

Abstract
ACAPHA, a botanical drug for the treatment of human esophageal cancer in China, is under investigation as a lung cancer chemoprevention agent at the BC Cancer Agency (Vancouver, BC, Canada). Little or no information is available on the pharmacokinetics of ACAPHA in animals. The objectives of this study were as follows: to examine the disposition kinetics of matrine, a bioactive marker of ACAPHA in the rat; to develop a physiologically based pharmacokinetic (PBPK) model for pure matrine; and to characterize the absorption and clearance of crude matrine in ACAPHA-treated rats using the PBPK model. Pure matrine (15 mg/kg) or crude matrine in the form of ACAPHA (0.38 or 3.8 g/kg) was administered to the rat by gavages. The rats were sacrificed at different time points postdosing. Blood and major organs were removed from the rat, extracted with toluene/butanol, and quantified for matrine using gas chromatography-mass spectrometry. An 11-compartment, flow-limited PBPK model of matrine was developed. The PBPK model was able to simulate closely the empirical data of rats treated with pure matrine. Because the absorption and clearance of crude matrine in ACAPHA-treated rats could not be parameterized a priori, they were estimated by fitting the experimental data to the PBPK model. Results of the study show that pure matrine is absorbed and eliminated by the rat at faster rates than crude matrine. Moreover, the ACAPHA matrix may change the pharmacokinetics of matrine in the rat significantly. The PBPK model is a valuable tool to gain insights into the disposition kinetics of a botanical drug.
ACAPHA (also known as antitumor A) is a botanical drug prepared from six different Chinese herbs, including Sophora tonkinensis, Polygonum bistorta, Prunella vulgaris, Sonchus brachyotus, Dictamnus dasycarpus, and Dioscorea bulbifera. Previous clinical trial studies have shown that ACAPHA treatment can reduce cancer progression in patients with marked esophageal dysplasia by 50% (Lin et al., 1990). ACAPHA also decreases chemically induced lung tumor multiplicity and tumor load by 40 and 70%, respectively, in p53 transgenic mice that lack the Ink4a/Arf tumor suppressor genes (Zhang et al., 2004). ACAPHA is currently under investigation as a potential chemoprevention agent for lung cancer in former smokers at the BC Cancer Agency (Vancouver, BC, Canada). Chemoprevention refers to the use of food, beverages, or pharmacological agents in inhibiting, delaying, or reversing the progression of carcinogenesis (Hong and Sporn, 1997).
In the present study, the pharmacokinetics (PK) of ACAPHA is studied in the rat using matrine, a quinolizidine alkaloid of the S. tonkinensis roots, as a bioactive marker (Srinivasan, 2006). Figure 1 shows the chemical structure of matrine. The rationale of using matrine as a bioactive marker of ACAPHA is as follows: 1) Matrine and related chemicals have been shown to possess antineoplastic and anti-inflammatory activities toward leukemia cells (Zhu, 2001). These chemicals also are capable of stimulating immunological activity and inhibiting tumor growth in humans (Chang, 1992; Xu and Jiang, 1998). 2) Although matrine constitutes <2% (w/w) of the S. tonkinensis roots, it is the only alkaloid found in the plasma/tissues of rats after ACAPHA administration (Sit et al., 2004; Gao, 2007). 3) Specific and sensitive analytical methods, such as gas chromatography (GC)/mass spectrometry (MS) (Sit et al., 2004), high-performance liquid chromatography/UV (Wu et al., 2003), and high-performance liquid chromatography/MS (Wang et al., 2005), have been developed to detect and quantify matrine in biological samples. 4) Matrine standard of high chemical purity is readily available from commercial sources.
Fig. 1.

Chemical structure of matrine.
Little or no information was available on the PK of crude matrine in animals or humans after administering the extract of an herbal product, except the Sophora flavescens Ait roots (Wu et al., 2005; Zhang et al., 2008). In contrast, the PK of pure matrine was studied extensively in animals (Luo and Xia, 1991; Wang and Huang, 1992; Wu et al., 2003) and humans (Wang et al., 1994). Plasma matrine PK in the rat was linearly related to the intravenous dose from 4 to 40 mg/kg (Wu et al., 2003). Matrine was found to distribute widely to the plasma/tissues of the rat after administering a single oral dose of the chemical (Luo and Xia, 1991). The time course of matrine concentrations in human (Wang et al., 1994) and rabbit (Wang and Huang, 1992) plasma could be described by a two-compartment pharmacokinetic model after intravenous administration.
We have developed a physiologically based pharmacokinetic (PBPK) model (Gerlowski and Jain, 1983) to study the pharmacokinetic properties and tissue distribution of matrine in the rat after administration of pure matrine. The PBPK model is based largely on the actual anatomy and physiology of the rat and the physicochemical properties of matrine. Although the PBPK model has been used widely to study the PK of individual environmental chemicals (Clewell and Andersen, 1985) and/or their mixtures (Haddad et al., 1999), relatively few PBPK models have been developed for the prescription drug (Theil et al., 2003). Even fewer PBPK models are developed for the botanical drug. As far as we know, only the following botanical drug PBPK models have been reported to date: soy isoflavones (Schlosser et al., 2006; Law, 2007a), tea catechins (Law, 2006, 2007b), caffeine (Ginsberg et al., 2004), sophoridine (Hu and Huang, 1995), and glycyrrhizic acid (Ploeger et al., 2000). PBPK models had been used to extrapolate dose-response relationships among species, routes of administration, and dosage regimens (Nestorov, 2003). They also were used to predict the concentration-time profiles of several bioactive chemicals simultaneously in humans after administration of an herbal product (Law, 2007a,b). The objectives of the present study were to study matrine tissue distribution in the rat after administering pure matrine or crude matrine in the form of ACAPHA, to develop a PBPK model of matrine based on the kinetic data derived from pure matrine-treated rats, and to characterize crude matrine absorption and clearance in ACAPHA-treated rats using the PBPK model.
