

Abstract
P-glycoprotein (P-gp) plays an important role in determining net brain uptake of fexofenadine. Initial in vivo experiments with 24-h subcutaneous osmotic minipump administration demonstrated that fexofenadine brain penetration was 48-fold higher in mdr1a(–/–) mice than in mdr1a(+/+) mice. In contrast, the P-gp efflux ratio at the blood-brain barrier (BBB) for fexofenadine was only ∼4 using an in situ brain perfusion technique. Pharmacokinetic modeling based on the experimental results indicated that the apparent fexofenadine P-gp efflux ratio is time-dependent due to low passive permeability at the BBB. Fexofenadine brain penetration after terfenadine administration was ∼25- to 27-fold higher than after fexofenadine administration in both mdr1a(+/+) and mdr1a(–/–) mice, consistent with terfenadine metabolism to fexofenadine in murine brain tissue. The fexofenadine formation rate after terfenadine in situ brain perfusion was comparable with that in a 2-h brain tissue homogenate in vitro incubation. The fexofenadine formation rate increased ∼5-fold during a 2-h brain tissue homogenate incubation with hydroxyl-terfenadine, suggesting that the hydroxylation of terfenadine is the rate-limiting step in fexofenadine formation. Moreover, regional brain metabolism seems to be an important factor in terfenadine brain disposition and, consequently, fexofenadine brain exposure. Taken together, these results indicate that the fexofenadine BBB P-gp efflux ratio has been underestimated previously due to the lack of complete equilibration of fexofenadine across the blood-brain interface under typical experimental paradigms.
The blood-brain barrier (BBB) and blood-cerebrospinal fluid barrier (BCSFB) are the major interfaces and barriers between the central nervous system (CNS) and the peripheral circulation. The BBB and BCSFB are formed by a continuous layer of brain capillary endothelial cells (BBB) or epithelial cells that line the choroid plexus (BCSFB). The highly developed tight junctions and expression of numerous efflux transporters and metabolizing enzymes limit brain penetration of many endogenous compounds and therapeutic agents (Begley, 2003; Graff and Pollack, 2004).
Brain penetration can be beneficial or detrimental, depending on the target site of therapy and the potential for untoward side effects. Adequate exposure in the brain is essential for the treatment of cerebral diseases. However, brain exposure would ideally be minimized for compounds intended to treat peripheral disorders. BBB permeability, a primary determinant of substrate exposure in brain tissue, is influenced by many physicochemical properties such as molecular weight, pKa, lipophilicity, polar surface area, and the number of hydrogen bonds (Brodie et al., 1960; Liu et al., 2004). The extent of brain exposure, and thus the CNS pharmacologic or toxic effect, is determined by passive permeability, plasma and brain tissue binding, uptake or efflux transport activity, cerebrospinal fluid (CSF) bulk flow, and biotransformation in the CNS (Fenstermacher et al., 1981; Schinkel et al., 1994; Kalvass and Maurer, 2002; Liu and Chen, 2005; Kalvass et al., 2007a). Accurate prediction of CNS penetration is difficult because of these complex mechanisms and parameters.
In a recent study, nonspecific protein binding (i.e., capacity-limited binding to proteins other than the target receptor) in brain tissue and plasma was first used to predict in vivo CNS drug penetration (Kalvass and Maurer, 2002). These predictions were based in part on the assumption that, when compounds distribute between brain and blood solely by passive diffusion, the unbound brain and plasma concentrations are equal at distribution equilibrium. Under these conditions, the brain-to-plasma concentration ratio (brain partition coefficient; Kp,brain) is simply a function of the relative plasma and tissue unbound fractions Kp,brain = fu,plasma/fu,brain. For many compounds that are subject to P-glycoprotein (P-gp)-mediated efflux at the BBB, brain penetration has been predicted successfully based on this approach as modified to account for the P-gp-mediated efflux activity (Summerfield et al., 2006; Kalvass et al., 2007a).
Nonsedating antihistamines represent an interesting class of compounds because the lack of a sedative effect is primarily due to exclusion from the brain at the blood-brain interface. Fexofenadine, for example, has been identified as a substrate of P-gp in Caco-2 cells and MDR1-overexpressing LLC-PK1 cells (Cvetkovic et al., 1999; Petri et al., 2004). P-gp subsequently was demonstrated to decrease intestinal absorption and brain exposure of fexofenadine by comparing wild-type mice with mdr1a/1b(–/–) animals (Tahara et al., 2005).
The extent of fexofenadine brain penetration in normal rats and mice is minimal (Mahar Doan et al., 2004; Tahara et al., 2005). In contrast, terfenadine enters the brain rapidly (Mahar Doan et al., 2004; Obradovic et al., 2007). Terfenadine is metabolized to hydroxyl-terfenadine and then to the active metabolite fexofenadine by hepatic CYP3A4 in human (Ling et al., 1995; Markham and Wagstaff, 1998). Metabolic enzymes such as cytochrome P450s (P450s) are also expressed in the human and rodent brain (Strobel et al., 2001; Miksys and Tyndale, 2002; Meyer et al., 2007). Brain P450s play an important role in local metabolism of endogenous compounds and xenobiotics, although brain P450s is only approximately 1 to 10% of liver content and not likely to influence the overall metabolism of the body. Mouse CYP3A11 and CYP3A13, the counterpart of human CYP3A4, were responsible for 6β-hydroxylation of testosterone in the brain and were induced by antiepileptic drug phenytoin treatment (Hagemeyer et al., 2003; Meyer et al., 2006). In the current study, the brain metabolism of terfenadine to fexofenadine was examined.
