

Abstract
Histone deacetylase 6 (HDAC6), a microtubule-associated tubulin deacetylase, plays a significant role in the formation of protein aggregates in many neurodegenerative disorders. Parkin, a protein-ubiquitin E3 ligase linked to Parkinson's disease, accumulates at the centrosome in a microtubule-dependent manner in response to proteasome inhibition. Here, we show that the centrosome recruitment of parkin was mediated by its direct binding to HDAC6 through multiple interaction domains. The tubulin deacetylase activity of HDAC6 was required for the accumulation of parkin as well as its dispersion upon the reversal of proteasome inhibition. The bidirectional movements of parkin required intact microtubule network and were dependent on dynein and kinesin 1, respectively. Tubulin deacetylation increases microtubule dynamicity and may thus facilitate microtubule-based trafficking of the parkin-HDAC6 complex. The results suggest that HDAC6 acts as a sensor of proteasome inhibition and directs the trafficking of parkin by using different motor proteins.
Keywords: parkin, HDAC6, proteasome, aggresome, microtubule, Parkinson's disease
Introduction
Mutations of parkin, a protein-ubiquitin E3 ligase, are linked to Parkinson's disease (PD). While the intracellular inclusion termed Lewy body is generally observed in Parkinson's disease, parkin-mutated PD patients do not have them, suggesting a critical role of parkin in Lewy body formation (Savitt et al., 2006). Previous studies have shown that parkin accumulates at the centrosome when the 26S proteasome is inhibited (Junn et al., 2002; Ardley et al., 2003; Zhao et al., 2003; Muqit et al., 2004). It is unclear how parkin, which binds to tubulin and microtubules with very high affinity (Ren et al., 2003; Yang et al., 2005), moves to the centrosome. It also remains elusive whether the centrosomal recruitment of parkin represents an active cellular response for parkin to ubiquitinate misfolded proteins more efficiently, or whether it is a passive process in which autoubiquitinated parkin is accumulated along with other ubiquitinated proteins. Thus, it is important to understand whether the centrosomal recruitment of parkin is reversible and if so, the underlying mechanisms for both processes.
Ubiquitin-dependent proteolysis is a fundamental cellular response in which misfolded proteins are ubiquitinated and degradation by the 26S proteasome. When the proteasome is overwhelmed by misfolded proteins, a large intracellular inclusion called the aggresome is formed near the centrosome (Kopito, 2000). Previous studies have shown that histone deacetylase 6 (HDAC6) is associated with microtubules and accumulates in the aggresome through its interaction with p150Glued (Kawaguchi et al., 2003), a component of the dynein motor complex (Schroer, 2004). Through a C-terminal ubiquitin-binding zinc finger domain (ZnF-UBP), HDAC6 binds to ubiquitinated proteins and plays a critical role in their accumulation at the aggresome (Kawaguchi et al., 2003).
HDAC6 is a unique member of the histone deacetylase family; it deacetylates not only histones (Grozinger et al., 1999), but also α-tubulin (Hubbert et al., 2002), Hsp90 (Kovacs et al., 2005) and cortactin (Zhang et al., 2007). In contrast to other HDACs, HDAC6 has two catalytically active deacetylase domains (DD1 and DD2). Both deacetylase domains seem to be required for its histone deacetylase activity, which is inhibited by broad spectrum HDAC inhibitors such as trichostatin A (TSA) (Haggarty et al., 2003). The second deacetylase domain (DD2) is responsible for the tubulin deacetylase activity, which is selectively inhibited by tubacin (TBC) (Haggarty et al., 2003). In contrast, tubacin does not inhibit the histone deacetylase activity of HDAC6 (Haggarty et al., 2003). Deacetylation of the lysine 40 residue on α-tubulin has been shown to increase the dynamicity of microtubules (Matsuyama et al., 2002; Tran et al., 2007) and may thus facilitate cellular processes that require microtubule-based transport. Here we provide evidence that HDAC6 mediated the reversible recruitment of parkin to the centrosome in response to proteasome inhibition or disinhibition. HDAC6 formed a tight complex with parkin and used dynein or kinesin motor proteins in the accumulation or dispersion of parkin. Both processes required the tubulin deacetylase activity of HDAC6, suggesting a key role of this activity for HDAC6 to act as a sensor of proteasome inhibition.
Materials and Methods
Antibodies, siRNA and cDNA constructs.
The polyclonal antibody against parkin was generated previously (Ren et al., 2003). Polyclonal antibodies against HDAC6 and Myc were from Upstate Biotechnology. Monoclonal antibodies against dynein heavy chain (DHC) (clone 440.4), γ-tubulin, α-tubulin, FLAG (M2), and anti-FLAG-conjugated (M2) agarose were from Sigma. Monoclonal antibody against GST was from Santa Cruz Biotechnology. Polyclonal antibody against kinesin 1 (KIF5B) was from Abcam. Alexa Fluor 488-conjugated anti-rabbit, Alexa Fluor 594-conjugated anti-mouse, Alexa Fluor 594-conjugated anti-rabbit secondary antibodies were from Invitrogen. Small interfering RNAs (siRNAs) against human HDAC6 (CUGCAAGGGAUGGAUCUGAtt), DHC (GAACGUGCGUUAUACCGCAtt) or KIF5B (GCACAUCUCAAGAGCAAGUtt) were purchased from Ambion. In a previous study, we cloned cDNAs of human parkin and its domains Ubl (U, 1–76 aa), Linker (L, 77–237 aa), RING1 (R1, 217–310 aa), IBR (I, 307–405 aa), RING2 (R2, 395–465), RING1-IBR (RI, 217–405 aa); IBR-RING2 (IR, 307–465 aa) (Yang et al., 2005). These cDNAs were subcloned into pCMV-Tag3A or 3B (Stratagene) to add Myc tag at the N terminus and subcloned again with the Myc tag into pcDNA3.1 (Invitrogen). Glutathione S-transferase (GST) fusions of parkin and its domains were generated by subcloning these cDNAs into pGEX-4T-1 (GE Healthcare). C-terminal FLAG-tagged human HDAC6 in pcDNA3.1 (from Eric Verdin at University of California, San Francisco, San Francisco, CA) (North et al., 2003) was used to generate the following HDAC6 constructs. The catalytically inactive double mutant (H216A/H611A) was generated with QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene). C-terminal FLAG-tagged HDAC6 domains at amino acids 1–105 (N-terminal, N), 106–439 (DD1), 440–840 (deacetylase domain 2, DD2) and 841–1215 C-terminal (C) were generated by PCR, subcloning into pcMV-tag4A (Stratagene) and subcloning the FLAG-tagged domains into pcDNA3.1. N-terminal His×6 and C-terminal FLAG-tagged HDAC6 (His×6-HDAC6-FLAG) or DD1 (His×6-DD1-FLAG) or DD2 (His×6-DD2-FLAG) were generated by subcloning the C-terminal FLAG-tagged cDNA into pQE-80L (Qiagen). All constructs were verified by sequencing. GFP plasmid (pEGFP-C1) was purchased from BD Biosciences. Dominant-negative kinesin 1 construct was generated by PCR amplification of the tail domain of mouse KIF5B (last 151 aa), which binds to cargos and compete with endogenous KIF5B (Setou et al., 2002). The construct was subcloned into pEGFP-C1.
Cell lines, transfection, drug treatment and immunostaining.
