

Abstract
Background/Aims
Chronic ethanol exposure impairs liver regeneration due to inhibition of insulin signaling and oxidative injury. PPAR agonists function as insulin sensitizers and anti-inflammatory agents. We investigated whether treatment with a PPARδ agonist could restore hepatic insulin sensitivity, survival signaling, and regenerative responses vis-a-vis chronic ethanol feeding.
Methods
Adult rats were fed isocaloric liquid diets containing 0% or 37% ethanol, and administered a PPARδ agonist by i.p. injection. We used liver tissue to examine histopathology, gene expression, oxidative stress, insulin signaling, and regenerative responses to 2/3 hepatectomy.
Results
Chronic ethanol feeding caused insulin resistance, increased oxidative stress, lipid peroxidation, DNA damage, and hepatocellular injury in liver. These effects were associated with reduced insulin receptor binding and affinity, impaired survival signaling through PI3K/Akt/GSK3β, and reduced expression of insulin responsive genes mediating energy metabolism and tissue remodeling. PPARδ agonist treatment reduced ethanol-mediated hepatic injury, oxidative stress, lipid peroxidation, and insulin resistance, increased signaling through PI3K/Akt/GSK3β, and enhanced the regenerative response to partial hepatectomy.
Conclusions
PPARδ agonist administration may attenuate the severity of chronic ethanol-induced liver injury and ethanol’s adverse effects on the hepatic repair by restoring insulin responsiveness, even in the context of continued high-level ethanol consumption.
Keywords: Alcoholic liver disease, insulin receptor binding, insulin sensitizers
Introduction
Chronic high-level ethanol consumption impairs liver regeneration by inhibiting insulin signaling that mediates hepatocellular DNA synthesis. Insulin transmits pro-growth signals by activating complex intracellular pathways, beginning with ligand binding to cell surface receptors, followed by activation of intrinsic receptor tyrosine kinases that phosphorylate insulin receptor substrate type 1 (IRS-1). Consequently, intracellular signals are transmitted downstream to mediate growth, survival, energy metabolism, gene expression, and motility, all of which are needed for liver remodeling and regeneration (1-5). Ethanol’s inhibitory effects on insulin signaling result in decreased activation of phosphatidylinositol-3-kinase (PI3K), which mediates growth, survival, glucose utilization, and energy metabolism, and the Ras/Raf/MAPK/ERK pathway, which promotes hepatocyte proliferation. Since PI3K has a critical role in cell survival and energy metabolism, ethanol-induced hepatic insulin resistance also results in increased DNA damage, mitochondrial dysfunction, and activation of pro-apoptosis mechanisms (6-9)
Peroxisome proliferator-activated receptors (PPARs) are nuclear hormone receptors that bind to DNA and regulate gene transcription in a broad range of cells and tissues. PPARs are regulated by ligand binding and mediate their effects by heterodimerizing with the retinoid × receptor (10). Three distinct isoforms of PPARs exist: PPARα, PPARδ (also referred to as PPARβ), and PPARγ. PPARα is most abundantly expressed in brown adipose tissue and liver, followed by kidney, heart and skeletal muscle. PPAR-α is activated by polyunsaturated fatty acids and fibrates. PPARα regulates adipocyte growth and differentiation, lipid metabolism, lipoprotein synthesis, and tissue inflammatory responses. PPARδ is widely expressed, but most abundant in gut, kidney and heart. PPARδ regulates lipid catabolism and oxidative phosphorylation in fat and muscle (11, 12), and it has a functional role in activating hepatic stellate cells during liver injury, which contributes to the pro-inflammatory response (13). PPARγ is primarily expressed in adipose tissue, followed by colon, immune cells, and retina. PPARγ influences storage of fatty acids in adipose tissue and regulates insulin sensitivity (14, 15).
Previous studies demonstrated that chronic ethanol-induced liver injury is partly mediated by hepatic insulin resistance and oxidative stress with increased DNA damage and lipid peroxidation (16). In light of the known therapeutic actions of PPAR agonists, we evaluated the effectiveness of PPAR agonists in restoring insulin responsiveness, insulin signaling, histology, and regenerative responses in livers of chronic ethanol-fed rats. We focused on the δ class of PPAR agonists because it was already shown to have positive effects on hepatic insulin sensitivity (17). Our hypothesis was that by improving insulin signaling and insulin responsive gene expression, ethanol‘s adverse effects on liver injury and repair (7, 16, 18) would be partially attenuated. The results demonstrated that treatment with a PPARδ agonist can effectively reduce injury, oxidative stress, and DNA damage, and substantially improved the regenerative response in livers of chronic ethanol-fed rats.
Methods
Chronic ethanol exposure model
Adult male (~200-250 g) Long Evans (LE) rats (Harlan Sprague Dawley, Inc., Indianapolis, Indiana) were pair-fed with isocaloric liquid diets (BioServ, Frenchtown, NJ) containing 0% or 37% ethanol by caloric content for 8 weeks (7). During the last 3 weeks of liquid diet feeding, rats administered twice weekly intra-peritoneal (i.p.) injections of saline or a PPARδ (L-165,041; 2 μg/Kg) agonist (CalBiochem, Carlsbad, CA). L-165,041 displays >100-fold selectivity for PPARδ compared with other PPAR receptors, and has biological actions on insulin, glucose and lipid metabolism (19). After the feeding regimen, rats were subjected to 2/3 hepatectomy (5) to measure regenerative responses in the liver (5, 7). Serum and liver were harvested at surgery (0 h), and 24 h later, during the peak regeneration (5). Serum was used to measure adiponectin and leptin, as indices of systemic insulin resistance. All experiments were conducted in accordance with guidelines established by the National Institutes of Health and approved by the institutional Animal Care and Use Committee at the Lifespan-Rhode Island Hospital.
Receptor binding assays
Competitive saturation binding studies were used to determine if PPARδ agonist treatments abrogated ethanol-impaired insulin or IGF-1 binding to their corresponding receptors in liver. Fresh frozen tissue was homogenized in lysis buffer (7) and optimally diluted in binding buffer to achieve 20% specific binding (20). We generated binding curves with 8 pooled liver samples per group. For total binding, samples were incubated in 100 μl reactions containing binding buffer and 0.0031 to 1 μCi/ml of [125I] (2000 Ci/mmol) insulin or IGF-1. For non-specific binding, samples were identically prepared with the addition of 0.1 μM unlabeled ligand. Incubations were carried out for 16 hours at 4°C in non-binding 96-well plates (Corning Incorporated Life Science, Lowell, MA). Bound [125I] insulin or IGF-1 was captured onto polycarbonate filter plates and measured in a TopCount (Packard Instrument Company, Meriden, CT) (16). Specific binding was calculated by subtracting fmol/mg of non-specifically bound isotope from the total bound isotope. The results were plotted and analyzed using GraphPad Prism 5 software (San Diego, CA).