Materials and Methods
Chemicals. Matrine was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Chemical purity of matrine, determined by GC/MS and high-performance liquid chromatography, was >99%. Pure matrine was fine white powders that dissolved readily in water and common organic solvents. Matrine-14, 14-d2 was synthesized in our laboratory using a chemical reaction that could replace >97% of the hydrogen at position 14 of the matrine molecule with deuterium (Sit et al., 2004). ACAPHA was a gift from Global Cancer Strategies Ltd. (British Columbia, Canada); it was a mixture of dark brown powders and amorphous solids. ACAPHA was standardized to 0.4% (w/w) of pure matrine by the manufacturer. No oxymatrine was present in the final product.
Animals. Male Sprague-Dawley rats (325–375 g) were purchased from Charles River Canada (Montreal, QC, Canada). The rats were maintained on a constant light/dark cycle with light from 7:00 AM to 7:00 PM and darkness from 7:00 PM to 7:00 AM. Tap water and food were provided ad libitum. The rats were used in the tissue distribution studies after a 7-day acclimation period. The procedure associated with animal care and experimentation was carried out according to the Canadian Council on Animal Care Guidelines and with formal approval of the Simon Fraser University Animal Care Committee.
Preparation of ACAPHA Suspension and Matrine Solution. Pure matrine was dissolved in distilled water as a 1 mg/ml solution before being administered to the rat. ACAPHA did not dissolve in water completely; it was prepared as aqueous slurry (25 or 250 mg/ml), which was dispersed by ultrasonication before use.
Animal Studies. Rats were fasted overnight but allowed free access to water before pure matrine or ACAPHA administration. Groups of rats (n = 18) were given a single dose of ACAPHA (3.8 or 0.38 g/kg) or pure matrine (15 mg/kg) by gavages. Because ACAPHA was standardized to 0.4% matrine (w/w), a 3.8 g/kg ACAPHA dose was equivalent to a dose of 15 mg/kg pure matrine. After dosing, three rats from each group were randomly selected and sacrificed by CO2 asphyxiation at different time points: 0, 0.25, 1, 2, 4, and 6 h for the 0.38 g/kg ACAPHA study and 0, 0.25, 1, 3, 6, and 12 h for the 3.8 g/kg ACAPHA and the 15 mg/kg pure matrine studies. A blood sample was withdrawn immediately from the rat by cardiac puncture and centrifuged at 3000g for 5 min to collect the plasma. Major organs such as the heart, liver, spleen, lung, kidney, fat, muscle, and brain also were collected from the rat. The organs were rinsed briefly in distilled water, wiped dry with paper, and stored in vials at –20°C until analysis. Matrine was stable chemically under these storage conditions for at least 1 month.
Blood/Plasma Ratio. Approximately 5 ml of freshly drawn rat blood was mixed with a standard matrine solution to a final concentration of 10 or 100 μg/ml. After incubation at 37°C for 30 min, aliquots of the blood samples were removed and centrifuged at 3000g for 5 min at room temperature to obtain the plasma. Preliminary studies indicated that matrine equilibrium was established between the blood and the plasma in 20 min under these experimental conditions. Matrine concentrations in the plasma samples were measured by GC/MS (Sit et al., 2004). The concentrations in blood were assumed to be the theoretical concentration. The blood/plasma concentration ratio was calculated by dividing matrine concentration in the whole blood with those in the plasma.
Extraction of Matrine from Plasma and Tissue Samples. Plasma/tissue samples were extracted and quantified for matrine according to the procedure of Sit et al. (2004). In brief, approximately 1.0 g of tissue sample was weighed and homogenized in 3 ml of distilled water with a Kinematica homogenizer (PCU-2-110; Kinematica, Littau-Lucerne, Switzerland). A 1-ml aliquot of the tissue homogenate or plasma was removed and mixed with 250 ng of matrine-14,14-d2 (the internal standard), 3 ml of toluene/butanol [7:3 (v/v)], and 0.5 ml of NaOH (1 M) in a centrifuge tube. The mixture was shaken on a mechanical shaker for 20 min and centrifuged at 3000 rpm to separate the layers. The organic layer was removed and extracted with 0.5 ml of HCl (0.25 M). The organic layer was discarded, and the remaining aqueous layer was neutralized with 0.5 ml of 1.0 M NaOH before being extracted by 200 μl of toluene/butanol [9:1 (v/v)]. The organic layer was separated by centrifugation and analyzed by GC/MS (Sit et al., 2004). Matrine concentration in the tissue sample was calculated after correcting for the dilution volume and tissue weight.
Gas Chromatography/Mass Spectrometric Determination of Matrine. A Hewlett-Packard 5890 series II gas chromatograph (Hewlett Packard, Palo Alto, CA) coupled to a 5971 mass spectrometric detector was used to quantify matrine in the toluene/butanol [9:1 (v/v)] extract. Chromatographic separation was performed with a 5% diphenyl-95% dimethylpolysiloxane capillary column (30 m × 0.25 mm × 0.25 μm, HP-5 MS). Helium was used as the carrier gas. The initial oven temperature was set at 110°C, maintained for 1 min, and then increased to 220°C at a rate of 30°C /min and maintained for 1 min. The temperature was further increased to 300°C at a rate of 15°C/min and maintained for 3 min. Ionization was performed under electron impact ionization with 70 eV. Matrine and matrine-14,14-d2 were quantified using m/z 248 m/z 250, respectively (Sit et al., 2004). A linear response was obtained with matrine standards over the range of 10 to 500 ng/ml. The lower limit of quantitation (LLOQ) of the GC/MS method for matrine was 12 ng/ml plasma or tissue homogenate. A plasma/tissue sample was assigned to half of the LLOQ when it was below 12 ng/ml in concentration. Thus, if two consecutive time points were below the LLOQ, the first and second time points would be given 6.0 and 0.0 ng/ml concentrations, respectively.