HEK293T and SH-SY5Y cells (from ATCC) were maintained in DMEM with 10% fetal bovine serum (Invitrogen). For immunocytochemistry, SH-SY5Y cells were transfected for 48 h with siRNAs (0.025 μg/cm2 culture) or plasmids (0.4 μg/cm2 culture). GFP plasmid (0.025 μg/cm2 culture) was cotransfected to mark transfected cells. For Western blotting, SH-SY5Y cells were transfected with siRNAs (0.25 μg/cm2 culture) for three times using Superfect (Qiagen). HEK293T cells were transfected for 48 h with various DNA constructs using the calcium phosphate method. Cells were treated with MG132 (carbobenzoxy-l-leucyl-l-leucyl-l-leucinal; EMD Chemicals) for 12 h at 5 μm, or treated with MG132 for 24 h at 5 μm and then incubated in media without MG132 for 12 or 24 h. In some experiments, 1 μm TSA (Sigma), 10 μm TBC or 10 μm niltubacin (nTBC) (from Ralph Mazitschek and Stuart Schreiber at Harvard University, Cambridge, MA) was added 3 h before MG132 and lasted till the end of MG132 treatment. In other experiments, 200 μm adenylyl imidodiphosphate (AMP-PNP, Sigma), 10 μm aurintricarboxylic acid (AA, Sigma), 0.1 μm taxol (Tax, Sigma) or 0.1 μm nocodazole (Noc, Sigma) was added during MG132 withdrawal. After drug treatment, cells were fixed with cold methanol at 4°C for 20 min and blocked with 3% of BSA for 1 h. This was followed by incubation in primary antibodies overnight at 4°C and secondary antibodies at room temperature for 2 h. Fluorescence images were acquired on a Zeiss LSM 510 META confocal microscope, using 40× N.A. 1.3 EC-Plan NeoFluar objective lenses. Monochrome images (512 × 512 pixels) were pseudocolored and merged with the software Zeiss LSM Image Examiner. Parkin accumulation was quantified by measuring the background-subtracted sum of pixel intensity within a constantly defined circle around the aggregate using the software NIH ImageJ. The results were corroborated by another quantification method that measures the size of the parkin aggregate using a manually drawn circle around the aggregate (data not shown). Statistical analysis was performed using the software Origin (Origin Lab). Data are expressed as mean ± SEM.
Immunoprecipitation and Western blotting.
Cells in 10 cm dishes were lysed on ice in 1 ml cold lysis buffer A containing 1% Triton X-100, 10 mm Tris, pH 7.6, 300 mm NaCl, 30 mm sodium pyrophosphate, 50 mm NaF, 5 mm EDTA, 0.1 mm Na3VO4, 0.5 mm PMSF and protease inhibitor mixture (Roche). After centrifugation at 16,000 × g, supernatants were incubated for 2 h at 4°C with anti-FLAG conjugated agarose or various antibodies. The immunoprecipitates were analyzed by Western blotting using the ECL method (Amersham).
Protein purification and pull-down assays.
E. coli (BL21 strain) transformed with various GST- or His×6-tagged constructs were induced for 2 h at 37°C with 0.5 mm IPTG to express GST fusions of U, I or R2, or for 4 h at 37°C with 0.05 mm IPTG to express GST fusions of P, L or R1, or for 4 h at 28°C with 0.5 mm IPTG to express His×6-HDAC6-FLAG, or for 4 h at 37°C with 0.5 mm IPTG to express His×6-DD1-FLAG or His×6-DD2-FLAG. After induction, cells were lysed in 20% culture volume of buffer B, which contains BugBuster protein extraction reagent with Benzonase Nuclease (1 μl/ml), lysozyme (1 kU/ml) (EMD Chemicals) and 0.5 mm PMSF. GST fusions were affinity purified using GST-Bind resin (EMD Chemicals) and His×6-tagged proteins were affinity purified using Ni-NTA agarose (Qiagene). Equimolar amounts of purified GST fusion proteins were immobilized on GST-Bind resin, which were then incubated with 4 nm purified His×6-tagged proteins. Alternatively, equal amounts of purified His×6-tagged proteins were immobilized on Ni-NTA agarose, which were then incubated with 4 nm of GST fusion proteins. All incubations were performed in buffer C, which contained 1% Triton X-100, 10 mm Tris, pH 7.6, 500 mm NaCl and 0.5 mm PMSF. The beads were washed three times in buffer C and analyzed by Western blotting with anti-FLAG or anti-GST.
Results
HDAC6 inhibitors block the centrosomal recruitment of parkin induced by proteasome inhibition
A number of studies have consistently shown that parkin accumulates at the centrosome in response to proteasomal inhibition (Junn et al., 2002; Ardley et al., 2003; Zhao et al., 2003; Muqit et al., 2004). Since HDAC6 plays a critical role in the formation of the aggresome (Kawaguchi et al., 2003), we investigated the role of HDAC6 in the centrosomal recruitment of parkin. The human dopaminergic neuroblastoma cell line SH-SY5Y was treated with the proteasome inhibitor MG132 (5 μm for 12 h) in the presence or absence of HDAC6 inhibitors, which were added from 3 h before till the end of MG132 treatment. We costained the cells for parkin (green) and the centrosome marker γ-tubulin (red) to examine the subcellular distribution of parkin in relation to the centrosome. The corresponding single color images are shown in supplemental Fig. S1, available at www.jneurosci.org as supplemental material. Without any treatment, only a small amount of parkin was localized at the centrosome (Fig. 1A), while most of parkin is distributed along microtubules (Ren et al., 2003). MG132-induced parkin accumulation at the centrosome (Fig. 1B) was markedly attenuated by the broad-spectrum HDAC inhibitor TSA (1 μm) (Fig. 1C), or the tubulin-specific HDAC6 inhibitor tubacin (10 μm) (Fig. 1D), but not its inactive analog niltubacin (10 μm) (Fig. 1E). None of these inhibitors per se had any significant effect on the subcellular distribution of parkin without MG132 treatment (Fig. 1F–H). We quantified centrosomal recruitment of parkin by measuring background-subtracted fluorescence intensity within a constantly defined circle around the aggregate using the software NIH ImageJ. As shown in Figure 1H, MG132 induced 11.68 ± 1.15-folds increase in the intensity of parkin accumulation, compared with the vehicle treatment, which was normalized at 1 (p < 0.001, n = 30). The effect was significantly reduced by TSA (4.48 ± 0.51, p < 0.001, n = 30) or tubacin (4.96 ± 0.49, p < 0.001, n = 30), but not by niltubacin (11.83 ± 1.09, p > 0.92, n = 30). Since tubacin selectively inhibits the deacetylase activity of HDAC6 toward tubulin, but not histones (Haggarty et al., 2003), these results suggest that the tubulin deacetylase activity of HDAC6 is required for the centrosomal accumulation of parkin in response to proteasome inhibition.
Figure 1.

MG132-induced centrosomal accumulation of parkin is blocked by HDAC6 inhibitors. A–G, SH-SY5Y cells were treated as indicated. Veh, vehicle; MG, the proteasome inhibitor MG132 (5 μm); TSA, the broad-spectrum HDAC inhibitor (1 μm); TBC, the tubulin-specific HDAC6 inhibitor (10 μm); nTBC, the inactive analog of tubacin (10 μm). MG132 was applied for 12 h, while inhibitors were applied from 3 h before and lasted for 15 h. Cells were costained for parkin (PKN, green) and the centrosome marker γ-tubulin (γ-Tub, red). Scale bar, 10 μm. MG132 induced marked accumulation of parkin at the centrosome (B). The effect was inhibited by TSA (C) or TBC (D), but not nTBC (E). TSA (F), TBC (G), or nTBC (H) alone had no significant effect on parkin accumulation. H, Statistical summary of the intensity of parkin accumulation at the centrosome. *p < 0.001 versus Veh; #p < 0.001 versus MG132; n = 30 cells randomly selected from three independent experiments for each condition.
