

Abstract
Neuronal activity largely depends on two key components on the membrane: the Na,K-ATPase (NKA) that maintains the ion gradients and sets the foundation of excitability, and the ionotropic glutamatergic AMPA receptors (AMPARs) through which sodium influx forms the driving force for excitation. Because the frequent sodium transients from glutamate receptor activity need to be efficiently extruded, a functional coupling between NKA and AMPARs should be a necessary cellular device for synapse physiology. We show that NKA is enriched at synapses and associates with AMPARs. NKA dysfunction induces a rapid reduction in AMPAR cell-surface expression as well as total protein abundance, leading to a long-lasting depression in synaptic transmission. AMPAR proteolysis requires sodium influx, proteasomal activity and receptor internalization. These data elucidate a novel mechanism by which NKA regulates AMPAR turnover and thereby synaptic strength and brain function.
Introduction
AMPA-type glutamatergic receptors (AMPARs) are heterotetromeric, sodium-permitting channels composed of GluR1–4 subunits acting as the major mediator of central synaptic transmission. Synaptic expression of AMPARs is subject to activity-dependent regulation, which is critical for the formation of synaptic plasticity such as long-term potentiation (LTP) and depression (LTD). Although receptor trafficking is efficient for transient changes of AMPAR synaptic localization, alterations in total receptor amount, which is determined by a balance of protein synthesis and degradation, may be more important for long-term modulation of synaptic efficacy (Bingol and Schuman, 2005; Yi and Ehlers, 2005; Hou et al., 2008). Therefore, regulation of receptor biogenesis and disposal can serve as a novel cellular means for synaptic function. It has been shown that AMPARs have a half-life of ∼20–30 h (O'Brien et al., 1998; Huh and Wenthold, 1999), indicating the involvement of active degradation processes. However, whether the protein abundance of AMPARs is dynamically regulated as a cellular response remains to be studied.
Na,K-ATPase (NKA) is a plasma membrane-embedded sodium pump that functions against the gradients to maintain the asymmetric distribution of sodium and potassium (Kaplan, 2002). The sodium gradient forms the foundation for synaptic transmission and neuronal excitation. Because of the frequent perturbation of ion homeostasis resulting from constant neuronal activity, the workload of NKA is so high that it consumes nearly half of the ATP in the brain. NKA is a heterodimer composed of two subunits: the catalytic α subunit that contains ATPase activity and the regulatory β subunit that is required for the enzymatic activity of NKA. At the single-neuron level, immunostainings have shown widespread localization of NKA in the soma and the dendrites, but its subcellular distribution has not been carefully examined (Brines and Robbins, 1993; Anupama Adya and Mallick, 1998).
During synaptic transmission, AMPAR-mediated currents are carried by sodium ions that flow into the interior of the neuron, typically within a microspace of the spine <1 μm3. In hippocampal neurons, one action potential can cause several-fold increase in intraspinal sodium. A typical LTP protocol of 100 Hz stimulation increases spine sodium from 10 mm to >100 mm (Rose and Konnerth, 2001). The frequent and often large rises of intraspinal sodium must be exuded efficiently to maintain synapse electrophysiology, a task achieved via the activity of NKA. Therefore, cross talk between AMPARs and the NKA should be in place to coordinate their functions. Remarkably, little is known about the effect of NKA on AMPAR expression and localization. We report here that NKA is enriched at synaptic sites and colocalizes and associates with AMPARs. NKA suppression results in a rapid internalization and proteasome-mediated degradation of AMPARs, leading to a lasting suppression of AMPAR-mediated synaptic transmission. These findings demonstrate a novel cellular means of glutamate receptor regulation, and may provide new insight into our understanding of many neuropathological conditions.
Materials and Methods
Primary cortical neuron culture.
Cortices from E18 rat embryos were digested with papain (0.5 mg/ml in EBSS, 37°C for 20 min), washed and gently triturated by passing the tissue through a Pasteur pipette with a fire-polished tip. Neurons were counted and plated onto poly-l-lysine (Sigma, 0.5 mg/ml) precoated 60 mm Petri dishes (Becton Dickinson) at 4 × 106/dish for biochemistry or dishes containing five coverslips (0.3 × 106 per 60 mm dish) for immunostainings. To ensure high-quality cell adhesion and growth, coverslips were first incubated in nitric acid overnight and thoroughly washed with four changes of large amounts of water every 2 h. Dried coverslips were then baked at 200°C for 2 h, coated with poly-l-lysine (Sigma, 0.5 mg/ml) overnight and washed again before being incubated in plating medium for cell plating. The plating medium is MEM containing 10% fetal bovine serum (FBS), 5% horse serum (HS), 31 mg of cystine and 1% P/S/G. 24 h after plating, the culture medium was completely replaced with feeding medium (Neurobasal medium supplemented with 1% HS, 2% B-27 and 1% P/S/G). Thereafter, neurons were fed twice a week with 2 ml feeding medium/dish until use. Since glia are important in neuron growth and synaptogenesis, glia cells were inhibited by adding FDU at DIV 5, but not completely eliminated from the culture.
Western blot analysis of protein abundance.
Two-week-old cultured cortical neurons in 60 mm dishes (4 × 106/dish) were incubated with ouabain (50 μm) in culture medium for 1 h or as indicated. For calcium-free or sodium replacement experiments, cells were incubated with ACSF containing 1 mm EGTA and 0 calcium, or sodium being replaced with NMDG or LiCl, supplemented with ouabain 10 min later for 1 h. After treatment cells were rinsed with ACSF, harvested in ice-cold lysis buffer (PBS supplemented with 1% Triton X-100, 0.5% deoxycholate, 0.1% SDS and 1:300 protease inhibitor cocktail containing AEBSF, Aprotinin, Bedysyin, E-64, Leupeptin and Pepstatin A, Sigma), and rotated at 4°C for 1 h. After centrifugation of the lysates at 14,000 rpm for 15 min, supernatants were boiled with 4× sample buffer followed by SDS-PAGE separation and immunoprobing with antibodies against the C terminus of GluR1 (anti-GluR1Ct, 1:100), GluR2/3 (1:500), NR1 (1:500), PSD-95 (1:800), glutamate receptor-interacting protein (GRIP; 1:500), and N-ethylmaleimide-sensitive factor (NSF; 1:1000). The blot was developed using enhanced chemiluminescence detection methods (Amersham). Films were scanned and the optical intensities of the protein bands were quantified using ImageJ NIH software.
Isolation of cell-surface AMPARs.
Cultured cortical neurons were incubated with antibodies against the extracellular N-terminal domain of AMPAR GluR1 subunits (anti-GluR1Nt from rabbit, 1:100) for 5 min in culture medium at 37°C. Antibodies against the intracellular C-terminal of GluR1 (GluR1Ct, polyclonal, 1:100) and nonspecific rabbit IgG (1:100) were also added to another two dishes, respectively, as controls. All antibodies and IgG have been adjusted to the same stock concentration of 0.5 mg/ml. After incubation neurons were washed three times with cold ACSF on ice to remove free antibodies. Cells were homogenized in lysis buffer, to which protein A beads (50 μl in each lysate of 500 μl) were added for 2 h on rotation at 4°C. The final yields were subjected to SDS-PAGE gels and probed with anti-GluR1Ct antibody. In some dishes, surface AMPARs were isolated by surface biotinylation. After ouabain treatment, culture dishes were placed on ice to stop receptor trafficking, washed with cold ACSF and incubated with biotin (1 mg/ml) for 30 min to label surface proteins. The surface AMPARs were purified by Neutroavidin beads (Pierce) as described previously (Man et al., 2007).
Glutathione S-transferase pulldown assays.
Solubilized hippocampal extracts (800 μg of protein) were incubated with glutathione-Sepharose beads (Amersham Biosciences) bound to the indicated glutathione S-transferase (GST)-fusion proteins (20 μg) at 4°C overnight with shaking. Beads were then washed three times with 500 μl of PBS containing 0.1% Triton X-100 before the bound proteins were eluted with glutathione elution buffer. Elutes were incubated in sample buffer and subjected to 10% SDS-PAGE.
Time course of surface and total AMPAR reduction.
Cortical neurons were cultured in six-well plates (1 × 106/well) to synchronize the procedures among different treatments. The same amount of ouabain (50 μm) was added to individual wells at different time points so that all wells with varied lengths of ouabain incubation were ready for surface labeling and lysis simultaneously. After treatment, plates were washed, incubated with anti-GluR1N (1:100) for 10 min at 37°C and washed with ACSF on ice to remove the remaining antibody. After cell lysis, a small portion of lysate was taken to examine total AMPAR levels, and the remainder of the same lysate was incubated with protein A Sepharose beads to isolate surface AMPARs. Time courses of the reduction in both surface-localized and total AMPARs were drawn from the optical intensity of Western blots.
Immunostaining.
Neurons were plated at low density (0.3 × 106 per dish with five coverslips) to reduce the overlap among neurons so that single dendrites could be readily selected and analyzed. After 1 h incubation with ouabain in culture medium, neurons were washed with ACSF, fixed with 4% paraformaldehyde plus 4% sucrose (10 min) and permeabilized with 0.3% Triton X-100 (10 min) on ice. After 1 h blocking with 10% normal goat serum, cells were incubated sequentially with antibodies against PSD-95 (mouse, 1:500) and NKA α1 subunits (rabbit, 1:300), or GluR2Ct (mouse, 1:100) and NKA α1 (rabbit, 1:300) for 2 h each at room temperature. Cells were then washed and incubated with fluorescence-conjugated secondary antibodies (Alexa Fluor 555 red anti-mouse, 1:700, and/or Alexa Fluor 488 green anti-rabbit, 1:700) for 1 h for visualization.
Image collection on immunolabeled neurons.
Immunostained coverslips were mounted onto slides using ProlongGold anti-fade reagent (Invitrogen) and kept in the dark for >4 h before imaging. Using 63× oil-immersion objective (numerical aperture, 1.4), a differential interference contrast snap was first taken for morphology purposes. The exposure time for fluorescence signal was set automatically by the software and adjusted manually so that the signal intensity was within the full dynamic range. Either the glow scale look-up table or the histogram was used to monitor the saturation level. Once the parameters were set they were fixed and used for all the imaging in an experiment. For double staining, dual filters were used for PSD-95 or GluR2 (red) and NKA α1 subunits (green). Two channels were imaged sequentially and a merged image was formed automatically by the Zeiss imaging software. Some immunostainings were also imaged by confocal microscopy.
Live imaging.
Two to three days after transfection of GFP-GluR1 or PH-GluR1 (1 μg per coverslip) with Lipofectamine 2000, cortical neurons were transferred to an imaging chamber with temperature controlled at 37°C. Receptors were visualized with excitation at 488 nm and imaged every 15 min. Ouabain (50 μm) was added directly to the chamber containing 1 ml of ACSF buffered with 10 mm HEPES. Some cells were pretreated with MG-132 for 30 min in culture medium then transferred for imaging in the presence of MG-132. Cells were checked before each imaging to ascertain there was no shift in focus. Fluorescence intensity at the soma was measured at different time points using Image-J and normalized to the starting value.
mEPSC whole-cell patch clamp recording.