Liver tissue assays
RNA was isolated, reverse transcribed, and used to measure PPARδ gene expression by quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) analysis as described (21). Protein expression was examined by Western blot, immunoprecipitation-Western blot analysis, and enzyme-linked immunosorbant assays (ELISAs) (7, 16, 22, 23). For immunoprecipitation studies, liver homogenates with equivalent amounts of protein were incubated with PTEN antibody and protein A/G agarose for 1 hour at 4°C. Washed samples were subjected to Western blot analysis with antibodies to p85α subunit of PI3K or PTEN (22). We used direct binding ELISAs to measure 4-hydroxynonenal (HNE) immunoreactivity as described (16, 23). Immunoreactivity was detected with horseradish peroxidase (HRP)-conjugated secondary antibody (Pierce, Rockford, IL) and Amplex Red soluble fluorophore (Molecular Probes, Eugene, OR). Fluorescence light units (FLU) were measured (Ex 579/Em 595) in a SpectraMax M5 microplate reader (Molecular Devices Corp., Sunnyvale, CA). Negative control assays included incubations with primary, secondary, or both antibodies omitted.
In Situ assays of oxidative stress
Paraffin-embedded histological sections were stained with hematoxylin-eosin, and adjacent sections were immunostained for 8-hydroxy-2’-deoxyguanosine (8-OHdG), or 4-hydroxy 2,3-nonenal (HNE) as indices of DNA damage and membrane lipid peroxidation. Tissues were immunostained with 0.5-1 μg/mL of primary antibody (7), and immunoreactivity was detected with HRP-conjugated polymer-tagged secondary antibodies (Abcam, Cambridge, MA, USA) and diaminobenzidine.
Source of reagents
Human recombinant [125I] Insulin and IGF-1 were purchased from Amersham Biosciences (Boston, MA). Unlabeled human insulin and recombinant IGF-1 were purchased from Bachem (Torrance, CA). Monoclonal antibodies to GAPDH and β-actin were purchased from Chemicon (Tecumsula, CA). The A85G6 mouse monoclonal antibody was used to detect AAH (24). Antibodies to pAkt (AF887) were purchased from B & D (Franklin Lakes, NJ); phospho-GSK3β (Ser9/21, #9331) from Cell Signaling Technology (Beverly, MA), GSK3β (MAB8689) from Chemicon (Millipore) and p85 subunit of PI3K (sc-423), PTEN (sc2350), Akt1 (sc-1518), and anti-rabbit, -mouse, and -goat HRP-conjugated IgG (SC-2374; SC-2350) were purchased from Santa Cruz (Santa Cruz, CA). Immunoprecipitating antibodies to PTEN were provided by Dr. Nicholas Leslie (University of Dundee). All other fine chemicals were purchased from CalBiochem (Carlsbad, CA) or Sigma-Aldrich (St. Louis, MO).
Statistical analysis
Data depicted in graphs represent the mean ± S.E.M. or mean ± 95% C.I.L. for each group. Inter-group comparisons were made using repeated measures analysis of variance (ANOVA) and the post-hoc Tukey-Kramer test for significance or Student T-tests (GraphPad Prism 5 software, San Diego, CA).
Results
Effects of chronic ethanol exposure and PPARδ agonist treatment on serum adipokines and hepatic PPARδ gene expression. Chronic ethanol abuse dysregulates glucose and lipid homeostasis, resulting in insulin resistance with altered adiponectin and leptin levels (25). Chronic ethanol feeding decreases serum adiponectin levels, and treatment with exogenous adiponectin ameliorates ethanol-induced liver injury (26-28). Studies in human subjects showed that acute exposure to ethanol impairs leptin secretion (29), while chronic abuse increases serum leptin levels (30), suggesting increased energy expenditure. However, in the setting of severe malnutrition, serum leptin may be reduced (31), possibly to conserve body mass. Herein, we observed significantly reduced serum adiponectin levels following chronic ethanol exposure, with no improvement effectuated by PPARδ agonist treatment (Fig 1A). These findings are consistent with previous reports and suggest chronic ethanol feeding perturbed lipid homeostasis. In contrast, serum leptin was not significantly altered by the chronic ethanol feeding or PPARδ agonist treatment (Fig 1B), consistent with observations in healthy humans (32), and suggesting that systemic energy expenditure was not compromised in our model.
Figure 1.
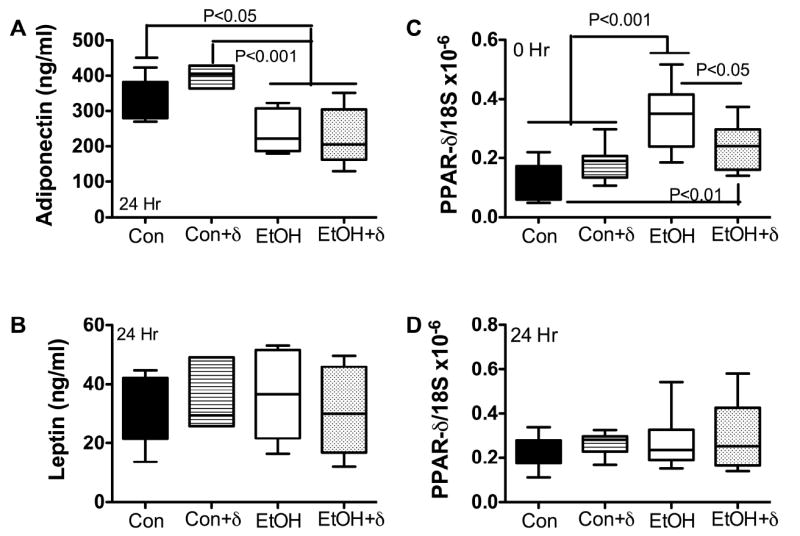
Chronic ethanol feeding reduces serum adiponectin and increased hepatic PPARδ mRNA levels. LE adult male rats were fed isocaloric liquid diets containing 0% or 37% ethanol by caloric content for 8 weeks, and then subjected to 2/3 hepatectomy. During the last 3 weeks on the liquid diets, rats were treated with the L-165,041 PPARδ agonist by i.p. injection. Serum and liver tissue were harvested at the time of surgery, and 24 hours later, during peak regeneration. Serum (A) adiponectin and (B) leptin levels were measured by ELISA using a commercially available assay kit. Liver tissue harvested (C) by 2/3 hepetactomy, or (D) 24 h later was used to measure PPARδ mRNA by qRT-PCR analysis. mRNA levels were normalized to 18S rRNA. Box plots display mean ± S.D. or results. Intergroup statistical comparisons were made by 1-way ANOVA with post hoc Tukey-Kramer significance tests. Significant P-values are shown within each panel.