HDAC6 is required for parkin to accumulate in the centrosome in response to proteasome inhibition
To substantiate the involvement of HDAC6 in the centrosomal recruitment of parkin, we examined whether parkin accumulation was affected by suppressing the expression of endogenous HDAC6. SH-SY5Y cells were cotransfected with GFP and siRNA against HDAC6 or scrambled sequence. After the cells were treated without or with MG132 (5 μm for 12 h), they were stained for parkin (green) and GFP (red). The corresponding single color images are shown in supplemental Fig. S2, available at www.jneurosci.org as supplemental material. HDAC6 siRNA did not change the subcellular distribution of parkin in the absence of MG132 (Fig. 2C), but markedly attenuated the centrosomal accumulation of parkin in response to MG132 (Fig. 2B,C). In contrast, the control siRNA with scrambled sequence had no significant effect on MG132-induced parkin accumulation (Fig. 2A,C). Transfection of HDAC6 siRNA greatly diminished the level of endogenous HDAC6, as evidenced by immunostaining and Western blotting (Fig. 2D). To provide more evidence, we transfected SH-SY5Y cells with GFP and wild-type HDAC6 (wt-H6) or the catalytically inactive HDAC6 double mutant (mut-H6, H216A/H611A), which loses the ability to deacetylate tubulin (Zhang et al., 2003). Ectopic expression of HDAC6 did not appreciably change the subcellular distribution of parkin in the absence of MG132 (Fig. 2H), but significantly increased parkin accumulation (17.55 ± 1.61) compared with untransfected cells in the same field (Fig. 2E) or cells transfected with the empty vector (11.86 ± 1.08, p < 0.001, n = 30) (Fig. 2H). The effect of HDAC6 was significantly blocked by tubacin (7.27 ± 0.74, p < 0.002, n = 30) (Fig. 2G), but not its inactive analog niltubacin (17.37 ± 1.54, p > 0.93, n = 30) (Fig. 2H). Expression of the mutant HDAC6 did not appreciably change the subcellular distribution of parkin in the absence of MG132 (Fig. 2H), nor did it significantly increase parkin accumulation in response to MG132 (11.78 ± 1.14-fold, p > 0.96 vs vector transfection) (Fig. 2F,H). Thus, the centrosomal recruitment of parkin is enhanced by HDAC6 overexpression and suppressed by HDAC6 knockdown.
Figure 2.
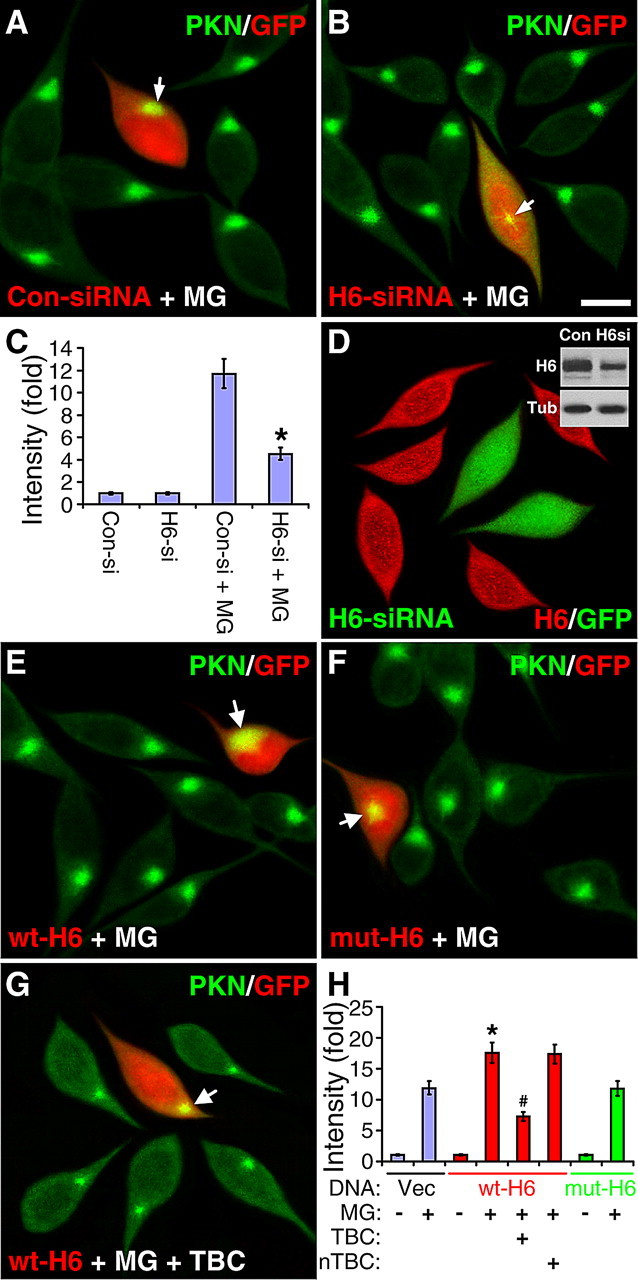
MG132-induced centrosome accumulation of parkin is decreased by HDAC6 siRNA and increased by HDAC6 overexpression. A–C, SH-SY5Y cells cotransfected with GFP and siRNA for HDAC6 or control siRNA with scrambled sequence were treated without or with MG132 (5 μm for 12 h). Cells were stained for parkin (PKN, green). GFP as a transfection marker was pseudocolored red. HDAC6 siRNA (H6-siRNA) markedly reduced centrosomal accumulation of parkin in response to MG132 (B), while the control siRNA (Con-siRNA) had no significant effect (A). Scale bar, 10 μm. Arrow, Parkin accumulation in transfected cells. C, Statistical summary of the intensity of parkin accumulation under the above conditions and in the absence of MG132. *p < 0.001 versus Con-siRNA + MG132, n = 30 cells randomly selected from three independent experiments for each condition. D, Transfection of HDAC6 siRNA (marked by GFP, green) greatly diminished HDAC6 expression as indicated in immunofluorescence (red) and immunoblot (inset). E–G, SH-SY5Y cells cotransfected with GFP and wild-type HDAC6 (wt-H6, E and G) or its catalytically inactive double mutant (mut-H6, F) were treated with MG132 (MG, 5 μm for 12 h) in the absence or presence of TBC (10 μm, G). Cells were stained for parkin (PKN, green) and GFP fluorescence (marking transfected cells) was pseudocolored red. As highlighted by arrows, MG132-induced parkin accumulation was markedly increased in cells transfected with wt-H6 (E), but not mut-H6 (F). The effect of wt-H6 was reversed by tubacin (G), but not niltubacin (H). H, Statistical summary of the intensity of parkin accumulation at the centrosome under the conditions in (E–G). *p < 0.002 versus empty vector (Vec) transfection plus MG132 treatment; #p < 0.001 versus wt-H6 plus MG132; n = 30 cells randomly selected from three independent experiments for each condition.
HDAC6 and parkin directly binds to each other through multiple interaction domains
Both parkin and HDAC6 normally associate with microtubules (Hubbert et al., 2002; Ren et al., 2003) and become accumulated in the centrosome in response to proteasomal inhibition (Kawaguchi et al., 2003; Zhao et al., 2003). It suggests that parkin and HDAC6 may exist in a complex, which is transported along microtubules to the centrosome when the proteasome is inhibited. To test whether parkin physically interacts with HDAC6, we performed coimmunoprecipitation assays in SH-SY5Y cells. As shown in Figure 3A, the parkin antibody coimmunoprecipitated with endogenous HDAC6. The effect was abolished when the parkin antibody was preincubated with its antigenic peptide. To identify the domains responsible for the binding between HDAC6 and parkin, we cotransfected HEK293T cells with HDAC6-FLAG and Myc-tagged parkin (P) or its U, L, R1, I, R2 domain (Yang et al., 2005). As shown in Figure 3B, HDAC6-FLAG coimmunoprecipitated with the Linker, RING1 or RING2 domain as well as the full-length parkin. In contrast, the Ubl or IBR domain of parkin did not significantly bind to HDAC6. The combined RING1-IBR (RI) or IBR-RING2 (IR) construct was also coimmunoprecipitated by HDAC6, because each of them contains a domain that interacted with HDAC6.
Figure 3.