A coverslip of 14- to 16-d-old cortical neurons was transferred to the recording chamber with extracellular solution containing (in mm) 140 NaCl, 3 KCl, 1.5 MgCl2, 2.5 CaCl2, 11 glucose and 10 HEPES, pH 7.4, which was supplemented with TTX (1 μm) to block action potentials, APV (50 μm) to block NMDARs and bicuculline (20 μm) to block GABAA receptor-mediated IPSCs. Whole-cell voltage-clamp recordings were made with patch pipettes filled with an intracellular solution containing (in mm) 100 Cs-methanesulfonate, 10 CsCl, 10 HEPES, 0.2 EGTA, 4 Mg-ATP, 0.3 Na-GTP, 5 QX-314 and 10 Na-phosphocreatine, pH 7.4, with the membrane potential clamped at –70 mV. Recordings started 10 min after establishing whole-cell configuration to ensure equilibration between the pipette solution and the cytosol. After 10 min recording of basal mEPSC, ouabain was added to the bath solution and recording continued for 30–40 min in the presence of ouabain. To examine long-term effects of ouabain treatment, coverslips were incubated with ouabain for 1 h, then transferred to ACSF immediately for recording (referred to as 1 h) or to normal culture medium to recover for varied periods of time until being recorded.
Data analysis.
ALL values are expressed as mean ± SEM, and analyzed using a Student's t test for comparison between two groups. Statistical significance is defined as p < 0.05.
Results
NKA colocalizes and interacts with AMPARs in neurons
To determine the subcellular distribution of NKA and more specifically its synaptic localization, we costained the α1 subunit of NKA together with PSD-95, a marker protein for excitatory synapses, in cultured cortical neurons. Consistent with previous studies indicating α1 as a major neuronal NKA subtype (McGrail et al., 1991; Pietrini et al., 1992), we found strong immunofluorescence throughout the neuron including the soma and the dendrites. Notably, NKA showed a nonuniform, punctate pattern in dendrites, reminiscent of synaptic distribution. Indeed, when the NKA signal was merged with that of PSD-95, we observed a high degree of codistribution (Fig. 1A). About half (55 ± 9%, n = 10 cells) of PSD-95 signals contained NKA α1, whereas approximately one third (26 ± 6%, n = 10 cells) of NKA α1 signals overlapped with PSD-95 (Fig. 1C). The colocalization of NKA with AMPARs was further confirmed by confocal microscopic examination (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Because AMPARs, the primary mediator of excitatory synaptic transmission, are a major contributor of intraspinal sodium transients, we reasoned that NKA should be intimately positioned with AMPARs to facilitate local sodium homeostasis. To test this, we double labeled NKA α1 and AMPAR GluR2 subunits, and a similar colocalization was observed (Fig. 1B), with a majority of GluR2 immunosignals overlapped with NKA α1 (59 ± 3%, n = 10 cells), and 40 ± 5% (n = 10 cells) of NKA α1 overlapped with GluR2 (Fig. 1C). Given that at individual synapses AMPAR puncta were often colocalized with NKA but with only partial overlapping territory, the degree of synaptic coexistence of NKA and AMPARs should be significantly higher than the rate of signal overlap. To further confirm the synaptic localization of NKA, synaptosomes were isolated from rat brain tissue after serial centrifugations. As known synaptic components, both PSD-95 and AMPARs were enriched in synaptosome (Fig. 1D). Consistent with its immunolocalization, a large amount of NKA was detected in synaptosome. Interestingly, NKA α1 appeared to have a stronger synaptic preference than the α3 subunit, although the latter is considered a neuron specific subtype. We next examined the possibility of physical interactions between NKA and glutamate receptors. Using rat cortex lysates, antibodies against NKA α1, but not α3 or β subunits, coimmunoprecipitated AMPARs (Fig. 1E), indicating a specific association of NKA α1 subunit with AMPARs. We failed to pulldown a significant amount of NKA by using an antibody against the C-terminal domain of GluR1 (data not shown), suggesting that the intracellular C terminus may be involved in the interaction. To test this, brain lysate was incubated with GST fusion protein of GluR C-terminals. Surprisingly, GST pulldown revealed an interaction of NKA with the C-terminal of GluR2, but not GluR1 (Fig. 1F), indicating that NKA via GluR2 C-tail associates with AMPAR complexes in neurons.
Figure 1.

NKA is enriched in synapses and associates with AMPARs. A, Cultured cortical neurons were fixed and immunostained under permeabilized conditions. The top panel is a representative neuron double-labeled with antibodies against the synaptic marker protein PSD-95 (monoclonal, 1:500) and the NKA α1 subunit (polyclonal, 1:300). PSD-95 showed a typical punctate pattern indicating its synaptic localization. The NKA α1 subunit immunofluorescence was distributed over the whole cell, with stronger intensity at the soma. Note that in the dendrites, NKA (green) forms clusters that were colocalized with PSD-95 (red) as indicated in the merged image (overlay), indicating synaptic enrichment of NKA. A selected region was enlarged (bottom) for clarity. Arrows indicate synaptic clusters with strong colocalization (yellow). B, Double staining of NKA and AMPARs. Under permeant conditions, cortical neurons were incubated with antibodies against NKA α1 subunits (polyclonal, 1:300), and GluR2 subunits (monoclonal, 1:300). A representative neuron was shown in separate channels (GluR2 in red, NKA α1 in green) and overlay (top). A boxed area was enlarged for clarity (bottom). Arrows indicate sites of colocalization. C, Quantification of pixel colocalization of NKA α1 with PSD-95 or GluR2. D, NKA in synaptosome. Synaptosome lysate prepared from rat cortical tissue was separated by SDS-PAGE and probed with different antibodies. Whereas both NKA α1 and α3 subunits were detected in synaptosome, α1 showed higher degree of enrichment. E, Coimmunoprecipitation of NKA and AMPARs. Rat brain cortical tissue was homogenized manually in lysis buffer and further solubilized for 1 h. Equal amounts of supernatant (500 μg/500 μl) were incubated with antibodies against the NKA α1, α3 or β subunits (3 μg each), or rabbit IgG (3 μg) as control. After protein A beads incubation and precipitation, the protein complexes were probed with anti-GluR1and reprobed with anti-GluR2 antibodies. F, GST pulldown assays. GST fusion proteins (20 μg) of C-terminals of GluR1 (GluR1CT) and GluR2 (GluR2CT), and of GluR2 N-terminal (GluR2NT) were incubated with cortical brain lysate (800 μg). GST alone was used as control. Pellets of GST pulldown were examined by Western blotting using anti-NKA α1 antibodies (top). A parallel gel was stained by Coomassie blue to confirm the loading of fusion proteins (bottom).
NKA inhibition causes a reduction in AMPAR abundance
During synaptic transmission, activation of glutamate receptors, including AMPARs and NMDARs, allows ions to flow in and out of the dendritic spines, leading to altered ion gradients that need to be efficiently reset by NKA. Therefore, a functional coupling or coordination between glutamate receptors and NKA is necessary to maintain intraspinal microenvironment and synaptic function. To explore this possible interplay, we examined glutamate receptor expression and localization after suppression of NKA activity. In cultured cortical neurons, we inhibited NKA activity using ouabain, a cardiac glycoside that specifically binds to and suppresses all types of NKAs (Scheiner-Bobis, 2002; Xiao et al., 2002). By Western blot analysis, we found that 1 h ouabain (50 μm) treatment induced a dramatic reduction in the protein levels of AMPAR GluR1 subunits (56 ± 7% of control, n = 8, p < 0.05), as well as GluR2/3 subunits (80 ± 6%, n = 4, p < 0.05) (Fig. 2A,B). The reduction started within 30 min of ouabain application, and advanced to a higher extent with longer treatment (Fig. 2C). As a control, tubulin amount was not affected by ouabain. Strikingly, no changes were found in NMDAR abundance by ouabain treatment (NR1, 102 ± 6%, n = 5) (Fig. 2D), indicating a selective regulation of AMPARs during NKA dysfunction. To investigate whether glutamate receptor-associated proteins were also regulated, we found a reduction of GRIP (72 ± 8%, n = 5, p < 0.05), but not PSD-95 (102 ± 3%, n = 5) or NSF (107 ± 5%, n = 5), after 1 h ouabain incubation (Fig. 2E–G). Since GRIP directly interacts with AMPARs, this may suggest that the AMPAR complexes are targeted as a whole by signals triggered from NKA inhibition.
Figure 2.

NKA inhibition induces a reduction in total AMPAR abundance. Cortical neurons were incubated with the NKA inhibitor ouabain (Oua, 50 μm) in culture medium for 1 h and cell lysates were used for detecting total protein amount. Ouabain treatment drastically reduced the protein amount of GluR1 (A) (n = 8) and GluR2/3 (B) (n = 3). As an internal control, the protein level of tubulin was not affected in all experiments. A time course of ouabain incubation (C) showed that GluR1 amount was reduced within 30 min, which reached plateau in 2 h (n = 3). In contrast, NMDAR NR1 was not affected by ouabain (D) (n = 5). Postsynaptic proteins that associate with AMPARs or NMDARs were differentially regulated. Ouabain treatment reduced the protein level of GRIP (F) (n = 5), but not PSD-95 (E) (n = 5) or NSF (G) (n = 5). Bar graph data present means ± SEM, *p < 0.05, t test.
Because NKA is the key housekeeping component in a cell, a concern is whether ouabain treatment affects the general heath of the neuron. Studies have demonstrated that ouabain induces neuronal death at high concentrations (>100 μm) and prolonged incubation time (Xiao et al., 2002; Yu, 2003). We used a relatively low concentration (50 μm, which inhibits NKA by 60% (Blanco and Mercer, 1998) for only a short period of time. Under this concentration even 24 h treatment only induces ∼15% cell death (Xiao et al., 2002). Using propidium iodide staining, we found no significant change in cell death after 1 h incubation of 50 μm ouabain (data not shown). Furthermore, we tested ouabain dose–response at concentrations of 0.1 μm, 5 μm, 20 and 50 μm. GluR1 reduction was observed by as low as 0.1 μm ouabain (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material), indicating that the ouabain-dependent effect on AMPAR abundance is not nonspecific effect of cell death, which is further supported by the lack of changes in other proteins including NMDAR, PSD-95 and tubulin. Moreover, we found that inhibition of NKA by potassium-free solution (supplemental Fig. S2B, available at www.jneurosci.org as supplemental material) and agrin (Hilgenberg et al., 2006) (supplemental Fig. S3, available at www.jneurosci.org as supplemental material) also induced AMPAR reduction, strongly indicating NKA as the cellular machinery that mediates the ouabain effect.