Despite the ability to predict brain exposure of many compounds, the P-gp effect coupled with relative protein binding in blood and brain tissue can only partially explain the impaired brain penetration of fexofenadine and cetirizine (Kalvass et al., 2007a), two nonsedating antihistamines with poor permeability at the BBB. Fexofenadine brain penetration is ∼35-fold overestimated based on the plasma-to-brain unbound fraction ratio assumption; this magnitude of overestimation cannot be explained by the ∼3-fold P-gp effect at the BBB that has been reported (Cvetkovic et al., 1999; Tahara et al., 2005). Efflux protein(s) other than P-gp that may serve as barrier transport systems at the blood-brain interface have been proposed (Kalvass et al., 2007a). However, the functional efficiency and biologic significance of other BBB efflux transporters remain unclear, and no evidence is available to suggest an interaction between fexofenadine and other efflux transporters in brain capillary endothelium. The purpose of this study, therefore, was to understand the underlying mechanism(s) governing fexofenadine exposure in brain tissue and to re-examine P-gp-mediated fexofenadine efflux at the BBB. The central hypothesis underlying this effort is that, to date, studies have underestimated the actual BBB P-gp effect on fexofenadine brain uptake.
In the current study, the rate and extent of fexofenadine brain uptake and P-gp-mediated efflux activity at the BBB was determined using an in situ brain perfusion technique and subcutaneous osmotic minipump containing fexofenadine or terfenadine in mdr1a(+/+) and mdr1a(–/–) mice, and the drugs were continuously released over 24 to 168 h. A pharmacokinetic model was used to simulate the time dependence of the P-gp efflux ratio for fexofenadine and other compounds that have different passive permeability at the BBB. In addition, the influence of local metabolism on terfenadine brain disposition and consequent fexofenadine brain exposure was investigated.
Materials and Methods
Adult CF-1 mdr1a(+/+) mice and their natural mutant mdr1a(–/–) counterparts (30–40 g, 6–8 weeks of age) were purchased from Charles River Laboratories, Inc. (Wilmington, MA). All mice were maintained on a 12-h light/dark cycle with access to water and food ad libitum. All experimental procedures were performed under full anesthesia induced with ketamine/xylazine (100/10 mg/kg, i.p.). All procedures were approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill and were conducted in accordance with Principles with Laboratory Animal Care (NIH publication no. 85-23, revised in 1985).
[14C]Diazepam (56.0 mCi/mmol) was purchased from GE Healthcare (Little Chalfont, Buckinghamshire, UK). [3H]Inulin (180.0 mCi/g) was purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). Terfenadine, fexofenadine hydrochloride, and loperamide were purchased from Sigma-Aldrich (St. Louis, MO). All of the other chemicals were commercially available and of reagent grade.
In Situ Mouse Brain Perfusion. The details of the in situ mouse brain perfusion have been described elsewhere (Dagenais et al., 2000). In brief, mice were anesthetized with ketamine/xylazine (100/10 mg/kg, i.p.). The perfusion buffer (Krebs-bicarbonate buffer, with 9 mM d-glucose, pH 7.4) was oxygenated with 95% O2 and 5% CO2 and maintained at 37°C. [14C]Diazepam and [3H]inulin were used as blood flow rate and vascular space markers, respectively. Fexofenadine or terfenadine at a final concentration of 2 μM was added to the perfusate. The right common carotid artery was cannulated with PE-10 tubing (Braintree Scientific Inc., Braintree, MA) after ligation of the external carotid artery. The cardiac ventricles were severed immediately before perfusion at 2.5 ml/min for 60 s via a syringe pump (Harvard Apparatus Inc., Holliston, MA). The experiment was terminated by decapitation and the right cerebral hemisphere was collected. Aliquots of perfusate were collected through the syringe and tubing for determination of substrate concentrations in perfusate. The perfusate and brain samples were stored at –20°C until analysis by high-performance liquid chromatography/tandem mass spectrometry (HPLC/MS/MS). The radioactive brain samples were digested with 0.7 ml Solvable (PerkinElmer Life and Analytical Sciences) at 50°C overnight. Five milliliters of UltimaGold scintillation cocktail (PerkinElmer Life and Analytical Sciences) was added and vortex-mixed. Total radioactivity (3H and/or 14C) was determined in a Packard Tri-carb TR 1900 liquid scintillation analyzer (PerkinElmer Life and Analytical Sciences).
Parameters related to the in situ brain perfusion, i.e., the cerebral vascular volume (ml/100 g brain), were calculated using the following equation:
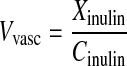 |
the initial brain uptake clearance (Clup, ml/min/100 g brain) was calculated as:
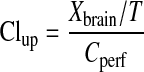 |
and the apparent distributional volume (Vd, ml/100 g brain) was calculated as:
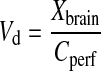 |
where the amount of substrate in brain Xbrain was corrected for blood vessel contamination by subtraction of vascular volume or a final 15-s washout with drug-free saline. The permeability surface product (PS) values were calculated based on the Crone-Renkin equation (Takasato et al., 1984):
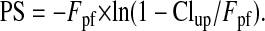 |
Fpf is the perfusion flow rate measured using [14C]diazepam as the flow rate marker.
Mathematical Modeling and Simulations. The time to equilibrium of brain penetration is dependent on the passive permeability, brain unbound fraction, and BBB P-gp-mediated efflux (Liu et al., 2005; Kalvass et al., 2007b). The time course of P-gp efflux ratio after continuous infusion was simulated based on a similar model structure with additional P-gp-mediated efflux activity. As the structure shows in Fig. 1, the model contains one central (blood) compartment and one brain compartment; Vc and Vbr are the physiologic blood and brain volumes, respectively. Cld represents the passive permeability between blood and brain compartments, Cleff is the P-gp-mediated efflux clearance, Cl is the systemic clearance, and fu,p and fu,br are plasma and brain unbound fraction, respectively. Modeling was based in part on the assumption that only unbound drug is able to cross the BBB. The passive permeability Cld for fexofenadine is very low relative to the blood flow rate and, therefore, was assumed to be equal to the permeability coefficient PS determined by the in situ brain perfusion technique. Numerical values for these parameters were assigned as follows: Vp = 49 ml/kg and Vbr = 17 ml/kg (Brown et al., 1997); Cl = 30 ml/min/kg (Tahara et al., 2005); fu,p = 0.35 and fu,br = 0.077 (Kalvass et al., 2007a). The infusion rate k0 was set to a value of 1 (unit dose). Fexofenadine PS = 1.32 ml/min/100 g brain based on the in situ brain perfusion data. Three pairs of Cld and Cleff were assigned: Cld1 = 0.022, Cld2 = 0.22, Cld3 = 2.22 ml/min/kg; the corresponding Cleff1 = 0.54; Cleff2 = 5.39, Cleff3 = 53.90 ml/min/kg for P-gp-competent mice assuming a steady-state P-gp efflux ratio of 25. Cleff = 0 for all P-gp-deficient mice. The P-gp efflux ratio was calculated as the ratio of brain partition coefficient in P-gp-deficient mice to P-gp-competent mice. The differential equations used for simulations were:
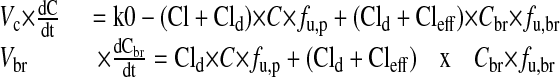 |
where C and Cbr are the total plasma and brain concentrations, respectively.