PPARδ mRNA levels were higher in ethanol-exposed relative to control livers, but treatment with L-165,041 restored PPARδ expression to control levels (Fig 1C). However, 24 h post 2/3 hepatectomy, the PPARδ mRNA levels were similar in all groups, including ethanol-exposed rats that had not been treated with L-165,041 (Fig 1D). Since PPARδ mediates cellular proliferation, survival, and lipid homeostasis (13, 33, 34), the increased mRNA levels in ethanol-exposed livers could represent a compensatory response to increased injury and cell turnover (see below), whereas during liver regeneration, i.e. 24 h after partial hepatectomy, the higher levels of PPARδ in all groups correlate with the massive activation of pro-growth and pro-survival mechanisms.
PPARδ agonist reverses ethanol-induced liver pathology
Livers from rats fed with the control liquid diet or chow had well-organized lobular architectures with minimal evidence of steatosis, variation in nuclear size, or hepatocyte dropout (data not shown). In contrast, ethanol exposed livers exhibited microvesicular and macrovesicular steatosis with multi-focal intralobular lymphomononuclear cell inflammation and scattered areas of apoptosis or hepatocyte drop-out (Fig. 2). In addition, ethanol feeding caused liver architectural disarray with loss of regular chords and increased variability in hepatocyte nuclear size. In ethanol-fed rats, treatment with L-165,041 reduced the liver architectural disarray, micro- and macrosteatosis, and apoptotic cell death. Although small foci of inflammation (Fig. 2E, arrow) were detected, such lesions were less conspicuous than in vehicle-treated, ethanol-exposed livers.
Figure 2.

PPARδ agonist partially reverses ethanol-induced liver histopathology. Chronic ethanol feeding caused (A, B) hepatic architectural disarray with (C, D) increased steatosis, (D) apoptosis (black arrow), and Mallory’s hyaline (white arrow) (insets). (E-H) PPARδ agonist treatment (E, F) improved hepatic chord-like architecture and reduced (E-H) steatosis and (H) apoptosis in ethanol-exposed livers. However, (G, inset) scattered small foci of inflammation and apoptosis were still detectable. (Original magnification: A, E- 50x; B, F - 200x; C, G - 400x; D, H - 600x).
Effects of ethanol on insulin and IGF-1 receptor binding
We measured insulin and IGF-1 receptor binding and affinity in liver tissue excised at the time of 2/3 hepatectomy to assess hepatic insulin resistance after 8 weeks on the liquid diets, and 3 weeks of L-165,041 or vehicle treatment. Top-level insulin binding to its own receptor (BMAX ± SD) was 7.23 ± 2.66 in controls, compared with 2.59 ± 0.61 in ethanol-exposed rats (P<0.001 relative to control) (Fig 3). Insulin receptor binding affinity (Kd ± SEM) was significantly higher in control (51.38 ± 18.63) compared with ethanol-exposed (165.5 ± 73) livers (P<0.001). (Note that higher affinity binding is associated with a lower Kd). L-165,041 treatment increased the BMAX (4.21 ± 0.45) and decreased the Kd (33.85 ± 9.91) in ethanol-exposed livers (both P<0.001 relative to corresponding vehicle-treated rats), consistent with the hypothesis that PPARδ improves hepatic insulin sensitivity.
Figure 3.
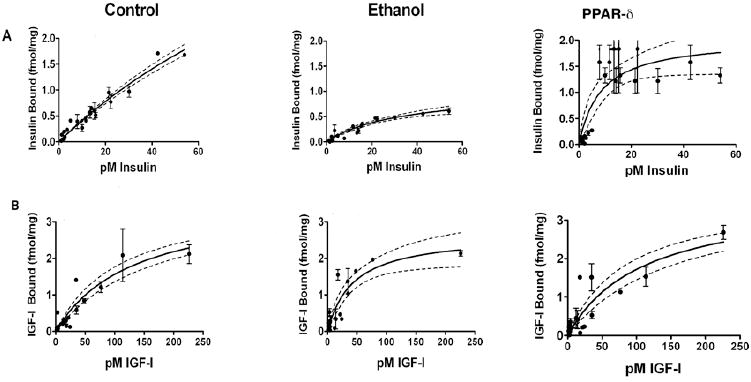
Effects of ethanol and PPARδ agonist (L-165,041) treatment on Insulin and IGF-1 receptor binding. Competitive saturation binding assays were performed by incubating liver membrane protein extracts (8 rats per group) with 0.5-100 pM [125I]-labeled insulin or IGF-1 in the presence or absence of 100 nM unlabeled ligand. Specific binding, saturation binding (BMAX), dissociation constants/binding affinity (Kd), and corresponding S.D. and 95% CI were calculated using the GraphPad Prism 5 software. Graphs depict specific (A) insulin or (B) IGF-1 receptor binding (fmol/mg protein) ± 95% CI and S.E.M. measured in livers from control, ethanol fed+vehicle-treated, and ethanol fed+PPARδ agonist-treated rats. See Results section for statistical analysis data.
Chronic ethanol feeding impaired IGF-1 binding to a lesser degree than insulin binding. The IGF-1 BMAX in control livers was 3.63 ± 0.77 and the Kd was 131.9 ± 49.7, while in ethanol-fed rats, the BMAX was 2.66 ± 0.38 and the Kd 43.57 ± 13.71 (NS and P<0.001 respectively) (Fig 3). Therefore, although the BMAX was somewhat lower, IGF-1 receptor binding affinity was significantly higher in the ethanol-exposed livers. L-165,041 treatment increased the IGF-1 BMAX to 3.31 ± 0.89, but also increased the Kd to 120.6 ± 52.1 in ethanol-exposed livers, rendering the receptor binding properties not significantly different from control.