Proteasome-mediated proteolysis underlies NKA-dependent AMPAR reduction
AMPARs undergo constant turnover with a half-life of ∼30 h (O'Brien et al., 1998). Since a stable receptor pool is maintained by a balance between protein synthesis and degradation, the reduction in AMPAR abundance during NKA inhibition may be attributable to suppressed protein synthesis or enhanced proteolysis, or both. To distinguish between these possibilities, we first inhibited protein synthesis. We found that when the protein synthesis inhibitor anisomycin (30 μm) was applied 40 min before and during 1 h ouabain treatment, ouabain still reduced AMPAR levels comparable to that of ouabain alone (Fig. 3A,B), indicating that protein degradation, but not synthesis, is likely responsible for AMPAR reduction. Protein-bearing vesicles can fuse into the lysosome to be degraded nonspecifically by lysosomal proteases. When AMPARs are internalized after receptor activation, it has been shown that the endocytosed receptors are transported to lysosome for degradation (Ehlers, 2000). We incubated cortical neurons with a mixture of six protease inhibitors including leupeptin to inhibit lysosomal activity. Surprisingly, ouabain-induced AMPAR reduction was not affected (Fig. 3A,B), indicating the existence of divergent destinations for AMPAR degradation. In addition to lysosomal pathway, proteins can also be degraded by proteasome, during which target protein molecules are usually conjugated with ubiquitin and then transferred to proteasome for ATP-dependent degradation. As the ouabain effect is highly specific to AMPARs, we suspected an involvement of the proteasome pathway in this case. In supporting this, we found that AMPAR reduction was completely abolished by the proteasome specific inhibitor MG132 (10 μm) (Fig. 3A,B), strongly indicating NKA-dependent AMPAR degradation by proteasome.
Figure 3.

Ouabain reduces AMPAR abundance via proteasome-mediated proteolysis. A, B, Cortical neurons were first incubated with the protein synthesis inhibitor anisomycin (Aniso, 30 μm) for 1 h or a specific proteasome inhibitor MG-132 (MG, 10 μm), or mixed protease inhibitors (Pro inhi, 1:300) for 30 min, and then supplemented with ouabain (Oua, 50 μm) for 1 h. Compared with the control (Con), ouabain treatment significantly reduced GluR1 amount. Suppression of protein synthesis did not affect ouabain-induced GluR1 reduction, indicating the change in AMPAR amount was not caused by inhibition of AMPAR synthesis. In contrast, blockage of proteasome activity by MG-132 completely abolished the ouabain effect, whereas a mixture of protease inhibitors had no effect, indicating that the NKA-dependent decrease of AMPARs was caused by proteasome-mediated protein degradation (n = 4). *p < 0.05, t test. C, D, Cortical neurons were treated with ouabain for 30 min and 60 min and immunostained with antibodies against GluR1 under permeant conditions. Ouabain treatment reduced immunofluorescence intensity at both the soma (n = 12–23) and the dendritic puncta (n = 400–500 puncta). E, F, Application of MG-132 30 min before and during 1 h ouabain treatment largely blocked the ouabain effect on dendritic GluR1 immunointensity (n = 23), consistent with the notion of AMPAR degradation via proteasome. G, H, Live imaging of GluR1 degradation in transfected neurons. A coverslip of neurons transfected with GFP-GluR1 was transferred to a chamber in ACSF under a fluorescence microscope and imaged every 15 min. Control neurons showed stable GFP fluorescence in 1.5 h. After addition of ouabain in the bath at time 0, GFP-GluR1 fluorescence intensity dropped to 60% of the control before stabilization at low levels. However, in the presence of the proteasome inhibitor MG-132 (10 μm), no changes in fluorescence intensity were observed during ouabain treatment. Images were representative neurons imaged under control and ouabain conditions, part of which was enlarged for clarity (G). The rainbow bar on the right indicates the relative intensity of the fluorescence signal. Quantitative data were from measurements of the soma (n = 7 for each experiment) (H). Bar graph data present means ± SEM, *p < 0.05, t test.
To examine AMPAR degradation with subcellular resolution, we performed immunostainings of GluR1 under permeant conditions in ouabain-treated neurons. Consistent with biochemical results, a marked reduction in GluR1 immunointensity was observed across the cell. Immunointensity in the soma was reduced to ∼60% (n = 23) and 30% (n = 12) of the control (n = 17) after 30 and 60 min ouabain treatment, respectively. Interestingly, when synaptic GluR1 clusters were analyzed, we found that GluR1 levels were reduced at a rate comparable to that at the soma (n = 400–500) (Fig. 3C,D). This result suggests that the dendritic AMPARs may not be trafficked to the soma for degradation. Rather, they were more likely degraded by proteasomes localized in the spine (Bingol and Schuman, 2006). In agreement with Western blot analysis, the reduction in GluR1 immunofluorescence was blocked by proteasome inhibitor MG132 (Fig. 3E,F). Furthermore, to directly visualize NKA-dependent AMPAR degradation and to examine its dynamics, we transfected cortical neurons with GFP-tagged GluR1 subunits (GFP-GluR1) and live imaged the GFP signal in a temperature-controlled recording chamber. Images of a transfected (green) neuron were taken every 15 min, an interval we found optimal to avoid photobleaching. During the 75 min time course, transfected cells showed stable GFP intensity. However, addition of ouabain (50 μm) to the bath decreased GFP signal in as little as 15 min, which peaked at 60 min with a maximal 40% reduction (n = 7) (Fig. 3G,H). A decrease in fluorescence intensity was observed at both the soma and the dendrites, with a higher rate at the dendritic region. Addition of MG132 (10 μm) 30 min before and during ouabain treatment completely blocked the ouabain effect, indicating that the exogenously introduced receptor subunits were subject to the same degradation process as endogenous ones.
The requirement of calcium and sodium in NKA-dependent AMPAR degradation
Loss of NKA function causes accumulation of intracellular sodium, leading to depolarization of membrane potentials. In cultured hippocampal neurons, ouabain has been shown to induce an increase of intracellular sodium (Rose and Ransom, 1997). Furthermore, after sodium accumulation, membrane depolarization leads to activation of voltage-gated calcium channels and rises in intracellular calcium concentration. Lasting changes in membrane potential can also lead to activation of signaling membrane proteins such as adenylyl cyclase (Cooper et al., 1998). By whole-cell recording in cultured cortical neurons, we observed a membrane depolarization of ∼15 mV by 50 μm ouabain that plateaued in 10 min, leading to a transient high-frequency firing of action potentials that diminished to total silence because of sodium channel inactivation by high membrane potential (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). To investigate whether the ouabain effect on AMPAR degradation is caused by membrane depolarization, we treated cortical neurons with 20 mm KCl in medium, a concentration shown to depolarize the membrane potential by ∼30 mV (Herberth et al., 2002), which is comparable to or stronger than the ouabain effect in our preparation. Western blotting demonstrated that 1 h KCl treatment did not change total AMPAR protein levels, whereas the ouabain effect remained in the presence of high KCl (KCl, 99 ± 5%, n = 3, p > 0.05; KCl+ouabain 65 ± 5%, n = 3, p < 0.05) (Fig. 4A), excluding membrane depolarization per se as the cause of the ouabain effect.
Figure 4.
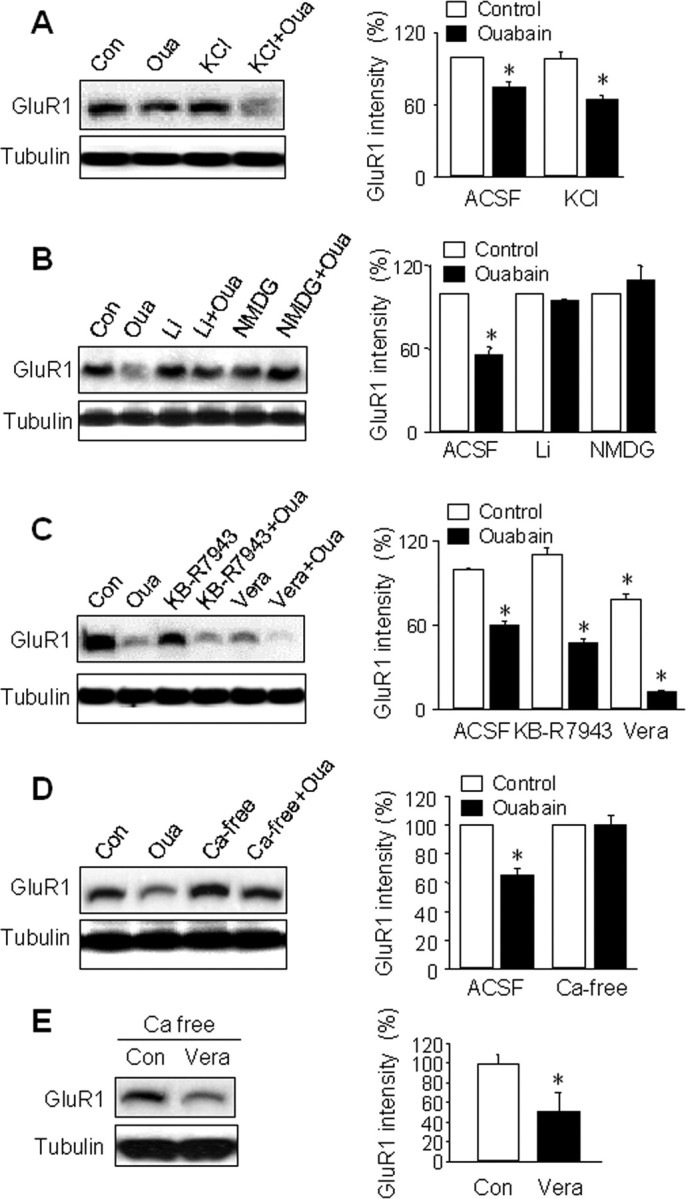
Involvement of sodium and calcium in NKA-dependent AMPAR proteolysis. A, Cortical neurons were treated for 1 h with ouabain (50 μm), or KCl (20 mm) to mimic ouabain-induced membrane depolarization. High potassium did not change GluR1 protein levels, whereas ouabain alone or in combination with KCl significantly reduced GluR1 amounts. Tubulin shows equal amount of loading. Bar graphs (right) represent the average from three independent experiments. B, Extracellular sodium on ouabain effect. Sodium in ACSF was replaced with the membrane-impermeable substitute NMDG or channel permeant substitute lithium (LiCl). When neurons were incubated with ouabain under either sodium-free condition, the ouabain effect on GluR1 abundance was blocked (n = 3). C, Neurons were treated with the sodium channel opener veratridine (Vera, 20 μm) to enhance sodium influx, and KB-R7943 (5 μm) to block the reversed mode of calcium-sodium exchanger, respectively, alone or together with ouabain for 1 h. Veratridine caused a reduction in GluR1 and markedly enhanced the ouabain effect when used together (Vera+Oua). KB-R7943 showed no effect when used alone and did not affect the ouabain effect when used in combination (n = 3). D, Extracellular calcium on ouabain effect. Neurons were rinsed with and incubated in calcium-free ACSF (ACSF containing 0 calcium plus 1 mm EGTA, Ca-free) for 10 min, then supplemented with ouabain for 1 h. Compared with the ouabain effect in normal solution (Oua), the calcium-free condition largely abolished ouabain-induced GluR1 reduction (n = 4). E, Neurons were incubated in calcium-free solution for 10 min followed by addition of veratridine (Vera, 20 μm) for 1 h. Under this condition, veratridine still caused reduction in GluR1 amount (n = 4). All the bar graph data present means ± SEM, *p < 0.05, t test.