Fig. 1.

Scheme for the mathematical model used to simulate the P-gp efflux ratio over time. Parameter definitions are included under Materials and Methods.
In Vivo Osmotic Minipump Studies. CF-1 mice (n = 3 per group) were anesthetized with ketamine/xylazine (100/10 mg/kg, i.p.). An Alzet osmotic minipump (release rate 1 μl/h over 1 week; Alza, Palo Alto, CA) was selected. Initial experiments were directed at determining fexofenadine brain penetration and P-gp efflux ratio after fexofenadine and terfenadine administration. To produce similar plasma concentrations of fexofenadine for comparison, pumps were filled with fexofenadine hydrochloride (80 μg/pump) or terfenadine (900 μg/pump) dissolved in 50% dimethyl sulfoxide (200 μl/pump) and placed subcutaneously in the back of mdr1a(+/+) and mdr1a(–/–) mice. The experiment was terminated by decapitation 24 h after implantation. Additional experiments were performed to study the time course of fexofenadine P-gp efflux ratio at the BBB. To accomplish this goal, additional groups of mdr1a(+/+) and mdr1a(–/–) mice received fexofenadine hydrochloride (872 μg/pump) to produce plasma concentrations similar to those reported by Tahara et al. (2005). The mice were sacrificed at 24, 72, and 168 h postimplantation. Trunk blood was collected in heparinized 1.5-ml microcentrifuge tubes, and plasma was harvested after centrifugation at 3000 rpm for 5 min. The plasma and brain samples were stored at –20°C until analysis by HPLC/MS/MS. The contribution of residual blood in the brain tissue to the brain partition coefficient was corrected by subtracting 1.8% from the calculated brain-to-plasma concentration ratio (Brown et al., 1997; Dagenais et al., 2000).
Mouse Brain Metabolism. Terfenadine brain metabolism was determined under two different experimental conditions. First, mdr1a(+/+) and mdr1a(–/–) mouse brain hemispheres were perfused with 2 μM terfenadine for 1 min (n = 3, with a procedure identical to that described under In situ Mouse Brain Perfusion). The concentrations of fexofenadine formed in the perfused brain hemisphere were determined by HPLC/MS/MS. Second, fresh mdr1a(+/+) mouse brains were harvested, washed with 0.15 M KCl, and homogenized in two volumes of phosphate buffer (pH 7.4, 1.15% KCl). Terfenadine or hydroxyl-terfenadine at a final concentration of 200 nM was incubated with brain homogenate (10 mg/ml) in the presence of potassium phosphate buffer (0.1 M, pH 7.4) and magnesium chloride (5 mM). The reaction was initiated by addition of freshly prepared NADPH (2 mM) (Ling et al., 1995; Holleran et al., 2004). After 2-h incubation at 37°C in a shaking water bath, the reaction was terminated by addition of ice-cold methanol to precipitate protein. The samples were centrifuged at 10,000g for 10 min. The supernatant was transferred to a fresh tube and evaporated under a stream of dry nitrogen. The residue was reconstituted with 50% methanol in water, and an internal standard was added. The formation of fexofenadine from terfenadine or hydroxyl-terfenadine was determined by HPLC/MS/MS.
Sample Preparation and Quantitation. The brain tissue was homogenized with addition of two volumes of distilled water after brief probe sonication. The perfusate was diluted with two volumes of methanol. A 25-μl aliquot of plasma or brain homogenate was transferred to an HPLC vial, and protein was precipitated with 100 μl of methanol containing internal standard (10 ng/ml loperamide) followed by a 25-μl aliquot of dimethyl sulfoxide. The sample was vortex-mixed and centrifuged. Standard solutions ranging from 0.5 to 5000 nM were prepared in a similar manner. In brief, 25 μl of blank plasma or brain homogenate, 100 μl of methanol containing internal standard, and 25 μl of serially diluted standard solution were mixed and centrifuged. The supernatant was analyzed by HPLC-tandem mass spectrometry (API 4000 triple quadrupole with TurboIonSpray interface; Applied Biosystems/MDS Sciex, Concord, ON, Canada). Three microliters of sample solutions were injected via an autosampler (Leap, Carrboro, NC). Fexofenadine, terfenadine, terfenadine-OH, and the internal standard loperamide were eluted from an Aquasil C18 column (2.1 × 50 mm, dp = 5 μm; Thermo Fisher Scientific, Waltham, MA) using a mobile phase gradient [A, 0.1% formic acid in water; B, 0.1% formic acid in methanol; 0–0.70-min hold at 0% B, 0.70–3.12-min linear gradient to 90% B, 3.12–4.10-min hold at 90% B, 4.10–4.20-min linear gradient to 0% B, 4.20–4.90-min hold at 0% B; solvent delivery system (Shimadzu, Kyoto, Japan); flow rate = 0.75 ml/min; 0.8–4 min directed to mass spectrometer] and were detected in positive-ion mode using multiple reaction monitoring: fexofenadine: 502.4→466.4 m/z, terfenadine, 472.3→ 436.5 m/z; terfenadine-OH, 488.3→452.5 m/z; loperamide, 477.4→266.0 m/z. All analytes were quantified with standard curves in the linear range of the relationship between detector response and analyte concentration prepared in the appropriate matrix. The lower limit of detection was 0.5 ng/ml; inter- and intraday coefficients of variations were <15%.