Multiple interaction domains mediate direction binding between HDAC6 and parkin. A, Coimmunoprecipitation of endogenous parkin and HDAC6 in SH-SY5Y cells. Cleared lysates were immunoprecipitated with an irrelevant antibody (Con), parkin antibody (PKN) or the parkin antibody with its antigenic peptide (PKN+pep). Immunoprecipitates and 1% of input were Western blotted with anti-HDAC6. B, Domains of parkin responsible for binding HDAC6. Myc-tagged parkin constructs as shown in the diagram were transfected with HDAC6-FLAG in HEK293T cells. FLAG immunoprecipitates and 1% input from cleared cell lysates were blotted with anti-Myc. HDAC6-FLAG coimmunoprecipitated with full-length parkin (P) or its L, R1 or R2 domains, but not with U or IRB (I) domain. The RI (R1+IBR) or IR (IBR+R2) construct was also coimmunoprecipitated with HDAC6. C, Domains of HDAC6 responsible for binding parkin. FLAG-tagged HDAC6 constructs as shown in the diagram were transfected with Myc-parkin in HEK293T cells. FLAG immunoprecipitates were blotted with anti-Myc. Parkin coimmunoprecipitated with either DD1 or DD2 of HDAC6, but not the N-terminal or C-terminal domain of HDAC6. Total cell lysates were also blotted with anti-FLAG or anti-Myc to show expression levels of HDAC6 or parkin constructs, respectively. D, Coimmunoprecipitation between interaction domains of HDAC6 and parkin as indicated in the diagram. FLAG-tagged DD1 or DD2 domain of HDAC6 was transfected without (−) or with Myc-tagged HDAC6-binding domains of parkin (L, R1 or R2) or full-length parkin (P). FLAG immunoprecipitates were blotted with anti-Myc. DD1 domain of HDAC6 coimmunoprecipitated with L or R1 domain of parkin, while DD2 domain of HDAC6 coimmunoprecipitated with the R2 domain of parkin. E, Direct binding of purified parkin domains with purified HDAC6 in vitro. GST-tagged parkin domains were purified from E. coli and immobilized on glutathione beads, which was incubated with bacterially purified Hisx6-HDAC6-FLAG (His-H6-FLAG) protein. After extensive washing in a buffer containing 1% Triton X-100 and 500 mm NaCl, the beads were analyzed by Western blotting with anti-FLAG (top). The L, R1 or R2 domain or the full-length parkin (P), but not the U, I or GST alone (−), pulled down HDAC6. Expression levels of GST or GST-parkin constructs were shown in a separate GST blot in the bottom panel. F, Direct binding between interaction domains of HDAC6 and parkin in vitro. Purified GST (−) or GST-parkin constructs (L, R1, R2 or P) were immobilized on glutathione beads, which were incubated with purified DD1 or DD2 domain of HDAC6 (His-DD1-FLAG or His-DD2-FLAG). The beads were analyzed by Western blotting with anti-FLAG. The L or R1 domain of parkin pulled down DD1 domain of HDAC6 (top panel), while the R2 domain of parkin pulled down DD2 domain of HDAC6 (middle). GST blot of the inputs showed expression level of the GST constructs (bottom). All experiments were repeated at least three times.
To map out the domains of HDAC6 responsible for binding parkin, we generated FLAG-tagged constructs covering the N region, DD1, DD2 or the C region of HDAC6 and cotransfected these constructs with Myc-parkin in HEK293T cells. As shown in Figure 3C, only DD1 or DD2, but not the N-terminal or C-terminal domain, of HDAC6 was coimmunoprecipitated with parkin. Since the ubiquitin-binding motif ZnF-UBP is in the C-terminal domain of HDAC6, the results suggest that HDAC6 binds to parkin directly, not through the ubiquitin chain on potentially autoubiquitinated parkin.
To examine how these domains on parkin and HDAC6 interact with each other, we cotransfected FLAG-tagged DD1 or DD2 domain of HDAC6 (which could bind to parkin) with Myc-tagged Linker, RING1 or RING2 domain of parkin (which could bind to HDAC6). As shown in Figure 3D, the DD1 domain of HDAC6 coimmunoprecipitated with the Linker or RING1 domain of parkin, but not its RING2 domain. The DD2 domain coimmunoprecipitated with RING2, but not the Linker or RING1 domain. Thus, the strong binding between parkin and HDAC6 was mediated by multiple interaction domains: the DD1 domain of HDAC6 interacted with Linker or RING1 domain of parkin, while the DD2 domain of HDAC6 bound to RING2 domain of parkin (Fig. 3D, diagram).
Since both parkin (Ren et al., 2003) and HDAC6 (Hubbert et al., 2002) binds to tubulin α/β heterodimers and microtubules, it is important to know whether the interaction between parkin and HDAC6 is direct or whether it is mediated through their interactions with tubulin or microtubules. We expressed and purified full-length and individual domains of HDAC6 and parkin in E. coli, which does not have tubulin. As shown in Figure 3E, GST-tagged parkin or its domains were immobilized on glutathione beads to pull down His×6-HDAC6-FLAG. Like full-length parkin (P), L, R1 and R2 domains pulled down similar amounts of HDAC6, while U and I domains did not retain any HDAC6. To test the direct binding between interaction domains of parkin and HDAC6, we immobilized purified GST-tagged parkin (P) or its L, R1 or R2 domain on glutathione beads, which were incubated with purified His×6-tagged DD1 or DD2 domain of HDAC6. As shown in Figure 3F, the DD1 domain of HDAC6 directly bound to the Linker or RING1 domain of parkin in vitro, while the DD2 domain of HDAC6 directly bound to the RING2 domain of parkin in vitro, confirming the coimmunoprecipitation data and the diagram in Figure 3D.
Direct binding to HDAC6 mediates the centrosome recruitment of parkin in response to proteasome inhibition
Since HDAC6 accumulates in the centrosome when the proteasome is inhibited (Kawaguchi et al., 2003), the centrosomal recruitment of parkin might be mediated by its direct binding to HDAC6. To test this, we expressed parkin or its domains without or with HDAC6 in SH-SY5Y cells. MG132 (5 μm for 12 h) did not induce significant accumulation of transfected parkin or its domains if HDAC6 was not cotransfected (Fig. 4A–F,S). In the presence of HDAC6, parkin or its linker, RING1 or RING2 domain significantly accumulated at the centrosome following MG132 treatment (p < 0.001, n = 20). In contrast, neither Ubl nor IBR domain exhibited any significantly accumulation at the centrosome (Fig. 4G–L,S). HDAC6-mediated accumulation of parkin or its linker, RING1 or RING2 domain was significantly blocked by tubacin (Fig. 4M–S) (p < 0.001, n = 20). MG132-induced centrosomal accumulation of HDAC6 was not affected by parkin or its domains (supplemental Fig. S3A′–F′, available at www.jneurosci.org as supplemental material), but was totally blocked by tubacin (supplemental Fig. S3M″–R″, available at www.jneurosci.org as supplemental material).
Figure 4.