Because intracellular sodium accumulation is one of the most prominent consequences after NKA inhibition, we wanted to determine the involvement of sodium. We first confirmed intracellular sodium accumulation by ouabain using a non-ratiometric fluorescence sodium dye, CoroNa Green (CoroNa) (Meier et al., 2006). We found that the average CoroNa intensity increased by 15 min ouabain treatment, with a further rapid increase in the next 15 min followed by slower changes (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). To investigate the role of sodium influx in AMPAR reduction, we replaced sodium in ACSF with N-methyl-d-glucamine (NMDG), a commonly used sodium substitute. In hippocampal neurons, it has been shown that application of Na-free NMDG solution reduces intracellular sodium levels to zero in 10 min (Rose and Ransom, 1997). When neurons were incubated with ouabain in NMDG solution, we found a complete blockade of AMPAR reduction compared with an NMDG solution control (n = 3) (Fig. 4B). Because NMDG is not membrane permeable and thus does not carry the same amount of charge across the membrane as sodium ions, one concern is that the difference may be caused by a lack of charge influx, rather than lack of sodium accumulation. To address this, we replaced the extracellular NaCl with the same amount of LiCl. Like sodium, lithium is also a monovalent ion that passes through sodium channels. Similar to NMDG, lithium also abolished ouabain-induced AMPAR degradation (n = 3) (Fig. 4B), strongly indicating the importance of sodium influx. We hypothesized that if sodium is indeed the mediator of the ouabain effect, an increase in intracellular sodium by other means should mimic the ouabain effects. Veratridine is a lipophilic toxin that binds to sodium channels in the plasma membrane and causes persistent channel opening by impairing channel inactivation (Strichartz et al., 1987). In hippocampal neurons it has been shown that intracellular sodium concentration increases from a resting level of ∼10 to 25 mm by 3 min veratridine treatment (Rose and Ransom, 1997). We therefore incubated neurons with veratridine (20 μm) in normal ACSF to allow inward flow of sodium. Consistent with our hypothesis, 1 h veratridine treatment significantly reduced AMPAR abundance (78 ± 4%, n = 3, p < 0.05) (Fig. 4C). Notably, GluR1 amount was reduced to a lower level when veratridine was given together with ouabain (11 ± 1%, n = 3, p < 0.05), consistent with an overwhelming sodium buildup resulting from a combination of facilitated influx and a lack of extrusion. Furthermore, we found that even in the presence of a glutamate receptor antagonist mixture including APV, CNQX and MCPG, veratridine remained capable of inducing a reduction in GluR1 (supplemental Fig. S5, available at www.jneurosci.org as supplemental material), indicating that the veratridine effect is not caused by depolarization-dependent massive transmitter release and glutamate receptor activation.
A direct consequence of intracellular sodium accumulation is calcium influx through voltage-gated calcium channels. Calcium has been implicated in glutamate-induced AMPAR internalization (Beattie et al., 2000; Man et al., 2000b; Zhou et al., 2001). Calcium also activates, directly or via signaling cascades, proteases and intracellular machinery for protein degradation (Mellgren, 1987; Koohmaraie, 1992; Nixon et al., 1994; Edelstein et al., 1996; Squier and Cohen, 1996; Gacko, 1997). To determine the involvement of calcium, we used calcium-free ACSF containing 1 mm calcium chelator EGTA. Under calcium-free conditions, 1 h ouabain treatment did not change AMPAR levels compared with the calcium-free ACSF control (n = 4) (Fig. 4D), indicating a critical role for calcium in the ouabain-dependent effect. To examine whether calcium channels were used as the route of entry, we used the nonselective calcium channel blocker cadmium (CdCl, 50 μm) to block all voltage-gated calcium channels. Similar to calcium withdrawal, CdCl completely abolished AMPAR reduction by ouabain (data not shown). Furthermore, we investigated whether the sodium-calcium exchanger (NCX) was involved in the ouabain effect. At normal conditions, NCX extrudes intracellular calcium driven by coupled sodium influx. However, high intracellular sodium concentrations during conditions like ouabain treatment can reverse the direction of the exchanger, causing an inward flow of calcium (Tortiglione et al., 2002; Annunziato et al., 2004). We found that when neurons were treated with ouabain in the presence of KB-R7943 (10 μm), an inhibitor that only blocks the reversed mode of NCX (Sheldon et al., 2004; Kim et al., 2007), the ouabain-induced AMPAR degradation remained (KB, 109 ± 5%, n = 3, p > 0.05; KB+ouabain, 47 ± 2%, n = 3, p < 0.05) (Fig. 4C), indicating that the reversed NCX activity does not likely contribute to AMPAR degradation.
Although calcium removal blocked ouabain effect, calcium entry may not be a necessary step downstream of sodium for AMPAR degradation. For instance, lack of calcium will suppress transmitter release and glutamate receptor activity, leading to diminished loading of intracellular sodium thereby blocking the ouabain effect. To examine this, we treated neurons with the sodium channel opener veratridine (50 μm) in calcium-free ACSF. We found that in the absence of calcium veratridine incubation still induced a significant reduction of GluR1 amount (n = 4) (Fig. 4E), indicating a direct role of sodium. In supporting this notion, when extracellular sodium was replaced with lithium, which should also depolarize the membrane and open calcium channels during NKA inhibition, the ouabain effect was completely blocked (Fig. 4B).
AMPAR activation is necessary for the NKA effect
We next investigated the route of entry through which sodium accumulated intracellularly during NKA inhibition. Under physiological conditions, intracellular sodium comes primarily from two sources: voltage-gated sodium channels and ligand-gated glutamate receptors (Bhattacharjee and Kaczmarek, 2005). To determine the contribution of the sodium channels, we incubated cortical neurons with TTX (1 μm) 15 min before and during 1 h ouabain treatment. To our surprise, in the presence of sodium channel blockade, ouabain still induced a significant reduction in AMPAR abundance, although to a less dramatic level (71 ± 6%, n = 4, p < 0.05) (Fig. 5A,B), suggesting that voltage-gated sodium channels may not be the major contributor for intracellular sodium rise. Indeed, a study has demonstrated that in the presence of TTX, ouabain causes a rise of intracellular sodium concentration to levels similar to the ouabain-only control (Rose and Ransom, 1997). We next investigated the role of AMPARs and NMDARs, the major subtypes of ionotropic glutamate receptors. Glutamatergic receptors are activated upon binding of glutamate that is released during active synaptic transmission by action potentials, or during resting conditions through random vesicle fusion with the presynaptic membrane resulting in miniature postsynaptic currents (mEPSCs) (Liao et al., 2001; Lu et al., 2001; Man et al., 2003; Rumbaugh et al., 2003). AMPARs permit monovalent ions and function primarily as sodium channels. Although NMDARs show high permeability to calcium and are often mistakenly considered a calcium channel, >80% of NMDA currents are actually carried also by sodium (Skeberdis et al., 2006). To differentiate the contribution of specific glutamate receptors, we incubated cortical neurons with 50 μm ouabain, together with the NMDAR antagonist APV (50 μm), or the AMPAR antagonist CNQX (20 μm), respectively. To ensure blockade, drugs were given 15 min before the addition of ouabain. Western analysis demonstrated that ouabain-induced AMPAR degradation was completely prevented by CNQX, but blocking of NMDARs by APV showed no significant difference (n = 4 each) (Fig. 5A,B). Similar experiments were performed on cultured cortical neurons by immunostaining of GluR1. Consistent with the biochemical results, whereas ouabain alone dramatically reduced GluR1 fluorescence intensity at dendrite, coincubation of ouabain with CNQX, but not APV, abolished the change (n = 40 dendrites) (Fig. 5C,D). These data suggest that the cytosolic sodium accumulated during pump suppression comes primarily from AMPAR channels.
Figure 5.

AMPAR activity is required for ouabain-induced AMPAR degradation. Cortical neurons were treated with the sodium channel blocker TTX (1 μm), NMDAR antagonist APV (50 μm), AMPAR antagonist CNQX (20 μm) for 70 min alone, or together with ouabain which was added 15 min after the antagonists. Cells were either lysed for Western blot (A and B, n = 4) or immunolabeled with anti-GluR1 antibodies under permeant conditions (C and D, n = 40). Ouabain treatment remained effective in causing GluR1 reduction in the presence of APV and TTX. In contrast, CNQX completely abolished the ouabain effect, indicating the importance of AMPAR-permitted sodium in mediating the ouabain effect. *p < 0.05, t test.
AMPAR internalization is a prerequisite to receptor degradation during NKA inhibition
We wanted to know whether AMPAR reduction affected receptor cell-surface expression. To specifically label AMPARs on the plasma membrane, we incubated cultured neurons with an antibody against the extracellular N terminus of GluR1 (anti-GluR1N). Cell lysates were incubated with protein A beads to isolate surface AMPARs. Western analysis showed a dramatic reduction of surface GluR1 in ouabain-treated cells compared with the control (Fig. 6A,B). Similar results were also obtained using surface biotinylation assays (Fig. 6C) (Man et al., 2007). AMPARs have been shown to be highly dynamic, trafficking constantly between the plasma membrane and intracellular compartments (Song and Huganir, 2002; Bredt and Nicoll, 2003; Malinow, 2003; Newpher and Ehlers, 2008). At basal conditions, a balance between the two opposing processes, receptor insertion and internalization, maintains a stable level of AMPARs at the cell-surface. AMPARs internalize both constitutively and in a regulated manner (Man et al., 2000b; Lee et al., 2002), and the latter often results in a reduction in AMPAR surface expression and depression in synaptic transmission (Man et al., 2000b). Therefore, the ouabain-induced surface AMPAR reduction may be attributable to receptor endocytosis. To directly visualize receptor internalization, we used pHluorin-tagged GluR1 (PH-GluR1). pHluorin is a pH-sensitive green fluorescent protein with decreased intensity as pH decreases. PH-GluR1 is visible only when expressed at the cell surface where the pH is ∼7, but is completely invisible when internalized into vesicles with acidic pH (Ashby et al., 2004). We have confirmed its pH sensitivity by reducing the extracellular pH to 5.5, causing a dramatic reduction of PH-GluR1 fluorescence intensity (data not shown). In neurons transfected with PH-GluR1, stable fluorescence intensity was observed when imaged every 15 min for 75 min (n = 5) (Fig. 6D). In contrast, application of ouabain in the bath solution triggered a rapid decrease in fluorescence signal, which was reduced to 70% of control at 15 min, 60% at 30 min till the end of the 60 min recording period (n = 7) (Fig. 6D), indicating a rapid receptor internalization by NKA inhibition.