Ethanol-induced defects in insulin signaling are partially reversed by PPARδ agonist treatment. Insulin binding to its own receptor results in tyrosine phosphorylation of IRS-1, and activation of the Ras/Raf/MAPK/ERK and PI3K/Akt/GSK3β pathways, which respectively mediate hepatocyte proliferation, and cell survival, motility, and energy metabolism. The PI3K/Akt/GSK3β pathway is negatively regulated by PTEN’s interaction with the p85α subunit of PI3K, as occurs with chronic ethanol exposure (7, 18, 22). Herein, we observed that chronic ethanol feeding reduced pAkt and pGSK3β in liver, but that these adverse effects of ethanol were partially reversed by PPARδ agonist treatments (Fig. 4). Therefore, in ethanol-fed rats, PPARδ agonist improvements in insulin receptor binding and affinity translated to enhanced survival signaling through PI3K/Akt. On the other hand, the relatively modest recovery in pGSK-3β following PPARδ agonist treatment may have been due to persistently elevated levels of oxidative stress and DNA damage, since besides insulin resistance, oxidative stress can also activate GSK-3β (35).
Figure 4.
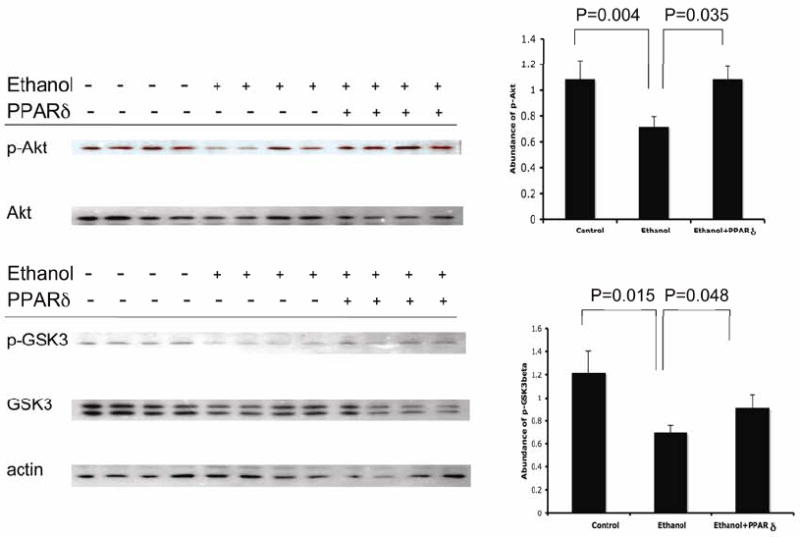
PPARδ agonist restores insulin signaling through the Akt pathway in chronic ethanol-fed rats. (Left) Representative Western blot analysis of pAkt, Akt, pGSK3β, and GSK3 levels in livers from control (-) or chronic ethanol-fed (+) rats treated with vehicle (-) or a PPARδ agonist (+) (N=10/group). (Right) Digital image quantification of the Western blot signals showing reduced levels of pAkt (upper panel) and pGSKβ (lower panel) in chronic ethanol exposed relative to control livers. Note increased levels of pAkt (nearly normalized) and pGSKβ (modest) in PPARδ agonist treated ethanol-fed relative to control livers.
Ethanol increases PTEN protein and phosphatase activity (7), thereby facilitating interactions between p85α and PTEN, and attendant attenuation of Akt, GSK3β, and BAD phosphorylation (22). Herein, we confirmed that chronic ethanol feeding increases the associations between p85α and PTEN in liver, but found that the degree to which PPARδ agonist treatment inhibited these interactions was relatively modest, and did not reach statistical significance (P=0.08; Fig 5). Therefore, PPARδ agonist improvements in insulin signaling were more likely mediated by enhanced insulin receptor binding than reduced associations between p85α and PTEN.
Figure 5.
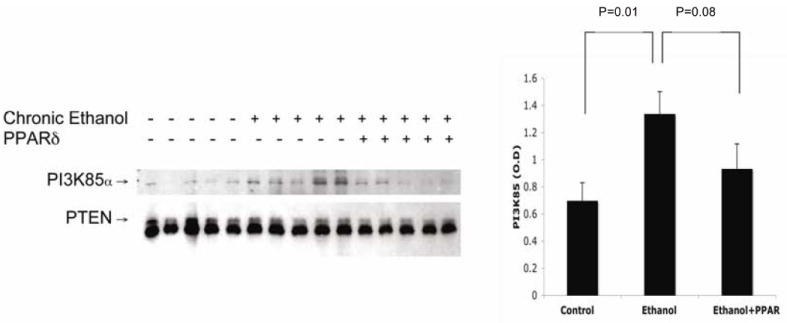
Chronic ethanol feeding increases association of PTEN with P85α subunit of PI3K. (Left) Liver homogenates (10 rats per group) were immunoprecipitated with anti-PTEN and subjected to Western blot analysis with (upper) anti-P85α or (lower) anti-PTEN. (Right) Results were quantified by digital imaging. Graphs depict mean ± S.D. of PI3K85α bound to PTEN. Chronic ethanol feeding increased the association of P85α subunit with PTEN, which inhibits survival signaling by reducing pAkt and pGSK3β (30). PPARδ agonist treatment marginally reduced the association between PTEN and P85α subunit (p=0.08) in ethanol-fed rat livers.
PPARδ agonist enhances insulin-responsive gene expression
We measured two insulin responsive genes in liver, namely glyceraldehydes-3-phosphate dehydrogenase (GAPDH) and aspartyl-(asparaginyl)-β-hydroxylase (AAH). Corresponding with previous reports (16), chronic ethanol exposure inhibited GAPDH and AAH protein expression in both basal and regenerating liver (Fig. 6). The significance of these results is that, since AAH has a functional role in cell migration (7) which is needed for remodeling after injury and during regeneration, ethanol’s inhibition of AAH could account for the associated hepatic architectural disarray. In addition, GAPDH is a key enzyme involved in energy metabolism which is also needed for repair and regeneration. Treatment with the PPARδ agonist enhanced GAPDH and AAH immunoreactivity in ethanol-exposed livers (Fig 6), and correspondingly, we observed a striking restoration of the hepatic architecture both prior to and during regeneration. In essence, these results link insulin-responsiveness and AAH/GAPDH expression to restoration of hepatic architecture during regeneration and repair.