Model Structure. The PBPK model of matrine for the rat consisted of 11 flow-limited compartments including the lung, heart, kidney, fat, muscle, brain, liver, spleen, gut, blood, and the rest of body (Fig. 2). The rest of the body compartment includes all other tissues that had not been identified in the model, i.e., the skin, bone, eye, prostate gland, etc. An 11-compartment PBPK model was used in the present study because ACAPHA was a multitarget therapeutic/prophylactic agent with different disease treatment endpoints. A linear, first order input function was used to describe oral absorption of matrine (see Appendix). Matrine metabolism was not considered in the present PBPK model because no matrine was metabolized by the rat (Xie et al., 1983; Gao, 2007). Matrine was excreted mainly by the kidney of the PBPK model (Fig. 2) because the rat excreted approximately 52 and 0.36% of the dose in the urine and feces, respectively, within 24 h after matrine administration (Luo and Xia, 1991).
Fig. 2.

Schematic diagram of the physiologically based pharmacokinetic model used to simulate the disposition of matrine in plasma and tissues of rats after administration of pure matrine or ACAPHA. C, matrine concentrations (ng/g or ml); Q, plasma flow rates (l/h). Subscripts refer to tissues (see Appendix).
Model Parameters. Physiological parameters. All physiological parameters are parameterized a priori (Table 1). The tissue volume and blood flow were expressed as a fraction of total body volume and cardiac output, respectively, and were taken from the literature (Luttringer at al., 2003). The total blood volume was divided into a two-thirds venous pool and one-third arterial pool. Gut contents was assumed to be 0.014 liters for rats weighing 0.25 kg (Angelo and Pritchard, 1987).
TABLE 1.
Parameters used in the physiologically based pharmacokinetic model of matrine for the rata
| Tissues | Blood Flow Rates (Percentage of Cardiac Outputb) | Volume (Percentage of Body Weight) | Tissue/Plasma Partition Coefficients |
|---|---|---|---|
| Adipose | 7.0 | 7.6 | 0.45 |
| Brain | 2.0 | 0.57 | 2.0 |
| Heart | 4.9 | 0.33 | 1.5 |
| Kidney | 14.1 | 0.73 | 10 |
| Liver | 17.5 | 3.66 | 5.5 |
| Gastrointestinal tract | 13.1 | 2.7 | 3.0 |
| Lung | 0.5 | 1.5 | |
| Muscle | 27.8 | 40.4 | 1.3 |
| Spleen | 2.0 | 0.2 | 6.3 |
| Blood | 8.2 | ||
| Rest of the body | 26.7 | 35.1 | 5.0 |
Mean data on tissue blood flow rate and volume were adapted from Luttringer et al. (2003).
Cardiac output was scaled from an allometric equation (14.1 × body weight0.75) based on 7.08 l/h for a 0.4-kg rat.
Tissue/plasma partition coefficients. In vivo tissue/plasma partition coefficients (Table 1) were estimated using the area method of Gallo et al. (1987) as follows: the kinetic data of rats after administering a single oral dose of 15 mg/kg pure matrine were analyzed using the noncompartmental approach of WinNonlin (version 1.5; Pharsight, Mountain View, CA). The area under the concentration-time curve (AUC) was calculated using the logarithmic trapezoidal rule. The tissue/plasma partition coefficient of a specific tissue was calculated by dividing the AUC of the tissue with the AUC of plasma.
Model parameterization for pure matrine. The blood/plasma ratio (BLPLR) of matrine in the rat was determined experimentally to be 0.97 ± 0.06 (mean ± S.D.) at our laboratory. An absorption rate constant (ka), 0.32 h–1, was estimated by fitting the plasma concentration-time data of Wu at al. (2003) to a one-compartment pharmacokinetic model with WinNonlin (version 1.5; Pharsight). Renal clearance (CL) was found to range from 1.4 to 1.6 l/h/kg. A CL of 1.4 l/h/kg was obtained by dividing the bolus intravenous dose with the plasma AUC reported by Wu et al. (2003). A second CL, 1.6 l/h/kg, was calculated from the renal excretion rate of matrine in the study of Luo and Xia (1991). Hepatic extraction ratio (EH) was assumed to be 0.3% of the administered dose because only 0.27% of an orally administered dose was excreted in the bile in 12 h (Luo and Xia, 1991). Moreover, matrine was not metabolized by the rats (Xie et al., 1983; Gao, 2007). The fecal excretion rate constant (kf) was assumed to be 1/transit time of the small intestine, and the transit time of small intestine was obtained from Davies and Morris (1993). The experimental bioavailability factor (F) was assumed to be 0.44 of the administered dose because approximately 44% of an orally administered dose was absorbed into the systemic circulation (Wu et al., 2003). It should be pointed out that F in the present study had no relationship to the absolute bioavailability of matrine (Anderton et al., 2004). Adjustments were made on the parameters with available experimental data during model development. However, once the parameter values were finalized (Table 2), no further adjustment was allowed.
TABLE 2.
Pharmacokinetic parameters of rats receive a single oral dose of pure matrine or ACAPHAa
| Parameter | Pure Matrine | ACAPHA |
|---|---|---|
| BLPLR | 1.0 | 1.0 |
| ka (h-1) | 6.5 | 4.0 (4.0)b |
| EH | 0.003 | 0.003 |
| F | 0.85 | 0.35 (0.85)b |
| tlag (h) | 0.1 | 0.1 |
| kf (h-1) | 0.67 | 0.67 |
| CL (l/h/kg) | 1.2 | 0.20 (0.35)b |
Rats were treated with a single oral dose of pure matrine (15 mg/kg) or ACAPHA (3.8 or 0.38 g/kg). ka, the absorption rate constant; CL, the renal clearance of matrine.
These parameters were optimized based on the experimental data of the ACAPHA studies; parameter values in brackets were for the 0.38 g/kg ACAPHA study.
Model validation. The PBPK model was validated by comparing model simulated results with the experimental data of Wistar rats after administering 40 mg/kg pure matrine as a single oral dose or as an intravenous bolus injection (Wu et al., 2003). The PBPK model was assumed to be validated if model simulation described closely the experimental data. Because we were unsuccessful in obtaining the original data from these authors, the concentration-time data were read digitally with ImageJ (Rasband, 2008). The parameter values of pure matrine (Table 2) were used to run the PBPK model in the validation study.