Statistical Analysis. Data are reported as mean ± S.D. for 3 mice per condition. A two-tailed Student's t test, or either a one-way or two-way analysis of variance (ANOVA), where appropriate, was used to determine the statistical significance of differences among two or more groups. The level of significance was corrected for multiple comparisons (e.g., Bonferroni test) or adjusted for unequal variance when necessary. In all cases, p < 0.05 was considered to be statistically significant.
Results
In Situ Mouse Brain Perfusion. [3H]Inulin was used as the brain capillary space marker to measure the BBB physical integrity. [14C]Diazepam was perfused as a marker of functional perfusate flow rate. The vascular volume was 1.69 ± 0.10 ml/100 g, and functional flow rate was 250 ± 41 ml/min/100 g brain. P-gp gene deficiency had no influence on the vascular volume or functional perfusion rate. The initial brain uptake clearance (Clup, ml/min/100 g brain tissue) of fexofenadine and terfenadine in mdr1a(+/+) and mdr1a(–/–) mice is shown in Fig. 2. The P-gp efflux ratio, calculated as the ratio of Clup in mdr1a(–/–) mice to mdr1a(+/+) mice, was 4.4 and 1.1 for fexofenadine and terfenadine, respectively.
Fig. 2.

Initial brain uptake clearance (Clup, ml/min/100 g) for fexofenadine and terfenadine in mdr1a(+/+) (solid bar) and mdr1a(–/–) (open bar) mice. The mouse brain hemisphere was perfused via the common carotid artery at 2.5 ml/min for 60 s. Data are presented as mean ± S.D. (n = 3). A Student's t test was used to determine the statistical significance of P-gp effect between mdr1a(+/+) and mdr1a(–/–) mice. The asterisk (*) represents the statistical difference (p < 0.05) between mdr1a(+/+) and mdr1a(–/–) mice.
Mathematical Modeling and Simulation. Simulations were conducted with three different sets of values for Cld and Cleff (Fig. 3). The time course of the apparent fexofenadine P-gp efflux ratio was simulated when Cld was equal to the actual value determined from the in situ brain perfusion experiment (1 × PS). All of the other variables were physiologic or biologic parameters (i.e., Vc, Vbr, fu,p, and fu,br) or estimates of fexofenadine pharmacokinetic parameters (i.e., Cl) obtained from the literature (Brown et al., 1997; Tahara et al., 2005; Kalvass et al., 2007a). The simulations predicted that 12 h and 36 h would be required for fexofenadine to achieve 50 and 90% of the steady-state P-gp efflux ratio, respectively. When Cld was increased 10-fold, the time to achieve the maximum P-gp efflux ratio shortened. In contrast, when Cld was 10-fold lower than the actual experimental value obtained for fexofenadine, the P-gp efflux ratio did not plateau during the time frame of the simulation.
Fig. 3.

Simulated P-gp efflux ratios over time for varying Cld and Cleff (—, Cld = 2.24, Cleff = 53.90; —–, Cld = 0.22, Cleff = 5.39; ······, Cld = 0.022, Cleff = 0.54; Cleff = 0 for P-gp-deficient mice). The P-gp efflux ratio was calculated as the ratio of brain-to-plasma concentration ratio in mdr1a(–/–) mice to mdr1a(+/+) mice. The solid circles (•) represent fexofenadine P-gp efflux ratios determined at 24, 72, and 168 h during subcutaneous osmotic minipump administration. The open circle (○) is the P-gp efflux ratio determined at 2.5 h in Tahara et al. (2005).
In Vivo Osmotic Minipump Studies. Fexofenadine brain-to-plasma concentration ratios at 24 h after administration of either fexofenadine or terfenadine in mdr1a(+/+) and mdr1a(–/–) mice are displayed in Fig. 4. Steady-state fexofenadine plasma concentrations were 28.5 ± 8.3 and 45.5 ± 3.1 nM in mdr1a(+/+) and mdr1a(–/–) mice during fexofenadine administration, respectively. During terfenadine administration, concentrations of terfenadine in plasma and brain tissue were below the limit of quantitation. However, plasma concentrations of fexofenadine derived from terfenadine were identical in mdr1a(+/+) (7.1 ± 2.8 nM) and mdr1a(–/–) (7.1 ± 1.7 nM) mice. Fexofenadine brain-to-plasma concentration ratios after 24 h of fexofenadine administration were 0.0056 ± 0.010 and 0.27 ± 0.08 in mdr1a(+/+) and mdr1a(–/–) mice, respectively. In contrast, the fexofenadine brain-to-plasma concentration ratios after 24 h of terfenadine administration were 0.14 ± 0.03 and 7.44 ± 2.12 in mdr1a(+/+) and mdr1a(–/–) mice, respectively. The fexofenadine brain-to-plasma ratios therefore were ∼25- and 27-fold higher in mdr1a(+/+) and mdr1a(–/–) mice, respectively, after terfenadine administration compared with after fexofenadine administration. The calculated fexofenadine P-gp efflux ratios after fexofenadine and terfenadine administration are 48 and 55, respectively.
Fig. 4.

Fexofenadine brain-to-plasma concentration ratio at 24 h in mdr1a(+/+) (solid bar) and mdr1a(–/–) (open bar) mice after subcutaneous osmotic minipump administration of fexofenadine or terfenadine. Data are presented as mean ± S.D. (n = 3). Student's t tests were used to compare brain-to-plasma concentration ratios between mdr1a(+/+) and mdr1a(–/–) mice. The asterisk (*) represents a statistical difference (p < 0.05) between mdr1a(+/+) and mdr1a(–/–) mice.