Figure 6.
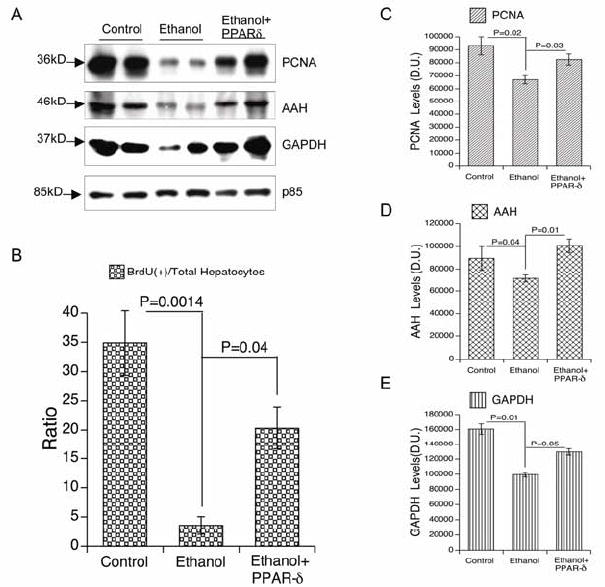
PPARδ agonist Improves ethanol-impaired liver regeneration. Adult male rats were fed with liquid diets containing 0% or 37% ethanol, treated with a PPARδ agonist, and then subjected to 2/3 hepatectomy (see Figure 1 legend). 24 h later, remnant regenerating livers were used to measure (A, C) proliferating cell nuclear antigen-PCNA, (A, D) aspartyl-(asparaginyl)-β-hydroxylase-AAH, (A, E) glyceraldehyde-3-phosphate dehydrogenase-GAPDH, and the (A) p85 subunit of PI3 kinase (negative control) expression by (A) Western blot analysis and (C-E) digital image quantification (arbitrary densitometry units; D.U.). 2 h prior to sacrifice, rats were injected with BrdU. Livers were immersion fixed and paraffin-embedded. Histological sections were immunostained to detect nuclear BrdU. Under code, BrdU positive and negative hepatocytes were enumerated in 10 adjacent 100x magnification fields per slide. (B) The graph depicts the percentages of BrdU-labeled nuclei in regenerating livers from control, ethanol exposed, and ethanol exposed+PPARδ agonist treated rats. For all studies, inter-group comparisons were made using one-way repeated measures ANOVA and the Tukey-Kramer post-hoc test of significance. P-values are indicated within each panel. Note that PPARδ agonist treatments significantly increased DNA synthesis, and PCNA, AAH, and GAPDH expression during liver regeneration.
PPARδ agonist augments hepatic regenerative response during chronic ethanol consumption
Since the PPARδ agonist treatments reduced hepatic insulin resistance in the setting of chronic ethanol exposure, we extended our investigations to determine if they would also enhance liver regeneration. DNA synthesis was assessed by BrdU incorporation into nuclear DNA. Twenty-four hours after 2/3 hepatectomy, approximately 35% of hepatocytes were BrdU labeled in control rats, whereas only 4% of hepatocytes were labeled in the ethanol fed group (Fig 6). Correspondingly, Western blot analysis demonstrated lower levels of PCNA in ethanol-fed relative to control livers, 24 hours after 2/3 hepatectomy (Fig 6). Interestingly, PPARδ agonist treatments partially reversed ethanol’s inhibitory effects on DNA synthesis as demonstrated by the 4- to 5-fold increases in BrdU immunohistochemical staining relative to vehicle-treatment (Fig 6).
Lipid peroxidation and DNA damage in ethanol-fed rat livers
In addition to impairing insulin responsiveness, ethanol mediates its adverse effects in liver by causing oxidative stress, lipid peroxidation, and DNA damage (16, 18). PPAR agonists enhance insulin sensitivity and also reduce inflammation/oxidative stress (15). We determined the effects of PPARδ agonist treatment on ethanol-induced lipid peroxidation and DNA damage by respectively examining HNE and 8-OHdG immunoreactivity by direct binding ELISA and/or immunohistochemical staining. The studies demonstrated higher levels of both HNE and 8-OHdG in ethanol-exposed relative to control livers by ELISA (Fig 7) and immunohistochemical staining (data not shown). PPARδ agonist treatments strikingly reduced HNE and 8-OHdG immunoreactivities in ethanol-exposed livers (Fig 7). During peak regeneration (24 h point), control livers exhibited only rare nuclear 8-OHdG labeling, while ethanol-exposed livers had abundant nuclear 8-OHdG immunoreactivity. PPARδ agonist treatments substantially reduced, but failed to abolish hepatic nuclear DNA damage in chronic ethanol-fed rats (Fig 7). Therefore, PPARδ agonist administration reduced but failed to abolish ethanol-mediated DNA damage and lipid peroxidation. This therapeutic effect of the PPARδ agonist most likely aided in restoring the hepatic regenerative response, despite continued chronic exposure to ethanol.
Figure 7.
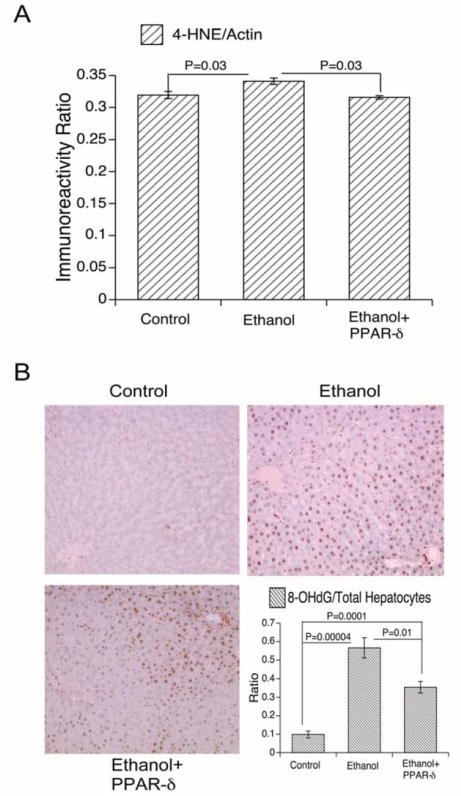
PPARδ agonist treatment partially reversed ethanol-induced oxidative stress. Rats were treated as described in the legend to Figure 6. Regenerating livers were used to measure (A) 4-HNE immunoreactivity (lipid peroxidation) by direct binding ELISA. (B) In addition, histological sections were immunostained to detect 8-OHdG as an index of DNA damage (see Methods; Original magnifications 100x). The ratios of 8-OHdG-positive nuclei to total nuclei in 10-100x microscopic fields were determined The graph (lower right panel) shows the mean relative densities of 8-OHdG immunoreactive nuclei in regenerating livers from control, ethanol exposed, and ethanol exposed+PPARδ agonist treated rats. Inter-group comparisons were made using one-way repeated measures ANOVA and the Tukey-Kramer post-hoc significance test. P-values are indicated within each panel.