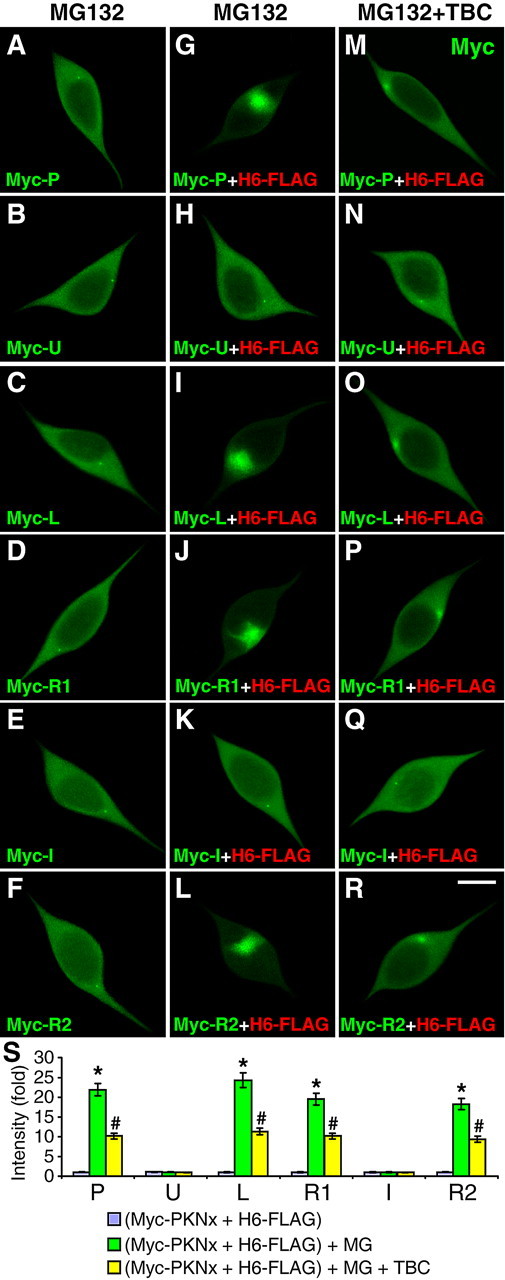
Direct binding with HDAC6 mediates the centrosome recruitment of parkin in response to proteasome inhibition. A–R, SH-SY5Y cells were transfected with Myc-tagged parkin or its domains as indicated [P, full-length parkin (A, G, M); U (B, H, N); L (C, I, O); R1 (D, J, P); I (E, K, Q); or R2 (F, L, R)] without (A–F) or with FLAG-tagged HDAC6 (H6-FLAG) (G–R). Cells were treated with MG132 (MG, 5 μm for 12 h), without (G–L) or with TBC (10 μm for 15 h) (M–R). Cells were costained for Myc (green) and FLAG (red, shown in supplemental Fig. S3, available at www.jneurosci.org as supplemental material) Overexpressed parkin or its domains did not show significant centrosome accumulation in response to MG132 (A–F). Coexpression with HDAC6 resulted in marked centrosome recruitment of P, L, R1 or R2, but not U or I, in response to MG132 (G-L). The recruitments of parkin or its HDAC6-binding domains were blocked by TBC (M, O, P and R), while the subcellular distribution of U or I was not significantly affected. MG132-induced centrosome accumulation of H6-FLAG and the blockade of the effect by tubacin were not affected by transfected parkin constructs (supplemental Fig. S3, available at www.jneurosci.org as supplemental material) S, Statistical summary of the intensity of parkin accumulation under the above conditions. *p < 0.001 versus the preceding bar; #p < 0.001 versus the preceding bar; n = 20 cells randomly selected from three independent experiments for each condition.
Reversal of proteasome inhibition induces HDAC6-dependent dispersion of parkin from the centrosome
To examine whether the centrosomal accumulation of parkin is reversible, we pretreated SH-SY5Y cells with MG132 (5 μm) for 24 h and then incubated the cells in media without the drug for various durations. Parkin dispersed from the centrosome to a diffused pattern in the original condition within 24 h (Fig. 5A–C,G). This effect was significantly blocked by tubacin (p < 0.001, n = 30) (Fig. 5D), but not niltubacin (Fig. 5G), suggesting that the tubulin deacetylase activity of HDAC6 is required for the dispersion of parkin as well. To confirm the dependence on HDAC6, we transfected siRNA against HDAC6 or scrambled sequence and found that knocking down HDAC6 expression significantly attenuated the dispersion of parkin upon MG132 withdrawal (p < 0.001, n = 30) (Fig. 5F,G). In contrast, the control siRNA had no significant effect (Fig. 5E,G). The corresponding single color images are shown in supplemental Fig. S4, available at www.jneurosci.org as supplemental material.
Figure 5.
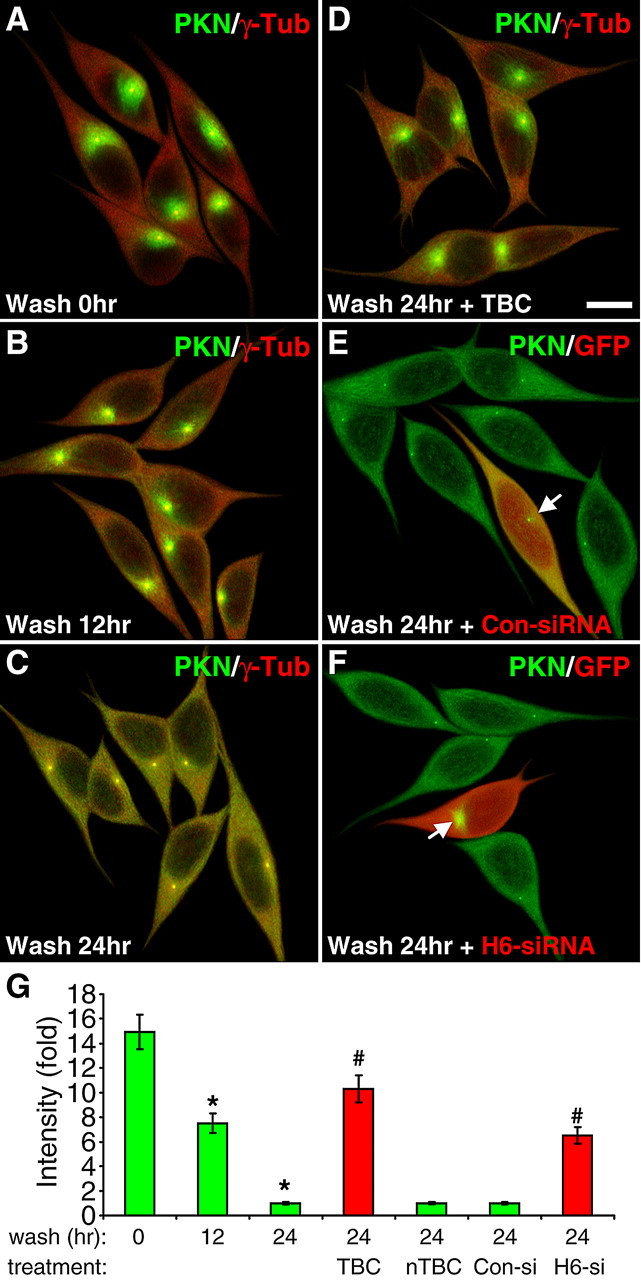
The dispersion of parkin upon MG132 withdrawal is also dependent on HDAC6. A–D, SH-SY5Y cells were treated with MG132 for 24 h, after which the cells were incubated without MG132 for the indicated durations without (B and C) or with TBC (10 μm, D) or nTBC (10 μm, G). Cells were costained for parkin (PKN, green) and γ-tubulin (γ-Tub, red). MG132-induced centrosome accumulation of parkin was completely reversed in a time-dependent manner by 24 h of MG132 withdrawal. The dispersion of parkin was greatly inhibited by tubacin (D), but not significantly affected by niltubacin (G). Scale bar, 10 μm. E, F, The dispersion of parkin upon MG132 withdrawal was greatly attenuated by HDAC6 siRNA (F), but not by control siRNA (E). Arrow, Parkin accumulation in transfected cells (marked by GFP in red pseudocolor). G, Statistical summary of the intensity of parkin accumulation under the above conditions. *p < 0.001 versus wash for 0 h; #p < 0.001 versus the preceding bar; n = 30 cells randomly selected from three independent experiments for each condition.
The centrosomal recruitment of parkin, but not its dispersion, is dependent on the dynein motor protein
Since the accumulation of HDAC6 at the centrosome is mediated through its interaction with the dynein motor protein (Kawaguchi et al., 2003), we tested whether the centrosomal recruitment of parkin was also dependent on dynein. We transfected SH-SY5Y cells with siRNA against dynein heavy chain, the central component of the dynein motor protein (Asai and Koonce, 2001). MG132-induced centrosomal accumulation of parkin was significantly reduced by siRNA against dynein heavy chain (Fig. 6B,G), but not the control with scrambled sequence (Fig. 6A). The expression level of dynein heavy chain was markedly reduced by its siRNA, but not the scrambled siRNA, as evidenced by immunostaining (Fig. 6E) and Western blotting (Fig. 6F). We also examined whether the dispersion of parkin after MG132 withdrawal was mediated by dynein or not. There was no significant difference in SH-SY5Y cells transfected with siRNA against DHC (Fig. 6D) or the scrambled control (Fig. 6C), or untransfected cells in the same field, in term of parkin dispersion after MG132 removal (Fig. 6G). By itself, neither siRNA had any significant effect on the subcellular distribution of parkin (Fig. 6G). The corresponding single color images are shown in supplemental Fig. S5, available at www.jneurosci.org as supplemental material.
