: nuclear receptor coactivators-an update. Endocrinology. 2002;143:2461–2465. doi: 10.1210/endo.143.7.8892. [DOI] [PubMed] [Google Scholar]
- 16.Hall Julie M, McDonnell Donald P. Coregulators in nuclear estrogen receptor action: from concept to therapeutic targeting. Mol Interv. 2005;5:343–357. doi: 10.1124/mi.5.6.7. [DOI] [PubMed] [Google Scholar]
- 17.Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the estrogen receptor. Nature (London) 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 18.Swain SM. Tamoxifen: the long and short of it. J Natl Cancer Inst. 1996;88:1510– 1512. doi: 10.1093/jnci/88.21.1516. [DOI] [PubMed] [Google Scholar]
- 19.Fisher B, Dignam J, Bryant J, DeCillis A, Wickerham DL, Wolmark N, Costantino J, Redmond C, Fisher ER, Bowman DM, Deschenes L, Dimitrov NV, Margolese RG, Robidoux A, Shibata H, Terz J, Paterson AHG, Feldman MI, Farrar W, Evans J, Lickley HL. Five versus more than five years of tamoxifen therapy for breast cancer patients with negative lymph nodes and estrogen receptor-positive tumors. J Nat Cancer Inst. 1996;88:1529–1542. doi: 10.1093/jnci/88.21.1529. [DOI] [PubMed] [Google Scholar]
- 20.Normanno N, Di Maio M, De Maio E, De Luca A, de Matteis A, Giordano A, Perrone F. Mechanisms of endocrine resistance and novel therapeutic strategies in breast cancer. Endocr Rel Cancer. 2005;12:721–747. doi: 10.1677/erc.1.00857. [DOI] [PubMed] [Google Scholar]
- 21.Schiff R, Osborne CK. New insight into estrogen receptor-a function and its implication for endocrine therapy resistance in breast cancer. Breast Cancer Res. 2005;7:205–211. doi: 10.1186/bcr1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez AL, Tamrazi A, Collins ML, Katzenellenbogen JA. Design, Synthesis, and in Vitro Biological Evaluation of Small Molecule Inhibitors of Estrogen Receptor a Coactivator Binding. J Med Chem. 2004;47:600–611. doi: 10.1021/jm030404c. [DOI] [PubMed] [Google Scholar]
- 23.Shao D, Berrodin TJ, Manas E, Hauze D, Powers R, Bapat A, Gonder D, Winneker RC, Frail DE. Identification of novel estrogen receptor a antagonists. J Steroid Biochem Mol Biol. 2004;88:351–360. doi: 10.1016/j.jsbmb.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Zhou HB, Collins ML, Gunther JR, Comninos JS, Katzenellenbogen JA. Bicyclo[2.2.2]octanes: Close structural mimics of the nuclear receptor-binding motif of steroid receptor coactivators. Bioorg Med Chem Lett. 2007;17:4118–4122. doi: 10.1016/j.bmcl.2007.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Becerril J, Hamilton AD. Helix mimetics as inhibitors of the interaction of the estrogen receptor with coactivator peptides. Ange Chem, Int Ed Engl. 2007;46:4471–4473. doi: 10.1002/anie.200700657. [DOI] [PubMed] [Google Scholar]
- 26.Gunther JR, Moore TW, Collins ML, Katzenellenbogen JA. Amphipathic Benzenes Are Designed Inhibitors of the Estrogen Receptor a/Steroid Receptor Coactivator Interaction. ACS Chem Biol. 2008;3:282–286. doi: 10.1021/cb800056r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell (Cambridge, Massachusetts) 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 28.Scheiper B, Bonnekessel M, Krause H, Fuerstner A. Selective Iron-Catalyzed Cross-Coupling Reactions of Grignard Reagents with Enol Triflates, Acid Chlorides, and Dichloroarenes. J Org Chem. 2004;69:3943–3949. doi: 10.1021/jo0498866. [DOI] [PubMed] [Google Scholar]
- 29.Tan J, Chang J, Deng M. The facile route to stereo-defined alkenyl-substituted pyrimidines. Synth Commun. 2004;34:3773–3783. [Google Scholar]