Discussion
PPARδ is expressed in liver, muscle, brain, and fat (36, 37), yet its potential role in treating hepatic insulin resistance has only been explored recently (17), when it was shown that treatment of diabetic mice with GW501516, a high affinity PPARδ agonist, improved hepatic insulin sensitivity (17). Herein, we demonstrate that PPARδ agonist mediated enhancement of hepatic insulin sensitivity is mediated by increased insulin receptor binding, increased signaling downstream through PI3K/Akt, which promotes cell survival, and reduced DNA damage and lipid peroxidation. Our findings are concordant with previous studies demonstrating protective effects of PPARδ expression and function in liver (38) due to reduced hepatic steatosis, steatohepatitis, increased fatty acid β-oxidation, and reduced activation of TNFα and NFκB (39). However, a few studies have generated conflicting data indicating that L-165,041 treatments could enhance hepatotoxicity due to increased stellate cell activation (13). Therefore, therapeutic effects of PPARδ agonists may vary with cell type and nature of tissue injury.
LE rats are highly sensitive to chronic alcohol exposure because, within a period of five or six weeks of ethanol feeding, their livers exhibit prominent macrosteatosis, inflammation, DNA damage, and apoptosis (7, 16). Given the known inhibitory effects of ethanol on insulin signaling, insulin-responsive gene expression, and liver regeneration (3-5, 8, 9), we investigated whether chronic ethanol-induced liver injury in LE rats was associated with hepatic insulin resistance, and if PPARδ agonist treatments, vis-à-vis continued ethanol exposure, could abrogate the hepatic insulin resistance and enhance liver regeneration. Because insulin resistance can be mediated by impaired binding to the insulin receptor (1, 6, 7, 20), we performed competitive saturation binding assays to measure top level binding and binding affinity. Since IGF-1 activates IRS pathways, and its downstream effects are similar to those of insulin (40, 41), we also measured IGF-1 receptor binding and affinity.
The studies demonstrated that chronic ethanol exposure significantly impairs binding to both insulin and IGF-1 receptors, but with greater adverse effects on the BMAX and Kd of insulin compared with IGF-1 receptors. PPARδ agonist treatments significantly enhanced both parameters of insulin receptor binding, but had modest effects on IGF-1 receptor binding in ethanol-exposed livers. Previous studies showed that altered membrane lipid composition is a major factor contributing to ethanol’s impairment of insulin receptor binding (20, 42, 43). Conceivably, the improvements in insulin receptor binding effectuated by the PPARδ treatments were promoted by enhanced fatty acid synthesis and membrane lipid repletion (17), resulting in increased survival signaling through PI3K/Akt (increased pAkt and pGSK3β), and insulin-responsive gene expression including GAPDH and AAH, and culminating in restoration of hepatic architecture. Attendant increases in GAPDH and AAH were important for mediating increased energy metabolism and cell motility required for remodeling of tissue following injury or during regeneration (44, 45).
While some hepatotoxic effects of ethanol are due to impaired insulin signaling, others are mediated by ethanol’s cytotoxic metabolites, principally acetaldehyde, together with oxidative stress, mitochondrial dysfunction, lipid peroxidation, and protein and DNA adduct formation (46). In our LE model, the livers had ethanol-induced histopathological lesions as previously described (7, 16). Since PPAR agonists have dual actions in their capacity to enhance insulin responsiveness and reduced cellular inflammation (15), we hypothesized that PPARδ agonist treatments would, in addition to improving insulin sensitivity, abrogate inflammation and injury in liver. As predicted, the PPARδ agonist treatments conspicuously reduced the severity of ethanol-induced steatohepatitis, apoptosis, and architectural disarray, despite continued chronic ethanol exposure. These results potentially offer a new strategy for treating humans with chronic alcohol-induced liver injury and hepatic dysfunction. Moreover, given the similarities between alcoholic- and non-alcoholic steatohepatitis (NASH) in terms of their common histopathologic features and insulin resistance (47), PPARδ agonists could potentially be suitable for treating NASH as well.