Fexofenadine hydrochloride at a dose of 872 μg/pump, selected to produce plasma concentrations (380–480 nM) similar to those in a previous study by Tahara et al. (2005), was administered in mdr1a(+/+) and mdr1a(–/–) mice for 24, 72, and 168 h. Plasma concentrations of fexofenadine achieved with this approach were 305 ± 74 and 836 ± 138 nM in mdr1a(+/+) and mdr1a(–/–) mice, respectively, and did not vary with time during this period. The brain-to-plasma concentration ratios of fexofenadine during prolonged continuous infusion by subcutaneous osmotic minipumps are shown in Fig. 5. The P-gp efflux ratios were 17, 23, and 25, at 24, 72, and 168 h, respectively. The calculated P-gp efflux ratio values, together with the P-gp efflux ratio at 2.5 h from Tahara et al. (2005), are shown in Fig. 3. These experimentally derived values correspond closely with the predicted values based on the mathematical simulation.
Fig. 5.

Fexofenadine brain-to-plasma concentration ratio in mdr1a(+/+) (solid bar) and mdr1a(–/–) (open bar) mice at 24, 72, and 168 h after subcutaneous osmotic minipump administration of fexofenadine. Data are presented as mean ± S.D. (n = 3). Two-way ANOVA was used to compare brain-to-plasma concentration ratios between mdr1a(+/+) and mdr1a(–/–) mice, and among the time points, respectively. The asterisk (*) represents a statistical difference (p < 0.05) between mdr1a(+/+) and mdr1a(–/–) mice. There were no differences for fexofenadine brain partition coefficients at different time points.
Mouse Brain Metabolism. The fexofenadine formation rate in brain tissue during in situ brain perfusion with 2 μM terfenadine for 1 min in mdr1a(+/+) and mdr1a(–/–) mice, and after 2-h incubation of 200 nM terfenadine or hydroxyl-terfenadine with whole brain tissue homogenate, is displayed in Fig. 6. The fexofenadine formation rate during 1-min terfenadine brain perfusion was 17.0 ± 3.1 and 12.0 ± 5.4 nM/min/kg brain tissue in mdr1a(+/+) and mdr1a(–/–) mice, respectively. The fexofenadine formation rate during a 2-h incubation of whole brain homogenate with terfenadine or hydroxyl-terfenadine was 12.9 ± 3.5 or 74.9 ± 11.8 nM/min/kg brain tissue, respectively.
Fig. 6.

The fexofenadine formation rate in the brain hemisphere after 60-s terfenadine in situ mouse brain perfusion in mdr1a(+/+) and mdr1a(–/–) mice, or a 2-h incubation of terfenadine (Terf) or terfenadine-OH (Terf-OH) with whole brain tissue homogenate. Data are presented as mean ± S.D. (n = 3). One-way ANOVA statistical analysis corrected for multiple comparisons (Bonferroni test) demonstrated that fexofenadine formation rate in the brain was significantly higher when incubated with terfenadine-OH than incubation with terfenadine or 1-min terfenadine brain perfusion (*, p < 0.05).
Discussion
Lipophilicity and ionization at physiologic pH are important determinants of brain penetration (Brodie et al., 1960). Fexofenadine is ionized at physiologic pH and crosses the BBB to only a minimal extent. In contrast to preformed fexofenadine, the fexofenadine prodrug terfenadine, a very lipophilic molecule, evidenced a brain uptake that approximated perfusate flow. This very high uptake clearance was similar between mdr1a(+/+) and mdr1a(–/–) mice, even though terfenadine has been reported to be a P-gp substrate (Mahar Doan et al., 2004; Obradovic et al., 2007). The lack of a discernible P-gp effect for this substrate was consistent with the efficient unidirectional uptake of terfenadine during short-term brain perfusion.
P-gp has been reported to be involved in fexofenadine intestinal secretion and, thus, in the systemic disposition of this compound. [14C]Fexofenadine plasma concentrations in mdr1a knockout mice were ∼4.6-fold higher than that in wild-type mice after intravenous administration (Cvetkovic et al., 1999). In the current study, fexofenadine plasma concentrations in mdr1a(–/–) mice were approximately 60 to 206% higher than in mdr1a(+/+) mice. These observations are in contrast to a previous report by Tahara et al. (2005), which indicated that plasma concentrations of fexofenadine were similar in P-gp-competent and P-gp-deficient bile duct-cannulated mice.
P-gp also is involved in fexofenadine penetration of the BBB. The influence of P-gp on brain uptake of fexofenadine was demonstrated for the first time with the in situ brain perfusion technique, using the mdr1a(–/–) mouse model of P-gp deficiency. The P-gp effect during short-term brain perfusion (4.4-fold) was modest. In vivo administration under conditions that produce steady state provides a more sensitive approach to quantitating the BBB P-gp effect than short-term brain perfusion when substrate equilibration across the blood-brain interface is slow. Continuous infusion with a subcutaneous osmotic minipump provides useful steady-state information from a simple experimental system. This approach was used in the current study. As anticipated, the in vivo P-gp efflux ratio exceeded that determined by the in situ brain perfusion, which has been demonstrated experimentally in the present study and previously (Dagenais et al., 2001). The present results also are consistent with previous mathematical predictions based on a simple three-compartment model (representing plasma, endothelial cell layer, and brain) that showed ERα (in vivo P-gp efflux ratio) must be higher than ERB (in situ P-gp efflux ratio) for the same substrate and efflux transporter system (Kalvass and Pollack, 2007).
Fexofenadine brain-to-plasma concentration ratio at 24 h (i.e., during continuous minipump administration) was 25- to 27-fold higher after terfenadine administration than fexofenadine administration in both mdr1a(+/+) and mdr1a(–/–) mice. This difference might be due to the low permeability of fexofenadine. In the presence of such a low uptake clearance, a significantly longer period of time would be required to reach distribution equilibrium across the BBB, and so the apparent brain-to-plasma ratio at 24 h may be an underprediction of the true equilibrium value. Indeed, this situation has been demonstrated previously by Liu et al. (2005) using a simulation approach. Alternatively, the precursor of fexofenadine, terfenadine, is permeable at the BBB. If terfenadine is metabolized in the brain after crossing the BBB, an increase in fexofenadine brain exposure, relative to administration of preformed fexofenadine, would be predicted.