Computer Simulation and Data Fitting. Computer simulation. The differential and algebraic equations describing the movement of matrine through the rat were formulated as a computer program. Mass balance differential equations for the model and the definition of the algebraic terms were given in the Appendix. The set of differential and algebraic equations incorporating the parameters in Tables 1 and 2 were solved numerically with the aid of AcslXtreme 2.0.1.2 (AEgis Technologies Group, Inc., Huntsville, AL).
Data fitting. The ka, F, and CL values of crude matrine after ACAPHA administration could not be parameterized a priori; they were estimated by fitting the experimental data to the PBPK model. The optimization procedure was carried out with all the parameter values of the pure matrine PBPK model fixed (Tables 1 and 2), except ka, F, and CL. Thus, ka, F, and CL were adjusted manually at first to get a feeling of the parameter effects on the simulation results. The adjusted values were then fitted to the experimental data using the maximized log likelihood function of AcslXtreme OptStat (AEgis Technologies Group, Inc.). Upper and lower bound values were also assigned to each of the ka, F, and CL parameters to ensure the final parameter values were biologically and mathematically logical.
Statistical and Sensitivity Analyses. Statistical analysis. Mean deviations between tissue concentrations predicted by the PBPK model (Cpred) and the experimental data (Cexptl) were expressed as the mean prediction error (MPE) according to the following equation (Wu, 1995):
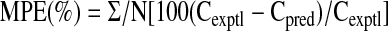 |
Prediction error was analogous to the calculation of “N-fold” deviations, and N was the number of time points on the tissue compartment.
Sensitivity analysis. Sensitivity was calculated by the following equation (Clewell et al., 1994):
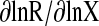 |
where R was a model output, and X was the parameter for which the sensitivity was assessed. The sensitivity coefficient was log-normalized to the parameter, and the log-normalized sensitivity coefficient (LNSC) was calculated by AcslXtreme 2.0.1.2 (AEgis Simulation, Inc.) using the central difference method. The LNSC represented the percentage change in an output value associated with the percentage change in the input parameter. Because matrine concentration in the lung and the blood compartments were the outputs of concern in our study, the sensitivities of lung and blood matrine concentrations were examined with respect to the following parameters: F, ka, tlag, EH, CL, BLPLR, and kf. Thus, if the absolute value of an LNSC was high, the parameter would have an important effect on the model prediction. If the absolute value of a LNSC was close to zero, the model parameter would have only a small effect on model prediction (Clewell et al., 1994).
Results
Matrine tissue distribution in the rat was studied using a single oral dose of pure matrine (15 mg/kg) or ACAPHA (0.38 or 3.8 g/kg). Preliminary results showed that the 3.8 g/kg ACAPHA study was limited by the volume of slurries that could be administered to the rat, whereas the 0.38 g/kg ACAPHA study was restricted by the low levels of matrine found in the plasma/tissues of the rat and the LLOQ of the GC/MS method. Because pure matrine was readily soluble in water, it was administered as an aqueous solution to the rat.
Figure 3 depicts the time course of model-simulated matrine concentrations and the mean experimental data of the rat after pure matrine or ACAPHA administration. The concentration-time data of individual plasma/tissue displayed the typical kinetic profile of an orally administered drug; matrine concentrations rose rapidly during the absorption phase and declined slowly in the postabsorptive phase. The 3.8 g/kg ACAPHA study had approximately 10-fold higher plasma/tissue matrine concentrations than the 0.38 g/kg ACAPHA study, of which the matrine concentrations were the lowest in the present studies. It is interesting that matrine concentrations in the 3.8 g/kg ACAPHA study were initially lower than the pure matrine study but became higher at approximately 2 (kidney) or 4 (plasma and other tissues) h postdosing (Fig. 3). In addition, the kinetic profiles of pure matrine and 3.8 g/kg ACAPHA studies were different, although both studies were conducted with the same matrine dose equivalents. Thus, the kinetic profile of a specific tissue in the 0.38 g/kg ACAPHA study but not the 3.8 g/kg ACAPHA study was somewhat parallel to that of the corresponding tissue in the pure matrine study (Fig. 3). Nevertheless, the PBPK model was able to simulate all pure matrine and ACAPHA data closely (Fig. 3).
Fig. 3.


Comparison of model-simulated and experimental matrine concentrations in the tissue/plasma of rats after receiving pure matrine or ACAPHA. a, plasma; b, kidney; c, spleen; d, brain; e, heart; f, muscle; g, lung; h, liver; i, fat. •, experimental data of rats receiving 15 mg/kg pure matrine; □, experimental data of rats receiving 3.8 g/kg ACAPHA; ▪, experimental data of rats receiving 0.38 g/kg ACAPHA. Data points, means ± S.D. of the experimental plasma/tissue concentrations from three different rats; –– –– ––, model-simulated matrine concentrations of rats receiving 15 mg/kg pure matrine; ———, model-simulated matrine concentrations of rats receiving 3.8 g/kg ACAPHA; —--—--, model-simulated matrine concentrations of rats receiving 0.38 g/kg ACAPHA.
Tables 1 and 2 list the final pharmacokinetic parameters that are used to implement the PBPK model. Two different sets of ka, F, and CL values (Table 2) were required to implement the PBPK model after pure matrine and ACAPHA administration because they were essentially two different matrine formulations. Two different sets of F and CL values (Table 2) also were needed to simulate the kinetic profiles of the rat after administration of 3.8 and 0.38 g/kg ACAPHA because they were the “best fitted” parameter values of the experimental data. An attempt was made to simulate both high- and low-dose ACAPHA data with a single set of F and CL values but with very little success because a good fit to the experimental data could be achieved only with the high- or low-dose study but not both studies concurrently (data not shown).