Finally, we hypothesized that ethanol’s inhibitory effects on insulin signaling through growth and survival pathways, together with its toxic effects on mitochondria leading to energy failure, oxidative stress, DNA damage, and lipid peroxidation, were responsible for ethanol’s impairment of liver regeneration. Consequently, we predicted that PPARδ agonist treatments, which ameliorate ethanol’s destructive pathway actions, would enhance liver regeneration in ethanol exposed rats. Indeed, the finding of substantially improved liver regeneration in PPARδ treated, ethanol-exposed rats supports these concepts, and further suggests that PPARδ agonists may aid in repair and regenerative processes both during and subsequent to liver injury caused by chronic alcohol abuse.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baumann CA, Ribon V, Kanzaki M, Thurmond DC, Mora S, Shigematsu S, et al. CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature. 2000;407:202–207. doi: 10.1038/35025089. [DOI] [PubMed] [Google Scholar]
- 2.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 3.Hsu MK, Qiao L, Ho V, Zhang BH, Zhang H, Teoh N, et al. Ethanol reduces p38 kinase activation and cyclin D1 protein expression after partial hepatectomy in rats. J Hepatol. 2006;44:375–382. doi: 10.1016/j.jhep.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 4.Poso H, Vaananen H, Salaspuro MP, Poso AR. Effects of ethanol on liver regeneration after partial hepatectomy in rats. Med Biol. 1980;58:329–336. [PubMed] [Google Scholar]
- 5.Wands JR, Carter EA, Bucher NL, Isselbacher KJ. Inhibition of hepatic regeneration in rats by acute and chronic ethanol intoxication. Gastroenterology. 1979;77:528–531. [PubMed] [Google Scholar]
- 6.Aroor AR, Shukla SD. MAP kinase signaling in diverse effects of ethanol. Life Sci. 2004;74:2339–2364. doi: 10.1016/j.lfs.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Yeon JE, Califano S, Xu J, Wands JR, De La Monte SM. Potential role of PTEN phosphatase in ethanol-impaired survival signaling in the liver. Hepatology. 2003;38:703–714. doi: 10.1053/jhep.2003.50368. [DOI] [PubMed] [Google Scholar]
- 8.Sasaki Y, Hayashi N, Ito T, Fusamoto H, Kamada T, Wands JR. Influence of ethanol on insulin receptor substrate-1-mediated signal transduction during rat liver regeneration. Alcohol Alcohol Suppl. 1994;29:99–106. [PubMed] [Google Scholar]
- 9.Sasaki Y, Wands JR. Ethanol impairs insulin receptor substrate-1 mediated signal transduction during rat liver regeneration. Biochem Biophys Res Commun. 1994;199:403–409. doi: 10.1006/bbrc.1994.1243. [DOI] [PubMed] [Google Scholar]
- 10.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 11.Choi KC, Lee SY, Yoo HJ, Ryu OH, Lee KW, Kim SM, et al. Effect of PPAR-delta agonist on the expression of visfatin, adiponectin, and resistin in rat adipose tissue and 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2007;357:62–67. doi: 10.1016/j.bbrc.2007.03.114. [DOI] [PubMed] [Google Scholar]
- 12.Escher P, Braissant O, Basu-Modak S, Michalik L, Wahli W, Desvergne B. Rat PPARs: quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology. 2001;142:4195–4202. doi: 10.1210/endo.142.10.8458. [DOI] [PubMed] [Google Scholar]
- 13.Hellemans K, Michalik L, Dittie A, Knorr A, Rombouts K, De Jong J, et al. Peroxisome proliferator-activated receptor-beta signaling contributes to enhanced proliferation of hepatic stellate cells. Gastroenterology. 2003;124:184–201. doi: 10.1053/gast.2003.50015. [DOI] [PubMed] [Google Scholar]
- 14.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moller DE, Berger JP. Role of PPARs in the regulation of obesity-related insulin sensitivity and inflammation. Int J Obes Relat Metab Disord. 2003;27(Suppl 3):S17–S21. doi: 10.1038/sj.ijo.0802494. [DOI] [PubMed] [Google Scholar]
- 16.de la Monte SM, Yeon JE, Tong M, Longato L, Chaudhry R, Pang MY, et al. Insulin resistance in experimental alcohol-induced liver disease. J Gastroenterol Hepatol. 2008;23:e477–e486. doi: 10.1111/j.1440-1746.2008.05339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee CH, Olson P, Hevener A, Mehl I, Chong LW, Olefsky JM, et al. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc Natl Acad Sci U S A. 2006;103:3444–3449. doi: 10.1073/pnas.0511253103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ronis MJ, Wands JR, Badger TM, de la Monte SM, Lang CH, Calissendorff J. Alcohol-induced disruption of endocrine signaling. Alcohol Clin Exp Res. 2007;31:1269–1285. doi: 10.1111/j.1530-0277.2007.00436.x. [DOI] [PubMed] [Google Scholar]
- 19.Berger J, Leibowitz MD, Doebber TW, Elbrecht A, Zhang B, Zhou G, et al. Novel peroxisome proliferator-activated receptor (PPAR) gamma and PPARdelta ligands produce distinct biological effects. J Biol Chem. 1999;274:6718–6725. doi: 10.1074/jbc.274.10.6718. [DOI] [PubMed] [Google Scholar]
- 20.Soscia SJ, Tong M, Xu XJ, Cohen AC, Chu J, Wands JR, et al. Chronic gestational exposure to ethanol causes insulin and IGF resistance and impairs acetylcholine homeostasis in the brain. Cell Mol Life Sci. 2006;63:2039–2056. doi: 10.1007/s00018-006-6208-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de la Monte SM, Tong M, Lester-Coll N, Plater M, Jr, Wands JR. Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: relevance to Alzheimer’s disease. J Alzheimers Dis. 2006;10:89–109. doi: 10.3233/jad-2006-10113. [DOI] [PubMed] [Google Scholar]
- 22.He J, de la Monte S, Wands JR. Acute ethanol exposure inhibits insulin signaling in the liver. Hepatology. 2007;46:1791–1800. doi: 10.1002/hep.21904. [DOI] [PubMed] [Google Scholar]
- 23.Cohen AC, Tong M, Wands JR, de la Monte SM. Insulin and Insulin-Like Growth Factor Resistance With Neurodegeneration in an Adult Chronic Ethanol Exposure Model. Alcohol Clin Exp Res. 2007;1573:1558. doi: 10.1111/j.1530-0277.2007.00450.x. [DOI] [PubMed] [Google Scholar]