Figure 6.
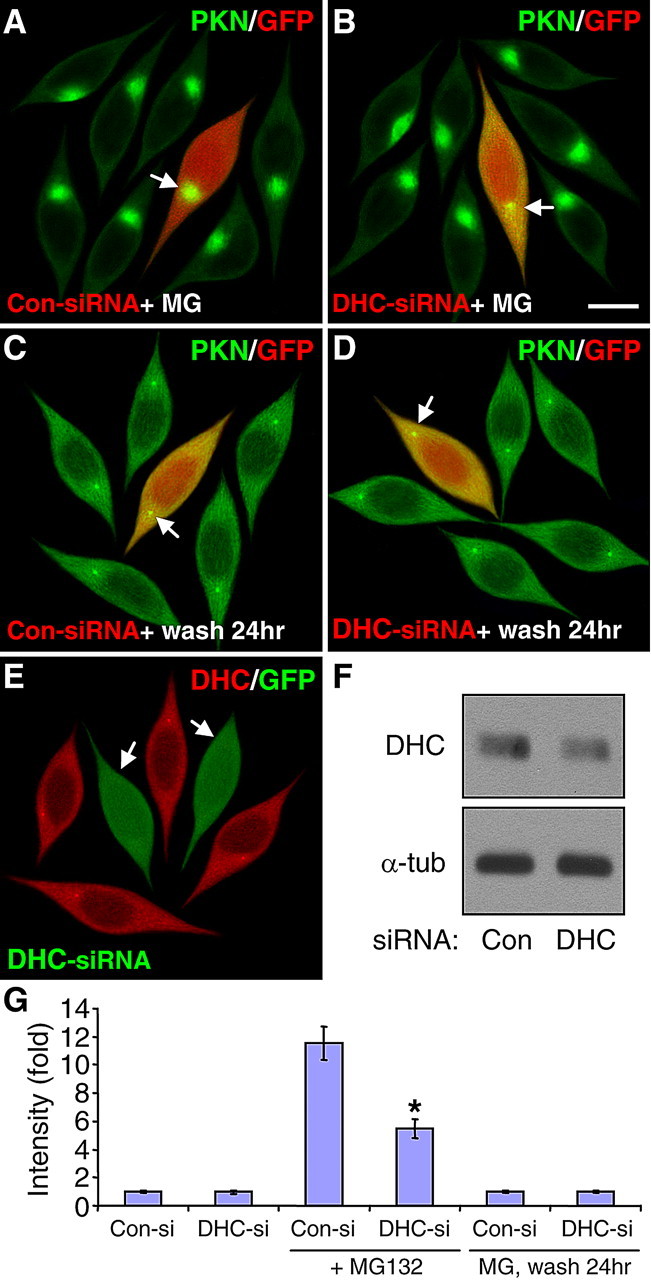
The centrosome recruitment of parkin, but not its dispersion, is dependent on the dynein motor protein. A, B, SH-SY5Y cells cotransfected with GFP (pseudocolored red) and siRNA for dynein heavy chain (DHC-siRNA) or control siRNA with scrambled sequence (Con-siRNA) were treated without (G) or with 5 μm MG132 for 12 h. Transfection of DHC-siRNA (B), but not Con-siRNA (A), markedly reduced MG132-induced centrosome accumulation of parkin (PKN, green). By itself, neither DHC-siRNA nor Con-siRNA had any significant effect on the subcellular distribution of parkin (G). Scale bar, 10 μm. Arrow, Parkin accumulation in transfected cells. C, D, After SH-SY5Y cells were treated with 5 μm MG132 for 24 h, they were incubated without MG132 (wash) for 24 h. At 12 h before the wash, cells were cotransfected with GFP and DHC-siRNA or Con-siRNA. Neither DHC-siRNA (D) nor Con-siRNA (C) had any significant effect on parkin dispersion at 24 h of wash. E, F, Transfection of DHC-siRNA (marked by GFP, arrow) greatly reduced the endogenous level of DHC as indicated by immunostaining (E) or Western blotting (F). The experiment was repeated three times with similar results. G, Statistical summary of the intensity of parkin accumulation under the conditions in (A–D). *p < 0.001 versus the preceding bar; n = 30 cells randomly selected from three independent experiments for each condition.
The dispersion of parkin from centrosome is dependent on kinesin 1 and intact microtubules
Microtubule-based transport from the centrosome to the cell periphery is largely mediated by kinesin motor proteins (Hirokawa and Takemura, 2005). To identify the mechanism responsible for the dispersion of parkin from the centrosome, we treated SH-SY5Y cells first with MG132 (5 μm) for 24 h, then with the broad spectrum ATPase inhibitors AMP-PNP (200 μm) or AA (10 μm) for 24 h during MG132 withdrawal. Parkin dispersion (1.0 ± 0.08, n = 30) (Fig. 7A) was significantly blocked by AMP-PNP (8.85 ± 0.82, n = 30, p < 0.001) (Fig. 7B) or AA (7.26 ± 0.70, n = 30, p < 0.001) (Fig. 7I). Based on the high sensitivity of kinesins to APTase inhibitors and the lack of involvement of dynein in the dispersion of parkin (Fig. 6), we examined the role of the major kinesin motor protein kinesin 1 (also known as kinesin heavy chain or KIF5). Parkin dispersion was significantly attenuated by siRNA against kinesin 1 (7.46 ± 0.63, n = 30, p < 0.001) (Fig. 7C), but not by the control siRNA with scrambled sequence (0.96 ± 0.08, n = 30) (Fig. 7I). The kinesin 1 siRNA markedly reduced the expression of endogenous kinesin 1, as evidenced by immunostaining and Western blotting (supplemental Fig. S7I–J, available at www.jneurosci.org as supplemental material). To substantiate the involvement of kinesin 1, we used a GFP-tagged dominant negative (dn) kinesin 1 tail construct, which disrupts kinesin 1-mediated transport by competing with endogenous kinesin 1 for cargos (Setou et al., 2002). Expression of the dn-kinesin 1 construct significantly reduced parkin dispersion (6.68 ± 0.66, n = 30, p < 0.001) (Fig. 7D), compared with transfection of the empty GFP vector (0.99 ± 0.08, n = 30) (Fig. 7I). Neither kinesin 1 siRNA (Fig. 7E) or dn-kinesin 1 (Fig. 7F) significantly affected the initial centrosomal recruitment of parkin in response to MG132 treatment (Fig. 7I, wash 0 h). Since our previous study has shown that the centrosomal recruitment of parkin is dependent on intact and dynamic microtubules (Zhao et al., 2003), we tested whether the dispersion of parkin following MG132 withdrawal was also dependent on microtubules. The microtubule-depolymerizing agent nocodazole significantly blocked parkin dispersion (11.60 ± 1.30, n = 30, p < 0.001) (Fig. 7G,I), so did the microtubule-stabilizing agent taxol (10.16 ± 0.99, n = 30, p < 0.001) (Fig. 7H,I). The requirement for intact, dynamic microtubule network is consistent with the involvement of kinesin 1 in HDAC6-mediated dispersion of parkin. The corresponding single color images are shown in supplemental Fig. S6, available at www.jneurosci.org as supplemental material. None of these drugs (AMP-PNP, AA, nocodazole, taxol) or kinesin 1 siRNA and dominant negative construct appreciably affected the subcellular distribution of parkin at the basal condition without MG132 (supplemental Fig. S7A–H, available at www.jneurosci.org as supplemental material).