The PBPK model of matrine in the rat was validated using the empirical data of Wu et al. (2003). As shown in Fig. 4, the PBPK model was able to predict closely the time course of matrine concentrations in the plasma of rats after administering 40 mg/kg pure matrine via the intravenous (Fig. 4a) or the p.o. (Fig. 4b) route.
Fig. 4.

Time course of matrine plasma concentrations in rats receive: a, bolus intravenous injection of 40 mg/kg pure matrine; or b, single oral dose of 40 mg/kg pure matrine. Solid line, model simulation. Data points, mean of experimental data. They were read digitally from the publication of Wu et al. (2003).
The MPE was used to confirm our initial observation that model predictions described closely the experimental data (see Fig. 3). Deviations within a factor of 2 between model prediction and experimental data, i.e., a MPE absolute value of <50%, were taken as the evidence that the PBPK model described the empirical data closely (Poulin and Theil, 2002). As shown in Table 3, all MPE absolute values were <50% between the high- and low-dose ACAPHA studies. Likewise, all MPE absolute values were <50% in the pure matrine study, except those of the spleen and lung.
TABLE 3.
Mean prediction errors over the entire simulation time ranges
| Tissue | Pure Matrine (15 mg/kg) | ACAPHA (0.38 g/kg) | ACAPHA (3.8 g/kg)a |
|---|---|---|---|
| Adipose | -19.0 | -42.4 | -14.5 |
| Kidney | -0.7 | -39.9 | 6.5 |
| Spleen | -59.9b | -13.9 | -11.7 |
| Liver | -34.6 | -33.8 | -25.5 |
| Brain | -28.6 | -10.8 | -31.6 |
| Heart | -36.6 | 30.4 | -45.3 |
| Plasma | -1.5 | 16.5 | -21.6 |
| Muscle | -7.2 | 8.7 | 26.9 |
| Lung | -57.0b | -35.2 | -14.9 |
Matrine equivalent dose was 15 mg/kg.
Mean prediction error absolute value >50%.
Table 4 summarizes the LNSC of the pure matrine study. The LNSC of the CL, ka, BLPLR, and EH parameters had negative values, whereas the LNSC of the F and tlag parameters had positive values. We assigned the tested parameters to high (>0.5)-, medium-, and low (<0.05)-impact groups according to the LNSC absolute values (Table 4). Because the LNSC of CL, BLPLR, and F were >0.5 in absolute values, they had the most impact on the predicted matrine concentrations in the lung and/or blood. Likewise, ka was found to have a moderate impact on the predicted matrine concentrations in the blood and lung. Because the LNSC absolute values of the EH, tlag, and kf parameters were <0.05, these parameters had only minor to negligible impacts on the predicted matrine concentrations in the blood and lung.
TABLE 4.
Normalized sensitivity coefficients for matrine concentrations in the lung and the plasma
|
Parametersa
|
Postdosing Time
|
|||||
|---|---|---|---|---|---|---|
| 2 h | 4 h | 6 h | 8 h | 10 h | 12 h | |
| High (>0.5) | ||||||
| CL | -0.93 (-0.62)b | -0.85 (-0.56) | -0.60 (-0.40) | -0.37 (-0.25) | -0.22 (-0.15) | -0.12 (-0.08) |
| BLPLR | 0.00 (-0.86) | 0.00 (-0.40) | 0.00 (-0.19) | 0.00 (-0.09) | 0.00 (-0.04) | 0.00 (-0.02) |
| F | 1.00 (0.86) | 0.61 (0.41) | 0.29 (0.19) | 0.14 (0.09) | 0.06 (0.04) | 0.03 (0.02) |
| Medium | ||||||
| ka | -0.14 (-0.09) | -0.06 (-0.04) | -0.03 (-0.02) | -0.01 (-0.01) | -0.01 (-0.00) | -0.00 (-0.00) |
| Low (<0.05) | ||||||
| EH | -0.01 (-0.01) | -0.01 (-0.01) | -0.01 (-0.00) | -0.00 (-0.00) | -0.00 (-0.00) | -0.00 (-0.00) |
| tlag | 0.05 (0.03) | 0.02 (0.02) | 0.01 (0.01) | 0.01 (0.00) | 0.00 (0.00) | 0.00 (0.00) |
| kf | 0.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) |
For explanation on abbreviations of the parameters, see Table 2.
Numbers without parentheses are sensitivity coefficients of the lung, and numbers in parentheses are sensitivity coefficients of the blood.
Discussion
Our PBPK model provides a simple description on the movement of matrine in the rat after pure matrine or ACAPHA administration (Fig. 2; Table 2). A comparison of the predicted versus actual concentration-time profiles (Fig. 3) show that the PBPK model is able to simulate the empirical data closely (Table 3). A surprise finding of our study is that different plasma/tissue kinetic profiles are found in the pure matrine and 3.8 g/kg ACAPHA studies although the matrine dose equivalents are the same in both studies (Fig. 3). An explanation for the different kinetic profiles in these studies is not readily available but may be related to the diminished matrine absorption (ka) and/or CL in the 3.8 g/kg ACAPHA study because of interactions between ACAPHA matrix and the gastrointestinal tract or the kidney: 1) For ka, as shown in Table 2, the final pure matrine ka value of the PBPK model is 6.5 h–1. It is approximately 20-fold higher than the 0.32 h–1 ka value derived by fitting the plasma concentration data of Wu et al. (2003) to a one-compartment pharmacokinetic model. Different pure matrine ka values are observed in these studies probably because they are conducted with different doses, rat species, model structures, and/or PK approaches. Table 2 also shows that pure matrine has a higher ka value than crude matrine in ACAPHA. This is probably due to the different water solubility of pure matrine and crude matrine. Linnankoski et al. (2006) have shown that drug absorption is determined mainly by the dissolution rate in the gastrointestinal fluids. Because pure matrine dissolves in water readily, it is rapidly absorbed by the rat. However, because crude matrine is physically trapped or bound to ACAPHA matrix, it does not dissolve in water easily. Thus, crude matrine is absorbed at a slower rate than pure matrine by the rat. An alternate explanation for a small crude matrine ka value may be that the ACAPHA matrix is able to inhibit active matrine transport and/or compete with matrine for absorption in the gastrointestinal tract. 2) For CL, the final pure matrine CL value of the PBPK model is shown to be 1.2 l/h/kg (Table 2). It is very close to the 1.4 and 1.6 l/h/kg CL values derived from the experimental data of Wu et al. (2003) and Luo and Xia (1991), respectively. In contrast, the final crude matrine CL values of the PBPK model are 0.35 and 0.20 l/h/kg, respectively, in rats treated with 0.38 and 3.8 g/kg ACAPHA (Table 2). The crude matrine CL values are very close to the 0.1 l/h/kg CL value reported for the S. flavescens Ait root extract (Zhang et al., 2008). Together, these studies show that pure matrine CL is consistently higher than crude matrine CL. Perhaps the matrices of ACAPHA and S. flavescens Ait root extract are able to inhibit active matrine secretion and/or induce matrine reabsorption by interacting with the canalicular transporters and/or P-glycoproteins of the kidney. Our assumption that matrine is being actively secreted into the kidney is supported by the 1.2 l/h/kg CL value of pure matrine (Table 2), which is approximately 4-fold higher than the 0.31 l/h/kg glomerulus filtration rate of the rat (Davies and Morris, 1993).