Figure 6.

AMPAR internalization during NKA inhibition. A, Isolation of surface AMPARs. Live neurons were incubated with antibodies against the extracellular N terminus of GluR1 (anti-GluR1N, rabbit, 1:100) to label surface AMPARs, or antibodies against the intracellular C terminus (anti-GluR1C, rabbit, 1:100) or rabbit IgG as a control, for 5 min, then washed to remove the remaining antibodies. Cell lysates were incubated with protein A beads to isolate surface GluR1. Western blot analysis (probed with anti-GluR1C) showed that GluR1 was isolated specifically after anti-GluR1N antibody incubation, compared with anti-GluR1C and IgG incubations that showed a negative detection. Arrow indicates the antibody band. B, Using surface pulldown assays as in A, ouabain treatment (1 h) dramatically reduced surface-localized AMPARs (GluR1-sur). Consistently, the total GluR1 amount from cell lysates (GluR1-lys) also decreased by ouabain (n = 3). C, Surface biotinylation. Neurons were treated with ouabain and then incubated with biotin (1 mg/ml) in ACSF on ice for 30 min. The biotinylated proteins in cell lysates were precipitated with immobilized streptavidin beads and analyzed by westerns. Similar to GluR1N antibody pulldown, surface biotinylation also showed GluR1 reduction by ouabain. Cell lysates were used for probing tubulin as a control. D, Live imaging of PH-GluR1. Neurons transfected with PH-GluR1 were imaged every 15 min. Fluorescence signal represents the amount of the surface-localized PH-GluR1. Under control conditions the fluorescence intensity remained stable during 75 min recording (n = 5). Addition of ouabain into the imaging bath ACSF induced a rapid reduction in PH-GluR1 fluorescence intensity (n = 7). E, Time courses of reduction of surface and total AMPARs. Neurons were incubated with ouabain for varied periods of time as indicated. Surface GluR1 was isolated as described in A and total GluR1 was derived from cell lysates. Optical intensity of GluR1 bands was plotted (below) showing an earlier and faster reduction of surface GluR1 compared with the change of total GluR1. F, Neurons were incubated in hypertonic sucrose (0.45 m) to block receptor endocytosis. No reduction in total GluR1 was observed after 1 h ouabain treatment (n = 3). G, Neurons were treated with MG-132 30 min before and during 1 h ouabain incubation. Surface GluR1 isolated as described in A still showed reduction when proteasome activity was inhibited (n = 3). Bar graph data present means ± SEM, *p < 0.05, t test.
The two events observed after ouabain treatment, AMPAR internalization as well as receptor degradation, could be either causal and sequential, or nonrelated independent processes. If the first scenario is true, i.e., receptor internalization and subsequent degradation, we expect (1) a delay in the onset of receptor degradation compared with receptor internalization, and (2) an abolishment in receptor degradation when internalization is blocked. To test these possibilities, we performed a time course of ouabain treatment from 10 min to 60 min. The surface AMPARs were isolated by anti-GluR1 N-terminal antibodies, and cell lysates were used for the total AMPAR amount. We found that at 10 min of ouabain incubation when the total AMPAR level remained largely unchanged, surface AMPARs already showed a marked reduction (Fig. 6E). The relative rate of decrease of surface AMPARs was faster than that of total receptor abundance (Fig. 6E), indicating that AMPARs are first endocytosed, then sorted for proteolysis. To further confirm this notion, we blocked receptor internalization with sucrose. Hypertonic sucrose (0.45 m) treatment inhibits clathrin-mediated endocytosis by disrupting the clathrin-coated pits (Hansen et al., 1993). AMPARs are known to undergo endocytosis via the clathrin-dependent pathway (Beattie et al., 2000; Lin et al., 2000; Man et al., 2000b), and previous studies have demonstrated that hypertonic sucrose reliably blocks AMPAR internalization in cultured neurons (Lin et al., 2000; Man et al., 2000b; Liu et al., 2006). We incubated cortical neurons with 0.45 M sucrose for 30 min, and then added ouabain in the presence of sucrose for another 60 min. Western analysis revealed that ouabain-dependent AMPAR degradation was completely abolished (n = 3) (Fig. 6F). To examine whether proteolysis is necessary for AMPAR endocytosis, we found that incubation with MG-132, which reliably blocked ouabain-induced AMPAR degradation as indicated by a stable level of total GluR1 (see also Fig. 3A,B), did not block ouabain-induced surface AMPAR internalization (n = 3) (Fig. 6G). These results strongly indicate that the receptor internalization process is independent of receptor degradation, and that the degradation machinery selectively targets the internalized, but not the preexisting intracellular pool of AMPARs. Since NKA associates with AMPARs at synapses, it is possible that the two molecular entities internalize together as a whole during NKA inhibition. Against this idea, we observed that although ouabain treatment reduced surface AMPAR expression, the abundance of surface NKA was not affected (supplemental Fig. S6, available at www.jneurosci.org as supplemental material), suggesting either a separation of the two complexes before AMPAR internalization, or a selective internalization of receptors that lack NKA association.
NKA inhibition induces long-lasting depression of AMPAR-mediated synaptic transmission
We found that NKA inhibition induces a reduction of surface-localized AMPARs. However, changes in membrane receptor abundance might not be of functional relevance if AMPARs are removed only from the extrasynaptic membrane domains. To investigate the effect of receptor trafficking in synaptic transmission, we performed whole-cell patch-clamp recording to examine AMPAR-mediated mEPSCs. Neurons were recorded in ACSF supplemented with TTX (1 μm) to block action potentials, APV (50 μm) and bicuculline (20 μm) to block NMDARs and GABAA receptor, respectively. Consistent with our imaging data showing reduction of synaptic AMPAR clusters, the mEPSC amplitudes decreased in neurons pretreated with ouabain compared with the control (Fig. 7A,B). The inhibition is long lasting; a 20% reduction remained even 10 h after ouabain washout (Fig. 7C). The long-term suppression in synaptic transmission matches our biochemical results that show little recovery in AMPAR reduction 2 h after ouabain treatment (supplemental Fig. S7, available at www.jneurosci.org as supplemental material). When mEPSC cumulative curve was plotted, interestingly we found that the ouabain-treated curve shifted to the left in an unparallel manner, with the initial segment overlapping with the control (Fig. 7D). When mEPSC amplitude histograms were compared, we observed a rise of the small and a concomitant drop of the moderate larger mEPSCs, but no significant changes to the extreme ends of the spectrum (Fig. 7E). This may suggest the existence of biased selection of synapses for inhibition. Although the cellular mechanisms remain elusive, ouabain-sensitive synapses might be defined by spine size which are not too small to limit AMPAR number and sodium flow, and not too large to dilute sodium accumulation.
Figure 7.

Long-lasting suppression of AMPAR-mediated synaptic transmission by NKA inhibition. A, B, AMPAR-mediated mEPSC was recorded in the presence of TTX (1 μm), APV (50 μm) and bicuculline (20 μm). Cortical neurons were incubated with ouabain in culture medium for 1 h and transferred to an ACSF-filled chamber for recording. Representative mEPSCs from nontreated (Control) and treated (Ouabain) neurons were shown. Bar graph (B) showed the average mEPSC amplitudes (n = 5 cells each). C, Neurons were treated with ouabain (1 h) and allowed to recover in culture medium for varied periods of time as indicated before being recorded. Average mEPSC amplitudes were normalized to the nontreated control. Decreases in mEPSC amplitudes remained even 10 h after ouabain treatment, indicating a long-lasting depression in synaptic activity (n = 4–7 cells for each time point). D, mEPSC amplitudes from control and ouabain-treated neurons were plotted cumulatively. A leftward shift was produced after ouabain treatment. E, Histogram of mEPSC amplitude.
Discussion
Physical association and functional coupling between NKA and AMPARs
Our data demonstrate a novel regulation in AMPAR synaptic localization and stability by NKA activity. We show that NKA colocalizes with AMPARs at synapses and is enriched in synaptosome preparation. The molecular mechanisms that serve the synaptic localization of NKA are not clear, but is could be achieved by interaction of NKA with synaptic molecules including PSD scaffolding proteins or glutamate receptors. Our coimmunoprecipitation and GST pulldown results demonstrate an association of NKA with AMPARs. The association appears to be mediated by NKA α1 subunits and the intracellular C-terminal of GluR2 subunits, but whether the interaction is direct remains unclear. NKA and AMPARs may associate indirectly via an unknown intermediate protein, such as the neuron specific NKA interacting protein NXYD7 (Béguin et al., 2002; Geering, 2006). We show that NKA inhibition by ouabain or other means including potassium-free conditions and agrin causes AMPAR internalization and subsequent degradation. Since sodium accumulation plays a critical role in ouabain-induced AMPAR turnover, this regulation may serve as a feedback mechanism to maintain intracellular sodium homeostasis. Given the importance of the sodium gradient for proper functioning of ion channels including glutamate receptors, a functional coupling of NKA and ionotropic glutamate receptors is presumably a necessary cellular device for synapse physiology. At the single synapse level, if NKA can act as a determinant of synaptic strength via regulating synaptic AMPAR numbers, specific synaptic plasticity may be achieved through regulating the release of NKA ligands, such as neuronal agrin at the presynaptic sites (O'Connor et al., 1995), whereas diffusively released or circulating NKA ligands may trigger global homeostatic plasticity across all synapses (Turrigiano and Nelson, 2004). In addition to NKA suppression, ouabain has been implicated in signaling processes, such as activation of tyrosine kinases (Tian et al., 2006) and NF-κB pathway (Aizman et al., 2001). However, we found that ouabain effect was not affected by suppression of tyrosine kinase activity (supplemental Fig. S8, available at www.jneurosci.org as supplemental material). Also, a recent study demonstrated that NF-κB knock-out in Drosophila caused GluR reduction (Heckscher et al., 2007) which is against the possible involvement of NF-κB signaling in our paradigm.