Figure 7.
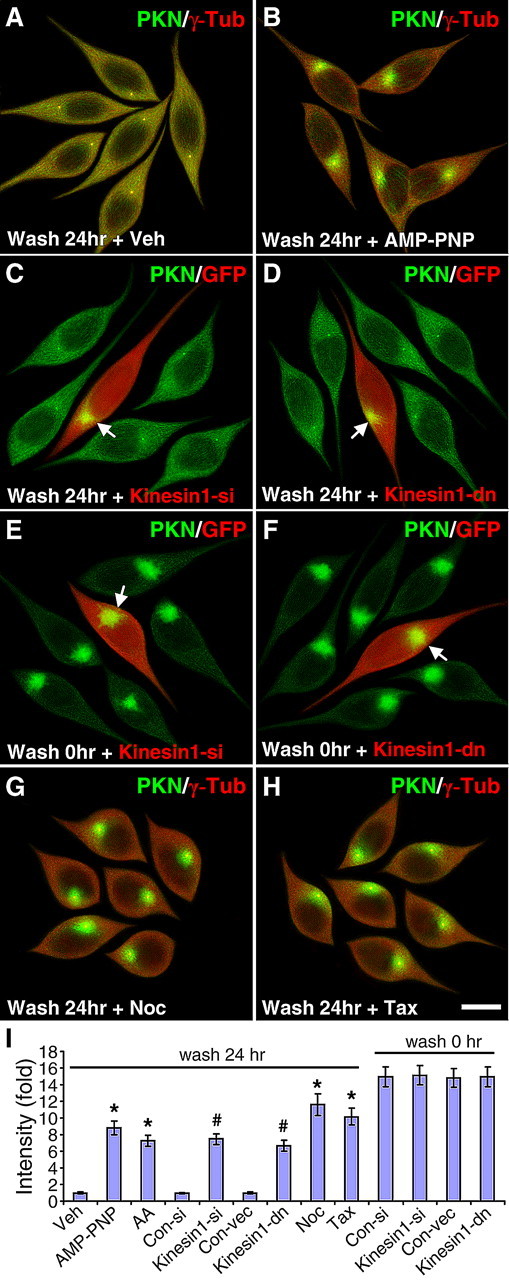
The dispersion of parkin from centrosome is dependent on kinesin-1 and intact microtubules. A, B, ATPase inhibitors blocked the dispersion of parkin in response to MG132 withdrawal. SH-SY5Y cells were treated with MG132 for 24 h and then incubated without MG132 (wash) for 24 h with vehicle (veh, A), 200 μm AMP-PNP (B) or 10 μm AA (I). C–F, Kinesin 1 siRNA (C) or dominant negative construct (D) significantly blocked the dispersion of parkin after MG132 withdrawal, but not the centrosome recruitment of parkin in response to MG132 (E, F). Arrow, Parkin accumulation in transfected cells (marked by GFP in red pseudocolor. G, H, Microtubule depolymerization (G) or stabilization (H) blocked the dispersion of parkin after MG132 withdrawal. Scale bar, 10 μm for all images. I, Statistical summary of the intensity of parkin accumulation under the above conditions. *p < 0.001 versus veh, #p < 0.001 versus the preceding bar; n = 30 cells randomly selected from three independent experiments for each condition.
Discussion
Parkinson's disease is characterized by the selective degeneration of dopaminergic neurons and frequently the presence of the intracellular protein aggregate termed Lewy body. Mutations of parkin, a protein-ubiquitin E3 ligase, represent one of the most prevalent genetic causes of the disease. However, Lewy bodies are generally not found in PD patients with parkin mutations, suggesting that parkin plays an essential role in the formation of protein aggregates in Parkinson's disease (Savitt et al., 2006). In this study, we identified the molecular and cellular mechanism by which parkin is recruited to the aggresome in response to proteasome inhibition. We found that parkin, through direct binding to HDAC6, accumulated at the centrosome when the proteasome was inhibited.
As a unique member of the histone deacetylase family, HDAC6 plays a central role in aggresome formation. It binds to the dynein motor complex through a motif between its two deacetylase domains and also associates with ubiquitinated proteins through a C-terminal ubiquitin-binding zinc finger domain (ZnF-UBP) (Kawaguchi et al., 2003). Thus, many ubiquitinated proteins, e.g., CFTR (Kawaguchi et al., 2003) and huntingtin (Iwata et al., 2005), are recruited to the aggresome by way of their association with HDAC6, which is transported to the minus-end of microtubules by the dynein motor protein (Kawaguchi et al., 2003). Parkin is very different in this regard. It directly bound to HDAC6 through multiple interaction domains that do not include the C-terminal ZnF-UBP motif of HDAC6. Both in vivo and in vitro results (Fig. 3) showed that parkin and HDAC6 formed a very tight complex that could withstand at least 500 mm NaCl and 1% Triton X-100. The in vitro data also conclusively showed that the binding was direct, not mediated by tubulin, to which both parkin (Ren et al., 2003) and HDAC6 (Hubbert et al., 2002) normally bind. As summarized in the diagram of Figure 3D, the first deacetylase domain (DD1) of HDAC6 bound to the linker or RING1 domain of parkin, while the second deacetylase domain (DD2) of HDAC6 only bound to the RING2 domain of parkin. It is interesting to note that the three domains of parkin that are responsible for binding HDAC6 are also the domains that are used to bind α/β tubulin heterodimers and microtubules (Yang et al., 2005). Thus, in the parkin/tubulin/HDAC6 ternary complex in vivo, both deacetylase domains of HDAC6 would be in close contact with parkin and tubulin, which raises the intriguing possibility that the deacetylation of tubulin might be coupled to its subsequent ubiquitination, as both modifications target the ε-NH2 group of a lysine residue (Caron et al., 2005).
The direct binding between parkin and HDAC6 mediates the centrosome recruitment of parkin. Any one of the three HDAC6-binding domains of parkin (Linker, RING1 or RING2) could be recruited to the centrosome by coexpressed HDAC6, while the two parkin domains that did not bind to HDAC6 (Ubl and IBR) could not be recruited to the centrosome by HDAC6 (Fig. 4). Without overexpressed HDAC6, there was no significant accumulation of parkin domains, which suggests that endogenous HDAC6 in SH-SY5Y cells is already saturated by endogenous parkin. Consistent with this, parkin recruitment was significantly abrogated when endogenous HDAC6 was knocked down by siRNA (Fig. 2B). In contrast, MG132-induced centrosome accumulation of HDAC6 was the same regardless whether parkin or its domains were overexpressed (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). These results indicate that parkin is passively directed by HDAC6 to the centrosome. Furthermore, the tubulin deacetylase activity of HDAC6 is required for the centrosome recruitment of both HDAC6 and parkin. When the activity was selectively inhibited by tubacin, the accumulation of HDAC6 (supplemental Fig. S3M″–R″, available at www.jneurosci.org as supplemental material) as well as parkin (Fig. 1D) or its domains (Fig. 4M–R) was significantly blocked. We confirmed that manipulations of HDAC6 activity or expression levels indeed changed the levels of α-tubulin acetylation (supplemental Fig. S8, available at www.jneurosci.org as supplemental material). HDAC6 inhibitors such as TSA or TBC or HDAC6 siRNA greatly increased tubulin acetylation, while overexpression of HDAC6, but not its catalytically inactive double mutant, markedly decreased tubulin acetylation. It is unclear why the tubulin deacetylase activity of HDAC6 is required for the bidirectional transport of parkin-HDAC6 complex along microtubules. One possibility is that deacetylation increases the dynamicity of microtubules (Matsuyama et al., 2002; Tran et al., 2007), which facilitates microtubule-based transport. However, a recent report shows that alterations in tubulin acetylation do not change microtubule dynamics in COS7 cells (Dompierre et al., 2007). Increased tubulin acetylation leads to recruitment of kinesin and dynein motors to microtubules to facilitate transport (Reed et al., 2006; Dompierre et al., 2007). These differences highlight the complex relationship between tubulin acetylation and microtubule-based transport, which is likely to be regulated in a dynamic manner depending on the cellular context (Verhey and Gaertig, 2007).