Distribution is another process that has an impact on the PK of matrine in the rat. The kinetic parameter used to describe matrine distribution is the volume of distribution at steady-state (Vdss), the equivalent plasma volume in which matrine is distributed to the rat body. We have employed two different methods to calculate the Vdss in the present study: 1) A Vdss of 1.45 liters is estimated for pure matrine using the noncompartmental approach to analyze the mean plasma concentration-time curve (Fig. 3a), and 2) A Vdss of 1.1 liters also is calculated by adding the products of each tissue volume and the corresponding partition coefficient and the plasma volume of the PBPK model (Benowitz et al., 1974). The closeness of these Vdss values suggests that the AUC-based partition coefficient estimation method (Gallo et al., 1987) provides reasonable estimates of the true value in the rats. It should be pointed out that both Vdss values are much larger than the average body weight of the rat used in the present study, which would be indicative of extensive matrine binding with the plasma/tissue proteins. However, because the BLPLR ratio is very close to one (Table 2), protein binding does not seem to be a logical explanation for the large Vdss in our study. A more likely explanation may be that the active transporters also are involved in the tissues/organ uptake of matrine. Thus, the liver and kidney, which accumulate high levels of matrine (Figs. 3, b and h), are also known to have high active transport activities. Other possible but less likely explanations may be that matrine is bound preferentially to the different tissue macromolecules, such as the DNA/RNA (Zhang et al., 2004).
The PBPK model requires 40 or more parameters for its implementation, and not all parameters affect the simulation results equally. We have used the analytical sensitivity analysis (Clewell et al., 1994) to identify the parameters that have significant impacts on the model prediction. Our results show that CL, BLPLR, ka, and F are sensitive parameters in predicting matrine concentrations in the lung and/or blood at different time intervals postdosing (Table 4). These results are in agreement with the finding that the LNSC of PBPK model parameters are complex functions of dosage, animal species, model structures, and dose surrogates of the target tissues (Clewell et al., 1994). Table 4 also shows the LNSC of CL, ka, BLPLR, and EH are negative values, whereas the LNSC of F and tlag are positive values. These indicate an increase in lung and/or blood matrine concentrations as F and tlag increase, or as CL, ka, BLPLR, and EH decrease. Because CL and ka are smaller values in the ACAPHA studies than the pure matrine study (Table 2), ACAPHA-treated rats would accumulate more matrine in the lung compared to the pure matrine-treated rats. Because mean matrine concentrations in the lung of the 3.8 g/kg ACAPHA study are higher than those of the pure matrine study at approximately 4 h postdosing and thereafter (Fig. 3), ACAPHA may be a better lung cancer chemoprevention agent than pure matrine. Further studies are required to determine whether the amount of matrine accumulated by the lung after ACAPHA administration can inhibit tumor growth while at the same time causing no intolerable toxicity to the normal tissues.
A PBPK model of sophoridine has been developed in the rat after a bolus intravenous injection of the chemical (Hu and Huang, 1995). Sophoridine is a stereoisomer of matrine. The BLPLR, CL, and Vdss parameter values of the sophoridine PBPK model are 1.0 and 1.35 l/h/kg and 0.89 liters, respectively (Hu and Huang, 1995), which are very close to the corresponding parameter values in our pure matrine study (Table 2). As a stereoisomer of matrine, sophoridine is expected to have similar physicochemical properties and PBPK model parameter values as matrine, but the Vdss of sophoridine (0.86 liters) is slightly less than the Vdss in our pure matrine study (see above). Nevertheless, in agreement with our study, the Vdss of sophoridine is much larger than the average body weight (0.23 kg) of the rat used in this study (Hu and Huang, 1995).
In summary, our investigations have demonstrated the usefulness of the PBPK model in gaining insights into the physiological, biochemical, and physical factors that play important roles in the observed kinetic behavior of matrine in the rat after pure matrine or ACAPHA administration. The PBPK model framework developed in this study also can be used to study the PK of dietary supplements, nutraceuticals, and functional foods.
Appendix
Mass balance equations for the PBPK model depicted in Fig. 2 describing matrine concentrations in arterial blood (CBA), venous blood (CBV), lung (CLG), heart (CHR), kidney (CKD), fat (CFT), muscle (CMS), brain (CBR), liver (CLV), spleen (CSP), gut tissue (CGT), gut contents (CGC), and the rest of the body (CRB) are presented.
Noneliminating Organs and Tissues
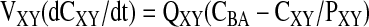 |
(1) |
where XY represents the heart, brain, spleen, fat, muscle, gut, and the rest of the body. The terms QXY, VXY, and PXY represent tissue blood flow, volume, and tissue/blood partition coefficient, respectively.