To test the first possible mechanism, simulation studies were conducted in parallel with animal experiments. Mathematical modeling demonstrated that the P-gp efflux ratio must be time-dependent. Moreover, these simulations successfully predicted the P-gp efflux ratios measured at the selected experimental time points. The interpretation of the simulation results is both straightforward and logical: because it takes longer for slowly permeable compounds to equilibrate across the BBB, the experimentally determined P-gp efflux ratio will also take a longer time to reach its equilibrium (theoretically maximum) condition. Experimentally, fexofenadine was administered for a prolonged time period up to 168 h. The present results demonstrate a clear time dependence of fexofenadine P-gp efflux ratio during this time range of 24 to 168 h. The determined P-gp efflux ratio values, together with the P-gp efflux ratio of ∼3.3 at 2.5 h taken from Tahara et al. (2005), were almost superimposable with the predicted values based on the model simulation.
To assess the influence of P-gp on fexofenadine brain exposure when fexofenadine is formed from terfenadine, versus when fexofenadine is administered directly, fexofenadine plasma concentrations must be similar between the two administration modalities. To achieve similar fexofenadine concentrations from preformed fexofenadine compared with fexofenadine concentrations after terfenadine administration, fexofenadine hydrochloride was administered via osmotic minipump at a continuous rate of 1.5 nM/h. The fexofenadine P-gp efflux ratios determined in vivo at distribution equilibrium were similar (55 versus 48), regardless of whether fexofenadine was formed from terfenadine or administered directly. The P-gp efflux ratio decreased when fexofenadine plasma concentration was ∼10-fold higher (305 nM) in the time course studies, consistent with at least partial saturation of the transport process.
Based on the impaired brain penetration of fexofenadine in both mdr1a(+/+) and mdr1a(–/–) mice at all time points after fexofenadine administration, the 25- to 27-fold increase of fexofenadine brain penetration after terfenadine administration could not be explained by simply attaining distribution equilibrium more rapidly with the prodrug. Thus, it was important to address the possibility that local metabolism of terfenadine to fexofenadine in the brain was responsible for the observed difference. Terfenadine is metabolized to hydroxyl-terfenadine then to the carboxylic acid metabolite fexofenadine, by hepatic CYP3A4 in humans. CYP3A11 and 3A13 are the murine counterparts of human 3A4 and are expressed in the mouse liver and brain (Hagemeyer et al., 2003; Meyer et al., 2006). After terfenadine administration, terfenadine plasma and brain concentrations were below the limit of quantitation, although fexofenadine concentrations in the plasma and brain were substantial. These observations confirm efficient metabolism of terfenadine in the mouse. Fexofenadine plasma concentrations were identical in mdr1a(+/+) and mdr1a(–/–) mice, indicating that the formation rate of fexofenadine from terfenadine did not differ between mouse strains. Terfenadine seemed to be metabolized to fexofenadine in mouse brain during a short (1 min) brain perfusion, as well as during a 2-h in vitro incubation with brain tissue homogenate (Fig. 6).
The fexofenadine formation rate in the brain during terfenadine brain perfusion was comparable between mdr1a(+/+) and mdr1a(–/–) mice, suggesting that the correspondent enzyme(s) activity in brain was not modulated by P-gp. These novel observations provided supporting evidence, consistent with the pharmacokinetic arguments, that terfenadine must be metabolized to fexofenadine both systemically and regionally in the brain. The local metabolism significantly increased fexofenadine brain exposure compared with what would be expected if fexofenadine was simply formed from terfenadine systemically.
The enzyme(s) responsible for terfenadine-to-fexofenadine biotransformation in murine brain is unknown, although it is reasonable to speculate that CYP3A11 and/or CYP3A13 may be responsible. This metabolic situation may have pharmacologic consequences. Fexofenadine is a nonsedating antihistamine, at least in part because it does not penetrate the BBB efficiently. In contrast, high-dose terfenadine administration has been associated with sedative effects (Mattila and Paakkari, 1999). It is possible that terfenadine itself is the causative agent for sedation. Alternatively, terfenadine may be inert but provides sufficient fexofenadine to the brain, via local metabolism, to allow fexofenadine to cause sedation.
Antihistamines represent an interesting pharmacologic class from the standpoint of BBB permeation and central nervous system responsivity. Fexofenadine and cetirizine are both nonsedating antihistamines caused by poor BBB permeability secondary to P-gp-mediated efflux at the BBB (Polli et al., 2003). Hydroxyzine is sedating, as is terfenadine at high doses (Mattila and Paakkari, 1999). A comprehensive pharmacokinetic study of cetirizine after intravenous administration of cetirizine and hydroxyzine has been conducted (Chen et al., 2003). Re-examination of these published results demonstrated that the cetirizine P-gp efflux ratio was ∼4.4 after administration of preformed cetirizine. However, the cetirizine P-gp efflux ratio after administration of hydroxyzine, the more permeable precursor of cetirizine, was ≥125 at 240 min. This experimentally determined efflux ratio is remarkably similar to the predicted value of 111 based on the unbound fraction of cetirizine in plasma versus brain and the intrinsic transport activity of cetirizine by P-gp (Kalvass et al., 2007a). Thus, the overall prediction of antihistamine brain penetration could be substantially improved by using accurately determined P-gp efflux ratios (that is, the efflux ratios determined after complete equilibration across the BBB). Such an approach would effectively address the two outliers—fexofenadine and cetirizine—identified in a previous compound set (Kalvass et al., 2007a).
An additional consideration for these compounds is that the cetirizine brain-to-plasma concentration ratio at 240 min was ∼7-fold higher after hydroxyzine administration compared with cetirizine administration. This situation is analogous to that for the terfenadine-fexofenadine precursor-metabolite pair as shown in the present series of experiments. The specific enzyme(s) responsible for hydroxyzine metabolism are unknown. However, it is conceivable that hydroxyzine enters the brain and is subsequently metabolized to cetirizine, thus increasing cetirizine local exposure after hydroxyzine administration. It is clear that additional experimentation would be required to confirm this speculation.