The centrosome recruitment of parkin is a reversible response, depending on the level of proteasome inhibition. Withdrawal of MG132 led to dispersion of parkin from the centrosome to the cytosol in a HDAC6-dependent manner (Fig. 5). Similar to the recruitment, the dispersion of parkin also required the tubulin deacetylase activity of HDAC6 (Fig. 5). The major difference in the bidirectional movements of parkin is the motor protein used in each direction. Parkin dispersion relied on the plus-end directed kinesin motor protein kinesin 1 (also known as kinesin heavy chain or KIF5) (Fig. 7), whereas parkin recruitment was dependent on the minus-end directed dynein motor protein (Fig. 6). Thus, HDAC6 acts as a sensor of proteasome inhibition. When proteasome is inhibited, HDAC6 couples to dynein to transport parkin-HDAC6 complex to the centrosome. Upon disinhibition of proteasome, HDAC6 switches to kinesin 1 to disperse the parkin-HDAC6 complex to the original cytosolic distribution pattern along microtubules. Further studies are necessary to define how HDAC6 senses the level of proteasome inhibition and switches motor proteins accordingly.
The reversible recruitment of parkin suggests that parkin remains catalytically active throughout the process and may serve to enhance the ubiquitination of substrate proteins being transported toward the aggresome. As the parkin-HDAC6 complex moves along microtubules to the centrosome, parkin ubiquitinates substrates such as CDCrel-1 and synphilin-1, which later accumulate in the aggresome (Chung et al., 2001; Zhao et al., 2003). During the process, these ubiquitinated substrates are recruited by HDAC6 through its C-terminal ubiquitin-binding zinc finger domain so that they can be retained in the parkin-HDAC6 complex for further ubiquitination and transport to the aggresome. Thus, the recruitment of parkin substrates to the aggresome has a positive feedback due to the synergistic actions of parkin and HDAC6 in the same complex. When parkin is mutated, its substrates cannot be effectively ubiquitinated and recruited by HDAC6, thus breaking the positive feedback and preventing the formation of Lewy bodies. Indeed, in parkin-deficit cells, misfolded DJ-1, which normally is a substrate of parkin, cannot be effectively recruited to the aggresome (Olzmann et al., 2007).
Because HDAC6 is required for the accumulation of many proteins such as CFTR (Kawaguchi et al., 2003) and huntingtin (Iwata et al., 2005) in the aggresome, the above-mentioned feedback paradigm may be generally applicable to other E3 ligases and their substrates. If this is true, then mutations of the corresponding E3 ligase may significantly affect the formation of aggresome containing specific substrates of that E3 ligase, in the same manner that mutations of parkin abrogates Lewy body formation.
Footnotes
This work was supported by National Institutes of Health Grant NS4172 and the New York State Center of Excellence in Bioinformatics and Life Sciences (J.F.). We thank Ralph Mazitschek and Stuart Schreiber at Harvard University (Cambridge, MA) (supported by Initiative for Chemical Genetics, National Cancer Institute, Bethesda, MD) for providing tubacin and niltubacin, and Eric M. Verdin at University of California at San Francisco (San Francisco, CA) for providing the HDAC6-FLAG construct.
References
- Ardley HC, Scott GB, Rose SA, Tan NG, Markham AF, Robinson PA. Inhibition of proteasomal activity causes inclusion formation in neuronal and non-neuronal cells overexpressing parkin. Mol Biol Cell. 2003;14:4541–4556. doi: 10.1091/mbc.E03-02-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai DJ, Koonce MP. The dynein heavy chain: structure, mechanics and evolution. Trends Cell Biol. 2001;11:196–202. doi: 10.1016/s0962-8924(01)01970-5. [DOI] [PubMed] [Google Scholar]
- Caron C, Boyault C, Khochbin S. Regulatory cross-talk between lysine acetylation and ubiquitination: role in the control of protein stability. Bioessays. 2005;27:408–415. doi: 10.1002/bies.20210. [DOI] [PubMed] [Google Scholar]
- Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med. 2001;7:1144–1150. doi: 10.1038/nm1001-1144. [DOI] [PubMed] [Google Scholar]
- Dompierre JP, Godin JD, Charrin BC, Cordelières FP, King SJ, Humbert S, Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozinger CM, Hassig CA, Schreiber SL. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci U S A. 1999;96:4868–4873. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci. 2005;6:201–214. doi: 10.1038/nrn1624. [DOI] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- Junn E, Lee SS, Suhr UT, Mouradian MM. Parkin accumulation in aggresomes due to proteasome impairment. J Biol Chem. 2002;277:47870–47877. doi: 10.1074/jbc.M203159200. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Matsuyama A, Shimazu T, Sumida Y, Saito A, Yoshimatsu Y, Seigneurin-Berny D, Osada H, Komatsu Y, Nishino N, Khochbin S, Horinouchi S, Yoshida M. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002;21:6820–6831. doi: 10.1093/emboj/cdf682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muqit MM, Davidson SM, Payne Smith MD, MacCormac LP, Kahns S, Jensen PH, Wood NW, Latchman DS. Parkin is recruited into aggresomes in a stress-specific manner: over-expression of parkin reduces aggresome formation but can be dissociated from parkin's effect on neuronal survival. Hum Mol Genet. 2004;13:117–135. doi: 10.1093/hmg/ddh012. [DOI] [PubMed] [Google Scholar]
- North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- Olzmann JA, Li L, Chudaev MV, Chen J, Perez FA, Palmiter RD, Chin LS. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J Cell Biol. 2007;178:1025–1038. doi: 10.1083/jcb.200611128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig J, Verhey KJ. Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol. 2006;16:2166–2172. doi: 10.1016/j.cub.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Ren Y, Zhao J, Feng J. Parkin binds to alpha/beta tubulin and increases their ubiquitination and degradation. J Neurosci. 2003;23:3316–3324. doi: 10.1523/JNEUROSCI.23-08-03316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitt JM, Dawson VL, Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest. 2006;116:1744–1754. doi: 10.1172/JCI29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroer TA. Dynactin. Annu Rev Cell Dev Biol. 2004;20:759–779. doi: 10.1146/annurev.cellbio.20.012103.094623. [DOI] [PubMed] [Google Scholar]
- Setou M, Seog DH, Tanaka Y, Kanai Y, Takei Y, Kawagishi M, Hirokawa N. Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature. 2002;417:83–87. doi: 10.1038/nature743. [DOI] [PubMed] [Google Scholar]
- Tran AD, Marmo TP, Salam AA, Che S, Finkelstein E, Kabarriti R, Xenias HS, Mazitschek R, Hubbert C, Kawaguchi Y, Sheetz MP, Yao TP, Bulinski JC. HDAC6 deacetylation of tubulin modulates dynamics of cellular adhesions. J Cell Sci. 2007;120:1469–1479. doi: 10.1242/jcs.03431. [DOI] [PubMed] [Google Scholar]
- Verhey KJ, Gaertig J. The tubulin code. Cell Cycle. 2007;6:2152–2160. doi: 10.4161/cc.6.17.4633. [DOI] [PubMed] [Google Scholar]
- Yang F, Jiang Q, Zhao J, Ren Y, Sutton MD, Feng J. Parkin stabilizes microtubules through strong binding mediated by three independent domains. J Biol Chem. 2005;280:17154–17162. doi: 10.1074/jbc.M500843200. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR, Yao TP, Lane WS, Seto E. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007;27:197–213. doi: 10.1016/j.molcel.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, Matthias P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22:1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Ren Y, Jiang Q, Feng J. Parkin is recruited to the centrosome in response to inhibition of proteasomes. J Cell Sci. 2003;116:4011–4019. doi: 10.1242/jcs.00700. [DOI] [PubMed] [Google Scholar]