Involvement of the ubiquitin-proteasome system in AMPAR trafficking and degradation
The ubiquitin-proteasome system (UPS) plays a key role in protein turnover and a variety of cellular functions (Hegde, 2004; d'Azzo et al., 2005). In neurons, proteasomes have been found to be enriched in the spine (Bingol and Schuman, 2006), and the UPS is involved in synapse development and maturation (DiAntonio et al., 2001), synaptic plasticity (Hegde, 2004), presynaptic vesicle release (Willeumier et al., 2006), and postsynaptic reorganization through proteolysis of postsynaptic proteins including PSD-95 and GRIP (Colledge et al., 2003; Ehlers, 2003; Bingol and Schuman, 2004; Guo and Wang, 2007). We showed that the ouabain-induced AMPAR degradation was completely abolished in the presence of proteasome inhibitors, indicating the involvement of protein ubiquitination and proteasome-dependent degradation. Indeed, the UPS has been intimately implicated in glutamate receptor turnover and trafficking. Inhibition of proteasomal activity affects glutamate-induced AMPAR internalization (Patrick et al., 2003). In Drosophila, inhibition of the proteasome by subunit mutation increases GluRIIB expression and enhances synaptic transmission at the neuromuscular junction (Haas et al., 2007). In rat cortical cultures, we observed a rapid increase in AMPAR levels during inhibition of proteasome activity, indicating the involvement of the UPS in constitutive AMPAR degradation in mammalian neurons (data not shown). How does the UPS participate in AMPAR turnover? The most straightforward process will be direct ubiquitination of AMPARs. In C. elegans ubiquitin molecules can be conjugated to AMPAR subunits, leading to alterations in GluR synaptic accumulation (Burbea et al., 2002). Consistently, our recent work demonstrates that mammalian AMPARs are also subject to direct ubiquitination. Because AMPAR degradation is sodium-dependent during NKA inhibition, we tested whether Nedd4, an E3 ligase whose activity is regulated by sodium (Harvey et al., 1999; Dinudom et al., 2001), is implicated in AMPAR turnover. We found that AMPAR subunits were indeed targeted by Nedd4 for ubiquitination (Lin and Man, unpublished observation). The molecular details of UPS-mediated AMPAR internalization remain to be investigated. A large body of evidence indicates that AMPARs use the clathrin-coated-pit machinery for endocytosis, which is initiated with the association of a clathrin adaptor protein AP2 to the intracellular C termini of AMPAR subunits (Man et al., 2000a; Sheng and Hyoung Lee, 2003). It is intriguing to note that the AP2 binding domain contains three lysine residues as potential ubiquitination targets. It is possible that ubiquitination at this domain enhances the binding of GluR with AP2 so as to facilitate AMPAR internalization.
Regulation of NKA function by endogenous inhibitors and pathological conditions
If NKA activity regulates AMPAR localization and stability, it is important to understand the cellular means by which the pump function is regulated under physiological/pathological conditions. Ouabain as a specific, high affinity inhibitor of NKA has long been used in the treatment of heart problems as well as in research. In addition, ouabain also exists endogenously in the body. Studies have confirmed that the same ouabain can be synthesized and released in the brain, especially by the hypothalamus, as well as in the peripheral adrenal cortex (Blaustein, 1996; Schoner, 2002, 2003). Ouabain is therefore now regarded as an endogenous steroid hormone circulating the body and the nervous system (Schoner, 2000, 2002). Another ouabain-like endogenous NKA inhibitor named Endobain is also present in the brain (Rodríguez de Lores Arnaiz et al., 1998, 2003). In addition, NKA may also be regulated via association with endogenous protein partners (Mao et al., 2005). So far the function of endogenous NKA inhibitors in the brain remains mysterious. It is intriguing to postulate that endogenous NKA inhibitors may function to regulate AMPAR synaptic distribution and synaptic transmission.
In addition to brain-released ouabain, a recent surprising finding has identified agrin as another endogenous NKA inhibitor (Hilgenberg et al., 2006). Agrin, originally found as a powerful regulator that recruits the acetylcholine receptors on postsynaptic sites in the neuromuscular junction (Wallace et al., 1991; Gillespie et al., 1996), is released not only from the nerve terminals of motor neurons but is also synthesized and released by neurons in the brain (Mantych and Ferreira, 2001; Hilgenberg et al., 2002; Smith and Hilgenberg, 2002). It has been demonstrated that agrin binds specifically to α3 subunits of NKA and inhibits neuronal NKA activity. A competition experiment confirms that endogenous neuron-released agrin indeed regulates NKA function (Hilgenberg et al., 2006). We suspected that agrin may also regulate AMPAR protein amount. In supporting this, when cortical neurons were incubated with agrin purified from transfected HEK cells, we found rapid membrane depolarization to levels similar or stronger than that induced by ouabain (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Importantly, agrin also caused a significant reduction in GluR1 abundance (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Therefore, through changing postsynaptic AMPAR turnover, activity-dependent agrin release may serve as a novel mechanism for synaptic plasticity.
As the primary energy user consuming half of the ATP in the brain, NKA activity is highly sensitive to ATP levels. Under energy deficient conditions such as hypoxia, ischemia and stroke, NKA dysfunction is often a major early pathological response (Mahadik et al., 1992; Mrsić-Pelcić et al., 2004), which, as a consequence, can cause long-term alterations in synaptic transmission. Reduced NKA activity has also been observed during aging and in aging-related neurodegenerative diseases such as Alzheimer's disease (Bahr et al., 1992; Nicoletti et al., 1995; Ossowska et al., 2001). Furthermore, dysfunction of synaptic NKA has also been implicated in certain metabolic disorders including maple syrup urine disease (Wajner et al., 2007), isovaleric acidemia (Ribeiro et al., 2007) and Lesch-Nyhan disease (Bavaresco et al., 2004). It is of interest to investigate whether the NKA-AMPAR cross talk plays any roles in the neuronal dysfunctions present in these diseases.
Sodium as a potential signaling molecule leads to AMPAR degradation
When NKA activity is inhibited, sodium will accumulate intracellularly, leading to membrane depolarization and subsequent calcium influx. We found that the removal of either extracellular sodium or calcium blocked ouabain-dependent AMPAR degradation. However, sodium and calcium may not act to activate two independent cascades. Rather, the dependency on extracellular calcium is likely caused by suppressed glutamate release at low calcium concentration leading to a reduction in receptor activity and sodium entry. In fact, sodium has increasingly been considered as a signaling molecule in addition to its traditional role as a charge carrier (Rose, 2002). Sodium is involved in regulation of potassium channels (Kameyama et al., 1984; Dryer, 1994; Yuan et al., 2003), epithelial sodium channels and glutamatergic NMDA receptor channel properties (Rose and Konnerth, 2001; Yu, 2006). Sodium influx has also shown to activate the calcium-independent adenylyl cyclase (Cooper et al., 1998). Furthermore, during synaptic transmission, the intraspine sodium transients are often large and long-lasting, suggesting an important role in synaptic plasticity (Rose, 2002). Our data demonstrate that AMPAR degradation is induced by sodium influxes mainly via AMPARs, not from voltage-gated sodium channels. This may be caused by a minor role of sodium channels at synaptic sites as indicated by a finding that action potential backpropagation does not affect spine sodium transients (Rose and Konnerth, 2001). We found that NMDAR activity played a minimal role in ouabain-dependent AMPAR degradation. Because NMDARs contribute to the same or even greater degree than that of AMPARs in intraspine sodium transients (Rose and Konnerth, 2001), it is possible that sodium signaling in the spine, like calcium-dependent cellular events (Pelkey et al., 2006), is also entry-dependent. The process by which sodium triggers proteolytic cascade remains unknown. Sodium has been shown to activate proteases (Banasiak et al., 2000, 2004) and regulate the ubiquitin E3 ligase (Harvey et al., 1999; Dinudom et al., 2001), which may target AMPARs for degradation. Alternatively, sodium accumulation may induce calcium release from intracellular stores, which then subsequently leads to receptor degradation. Obviously, further studies are needed to elucidate the detailed events bridging sodium accumulation and AMPAR removal at the synapse.
Footnotes
This work was supported in part by National Institutes of Health Grant MH 079407 (H.-Y.M.). We thank Dr. Jen-Wei Lin at Boston University for technical support in electrophysiology, Dr. Richard Huganir at Johns Hopkins University for pHluorin-tagged GluR1, and Dr. Jim Deshler at Boston University and Steve Amato of Man lab for critical reading.
References
- Aizman O, Uhlén P, Lal M, Brismar H, Aperia A. Ouabain, a steroid hormone that signals with slow calcium oscillations. Proc Natl Acad Sci U S A. 2001;98:13420–13424. doi: 10.1073/pnas.221315298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato L, Pignataro G, Di Renzo GF. Pharmacology of brain Na+/Ca2+ exchanger: from molecular biology to therapeutic perspectives. Pharmacol Rev. 2004;56:633–654. doi: 10.1124/pr.56.4.5. [DOI] [PubMed] [Google Scholar]
- Anupama Adya HV, Mallick BN. Comparison of Na-K ATPase activity in rat brain synaptosome under various conditions. Neurochem Int. 1998;33:283–286. doi: 10.1016/s0197-0186(98)00043-6. [DOI] [PubMed] [Google Scholar]
- Ashby MC, De La Rue SA, Ralph GS, Uney J, Collingridge GL, Henley JM. Removal of AMPA receptors (AMPARs) from synapses is preceded by transient endocytosis of extrasynaptic AMPARs. J Neurosci. 2004;24:5172–5176. doi: 10.1523/JNEUROSCI.1042-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahr BA, Godshall AC, Hall RA, Lynch G. Mouse telencephalon exhibits an age-related decrease in glutamate (AMPA) receptors but no change in nerve terminal markers. Brain Res. 1992;589:320–326. doi: 10.1016/0006-8993(92)91293-n. [DOI] [PubMed] [Google Scholar]