- 30.Zanda M, Talaga P, Wagner A, Mioskowski C. Efficient and regioselective 4-aminodechlorination of 2,4,6-trichloropyrimidine with N-sodium carbamates. Tetrahedron Lett. 2000;41:1757–1761. [Google Scholar]
- 31.Schroeter S, Stock C, Bach T. Regioselective cross-coupling reactions of multiple halogenated nitrogen-, oxygen-, and sulfur-containing heterocycles. Tetrahedron. 2005;61:2245–2267. [Google Scholar]
- 32.Harnden MR, Hurst DT. The chemistry of pyrimidinethiols. III. The synthesis of some substituted pyrimidinethiols and some thiazolo[5,4-d]pyrimidines. Aust J Chem. 1990;43:55–62. [Google Scholar]
- 33.Hurst DT. The synthesis and chemistry of 2-methylpyrimidine-4,6-dithiol and some related compounds. Aust J Chem. 1983;36:1477–82. [Google Scholar]
- 34.Fuerstner A, Leitner A, Mendez M, Krause H. Iron-Catalyzed Cross-Coupling Reactions. J Am Chem Soc. 2002;124:13856–13863. doi: 10.1021/ja027190t. [DOI] [PubMed] [Google Scholar]
- 35.Furstner A, Leitner A. Iron-catalyzed cross-coupling reactions of alkyl-Grignard reagents with aryl chlorides, tosylates, and triflates. Angew Chem, Int Ed Engl. 2002;41:609–612. [Google Scholar]
- 36.Galande AK, Bramlett KS, Trent JO, Burris TP, Wittliff JL, Spatola AF. Potent inhibitors of LXXLL-based protein-protein interactions. ChemBioChem. 2005;6:1991–1998. doi: 10.1002/cbic.200500083. [DOI] [PubMed] [Google Scholar]
- 37.Geistlinger TR, McReynolds AC, Guy RK. Ligand-Selective Inhibition of the Interaction of Steroid Receptor Coactivators and Estrogen Receptor Isoforms. Chem Biol. 2004;11:273–281. doi: 10.1016/j.chembiol.2004.01.016. [DOI] [PubMed] [Google Scholar]
- 38.Geistlinger TR, Guy RK. Novel Selective Inhibitors of the Interaction of Individual Nuclear Hormone Receptors with a Mutually Shared Steroid Receptor Coactivator 2. J Am Chem Soc. 2003;125:6852–6853. doi: 10.1021/ja0348391. [DOI] [PubMed] [Google Scholar]
- 39.Ellem SJ, Risbridger GP. Treating prostate cancer: a rationale for targeting local oestrogens. Nature Rev Cancer. 2007;7:621–627. doi: 10.1038/nrc2174. [DOI] [PubMed] [Google Scholar]
- 40.Murphy LC, Watson PH. Is oestrogen receptor-b a predictor of endocrine therapy responsiveness in human breast cancer? . Endocr Relat Cancer. 2006;13:327–334. doi: 10.1677/erc.1.01141. [DOI] [PubMed] [Google Scholar]
- 41.Saji S, Hirose M, Toi M. Clinical significance of estrogen receptor b in breast cancer. Cancer Chemother Pharmacol. 2005;56:s21–s26. doi: 10.1007/s00280-005-0107-3. [DOI] [PubMed] [Google Scholar]
- 42.Tamrazi A, Carlson KE, Daniels JR, Hurth KM, Katzenellenbogen JA. Estrogen Receptor Dimerization: Ligand Binding Regulates Dimer Affinity and DimerDissociation Rate. Mol Endocrinol. 2002;16:2706–2719. doi: 10.1210/me.2002-0250. [DOI] [PubMed] [Google Scholar]
- 43.Sun J, Meyers MJ, Fink BE, Rajendran R, Katzenellenbogen JA, Katzenellenbogen BS. Novel ligands that function as selective estrogens or antiestrogens for estrogen receptor-alpha or estrogen receptor-beta. Endocrinology. 1999;140:800–804. doi: 10.1210/endo.140.2.6480. [DOI] [PubMed] [Google Scholar]
- 44.Carlson KE, Choi I, Gee A, Katzenellenbogen BS, Katzenellenbogen JA. Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: Evidence that an open pocket conformation is required for ligand interaction. Biochemistry. 1997;36:14897–14905. doi: 10.1021/bi971746l. [DOI] [PubMed] [Google Scholar]
- 45.Katzenellenbogen JA, Johnson HJ, Jr, Myers HN. Photoaffinity labels for estrogen binding proteins of rat uterus. Biochemistry. 1973;12:4085–4092. doi: 10.1021/bi00745a010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.