Liver
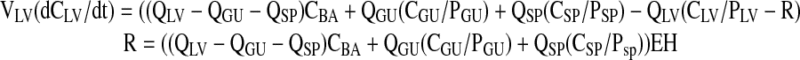 |
(2) |
where EH is the hepatic extraction ratio, and R represents biliary excretion of matrine.
Gut Tissue
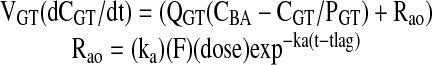 |
(3) |
where Rao is matrine input rate into the blood from the gut, ka is the absorption rate constant of matrine, dose represents matrine equivalents in ACAPHA or pure matrine, F is the apparent or empirical bioavailability factor, and tlag is the lag time for absorption.
Gut Contents
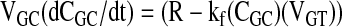 |
(4) |
where kf is the fecal excretion rate constant.
Kidney
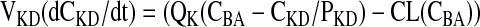 |
(5) |
where CL is the renal clearance of matrine.
Venous Blood
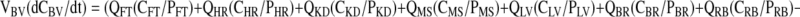 |
(6) |
Arterial Blood
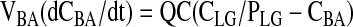 |
(7) |
Lung
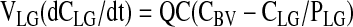 |
(8) |
This work was supported by the National Institutes of Health National Cancer Institute [Grant U01-CA96109].
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.108.023788.
ABBREVIATIONS: PK, pharmacokinetics; GC, gas chromatography; MS, mass spectrometry; PBPK, physiologically based pharmacokinetics; LLOQ, lower limit of quantification; AUC, area under the concentration-time curve; BLPLR, blood/plasma ratio; ka, absorption rate constant; CL, renal clearance; EH, hepatic extraction ratio; kf, fecal excretion rate constant; F, bioavailability factor; MPE, mean prediction error; LNSC, log-normalized sensitivity coefficient; tlag, lag time; Vdss, volume of distribution at steady-state.
References
- Anderton MJ, Manson MM, Verschoyle R, Gescher A, Steward WP, Williams ML, and Mager DE (2004) Physiological modeling of formulated and crystalline 3,3′-diindolylmethane pharmacokinetics following oral administration in mice. Drug Metab Dispos 32 632–638. [DOI] [PubMed] [Google Scholar]
- Angelo MJ and Pritchard AB (1987) Route-to-route extrapolation of dichloromethane exposure using a physiological pharmacokinetic model, in Pharmacokinetics in Risk Assessment: Drinking Water and Health, pp 254–264, National Academy Press, Washington, DC.
- Benowitz N, Forsyth FP, Melmon KL, and Rowland M (1974) Lidocaine disposition kinetics in monkey and man. I. Prediction by a perfusion model. Clin Pharmacol Ther 16 87–98. [DOI] [PubMed] [Google Scholar]
- Chang MY (1992) Anticancer Medicinal Herbs, Human Science and Technology Publishing House, Changsha, China.
- Clewell HJ and Andersen ME (1985) Risk assessment extrapolations and physiological modelling, in Advances in Health Risk Assessment for Systemic Toxicants and Chemical Mixtures: An International Symposium (Stara JF, Erdreich LS, eds), pp 111–131, Princeton Scientific Publishing Co., Princeton, NJ.
- Clewell HJ 3rd, Lee TS, and Carpenter RL (1994) Sensitivity of physiologically based pharmacokinetic models to variation in model parameters: methylene chloride. Risk Anal 14 521–531. [DOI] [PubMed] [Google Scholar]
- Davies B and Morris T (1993) Physiological parameters in laboratory animals and humans. Pharm Res 10 1093–1095. [DOI] [PubMed] [Google Scholar]
- Gallo JM, Lam FC, and Perrier DG (1987) Area method for the estimation of partition coefficients for physiological pharmacokinetic models. J Pharmacokinet Biopharm 15 271–280. [DOI] [PubMed] [Google Scholar]
- Gao GH (2007) Comparative Pharmacokinetics of Matrine: Pure Matrine vs. Crude Chemical in ACAPHA. Ph.D. thesis, Simon Fraser University, Burnaby, BC, Canada.
- Gerlowski LE and Jain RK (1983) Physiologically based pharmacokinetic modeling: principles and applications. J Pharm Sci 72 1103–1127. [DOI] [PubMed] [Google Scholar]
- Ginsberg G, Hattis D, Russ A, and Sonawane B (2004) Physiologically based pharmacokinetic (PBPK) modeling of caffeine and theophylline in neonates and adults: implications for assessing children's risks from environmental agents. J Toxicol Environ Health A 67 297–329. [DOI] [PubMed] [Google Scholar]
- Haddad S, Tardif R, Charest-Tardif G, and Krishnan K (1999) Physiological modeling of the toxicokinetic interactions in a quaternary mixture of aromatic hydrocarbons. Toxicol Appl Pharmacol 161 249–257. [DOI] [PubMed] [Google Scholar]
- Hong WK and Sporn MB (1997) Recent advances in chemoprevention of cancer. Science 278 1073–1077. [DOI] [PubMed] [Google Scholar]
- Hu YZ and Huang SK (1995) Physiological pharmacokinetic modeling of sophoridine. Chin J Pharmacol Toxicol 9 133–136. [Google Scholar]
- Law FCP (2006) A generic physiologically based pharmacokinetic model for TCM oncology research. Proceedings of the Conference of Traditional Chinese Medicine and Cancer Research: Fostering Collaborations; Advancing the Science; 2006 April 10–12; Bethesda, MD. National Cancer Institute, Bethesda, MD.
- Law FCP (2007a) Simulation of soy isoflavone kinetic profiles in rodents and humans using physiologically based pharmacokinetic (PBPK) model. Proceedings of the XI International Congress of Toxicology; 2007 July 15–19; Montreal, QC, Canada. Society of Toxicology of Canada, Montreal, QC, Canada.