- Banasiak KJ, Xia Y, Haddad GG. Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog Neurobiol. 2000;62:215–249. doi: 10.1016/s0301-0082(00)00011-3. [DOI] [PubMed] [Google Scholar]
- Banasiak KJ, Burenkova O, Haddad GG. Activation of voltage-sensitive sodium channels during oxygen deprivation leads to apoptotic neuronal death. Neuroscience. 2004;126:31–44. doi: 10.1016/S0306-4522(03)00425-1. [DOI] [PubMed] [Google Scholar]
- Bavaresco CS, Zugno AI, Tagliari B, Wannmacher CM, Wajner M, Wyse AT. Inhibition of Na+, K+-ATPase activity in rat striatum by the metabolites accumulated in Lesch-Nyhan disease. Int J Dev Neurosci. 2004;22:11–17. doi: 10.1016/j.ijdevneu.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci. 2000;3:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Béguin P, Crambert G, Monnet-Tschudi F, Uldry M, Horisberger JD, Garty H, Geering K. FXYD7 is a brain-specific regulator of Na, K-ATPase alpha 1-beta isozymes. EMBO J. 2002;21:3264–3273. doi: 10.1093/emboj/cdf330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A, Kaczmarek LK. For K+ channels, Na+ is the new Ca2+ Trends Neurosci. 2005;28:422–428. doi: 10.1016/j.tins.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. A proteasome-sensitive connection between PSD-95 and GluR1 endocytosis. Neuropharmacology. 2004;47:755–763. doi: 10.1016/j.neuropharm.2004.07.028. [DOI] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. Synaptic protein degradation by the ubiquitin proteasome system. Curr Opin Neurobiol. 2005;15:536–541. doi: 10.1016/j.conb.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature. 2006;441:1144–1148. doi: 10.1038/nature04769. [DOI] [PubMed] [Google Scholar]
- Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol. 1998;275:F633–650. doi: 10.1152/ajprenal.1998.275.5.F633. [DOI] [PubMed] [Google Scholar]
- Blaustein MP. Endogenous ouabain: role in the pathogenesis of hypertension. Kidney Int. 1996;49:1748–1753. doi: 10.1038/ki.1996.260. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Brines ML, Robbins RJ. Cell-type specific expression of Na+, K(+)-ATPase catalytic subunits in cultured neurons and glia: evidence for polarized distribution in neurons. Brain Res. 1993;631:1–11. doi: 10.1016/0006-8993(93)91179-v. [DOI] [PubMed] [Google Scholar]
- Burbea M, Dreier L, Dittman JS, Grunwald ME, Kaplan JM. Ubiquitin and AP180 regulate the abundance of GLR-1 glutamate receptors at postsynaptic elements in C. elegans. Neuron. 2002;35:107–120. doi: 10.1016/s0896-6273(02)00749-3. [DOI] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DM, Schell MJ, Thorn P, Irvine RF. Regulation of adenylyl cyclase by membrane potential. J Biol Chem. 1998;273:27703–27707. doi: 10.1074/jbc.273.42.27703. [DOI] [PubMed] [Google Scholar]
- d'Azzo A, Bongiovanni A, Nastasi T. E3 ubiquitin ligases as regulators of membrane protein trafficking and degradation. Traffic. 2005;6:429–441. doi: 10.1111/j.1600-0854.2005.00294.x. [DOI] [PubMed] [Google Scholar]
- DiAntonio A, Haghighi AP, Portman SL, Lee JD, Amaranto AM, Goodman CS. Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature. 2001;412:449–452. doi: 10.1038/35086595. [DOI] [PubMed] [Google Scholar]
- Dinudom A, Harvey KF, Komwatana P, Jolliffe CN, Young JA, Kumar S, Cook DI. Roles of the C termini of alpha-, beta-, and gamma-subunits of epithelial Na+ channels (ENaC) in regulating ENaC and mediating its inhibition by cytosolic Na+ J Biol Chem. 2001;276:13744–13749. doi: 10.1074/jbc.M011273200. [DOI] [PubMed] [Google Scholar]
- Dryer SE. Na(+)-activated K+ channels: a new family of large-conductance ion channels. Trends Neurosci. 1994;17:155–160. doi: 10.1016/0166-2236(94)90093-0. [DOI] [PubMed] [Google Scholar]
- Edelstein CL, Yaqoob MM, Schrier RW. The role of the calcium-dependent enzymes nitric oxide synthase and calpain in hypoxia-induced proximal tubule injury. Ren Fail. 1996;18:501–511. doi: 10.3109/08860229609052821. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Gacko M. Extralysosomal degradation of proteins. Rocz Akad Med Bialymst. 1997;42(Suppl 1):43–47. [PubMed] [Google Scholar]
- Geering K. FXYD proteins: new regulators of Na-K-ATPase. Am J Physiol Renal Physiol. 2006;290:F241–F250. doi: 10.1152/ajprenal.00126.2005. [DOI] [PubMed] [Google Scholar]
- Gillespie SK, Balasubramanian S, Fung ET, Huganir RL. Rapsyn clusters and activates the synapse-specific receptor tyrosine kinase MuSK. Neuron. 1996;16:953–962. doi: 10.1016/s0896-6273(00)80118-x. [DOI] [PubMed] [Google Scholar]
- Guo L, Wang Y. Glutamate stimulates glutamate receptor interacting protein 1 degradation by ubiquitin-proteasome system to regulate surface expression of GluR2. Neuroscience. 2007;145:100–109. doi: 10.1016/j.neuroscience.2006.11.042. [DOI] [PubMed] [Google Scholar]
- Haas KF, Miller SL, Friedman DB, Broadie K. The ubiquitin-proteasome system postsynaptically regulates glutamatergic synaptic function. Mol Cell Neurosci. 2007;35:64–75. doi: 10.1016/j.mcn.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SH, Sandvig K, van Deurs B. Clathrin and HA2 adaptors: effects of potassium depletion, hypertonic medium, and cytosol acidification. J Cell Biol. 1993;121:61–72. doi: 10.1083/jcb.121.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey KF, Dinudom A, Komwatana P, Jolliffe CN, Day ML, Parasivam G, Cook DI, Kumar S. All three WW domains of murine Nedd4 are involved in the regulation of epithelial sodium channels by intracellular Na+ J Biol Chem. 1999;274:12525–12530. doi: 10.1074/jbc.274.18.12525. [DOI] [PubMed] [Google Scholar]
- Heckscher ES, Fetter RD, Marek KW, Albin SD, Davis GW. NF-kappaB, IkappaB, and IRAK control glutamate receptor density at the Drosophila NMJ. Neuron. 2007;55:859–873. doi: 10.1016/j.neuron.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN. Ubiquitin-proteasome-mediated local protein degradation and synaptic plasticity. Prog Neurobiol. 2004;73:311–357. doi: 10.1016/j.pneurobio.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Herberth B, Pataki A, Jelitai M, Schlett K, Deák F, Spät A, Madarász E. Changes of KCl sensitivity of proliferating neural progenitors during in vitro neurogenesis. J Neurosci Res. 2002;67:574–582. doi: 10.1002/jnr.10155. [DOI] [PubMed] [Google Scholar]
- Hilgenberg LG, Ho KD, Lee D, O'Dowd DK, Smith MA. Agrin regulates neuronal responses to excitatory neurotransmitters in vitro and in vivo. Mol Cell Neurosci. 2002;19:97–110. doi: 10.1006/mcne.2001.1056. [DOI] [PubMed] [Google Scholar]
- Hilgenberg LG, Su H, Gu H, O'Dowd DK, Smith MA. Alpha3Na+/K+-ATPase is a neuronal receptor for agrin. Cell. 2006;125:359–369. doi: 10.1016/j.cell.2006.01.052. [DOI] [PubMed] [Google Scholar]
- Hou Q, Zhang D, Jarzylo L, Huganir RL, Man HY. Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proc Natl Acad Sci U S A. 2008;105:775–780. doi: 10.1073/pnas.0706447105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh KH, Wenthold RJ. Turnover analysis of glutamate receptors identifies a rapidly degraded pool of the N-methyl-D-aspartate receptor subunit, NR1, in cultured cerebellar granule cells. J Biol Chem. 1999;274:151–157. doi: 10.1074/jbc.274.1.151. [DOI] [PubMed] [Google Scholar]
- Kameyama M, Kakei M, Sato R, Shibasaki T, Matsuda H, Irisawa H. Intracellular Na+ activates a K+ channel in mammalian cardiac cells. Nature. 1984;309:354–356. doi: 10.1038/309354a0. [DOI] [PubMed] [Google Scholar]
- Kaplan JH. Biochemistry of Na, K-ATPase. Annu Rev Biochem. 2002;71:511–535. doi: 10.1146/annurev.biochem.71.102201.141218. [DOI] [PubMed] [Google Scholar]
- Kim JH, Sizov I, Dobretsov M, von Gersdorff H. Presynaptic Ca2+ buffers control the strength of a fast post-tetanic hyperpolarization mediated by the alpha3 Na(+)/K(+)-ATPase. Nat Neurosci. 2007;10:196–205. doi: 10.1038/nn1839. [DOI] [PubMed] [Google Scholar]
- Koohmaraie M. The role of Ca(2+)-dependent proteases (calpains) in post mortem proteolysis and meat tenderness. Biochimie. 1992;74:239–245. doi: 10.1016/0300-9084(92)90122-u. [DOI] [PubMed] [Google Scholar]
- Lee SH, Liu L, Wang YT, Sheng M. Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron. 2002;36:661–674. doi: 10.1016/s0896-6273(02)01024-3. [DOI] [PubMed] [Google Scholar]
- Liao D, Scannevin RH, Huganir R. Activation of silent synapses by rapid activity-dependent synaptic recruitment of AMPA receptors. J Neurosci. 2001;21:6008–6017. doi: 10.1523/JNEUROSCI.21-16-06008.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, Wang YT, Sheng M. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci. 2000;3:1282–1290. doi: 10.1038/81814. [DOI] [PubMed] [Google Scholar]
- Liu B, Liao M, Mielke JG, Ning K, Chen Y, Li L, El-Hayek YH, Gomez E, Zukin RS, Fehlings MG, Wan Q. Ischemic insults direct glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J Neurosci. 2006;26:5309–5319. doi: 10.1523/JNEUROSCI.0567-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Mahadik SP, Bharucha VA, Stadlin A, Ortiz A, Karpiak SE. Loss and recovery of activities of alpha+ and alpha isozymes of (Na(+) + K+)-ATPase in cortical focal ischemia: GM1 ganglioside protects plasma membrane structure and function. J Neurosci Res. 1992;32:209–220. doi: 10.1002/jnr.490320210. [DOI] [PubMed] [Google Scholar]
- Malinow R. AMPA receptor trafficking and long-term potentiation. Philos Trans R Soc Lond B Biol Sci. 2003;358:707–714. doi: 10.1098/rstb.2002.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Man HY, Ju W, Ahmadian G, Wang YT. Intracellular trafficking of AMPA receptors in synaptic plasticity. Cell Mol Life Sci. 2000a;57:1526–1534. doi: 10.1007/PL00000637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man HY, Lin JW, Ju WH, Ahmadian G, Liu L, Becker LE, Sheng M, Wang YT. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron. 2000b;25:649–662. doi: 10.1016/s0896-6273(00)81067-3. [DOI] [PubMed] [Google Scholar]
- Man HY, Wang Q, Lu WY, Ju W, Ahmadian G, Liu L, D'Souza S, Wong TP, Taghibiglou C, Lu J, Becker LE, Pei L, Liu F, Wymann MP, MacDonald JF, Wang YT. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 2003;38:611–624. doi: 10.1016/s0896-6273(03)00228-9. [DOI] [PubMed] [Google Scholar]