Based on the current study with the terfenadine/fexofenadine pair, together with published results for the hydroxyzine/cetirizine pair, administration of a permeable prodrug seems to provide useful information about the true P-gp efflux ratio for a poorly permeable metabolite. For many slow-equilibrating compounds, partitioning through the BBB is the rate-limiting step in brain uptake. In this situation, the assumption that simple diffusion of unbound substrate across the BBB leads to equivalent unbound concentrations in brain and plasma is violated because distribution equilibrium might not be achieved within the confines of a given experiment and thus may result in a lower extent of brain penetration than predicted based on the equilibrium assumption. Such a situation has been demonstrated for N[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine, which failed to reach equilibrium within a 24-h experiment (Liu et al., 2005).
After prodrug administration, a poorly permeable metabolite formed in the brain (i.e., in capillary endothelial cells, neurons, or glial cells) might evidence access to the P-gp binding site that is different from access that could be achieved from the luminal side (i.e., after systemic formation of the metabolite or direct administration of the preformed compound). The relative rate of metabolite formation in systemic circulation versus brain certainly would influence brain exposure, apparent brain-to-blood partitioning, and calculated P-gp efflux ratios. These experimental aspects warrant further investigation. Taken together, brain exposure is an integrative effect of passive permeability, protein binding in plasma and brain, interaction with active transporters at the BBB, and, in the special case of poorly permeable metabolites, systemic and regional brain metabolism. Finally, brain exposure for poorly permeable compounds is complicated further by CSF bulk flow, which in the mouse is ∼0.071 ml/min/100 g (Rudick et al., 1982). CSF bulk flow also poses a significant influence on the brain exposure of low-permeability compounds (Liu and Chen, 2005).
In conclusion, the current study investigated fexofenadine brain penetration and P-gp efflux ratio after fexofenadine and terfenadine administration. The results demonstrated that P-gp is an important determinant of fexofenadine efflux at the BBB and, consequently, the presentation of fexofenadine to brain tissue. Studies to date have underestimated the actual P-gp efflux ratio of fexofenadine at the BBB because the P-gp efflux ratio is time-sensitive, and previous experiments were not sufficiently long to uncover the true efflux ratio. The present experiments also demonstrated for the first time the metabolism of terfenadine to fexofenadine in murine brain. The enzyme(s) responsible for terfenadine metabolism in the brain require further investigation. These latter results underscore the fact that enzymes in the brain or the BBB not only represent a metabolic barrier for substrate presentation to the brain, but also might activate some drugs to cause CNS responses (either therapeutic or toxic). Modulation of brain metabolic enzymes by coadministered drugs might represent a source of drug-drug interaction.
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grant GM61191].
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.107.019893.
ABBREVIATIONS: BBB, blood-brain barrier; BCSFB, blood-cerebrospinal fluid barrier; CNS, central nervous system; CSF, cerebrospinal fluid; ER, efflux ratio; P-gp, P-glycoprotein; mdr1a(–/–), P-gp-deficient mice; mdr1a(+/+), P-gp-competent mice; P450, cytochrome P450; HPLC/MS/MS, high-performance liquid chromatography/tandem mass spectrometry; PS, surface product; ANOVA, analysis of variance; Cld, passive permeability between blood and brain compartments; Cleff, P-gp-mediated efflux clearance.
References
- Begley DJ (2003) Understanding and circumventing the blood-brain barrier. Acta Paediatr Suppl 92 83–91. [DOI] [PubMed] [Google Scholar]
- Brodie BB, Kurz H, and Schanker LS (1960) The importance of dissociation constant and lipid-solubility in influencing the passage of drugs into the cerebrospinal fluid. J Pharmacol Exp Ther 130 20–25. [PubMed] [Google Scholar]
- Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, and Beliles RP (1997) Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health 13 407–484. [DOI] [PubMed] [Google Scholar]
- Chen C, Hanson E, Watson JW, and Lee JS (2003) P-glycoprotein limits the brain penetration of nonsedating but not sedating H1-antagonists. Drug Metab Dispos 31 312–318. [DOI] [PubMed] [Google Scholar]
- Cvetkovic M, Leake B, Fromm MF, Wilkinson GR, and Kim RB (1999) OATP and P-glycoprotein transporters mediate the cellular uptake and excretion of fexofenadine. Drug Metab Dispos 27 866–871. [PubMed] [Google Scholar]
- Dagenais C, Rousselle C, Pollack GM, and Scherrmann JM (2000) Development of an in situ mouse brain perfusion model and its application to mdr1a P-glycoprotein-deficient mice. J Cereb Blood Flow Metab 20 381–386. [DOI] [PubMed] [Google Scholar]
- Dagenais C, Zong J, Ducharme J, and Pollack GM (2001) Effect of mdr1a P-glycoprotein gene disruption, gender, and substrate concentration on brain uptake of selected compounds. Pharm Res 18 957–963. [DOI] [PubMed] [Google Scholar]
- Fenstermacher JD, Blasberg RG, and Patlak CS (1981) Methods for Quantifying the transport of drugs across brain barrier systems. Pharmacol Ther 14 217–248. [DOI] [PubMed] [Google Scholar]
- Graff CL and Pollack GM (2004) Drug transport at the blood-brain barrier and the choroid plexus. Curr Drug Metab 5 95–108. [DOI] [PubMed] [Google Scholar]