- Law FCP (2007b) Incorporating physiologically-based pharmacokinetic (PBPK) model in clinical trials of Chinese medicine: a new approach for the product development processes. Proceedings of the 2007 International Conference and Exhibition of the Modernization of Chinese Medicine and Health Products; 2007 August 16–20; Hong Kong, SAR, China. Hong Kong Trade Development Council and the Modern Chinese Medicine International Association Limited, Hong Kong, SAR, China.
- Lin P, Zhang J, Rong Z, Han R, Xu S, Gao R, Ding Z, Wang J, Feng H, and Cao S (1990) Studies on medicamentous inhibitory therapy for esophageal precancerous lesions: 3- and 5-year inhibitory effects of antitumor-B, retinamide and riboflavin. Proc Chin Acad Med Sci Peking Union Med Coll 5 121–129. [PubMed] [Google Scholar]
- Linnankoski J, Mäkelä JM, Ranta VP, Urtti A, and Yliperttula M (2006) Computational prediction of oral drug absorption based on absorption rate constants in humans. J Med Chem 49 3674–3681. [DOI] [PubMed] [Google Scholar]
- Luo XY and Xia BN (1991) Pharmacokinetic study of matrine. Guiyang Med J 16 180–183. [Google Scholar]
- Luttringer O, Theil FP, Poulin P, Schmitt-Hoffmann AH, Guentert TW, and Lavé T (2003) Physiologically-based pharmacokinetic (PBPK) modeling of disposition of epiroprim in humans. J Pharm Sci 92 1990–2007. [DOI] [PubMed] [Google Scholar]
- Nestorov I (2003) Whole body pharmacokinetic models. Clin Pharmacokinet 42 883–908. [DOI] [PubMed] [Google Scholar]
- Ploeger B, Mensinga T, Sips A, Meulenbelt J, and DeJongh J (2000) A human physiologically-based model for glycyrrhizic acid, a compound subject to presystemic metabolism and enterohepatic cycling. Pharm Res 17 1516–1525. [DOI] [PubMed] [Google Scholar]
- Poulin P and Theil FP (2002) Prediction of pharmacokinetics prior to in vivo studies. II. Generic physiologically based pharmacokinetic models of drug disposition. J Pharm Sci 91 1358–1370. [DOI] [PubMed] [Google Scholar]
- Rasband W (2008) ImageJ, Image Processing and Analysis in Java Version 1.40, The Research Services Branch, National Institute of Mental Health, Bethesda, MD.
- Schlosser PM, Borghoff SJ, Coldham NG, David JA, and Ghosh SK (2006) Physiologically-based pharmacokinetic modeling of genistein in rats: I. Model development. Risk Anal 26 483–500. [DOI] [PubMed] [Google Scholar]
- Sit DS, Gao G, Law FC, and Li PC (2004) Gas chromatography-mass spectrometry determination of matrine in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 808 209–214. [DOI] [PubMed] [Google Scholar]
- Srinivasan VS (2006) Challenges and scientific issues in the standardization of botanicals and their preparations: United States pharmacopeia's dietary supplement verification program: a public health program. Life Sci 78 2039–2043. [DOI] [PubMed] [Google Scholar]
- Theil FP, Guentert TW, Haddad S, and Poulin P (2003) Utility of physiologically based pharmacokinetic models to drug development and rational drug discovery candidate selection. Toxicol Lett 138 29–49. [DOI] [PubMed] [Google Scholar]
- Wang XH and Huang SK (1992) Pharmacokinetics and pharmacodynamics of matrine and oxymatrine. Yao Xue Xue Bao 27 572–576. [PubMed] [Google Scholar]
- Wang PQ, Lu GH, Zhou XB, Shen JF, Chen SX, Mei SW, and Chen MF (1994) Pharmacokinetics of matrine in healthy volunteers. Yao Xue Xue Bao 29 326–329. [PubMed] [Google Scholar]
- Wang S, Wang G, Li X, Sun J, Ma R, and Sheng L (2005) Simultaneous determination of oxymatrine and its active metabolite matrine in dog plasma by liquid chromatography-mass spectrometry and its application to pharmacokinetic studies. J Chromatogr B Analyt Technol Biomed Life Sci 817 319–325. [DOI] [PubMed] [Google Scholar]
- Wu G (1995) Calculating predictive performance: a user's note. Pharmacol Res 31 393–399. [DOI] [PubMed] [Google Scholar]
- Wu X, Yamashita F, Hashida M, Chen X, and Hu Z (2003) Determination of matrine in rat plasma by high-performance liquid chromatography and its application to pharmacokinetic studies. Talanta 59 965–971. [DOI] [PubMed] [Google Scholar]
- Wu YJ, Chen JJ, and Cheng YY (2005) A sensitive and specific HPLC-MS method for the determination of sophoridine, sophocarpine and matrine in rabbit plasma. Anal Bioanal Chem 382 1595–1600. [DOI] [PubMed] [Google Scholar]
- Xie MZ, Zhou WZ, and Zhang Y (1983) Oxymatrine metabolic fate. Chin Med J (Engl) 94 145–150. [PubMed] [Google Scholar]
- Xu XR and Jiang JK (1998) Recent progress in anticancer bioactivity study of Sophora flavescens and its alkaloids. Chin J Integr Tradit Chin Western Med 4 235–239. [Google Scholar]
- Zhang L, Liu W, Zhang R, Wang Z, Shen Z, Chen X, and Bi K (2008) Pharmacokinetic study of matrine, oxymatrine and oxysophocarpine in rat plasma after oral administration of Sophora flavescens Ait: extract by liquid chromatography tandem mass spectrometry. J Pharm Biomed Anal 47 892–898. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wang Y, Yao R, Li J, Yan Y, La Regina M, Lemon WL, Grubbs CJ, Lubet RA, and You M (2004) Cancer chemopreventive activity of a mixture of Chinese herbs (antitumor B) in mouse lung tumor models. Oncogene 23 3841–3850. [DOI] [PubMed] [Google Scholar]
- Zhu NX (2001) Study on inducing and differentiating function and mechanism of matrine on leukaemia cells. Acta Tradit Chin Med Pharmacol (Shanghai) 15 43–44. [Google Scholar]