- Man HY, Sekine-Aizawa Y, Huganir RL. Regulation of {alpha}-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc Natl Acad Sci U S A. 2007;104:3579–3584. doi: 10.1073/pnas.0611698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantych KB, Ferreira A. Agrin differentially regulates the rates of axonal and dendritic elongation in cultured hippocampal neurons. J Neurosci. 2001;21:6802–6809. doi: 10.1523/JNEUROSCI.21-17-06802.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, Ferguson TS, Cibulsky SM, Holmqvist M, Ding C, Fei H, Levitan IB. MONaKA, a novel modulator of the plasma membrane Na, K-ATPase. J Neurosci. 2005;25:7934–7943. doi: 10.1523/JNEUROSCI.0635-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Nishimune A, Mellor JR, Henley JM. SUMOylation regulates kainate-receptor-mediated synaptic transmission. Nature. 2007;447:321–325. doi: 10.1038/nature05736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrail KM, Phillips JM, Sweadner KJ. Immunofluorescent localization of three Na, K-ATPase isozymes in the rat central nervous system: both neurons and glia can express more than one Na, K-ATPase. J Neurosci. 1991;11:381–391. doi: 10.1523/JNEUROSCI.11-02-00381.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier SD, Kovalchuk Y, Rose CR. Properties of the new fluorescent Na+ indicator CoroNa Green: comparison with SBFI and confocal Na+ imaging. J Neurosci Methods. 2006;155:251–259. doi: 10.1016/j.jneumeth.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Mellgren RL. Calcium-dependent proteases: an enzyme system active at cellular membranes? FASEB J. 1987;1:110–115. doi: 10.1096/fasebj.1.2.2886390. [DOI] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Menegola M, Trimmer JS. Calcium- and metabolic state-dependent modulation of the voltage-dependent Kv2.1 channel regulates neuronal excitability in response to ischemia. J Neurosci. 2005;25:11184–11193. doi: 10.1523/JNEUROSCI.3370-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrsić-Pelcić J, Pelcić G, Vitezić D, Antoncić I, Filipović T, Simonić A, Zupan G. Hyperbaric oxygen treatment: the influence on the hippocampal superoxide dismutase and Na+, K+-ATPase activities in global cerebral ischemia-exposed rats. Neurochem Int. 2004;44:585–594. doi: 10.1016/j.neuint.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Newpher TM, Ehlers MD. Glutamate receptor dynamics in dendritic microdomains. Neuron. 2008;58:472–497. doi: 10.1016/j.neuron.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoletti VG, Condorelli DF, Dell'Albani P, Ragusa N, Giuffrida Stella AM. AMPA-selective glutamate receptor subunits in the rat hippocampus during aging. J Neurosci Res. 1995;40:220–224. doi: 10.1002/jnr.490400210. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Saito KI, Grynspan F, Griffin WR, Katayama S, Honda T, Mohan PS, Shea TB, Beermann M. Calcium-activated neutral proteinase (calpain) system in aging and Alzheimer's disease. Ann N Y Acad Sci. 1994;747:77–91. doi: 10.1111/j.1749-6632.1994.tb44402.x. [DOI] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- O'Connor LT, Lauterborn JC, Smith MA, Gall CM. Expression of agrin mRNA is altered following seizures in adult rat brain. Brain Res Mol Brain Res. 1995;33:277–287. doi: 10.1016/0169-328x(95)00147-k. [DOI] [PubMed] [Google Scholar]
- Ossowska K, Wolfarth S, Schulze G, Wardas J, Pietraszek M, Lorenc-Koci E, Smiałowska M, Coper H. Decline in motor functions in aging is related to the loss of NMDA receptors. Brain Res. 2001;907:71–83. doi: 10.1016/s0006-8993(01)02601-4. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Bingol B, Weld HA, Schuman EM. Ubiquitin-mediated proteasome activity is required for agonist-induced endocytosis of GluRs. Curr Biol. 2003;13:2073–2081. doi: 10.1016/j.cub.2003.10.028. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Topolnik L, Lacaille JC, McBain CJ. Compartmentalized Ca(2+) channel regulation at divergent mossy-fiber release sites underlies target cell-dependent plasticity. Neuron. 2006;52:497–510. doi: 10.1016/j.neuron.2006.08.032. [DOI] [PubMed] [Google Scholar]
- Pietrini G, Matteoli M, Banker G, Caplan MJ. Isoforms of the Na, K-ATPase are present in both axons and dendrites of hippocampal neurons in culture. Proc Natl Acad Sci U S A. 1992;89:8414–8418. doi: 10.1073/pnas.89.18.8414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro CA, Balestro F, Grando V, Wajner M. Isovaleric acid reduces Na+, K+-ATPase activity in synaptic membranes from cerebral cortex of young rats. Cell Mol Neurobiol. 2007;27:529–540. doi: 10.1007/s10571-007-9143-3. [DOI] [PubMed] [Google Scholar]
- Rodríguez de Lores Arnaiz G, Reinés A, Herbin T, Peña C. Na+, K+-ATPase interaction with a brain endogenous inhibitor (endobain E) Neurochem Int. 1998;33:425–433. doi: 10.1016/s0197-0186(98)00046-1. [DOI] [PubMed] [Google Scholar]
- Rodríguez de Lores Arnaiz G, Herbin T, Peña C. A comparative study between a brain Na+, K(+)-ATPase inhibitor (endobain E) and ascorbic acid. Neurochem Res. 2003;28:903–910. doi: 10.1023/a:1023227510707. [DOI] [PubMed] [Google Scholar]
- Rose CR. Na+ signals at central synapses. Neuroscientist. 2002;8:532–539. doi: 10.1177/1073858402238512. [DOI] [PubMed] [Google Scholar]
- Rose CR, Konnerth A. NMDA receptor-mediated Na+ signals in spines and dendrites. J Neurosci. 2001;21:4207–4214. doi: 10.1523/JNEUROSCI.21-12-04207.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose CR, Ransom BR. Regulation of intracellular sodium in cultured rat hippocampal neurones. J Physiol. 1997;499:573–587. doi: 10.1113/jphysiol.1997.sp021951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh G, Sia GM, Garner CC, Huganir RL. Synapse-associated protein-97 isoform-specific regulation of surface AMPA receptors and synaptic function in cultured neurons. J Neurosci. 2003;23:4567–4576. doi: 10.1523/JNEUROSCI.23-11-04567.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiner-Bobis G. The sodium pump. Its molecular properties and mechanics of ion transport. Eur J Biochem. 2002;269:2424–2433. doi: 10.1046/j.1432-1033.2002.02909.x. [DOI] [PubMed] [Google Scholar]
- Schoner W. Ouabain, a new steroid hormone of adrenal gland and hypothalamus. Exp Clin Endocrinol Diabetes. 2000;108:449–454. doi: 10.1055/s-2000-8140. [DOI] [PubMed] [Google Scholar]
- Schoner W. Endogenous cardiac glycosides, a new class of steroid hormones. Eur J Biochem. 2002;269:2440–2448. doi: 10.1046/j.1432-1033.2002.02911.x. [DOI] [PubMed] [Google Scholar]
- Schoner W, Bauer N, Müller-Ehmsen J, Krämer U, Hambarchian N, Schwinger R, Moeller H, Kost H, Weitkamp C, Schweitzer T, Kirch U, Neu H, Grünbaum EG. Ouabain as a mammalian hormone. Ann N Y Acad Sci. 2003;986:678–684. doi: 10.1111/j.1749-6632.2003.tb07282.x. [DOI] [PubMed] [Google Scholar]
- Sheldon C, Diarra A, Cheng YM, Church J. Sodium influx pathways during and after anoxia in rat hippocampal neurons. J Neurosci. 2004;24:11057–11069. doi: 10.1523/JNEUROSCI.2829-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hyoung Lee S. AMPA receptor trafficking and synaptic plasticity: major unanswered questions. Neurosci Res. 2003;46:127–134. doi: 10.1016/s0168-0102(03)00040-3. [DOI] [PubMed] [Google Scholar]
- Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, Lin Y, Bennett MV, Yuste R, Castillo PE, Zukin RS. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci. 2006;9:501–510. doi: 10.1038/nn1664. [DOI] [PubMed] [Google Scholar]
- Smith MA, Hilgenberg LG. Agrin in the CNS: a protein in search of a function? Neuroreport. 2002;13:1485–1495. doi: 10.1097/00001756-200208270-00001. [DOI] [PubMed] [Google Scholar]
- Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002;25:578–588. doi: 10.1016/s0166-2236(02)02270-1. [DOI] [PubMed] [Google Scholar]
- Squier MK, Cohen JJ. Calpain and cell death. Cell Death Differ. 1996;3:275–283. [PubMed] [Google Scholar]
- Strichartz G, Rando T, Wang GK. An integrated view of the molecular toxinology of sodium channel gating in excitable cells. Annu Rev Neurosci. 1987;10:237–267. doi: 10.1146/annurev.ne.10.030187.001321. [DOI] [PubMed] [Google Scholar]
- Tian J, Cai T, Yuan Z, Wang H, Liu L, Haas M, Maksimova E, Huang XY, Xie ZJ. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol Biol Cell. 2006;17:317–326. doi: 10.1091/mbc.E05-08-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortiglione A, Pignataro G, Minale M, Secondo A, Scorziello A, Di Renzo GF, Amoroso S, Caliendo G, Santagada V, Annunziato L. Na+/Ca2+ exchanger in Na+ efflux-Ca2+ influx mode of operation exerts a neuroprotective role in cellular models of in vitro anoxia and in vivo cerebral ischemia. Ann N Y Acad Sci. 2002;976:408–412. doi: 10.1111/j.1749-6632.2002.tb04768.x. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Wajner A, Bürger C, Dutra-Filho CS, Wajner M, de Souza Wyse AT, Wannmacher CM. Synaptic plasma membrane Na(+), K (+)-ATPase activity is significantly reduced by the alpha-keto acids accumulating in maple syrup urine disease in rat cerebral cortex. Metab Brain Dis. 2007;22:77–88. doi: 10.1007/s11011-007-9046-5. [DOI] [PubMed] [Google Scholar]
- Wallace BG, Qu Z, Huganir RL. Agrin induces phosphorylation of the nicotinic acetylcholine receptor. Neuron. 1991;6:869–878. doi: 10.1016/0896-6273(91)90227-q. [DOI] [PubMed] [Google Scholar]
- Willeumier K, Pulst SM, Schweizer FE. Proteasome inhibition triggers activity-dependent increase in the size of the recycling vesicle pool in cultured hippocampal neurons. J Neurosci. 2006;26:11333–11341. doi: 10.1523/JNEUROSCI.1684-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao AY, Wei L, Xia S, Rothman S, Yu SP. Ionic mechanism of ouabain-induced concurrent apoptosis and necrosis in individual cultured cortical neurons. J Neurosci. 2002;22:1350–1362. doi: 10.1523/JNEUROSCI.22-04-01350.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JJ, Ehlers MD. Ubiquitin and protein turnover in synapse function. Neuron. 2005;47:629–632. doi: 10.1016/j.neuron.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Yu SP. Na(+), K(+)-ATPase: the new face of an old player in pathogenesis and apoptotic/hybrid cell death. Biochem Pharmacol. 2003;66:1601–1609. doi: 10.1016/s0006-2952(03)00531-8. [DOI] [PubMed] [Google Scholar]
- Yu XM. The role of intracellular sodium in the regulation of NMDA-receptor-mediated channel activity and toxicity. Mol Neurobiol. 2006;33:63–80. doi: 10.1385/MN:33:1:063. [DOI] [PubMed] [Google Scholar]
- Yuan A, Santi CM, Wei A, Wang ZW, Pollak K, Nonet M, Kaczmarek L, Crowder CM, Salkoff L. The sodium-activated potassium channel is encoded by a member of the Slo gene family. Neuron. 2003;37:765–773. doi: 10.1016/s0896-6273(03)00096-5. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Xiao M, Nicoll RA. Contribution of cytoskeleton to the internalization of AMPA receptors. Proc Natl Acad Sci U S A. 2001;98:1261–1266. doi: 10.1073/pnas.031573798. [DOI] [PMC free article] [PubMed] [Google Scholar]