- 24.Carter JJ, Tong M, Silbermann E, Lahousse SA, Ding FF, Longato L, et al. Ethanol impaired neuronal migration is associated with reduced aspartyl-asparaginyl-beta-hydroxylase expression. Acta Neuropathol. 2008;116:303–315. doi: 10.1007/s00401-008-0377-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sebastian BM, Kang L, Chen X, Nagy LE. Methods to investigate the effects of chronic ethanol on adipocytes. Methods Mol Biol. 2008;447:357–366. doi: 10.1007/978-1-59745-242-7_23. [DOI] [PubMed] [Google Scholar]
- 26.Chen X, Sebastian BM, Nagy LE. Chronic ethanol feeding to rats decreases adiponectin secretion by subcutaneous adipocytes. Am J Physiol Endocrinol Metab. 2007;292:E621–E628. doi: 10.1152/ajpendo.00387.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L, Nyomba BL. Glucose intolerance and resistin expression in rat offspring exposed to ethanol in utero: modulation by postnatal high-fat diet. Endocrinology. 2003;144:500–508. doi: 10.1210/en.2002-220623. [DOI] [PubMed] [Google Scholar]
- 28.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rojdmark S, Calissendorff J, Brismar K. Alcohol ingestion decreases both diurnal and nocturnal secretion of leptin in healthy individuals. Clin Endocrinol (Oxf) 2001;55:639–647. doi: 10.1046/j.1365-2265.2001.01401.x. [DOI] [PubMed] [Google Scholar]
- 30.Nicolas JM, Fernandez-Sola J, Fatjo F, Casamitjana R, Bataller R, Sacanella E, et al. Increased circulating leptin levels in chronic alcoholism. Alcohol Clin Exp Res. 2001;25:83–88. [PubMed] [Google Scholar]
- 31.Santolaria F, Perez-Cejas A, Aleman MR, Gonzalez-Reimers E, Milena A, de la Vega MJ, et al. Low serum leptin levels and malnutrition in chronic alcohol misusers hospitalized by somatic complications. Alcohol Alcohol. 2003;38:60–66. doi: 10.1093/alcalc/agg015. [DOI] [PubMed] [Google Scholar]
- 32.Yokoyama H, Hirose H, Ohgo H, Saito I. Associations between serum leptin levels and transaminase activities and the status of lifestyle in Japanese workers. Alcohol Clin Exp Res. 2004;28:159S–163S. doi: 10.1097/01.alc.0000133549.77624.42. [DOI] [PubMed] [Google Scholar]
- 33.Di-Poi N, Tan NS, Michalik L, Wahli W, Desvergne B. Antiapoptotic role of PPARbeta in keratinocytes via transcriptional control of the Akt1 signaling pathway. Mol Cell. 2002;10:721–733. doi: 10.1016/s1097-2765(02)00646-9. [DOI] [PubMed] [Google Scholar]
- 34.Han S, Ritzenthaler JD, Zheng Y, Roman J. PPARbeta/delta agonist stimulates human lung carcinoma cell growth through inhibition of PTEN expression: the involvement of PI3K and NF-kappaB signals. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1238–L1249. doi: 10.1152/ajplung.00017.2008. [DOI] [PubMed] [Google Scholar]
- 35.Chen GJ, Xu J, Lahousse SA, Caggiano NL, de la Monte SM. Transient hypoxia causes Alzheimer-type molecular and biochemical abnormalities in cortical neurons: potential strategies for neuroprotection. J Alzheimers Dis. 2003;5:209–228. doi: 10.3233/jad-2003-5305. [DOI] [PubMed] [Google Scholar]
- 36.Barak Y, Liao D, He W, Ong ES, Nelson MC, Olefsky JM, et al. Effects of peroxisome proliferator-activated receptor delta on placentation, adiposity, and colorectal cancer. Proc Natl Acad Sci U S A. 2002;99:303–308. doi: 10.1073/pnas.012610299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peters JM, Lee SS, Li W, Ward JM, Gavrilova O, Everett C, et al. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta) Mol Cell Biol. 2000;20:5119–5128. doi: 10.1128/mcb.20.14.5119-5128.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shan W, Nicol CJ, Ito S, Bility MT, Kennett MJ, Ward JM, et al. Peroxisome proliferator-activated receptor-beta/delta protects against chemically induced liver toxicity in mice. Hepatology. 2008;47:225–235. doi: 10.1002/hep.21925. [DOI] [PubMed] [Google Scholar]
- 39.Nagasawa T, Inada Y, Nakano S, Tamura T, Takahashi T, Maruyama K, et al. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARdelta agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. Eur J Pharmacol. 2006;536:182–191. doi: 10.1016/j.ejphar.2006.02.028. [DOI] [PubMed] [Google Scholar]
- 40.Cantarini MC, de la Monte SM, Pang M, Tong M, D’Errico A, Trevisani F, et al. Aspartyl-asparagyl beta hydroxylase over-expression in human hepatoma is linked to activation of insulin-like growth factor and notch signaling mechanisms. Hepatology. 2006;44:446–457. doi: 10.1002/hep.21272. [DOI] [PubMed] [Google Scholar]
- 41.Denley A, Carroll JM, Brierley GV, Cosgrove L, Wallace J, Forbes B, et al. Differential activation of insulin receptor substrates 1 and 2 by insulin-like growth factor-activated insulin receptors. Mol Cell Biol. 2007;27:3569–3577. doi: 10.1128/MCB.01447-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huo H, Guo X, Hong S, Jiang M, Liu X, Liao K. Lipid rafts/caveolae are essential for insulin-like growth factor-1 receptor signaling during 3T3-L1 preadipocyte differentiation induction. J Biol Chem. 2003;278:11561–11569. doi: 10.1074/jbc.M211785200. [DOI] [PubMed] [Google Scholar]
- 43.Matthews LC, Taggart MJ, Westwood M. Effect of cholesterol depletion on mitogenesis and survival: the role of caveolar and noncaveolar domains in insulin-like growth factor-mediated cellular function. Endocrinology. 2005;146:5463–5473. doi: 10.1210/en.2005-0236. [DOI] [PubMed] [Google Scholar]
- 44.Maeda T, Sepe P, Lahousse S, Tamaki S, Enjoji M, Wands JR, et al. Antisense oligodeoxynucleotides directed against aspartyl (asparaginyl) beta-hydroxylase suppress migration of cholangiocarcinoma cells. J Hepatol. 2003;38:615–622. doi: 10.1016/s0168-8278(03)00052-7. [DOI] [PubMed] [Google Scholar]
- 45.Dhar-Chowdhury P, Harrell MD, Han SY, Jankowska D, Parachuru L, Morrissey A, et al. The glycolytic enzymes, glyceraldehyde-3-phosphate dehydrogenase, triose-phosphate isomerase, and pyruvate kinase are components of the K(ATP) channel macromolecular complex and regulate its function. J Biol Chem. 2005;280:38464–38470. doi: 10.1074/jbc.M508744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dey A, Cederbaum AI. Alcohol and oxidative liver injury. Hepatology. 2006;43:S63–S74. doi: 10.1002/hep.20957. [DOI] [PubMed] [Google Scholar]
- 47.George J, Liddle C. Nonalcoholic fatty liver disease: pathogenesis and potential for nuclear receptors as therapeutic targets. Mol Pharm. 2008;5:49–59. doi: 10.1021/mp700110z. [DOI] [PubMed] [Google Scholar]