- Hagemeyer CE, Rosenbrock H, Ditter M, Knoth R, and Volk B (2003) Predominantly neuronal expression of cytochrome P450 isoforms CYP3A11 and CYP3A13 in mouse brain. Neuroscience 117 521–529. [DOI] [PubMed] [Google Scholar]
- Holleran JL, Fourcade J, Egorin MJ, Eiseman JL, Parise RA, Musser SM, White KD, Covey JM, Forrest GL, and Pan SS (2004) In vitro metabolism of the phosphatidylinositol 3-kinase inhibitor, wortmannin, by carbonyl reductase. Drug Metab Dispos 32 490–496. [DOI] [PubMed] [Google Scholar]
- Kalvass JC and Maurer TS (2002) Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm Drug Dispos 23 327–338. [DOI] [PubMed] [Google Scholar]
- Kalvass JC, Maurer TS, and Pollack GM (2007a) Use of plasma and brain unbound fractions to assess the extent of brain distribution of 34 drugs: comparison of unbound concentration ratios to in vivo P-glycoprotein efflux ratios. Drug Metab Dispos 35 660–666. [DOI] [PubMed] [Google Scholar]
- Kalvass JC, Olson ER, Cassidy MP, Selley DE, and Pollack GM (2007b) Pharmacokinetics and pharmacodynamics of seven opioids in P-gp-competent mice: assessment of unbound brain EC50 and correlation of in vitro, preclinical, and clinical data. J Pharmacol Exp Ther 323 346–355. [DOI] [PubMed] [Google Scholar]
- Kalvass JC and Pollack GM (2007) Kinetic considerations for the quantitative assessment of efflux activity and inhibition: implications for understanding and predicting the effects of efflux inhibition. Pharm Res 24 265–276. [DOI] [PubMed] [Google Scholar]
- Ling KH, Leeson GA, Burmaster SD, Hook RH, Reith MK, and Cheng LK (1995) Metabolism of terfenadine associated with CYP3A(4) activity in human hepatic microsomes. Drug Metab Dispos 23 631–636. [PubMed] [Google Scholar]
- Liu X and Chen C (2005) Strategies to optimize brain penetration in drug discovery. Curr Opin Drug Discov Devel 8 505–512. [PubMed] [Google Scholar]
- Liu X, Smith BJ, Chen C, Callegari E, Becker SL, Chen X, Cianfrogna J, Doran AC, Doran SD, Gibbs JP, et al. (2005) Use of a physiologically based pharmacokinetic model to study the time to reach brain equilibrium: an experimental analysis of the role of blood-brain barrier permeability, plasma protein binding, and brain tissue binding. J Pharmacol Exp Ther 313 1254–1262. [DOI] [PubMed] [Google Scholar]
- Liu X, Tu M, Kelly RS, Chen C, and Smith BJ (2004) Development of a computational approach to predict blood-brain barrier permeability. Drug Metab Dispos 32 132–139. [DOI] [PubMed] [Google Scholar]
- Mahar Doan KM, Wring SA, Shampine LJ, Jordan KH, Bishop JP, Kratz J, Yang E, Serabjit-Singh CJ, Adkison KK, and Polli JW (2004) Steady-state brain concentrations of antihistamines in rats: interplay of membrane permeability, P-glycoprotein efflux and plasma protein binding. Pharmacology 72 92–98. [DOI] [PubMed] [Google Scholar]
- Markham A and Wagstaff AJ (1998) Fexofenadine. Drugs 55 269–274; discussion 275–266. [DOI] [PubMed] [Google Scholar]
- Mattila MJ and Paakkari I (1999) Variations among non-sedating antihistamines: are there real differences? Eur J Clin Pharmacol 55 85–93. [DOI] [PubMed] [Google Scholar]
- Meyer RP, Gehlhaus M, Knoth R, and Volk B (2007) Expression and function of cytochrome p450 in brain drug metabolism. Curr Drug Metab 8 297–306. [DOI] [PubMed] [Google Scholar]
- Meyer RP, Hagemeyer CE, Knoth R, Kaufmann MR, and Volk B (2006) Anti-epileptic drug phenytoin enhances androgen metabolism and androgen receptor expression in murine hippocampus. J Neurochem 96 460–472. [DOI] [PubMed] [Google Scholar]
- Miksys SL and Tyndale RF (2002) Drug-metabolizing cytochrome P450s in the brain. J Psychiatry Neurosci 27 406–415. [PMC free article] [PubMed] [Google Scholar]
- Obradovic T, Dobson GG, Shingaki T, Kungu T, and Hidalgo IJ (2007) Assessment of the first and second generation antihistamines brain penetration and role of P-glycoprotein. Pharm Res 24 318–327. [DOI] [PubMed] [Google Scholar]
- Petri N, Tannergren C, Rungstad D, and Lennernas H (2004) Transport characteristics of fexofenadine in the Caco-2 cell model. Pharm Res 21 1398–1404. [DOI] [PubMed] [Google Scholar]
- Polli JW, Baughman TM, Humphreys JE, Jordan KH, Mote AL, Salisbury JA, Tippin TK, and Serabjit-Singh CJ (2003) P-glycoprotein influences the brain concentrations of cetirizine (Zyrtec), a second-generation non-sedating antihistamine. J Pharm Sci 92 2082–2089. [DOI] [PubMed] [Google Scholar]
- Rudick RA, Zirretta DK, and Herndon RM (1982) Clearance of albumin from mouse subarachnoid space: a measure of CSF bulk flow. J Neurosci Methods 6 253–259. [DOI] [PubMed] [Google Scholar]
- Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, Mol CA, van der Valk MA, Robanus-Maandag EC, and te Riele HP (1994) Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 77 491–502. [DOI] [PubMed] [Google Scholar]
- Strobel HW, Thompson CM, and Antonovic L (2001) Cytochromes P450 in brain: function and significance. Curr Drug Metab 2 199–214. [DOI] [PubMed] [Google Scholar]
- Summerfield SG, Stevens AJ, Cutler L, del Carmen Osuna M, Hammond B, Tang SP, Hersey A, Spalding DJ, and Jeffrey P (2006) Improving the in vitro prediction of in vivo central nervous system penetration: integrating permeability, P-glycoprotein efflux, and free fractions in blood and brain. J Pharmacol Exp Ther 316 1282–1290. [DOI] [PubMed] [Google Scholar]
- Tahara H, Kusuhara H, Fuse E, and Sugiyama Y (2005) P-glycoprotein plays a major role in the efflux of fexofenadine in the small intestine and blood-brain barrier, but only a limited role in its biliary excretion. Drug Metab Dispos 33 963–968. [DOI] [PubMed] [Google Scholar]
- Takasato Y, Rapoport SI, and Smith QR (1984) An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am J Physiol 247 H484–H493. [DOI] [PubMed] [Google Scholar]